Multifaceted Human Antigen R (HuR): A Key Player in Liver Metabolism and MASLD



Abstract
1. Introduction
2. Review Methodology
3. HuR Structure and Subcellular Localization
4. Regulation of HuR Expression and Protein Stability
5. Overview of HuR Function During Physiological and Pathological Conditions
5.1. Nuclear Functions of HuR Under Physiological Conditions
5.2. Cytoplasmic Roles of HuR During Stress and Disease

{kind=link}
{kind=link}
| HuR Target mRNA 1 | mRNA Function | HuR’s Effect on mRNA | Reference |
|---|---|---|---|
| CCNB1 | Cell cycle progression | Stabilization | [30] |
| CCND1 | Cell cycle progression | Stabilization | [40] |
| CCNE1 | Cell cycle progression | Stabilization | [103] |
| CDKN1A | Cell cycle inhibition | Stabilization | [39] |
| CSF2 | Pro-inflammatory | Stabilization | [88] |
| FOS | Rapid response gene | Stabilization | [22] |
| FOXQ1 | Tumorigenesis | Stabilization | [98] |
| NOS2 | Pro-inflammatory | Stabilization | [104] |
| PTGS2 | Pro-inflammatory | Stabilization | [79] |
| TGFB1 | Cell growth | Stabilization | [41] |
| TNF | Pro-inflammatory | Stabilization | [105] |
| XIAP | Anti-apoptotic | Enhanced Translation | [96] |
| CDKN1B | Cell cycle inhibition | Suppressed Translation | [97] |
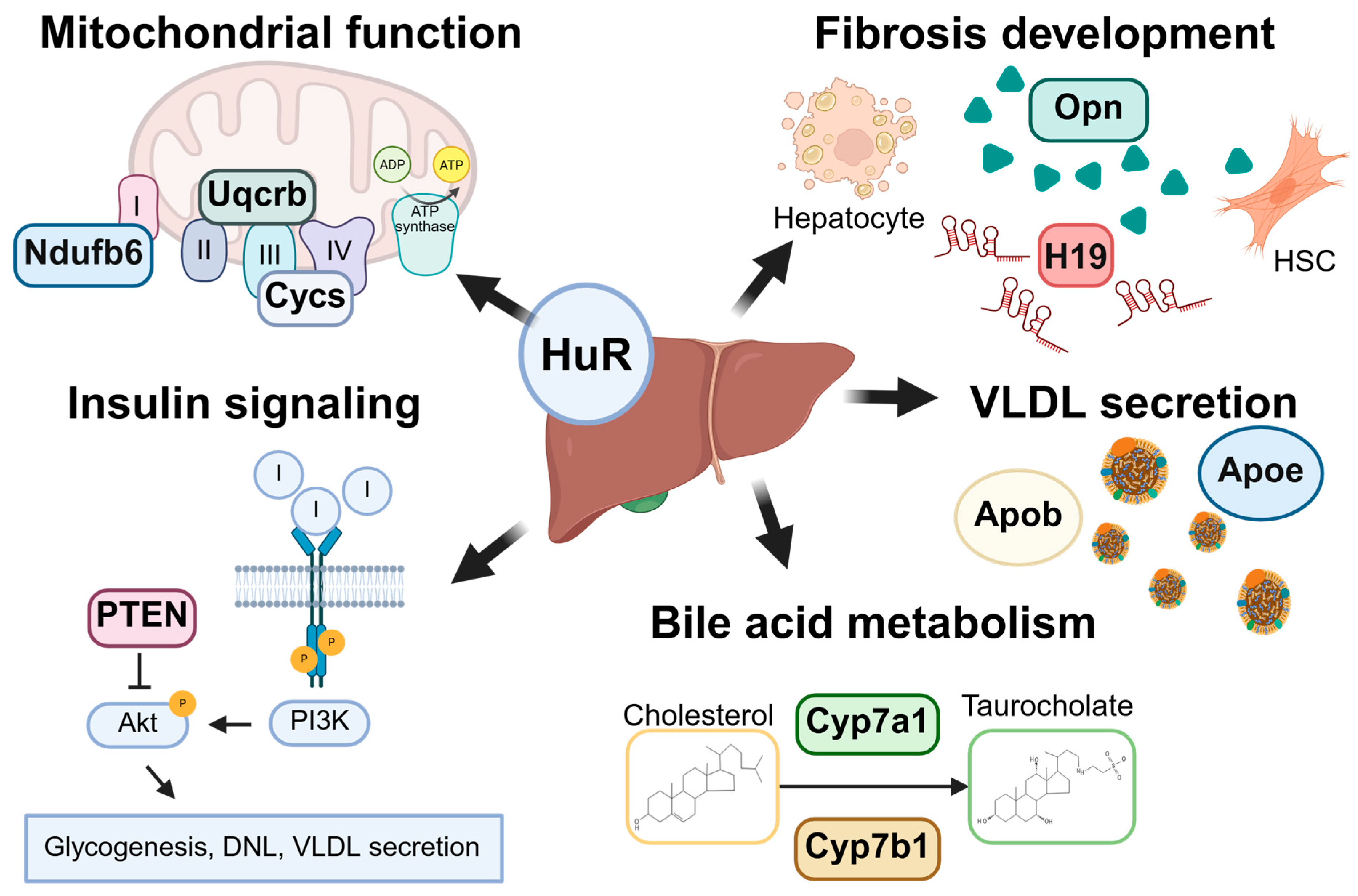
6. Role of HuR in Liver Homeostasis and MASLD
6.1. MASLD Pathogenesis and Progression
6.2. Role of HuR in Hepatic Steatosis
6.2.1. HuR Regulates Insulin Signaling and Hepatic Steatosis
6.2.2. HuR Regulation of Very Low-Density Lipoprotein Secretion
6.3. Role of HuR in Cholesterol Metabolism
6.4. Role of HuR in Bile Acid Metabolism
6.5. HuR Regulates Mitochondrial Function and Oxidative Stress
6.6. Role of HuR in Autophagy
6.7. HuR Regulates Cell Death and Survival
6.8. Role of HuR in Liver Inflammation
6.9. Role of HuR in Liver Fibrosis
7. Role of HuR in Extrahepatic Metabolic Comorbidities
7.1. Obesity
7.1.1. Overview of Obesity and Its Association with MASLD
7.1.2. Role of HuR in Adipocyte Differentiation and Obesity
7.2. Cardiovascular Disease and Type II Diabetes Mellitus
7.2.1. Overview of Cardiovascular Disease, Diabetic Cardiomyopathy, and Their Link to MASLD
7.2.2. Role of HuR in Metabolic Dysfunction-Associated Cardiovascular Disease
8. Conclusions and Future Perspectives
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Wong, V.W.S.; Ratziu, V.; Bugianesi, E.; Francque, S.; Zelber-Sagi, S.; Valenti, L.; Roden, M.; Schick, F.; Yki-Järvinen, H.; Gastaldelli, A.; et al. EASL-EASD-EASO Clinical Practice Guidelines on the management of metabolic dysfunction-associated steatotic liver disease (MASLD). J. Hepatol. 2024, 81, 492–542. [Google Scholar] [CrossRef]
- Huang, D.Q.; Wong, V.W.S.; Rinella, M.E.; Boursier, J.; Lazarus, J.V.; Yki-Järvinen, H.; Loomba, R. Metabolic dysfunction-associated steatotic liver disease in adults. Nat. Rev. Dis. Prim. 2025, 11, 14. [Google Scholar] [CrossRef] [PubMed]
- Wallace, C.; Gamkrelidze, I.; Estes, C.; Razavi, H.; Sanyal, A.J. Modeling the health and economic impact of pharmacologic therapies for MASLD in the United States. J. Hepatol. 2025, 83, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Miao, L.; Targher, G.; Byrne, C.D.; Cao, Y.-Y.; Zheng, M.-H. Current status and future trends of the global burden of MASLD. Trends Endocrinol. Metab. 2024, 35, 697–707. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.M.; Golabi, P.; Paik, J.M.; Henry, A.; Van Dongen, C.; Henry, L. The global epidemiology of nonalcoholic fatty liver disease (NAFLD) and nonalcoholic steatohepatitis (NASH): A systematic review. Hepatology 2023, 77, 1335–1347. [Google Scholar] [CrossRef] [PubMed]
- Eslam, M.; Newsome, P.N.; Sarin, S.K.; Anstee, Q.M.; Targher, G.; Romero-Gomez, M.; Zelber-Sagi, S.; Wong, V.W.-S.; Dufour, J.-F.; Schattenberg, J.M.; et al. A new definition for metabolic dysfunction-associated fatty liver disease: An international expert consensus statement. J. Hepatol. 2020, 73, 202–209. [Google Scholar] [CrossRef] [PubMed]
- Rinella, M.E.; Lazarus, J.V.; Ratziu, V.; Francque, S.M.; Sanyal, A.J.; Kanwal, F.; Romero, D.; Abdelmalek, M.F.; Anstee, Q.M.; Arab, J.P.; et al. A multisociety Delphi consensus statement on new fatty liver disease nomenclature. J. Hepatol. 2023, 79, 1542–1556. [Google Scholar] [CrossRef] [PubMed]
- Hagström, H.; Vessby, J.; Ekstedt, M.; Shang, Y. 99% of patients with NAFLD meet MASLD criteria and natural history is therefore identical. J. Hepatol. 2024, 56, E76–E77. [Google Scholar] [CrossRef] [PubMed]
- Zeng, J.; Fan, J.G.; Francque, S.M. Therapeutic management of metabolic dysfunction associated steatotic liver disease. United Eur. Gastroenterol. J. 2024, 12, 177–186. [Google Scholar] [CrossRef] [PubMed]
- Targher, G.; Byrne, C.D.; Tilg, H. MASLD: A systemic metabolic disorder with cardiovascular and malignant complications. Gut 2024, 73, 691–702. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Csermely, A.; Petracca, G.; Beatrice, G.; Corey, K.E.; Simon, T.G.; Byrne, C.D.; Targher, G. Non-alcoholic fatty liver disease and risk of fatal and non-fatal cardiovascular events: An updated systematic review and meta-analysis. Lancet Gastroenterol. Hepatol. 2021, 6, 903–913. [Google Scholar] [CrossRef] [PubMed]
- Deshpande, K.; Olynyk, J.; Ayonrinde, O.; Nosaka, K. Barriers to Exercise in Patients With Metabolic Dysfunction-Associated Steatotic Liver Disease: A Patient Survey. J. Clin. Med. Res. 2024, 16, 94–105. [Google Scholar] [CrossRef] [PubMed]
- Keam, S.J. Resmetirom: First Approval. Drugs 2024, 84, 729–735. [Google Scholar] [CrossRef] [PubMed]
- Sanyal, A.J.; Newsome, P.N.; Kliers, I.; Østergaard, L.H.; Long, M.T.; Kjær, M.S.; Cali, A.M.G.; Bugianesi, E.; Rinella, M.E.; Roden, M.; et al. Phase 3 Trial of Semaglutide in Metabolic Dysfunction-Associated Steatohepatitis. N. Engl. J. Med. 2025, 392, 2089–2099. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, M.J.; Gaunt, P.; Aithal, G.P.; Barton, D.; Hull, D.; Parker, R.; Hazlehurst, J.M.; Guo, K.; LEAN Trial Team; Abouda, G.; et al. Liraglutide safety and efficacy in patients with non-alcoholic steatohepatitis (LEAN): A multicentre, double-blind, randomised, placebo-controlled phase 2 study. Lancet 2016, 387, 679–690. [Google Scholar] [CrossRef] [PubMed]
- Newsome, P.N.; Buchholtz, K.; Cusi, K.; Linder, M.; Okanoue, T.; Ratziu, V.; Sanyal, A.J.; Sejling, A.-S.; Harrison, S.A.; NN9931-4296 Investigators. A Placebo-Controlled Trial of Subcutaneous Semaglutide in Nonalcoholic Steatohepatitis. N. Engl. J. Med. 2021, 384, 1113–1124. [Google Scholar] [CrossRef] [PubMed]
- Loomba, R.; Hartman, M.L.; Lawitz, E.J.; Vuppalanchi, R.; Boursier, J.; Bugianesi, E.; Yoneda, M.; Behling, C.; Cummings, O.W.; Tang, Y.; et al. Tirzepatide for Metabolic Dysfunction-Associated Steatohepatitis with Liver Fibrosis. N. Engl. J. Med. 2024, 391, 299–310. [Google Scholar] [CrossRef] [PubMed]
- Sanyal, A.J.; Bedossa, P.; Fraessdorf, M.; Neff, G.W.; Lawitz, E.; Bugianesi, E.; Anstee, Q.M.; Hussain, S.A.; Newsome, P.N.; Ratziu, V.; et al. A Phase 2 Randomized Trial of Survodutide in MASH and Fibrosis. N. Engl. J. Med. 2024, 391, 311–319. [Google Scholar] [CrossRef] [PubMed]
- Zafer, M.; Tavaglione, F.; Romero-Gómez, M.; Loomba, R. Review Article: GLP-1 Receptor Agonists and Glucagon/GIP/GLP-1 Receptor Dual or Triple Agonists-Mechanism of Action and Emerging Therapeutic Landscape in MASLD. Aliment. Pharmacol. Ther. 2025, 61, 1872–1888. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Sohal, A.; Batta, A. GLP-1, GIP/GLP-1, and GCGR/GLP-1 receptor agonists: Novel therapeutic agents for metabolic dysfunction-associated steatohepatitis. World J. Gastroenterol. 2024, 30, 5205–5211. [Google Scholar] [CrossRef] [PubMed]
- Michalopoulou, E.; Thymis, J.; Lampsas, S.; Pavlidis, G.; Katogiannis, K.; Vlachomitros, D.; Katsanaki, E.; Kostelli, G.; Pililis, S.; Pliouta, L.; et al. The Triad of Risk: Linking MASLD, Cardiovascular Disease and Type 2 Diabetes; From Pathophysiology to Treatment. J. Clin. Med. 2025, 14, 428. [Google Scholar] [CrossRef] [PubMed]
- Ma, W.-J.; Cheng, S.; Campbell, C.; Wright, A.; Furneaux, H. Cloning and characterization of HuR, a ubiquitously expressed Elav-like protein. J. Biol. Chem. 1996, 271, 8144–8151. [Google Scholar] [CrossRef] [PubMed]
- Campos, A.R.; Grossman, D.; White, K. Mutant alleles at the locus elav in Drosophila melanogaster lead to nervous system defects. A developmental-genetic analysis. J. Neurogenet. 1985, 2, 197–218. [Google Scholar] [CrossRef] [PubMed]
- Wutikeli, H.; Xie, T.; Xiong, W.; Shen, Y. ELAV/Hu RNA-binding protein family: Key regulators in neurological disorders, cancer, and other diseases. RNA Biol. 2025, 22, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Bakheet, T.; Hitti, E.; A Khabar, K.S. ARED-Plus: An updated and expanded database of AU-rich element-containing mRNAs and pre-mRNAs. Nucleic Acids Res. 2018, 46, D218–D220. [Google Scholar] [CrossRef] [PubMed]
- Clark, M.E.; Farinha, A.; Morrison, A.R.; Lisi, G.P. Structural, biological, and biomedical implications of mRNA interactions with the master regulator HuR. NAR Mol. Med. 2025, 2, ugaf002. [Google Scholar] [CrossRef] [PubMed]
- Myer, V.E.; Fan, X.H.C.; Steitz, J.A. Identification of HuR as a protein implicated in AUUUA-mediated mRNA decay. EMBO J. 1997, 16, 2130–2139. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Connick, M.C.; Vanderhoof, J.; Ishak, M.-A.; Hartley, R.S. MicroRNA-16 Modulates HuR Regulation of Cyclin E1 in Breast Cancer Cells. Int. J. Mol. Sci. 2015, 16, 7112–7132. [Google Scholar] [CrossRef] [PubMed]
- Haga, Y.; Bandyopadhyay, D.; Khatun, M.; Tran, E.; Steele, R.; Banerjee, S.; Ray, R.; Nazzal, M.; Ray, R.B. Increased expression of long non-coding RNA FIRRE promotes hepatocellular carcinoma by HuR-CyclinD1 axis signaling. J. Biol. Chem. 2024, 300, 107247. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.G.; Caldwell, M.C.; Lin, S.K.; Furneaux, H.; Gorospe, M. HuR regulates cyclin A and cyclin B1 mRNA stability during cell proliferation. EMBO J. 2000, 19, 2340–2350. [Google Scholar] [CrossRef] [PubMed]
- Gantt, K.; Cherry, J.; Tenney, R.; Karschner, V.; Pekala, P.H. An early event in adipogenesis, the nuclear selection of the CCAAT enhancer-binding protein β (C/EBPβ) mRNA by HuR and its translocation to the cytosol. J. Biol. Chem. 2005, 280, 24768–24774. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, M.; Aguila, H.L.; Michaud, J.; Ai, Y.; Wu, M.-T.; Hemmes, A.; Ristimaki, A.; Guo, C.; Furneaux, H.; Hla, T. Essential role of the RNA-binding protein HuR in progenitor cell survival in mice. J. Clin. Investig. 2009, 119, 3530–3543. [Google Scholar] [CrossRef] [PubMed]
- Mazroui, R.; Di Marco, S.; Clair, E.; von Roretz, C.; Tenenbaum, S.A.; Keene, J.D.; Saleh, M.; Gallouzi, I.-E. Caspase-mediated cleavage of HuR in the cytoplasm contributes to pp32/PHAP-I regulation of apoptosis. J. Cell Biol. 2008, 180, 113–127. [Google Scholar] [CrossRef] [PubMed]
- von Roretz, C.; Lian, X.J.; Macri, A.M.; Punjani, N.; Clair, E.; Drouin, O.; Dormoy-Raclet, V.; Ma, J.F.; Gallouzi, I.-E. Apoptotic-induced cleavage shifts HuR from being a promoter of survival to an activator of caspase-mediated apoptosis. Cell Death Differ. 2013, 20, 154–168. [Google Scholar] [CrossRef] [PubMed]
- Pang, L.J.; Tian, H.Y.; Chang, N.; Yi, J.; Xue, L.X.; Jiang, B.; Gorospe, M.; Zhang, X.W.; Wang, W.G. Loss of CARM1 is linked to reduced HuR function in replicative senescence. BMC Mol. Biol. 2013, 14, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Yi, J.; Chang, N.; Liu, X.W.; Guo, G.; Xue, L.X.; Tong, T.J.; Gorospe, M.; Wang, W.G. Reduced nuclear export of HuR mRNA by HuR is linked to the loss of HuR in replicative senescence. Nucleic Acids Res. 2009, 38, 1547–1558. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, U.; Adhya, S. Posttranscriptional regulation of cyclin D1 by ARE-binding proteins AUF1 and HuR in cycling myoblasts. J. Biosci. 2018, 43, 685–691. [Google Scholar] [CrossRef] [PubMed]
- Bai, D.N.; Gao, Q.; Li, C.X.; Ge, L.; Gao, Y.; Wang, H.C. A conserved TGFβ1/HuR feedback circuit regulates the fibrogenic response in fibroblasts. Cell. Signal. 2012, 24, 1426–1432. [Google Scholar] [CrossRef] [PubMed]
- Lafarga, V.; Cuadrado, A.; de Silanes, I.L.; Bengoechea, R.; Fernandez-Capetillo, O.; Nebreda, A.R. p38 Mitogen-activated protein kinase- and HuR-dependent stabilization of p21Cip1 mRNA mediates the G1/S checkpoint. Mol. Cell Biol. 2009, 29, 4341–4351. [Google Scholar] [CrossRef] [PubMed]
- Woodhoo, A.; Iruarrizaga-Lejarreta, M.; Beraza, N.; Juan, V.G.-D.; Embade, N.; Fernández-Ramos, D.; Martínez-López, N.; Gutiérrez-De Juan, V.; Arteta, B.; Caballeria, J.; et al. Human antigen R contributes to hepatic stellate cell activation and liver fibrosis. Hepatology 2012, 56, 1870–1882. [Google Scholar] [CrossRef] [PubMed]
- Ge, J.J.; Chang, N.; Zhao, Z.X.; Tian, L.; Duan, X.H.; Yang, L.; Li, L.Y. Essential Roles of RNA-binding Protein HuR in Activation of Hepatic Stellate Cells Induced by Transforming Growth Factor-β1. Sci. Rep. 2016, 6, 22141. [Google Scholar] [CrossRef] [PubMed]
- Li, J.Y.; Gong, L.; Liu, S.Z.; Zhang, Y.J.; Zhang, C.M.; Tian, M.; Lu, H.X.; Bu, P.L.; Yang, J.M.; Ouyang, C.H.; et al. Adipose HuR protects against diet-induced obesity and insulin resistance. Nat. Commun. 2019, 10, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Siang, D.T.C.; Lim, Y.C.; Kyaw, A.M.M.; Win, K.N.; Chia, S.Y.; Degirmenci, U.; Hu, X.; Tan, B.C.; Walet, A.C.E.; Sun, L.; et al. The RNA-binding protein HuR is a negative regulator in adipogenesis. Nat. Commun. 2020, 11, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Zeng, F.X.; Liu, Q.; Liu, H.H.; Liu, Z.X.; Niu, L.W.; Teng, M.K.; Li, X. The structure of the ARE-binding domains of Hu antigen R (HuR) undergoes conformational changes during RNA binding. Acta Crystallogr. Sect. D-Struct. Biol. 2013, 69, 373–380. [Google Scholar] [CrossRef] [PubMed]
- Pabis, M.; Popowicz, G.M.; Stehle, R.; Fernández-Ramos, D.; Asami, S.; Warner, L.; García-Mauriño, S.M.; Schlundt, A.; Martínez-Chantar, M.L.; Díaz-Moreno, I.; et al. HuR biological function involves RRM3-mediated dimerization and RNA binding by all three RRMs. Nucleic Acids Res. 2019, 47, 1011–1029. [Google Scholar] [CrossRef] [PubMed]
- Ma, W.-J.; Chung, S.; Furneaux, H. The Elav-like proteins bind to AU-rich elements and to the poly(A) tail of mRNA. Nucleic Acids Res. 1997, 25, 3564–3569. [Google Scholar] [CrossRef] [PubMed]
- Brennan, C.M.; Gallouzi, I.-E.; Steitz, J.A. Protein ligands to HuR modulate its interaction with target mRNAs in vivo. J. Cell Biol. 2000, 151, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Fan, X.C.; Steitz, J.A. Overexpression of HuR, a nuclear-cytoplasmic shuttling protein, increases the in vivo stability of ARE-containing mRNAs. EMBO J. 1998, 17, 3448–3460. [Google Scholar] [CrossRef] [PubMed]
- Fan, X.H.C.; Steitz, J.A. HNS, a nuclear-cytoplasmic shuttling sequence in HuR. Proc. Natl. Acad. Sci. USA 1998, 95, 15293–15298. [Google Scholar] [CrossRef] [PubMed]
- Lin, D.H.; Hoelz, A. The Structure of the Nuclear Pore Complex (An Update). Annu. Rev. Biochem. 2019, 88, 725–783. [Google Scholar] [CrossRef] [PubMed]
- Wing, C.E.; Fung, H.Y.J.; Chook, Y.M. Karyopherin-mediated nucleocytoplasmic transport. Nat. Rev. Mol. Cell Biol. 2022, 23, 307–328. [Google Scholar] [CrossRef] [PubMed]
- Gallouzi, I.-E.; Brennan, C.M.; Steitz, J.A. Protein ligands mediate the CRM1-dependent export of HuR in response to heat shock. RNA 2001, 7, 1348–1361. [Google Scholar] [CrossRef] [PubMed]
- Güttinger, S.; Mühlhäusser, P.; Koller-Eichhorn, R.; Brennecke, J.; Kutay, U. Transportin2 functions as importin and mediates nuclear import of HuR. Proc. Natl. Acad. Sci. USA 2004, 101, 2918–2923. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.G.; Yang, X.L.; Kawai, T.; de Silanes, I.L.; Mazan-Mamczarz, K.; Chen, P.L.; Chook, Y.M.; Quensel, C.; Köhler, M.; Gorospe, M. AMP-activated protein kinase-regulated phosphorylation and acetylation of importin α1. J. Biol. Chem. 2004, 279, 48376–48388. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.H.; Abdelmohsen, K.; Lal, A.; Pullmann, R.; Yang, X.L.; Galban, S.; Srikantan, S.; Martindale, J.L.; Blethrow, J.; Shokat, K.M.; et al. Nuclear HuR accumulation through phosphorylation by Cdk1. Genes Dev. 2008, 22, 1804–1815. [Google Scholar] [CrossRef] [PubMed]
- von Roretz, C.; Macri, A.M.; Gallouzi, I.-E. Transportin 2 Regulates Apoptosis through the RNA-binding Protein HuR. J. Biol. Chem. 2011, 286, 25983–25991. [Google Scholar] [CrossRef] [PubMed]
- Baños-Jaime, B.; Corrales-Guerrero, L.; Pérez-Mejías, G.; Rejano-Gordillo, C.M.; Velázquez-Campoy, A.; Martínez-Cruz, L.A.; Martínez-Chantar, M.L.; A De la Rosa, M.; Díaz-Moreno, I. Phosphorylation at the disordered N-end makes HuR accumulate and dimerize in the cytoplasm. Nucleic Acids Res. 2024, 52, 8552–8565. [Google Scholar] [CrossRef] [PubMed]
- Finan, J.M.; Sutton, T.L.; Dixon, D.A.; Brody, J.R. Targeting the RNA-Binding Protein HuR in Cancer. Cancer Res. 2023, 83, 3507–3516. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.Q.; Xu, L. The RNA-binding protein HuR in human cancer: A friend or foe. Adv. Drug Deliv. Rev. 2022, 184, 114179. [Google Scholar] [CrossRef] [PubMed]
- Dery, K.J.; Nakamura, K.; Kadono, K.; Hirao, H.; Kageyama, S.; Ito, T.; Kojima, H.; Kaldas, F.M.; Busuttil, R.W.; Kupiec-Weglinski, J.W. Human Antigen R (HuR): A Regulator of Heme Oxygenase-1 Cytoprotection in Mouse and Human Liver Transplant Injury. Hepatology 2020, 72, 1056–1072. [Google Scholar] [CrossRef] [PubMed]
- Beauchamp, P.; Nassif, C.; Hillock, S.; van der Giessen, K.; von Roretz, C.; Jasmin, B.J.; Gallouzi, I.-E. The cleavage of HuR interferes with its transportin-2-mediated nuclear import and promotes muscle fiber formation. Cell Death Differ. 2010, 17, 1588–1599. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.F.; Berkova, Z.; Mathur, R.; Sehgal, L.; Khashab, T.; Tao, R.-H.; Ao, X.; Feng, L.; Sabichi, A.L.; Blechacz, B.; et al. HuR Suppresses Fas Expression and Correlates with Patient Outcome in Liver Cancer. Mol. Cancer Res. 2015, 13, 809–818. [Google Scholar] [CrossRef] [PubMed]
- Raheja, H.; George, B.; Tripathi, S.K.; Saha, S.; Maiti, T.K.; Das, S.; Sarnow, P. Hepatitis C virus non-structural proteins modulate cellular kinases for increased cytoplasmic abundance of host factor HuR and facilitate viral replication. PLoS Pathog. 2023, 19, e1011552. [Google Scholar] [CrossRef] [PubMed]
- Al-Ahmadi, W.; Al-Ghamdi, M.; Al-Haj, L.; Al-Saif, M.; Khabar, K.S.A. Alternative polyadenylation variants of the RNA binding protein, HuR: Abundance, role of AU-rich elements and auto-Regulation. Nucleic Acids Res. 2009, 37, 3612–3624. [Google Scholar] [CrossRef] [PubMed]
- Govindaraju, S.; Lee, B.S. Krüppel-Like Factor 8 is a Stress-Responsive Transcription Factor that Regulates Expression of HuR. Cell. Physiol. Biochem. 2014, 34, 519–532. [Google Scholar] [CrossRef] [PubMed]
- Jeyaraj, S.C.; Dakhlallah, D.; Hill, S.R.; Lee, B.S.; Pullmann, R.; Rabb, H.; Gummadi, L.; Taylor, L.; Curthoys, N.P.; Mufti, J.; et al. Expression and distribution of HuR during ATP depletion and recovery in proximal tubule cells. Am. J. Physiol.-Ren. Physiol. 2006, 291, F1255–F1263. [Google Scholar] [CrossRef] [PubMed]
- Kang, M.J.; Ryu, B.K.; Lee, M.G.; Han, J.; Lee, J.H.; Ha, T.K.; Byun, D.S.; Chae, K.S.; Lee, B.H.; Chun, H.S.; et al. NF-κB Activates Transcription of the RNA-Binding Factor HuR, via PI3K-AKT Signaling, to Promote Gastric Tumorigenesis. Gastroenterology 2008, 135, 2030–2042.E3. [Google Scholar] [CrossRef] [PubMed]
- Ayupova, D.A.; Singh, M.; Leonard, E.C.; Basile, D.P.; Lee, B.S. Expression of the RNA-stabilizing protein HuR in ischemia-reperfusion injury of rat kidney. Am. J. Physiol.-Ren. Physiol. 2009, 297, F95–F105. [Google Scholar] [CrossRef] [PubMed]
- Pullmann, R.; Kim, H.H.; Abdelmohsen, K.; Lal, A.; Martindale, J.L.; Yang, X.L.; Gorospe, M. Analysis of turnover and translation regulatory RNA-Binding protein expression through binding to cognate mRNAs. Mol. Cell. Biol. 2007, 27, 6265–6278. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Xiao, L.; Wang, J.-Y. HuR and Its Interactions with Noncoding RNAs in Gut Epithelium Homeostasis and Diseases. Front. Biosci.-Landmark 2023, 28, 262. [Google Scholar] [CrossRef] [PubMed]
- Lal, A.; Mazan-Mamczarz, K.; Kawai, T.; Yang, X.L.; Martindale, J.L.; Gorospe, M. Concurrent versus individual binding of HuR and AUF1 to common labile target mRNAs. EMBO J. 2004, 23, 3092–3102. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.Q.; Chen, X.R.; Cheng, R.J.; Yang, F.; Yu, M.C.; Wang, C.; Cui, S.F.; Hong, Y.T.; Liang, H.W.; Liu, M.H.; et al. The Jun/miR-22/HuR regulatory axis contributes to tumourigenesis in colorectal cancer. Mol. Cancer 2018, 17, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.-J.; Wu, W.; Chen, Q.-Q.; Liu, S.-H.; Zheng, Z.-Y.; Cui, Z.-L.; Xu, J.-P.; Xue, Y.; Lin, D.-H. miR-29b-3p suppresses the malignant biological behaviors of AML cells via inhibiting NF-κB and JAK/STAT signaling pathways by targeting HuR. Bmc Cancer 2022, 22, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Abdelmohsen, K.; Srikantan, S.; Kuwano, Y.; Gorospe, M. miR-519 reduces cell proliferation by lowering RNA-binding protein HuR levels. Proc. Natl. Acad. Sci. USA 2008, 105, 20297–20302. [Google Scholar] [CrossRef] [PubMed]
- Gjorgjieva, M.; Ay, A.-S.; de Sousa, M.C.; Delangre, E.; Dolicka, D.; Sobolewski, C.; Maeder, C.; Fournier, M.; Sempoux, C.; Foti, M. MiR-22 Deficiency Fosters Hepatocellular Carcinoma Development in Fatty Liver. Cells 2022, 11, 2860. [Google Scholar] [CrossRef] [PubMed]
- Grammatikakis, I.; Abdelmohsen, K.; Gorospe, M. Posttranslational control of HuR function. Wiley Interdiscip. Rev. RNA 2017, 8, e1372. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Rao, J.N.; Zou, T.T.; Xiao, L.; Wang, P.-Y.; Turner, D.J.; Gorospe, M.; Wang, J.-Y.; Tansey, W.P. Polyamines Regulate c-Myc Translation through Chk2-dependent HuR Phosphorylation. Mol. Biol. Cell 2009, 20, 4885–4898. [Google Scholar] [CrossRef] [PubMed]
- Abdelmohsen, K.; Pullmann, R., Jr.; Lal, A.; Kim, H.H.; Galban, S.; Yang, X.; Blethrow, J.D.; Walker, M.; Shubert, J.; Gillespie, D.A.; et al. Phosphorylation of HuR by Chk2 regulates SIRT1 expression. Mol. Cell 2007, 25, 543–557. [Google Scholar] [CrossRef] [PubMed]
- Fernau, N.S.; Fugmann, D.; Leyendecker, M.; Reimann, K.; Grether-Beck, S.; Galban, S.; Ale-Agha, N.; Krutmann, J.; Klotz, L.-O. Role of HuR and p38MAPK in ultraviolet B-induced post-transcriptional regulation of COX-2 expression in the human keratinocyte cell line HaCaT. J. Biol. Chem. 2010, 285, 3896–3904. [Google Scholar] [CrossRef] [PubMed]
- Doller, A.; Huwiler, A.; Müller, R.; Radeke, H.H.; Pfeilschifter, J.; Eberhardt, W.; Wickens, M.P. Protein kinase Cα-dependent phosphorylation of the mRNA-stabilizing factor HuR: Implications for posttranscriptional regulation of cyclooxygenase-2. Mol. Biol. Cell 2007, 18, 2137–2148. [Google Scholar] [CrossRef] [PubMed]
- Fang, C.Q.; Pan, J.H.; Qu, N.; Lei, Y.T.; Han, J.J.; Zhang, J.Z.; Han, D. The AMPK pathway in fatty liver disease. Front. Physiol. 2022, 13, 970292. [Google Scholar] [CrossRef] [PubMed]
- David, P.S.; Tanveer, R.; Port, J.D. FRET-detectable interactions between the ARE binding proteins, HuR and p37AUF1. RNA 2007, 13, 1453–1468. [Google Scholar] [CrossRef] [PubMed]
- Fialcowitz-White, E.J.; Brewer, B.Y.; Ballin, J.D.; Willis, C.D.; Toth, E.A.; Wilson, G.M. Specific protein domains mediate cooperative assembly of HuR oligomers on AU-rich mRNA-destabilizing sequences. J. Biol. Chem. 2007, 282, 20948–20959. [Google Scholar] [CrossRef] [PubMed]
- Lebedeva, S.; Jens, M.; Theil, K.; Schwanhäusser, B.; Selbach, M.; Landthaler, M.; Rajewsky, N. Transcriptome-wide Analysis of Regulatory Interactions of the RNA-Binding Protein HuR. Mol. Cell 2011, 43, 340–352. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, N.; Corcoran, D.L.; Nusbaum, J.D.; Reid, D.W.; Georgiev, S.; Hafner, M.; Ascano, M.; Tuschl, T.; Ohler, U.; Keene, J.D. Integrative Regulatory Mapping Indicates that the RNA-Binding Protein HuR Couples Pre-mRNA Processing and mRNA Stability. Mol. Cell 2011, 43, 327–339. [Google Scholar] [CrossRef] [PubMed]
- Noh, J.H.; Kim, K.M.; Abdelmohsen, K.; Yoon, J.-H.; Panda, A.C.; Munk, R.; Kim, J.; Curtis, J.; Moad, C.A.; Wohler, C.M.; et al. HuR and GRSF1 modulate the nuclear export and mitochondrial localization of the lncRNA RMRP. Genes Dev. 2016, 30, 1224–1239. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Muñoz, M.D.; Bell, S.E.; Fairfax, K.; Monzon-Casanova, E.; Cunningham, A.F.; Gonzalez-Porta, M.; Andrews, S.R.; Bunik, V.I.; Zarnack, K.; Curk, T.; et al. The RNA-binding protein HuR is essential for the B cell antibody response. Nat. Immunol. 2015, 16, 415–425. [Google Scholar] [CrossRef] [PubMed]
- Srikantan, S.; Gorospe, M. HuR function in disease. Front. Biosci.-Landmark 2012, 17, 189–205. [Google Scholar] [CrossRef] [PubMed]
- Matsye, P.; Zheng, L.; Si, Y.; Kim, S.; Luo, W.Y.; Crossman, D.K.; Bratcher, P.E.; King, P.H. HuR promotes the molecular signature and phenotype of activated microglia: Implications for amyotrophic lateral sclerosis and other neurodegenerative diseases. Glia 2017, 65, 945–963. [Google Scholar] [CrossRef] [PubMed]
- Peng, W.D.; Furuuchi, N.; Aslanukova, L.; Huang, Y.-H.; Brown, S.Z.; Jiang, W.; Addya, S.; Vishwakarma, V.; Peters, E.; Brody, J.R.; et al. Elevated HuR in Pancreas Promotes a Pancreatitis-Like Inflammatory Microenvironment That Facilitates Tumor Development. Mol. Cell. Biol. 2018, 38, e00427-17. [Google Scholar] [CrossRef] [PubMed]
- Gallouzi, I.-E.; Brennan, C.M.; Stenberg, M.G.; Swanson, M.S.; Eversole, A.; Maizels, N.; Steitz, J.A. HuR binding to cytoplasmic mRNA is perturbed by heat shock. Proc. Natl. Acad. Sci. USA 2000, 97, 3073–3078. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Chen, Y.; Hu, W.R.; Liu, X.Y.; Hao, W.J.; Xing, J.; Ding, J.; Xu, Y.C.; Yao, F.; Zhao, Y.J.; et al. TRPM7 facilitates fibroblast-like synoviocyte proliferation, metastasis and inflammation through increasing IL-6 stability via the PKCα-HuR axis in rheumatoid arthritis. Int. Immunopharmacol. 2024, 132, 111933. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-X.; Zheng, H.-Y.; Zhou, K.; Xie, H.-L.; Ren, Z.; Liu, H.-T.; Liu, H.; Zhou, Z.-X.; Jiang, Z.-S. Multifaceted Nature of HuR in Atherosclerosis Development. Curr. Med. Chem. 2025, 32, 3423–3437. [Google Scholar] [CrossRef] [PubMed]
- Christodoulou-Vafeiadou, E.; Ioakeimidis, F.; Andreadou, M.; Giagkas, G.; Stamatakis, G.; Reczko, M.; Samiotaki, M.; Papanastasiou, A.D.; Karakasiliotis, I.; Kontoyiannis, D.L. Divergent Innate and Epithelial Functions of the RNA-Binding Protein HuR in Intestinal Inflammation. Front. Immunol. 2018, 9, 2732. [Google Scholar] [CrossRef] [PubMed]
- de Silanes, I.L.; Zhan, M.; Lal, A.; Yang, X.L.; Gorospe, M. Identification of a target RNA motif for RNA-binding protein HuR. Proc. Natl. Acad. Sci. USA 2004, 101, 2987–2992. [Google Scholar] [CrossRef] [PubMed]
- Durie, D.; Lewis, S.M.; Liwak, U.; Kisilewicz, M.; Gorospe, M.; Holcik, M. RNA-binding protein HuR mediates cytoprotection through stimulation of XIAP translation. Oncogene 2010, 30, 1460–1469. [Google Scholar] [CrossRef] [PubMed]
- Kullmann, M.; Göpfert, U.; Siewe, B.; Hengst, L. ELAV/Hu proteins inhibit p27 translation via an IRES element in the p27 5′UTR. Genes Dev. 2002, 16, 3087–3099. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.Q.; Gardashova, G.; Lan, L.; Han, S.; Zhong, C.C.; Marquez, R.T.; Wei, L.J.; Wood, S.; Roy, S.; Gowthaman, R.; et al. Targeting the interaction between RNA-binding protein HuR and FOXQ1 suppresses breast cancer invasion and metastasis. Commun. Biol. 2020, 3, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Joseph, R.; Srivastava, O.P.; Pfister, R.R. Downregulation of β-actin and its regulatory gene HuR affect cell migration of human corneal fibroblasts. Mol. Vis. 2014, 20, 593–605. [Google Scholar] [PubMed]
- Tian, M.; Wang, J.J.; Liu, S.M.; Li, X.Y.; Li, J.Y.; Yang, J.M.; Zhang, C.; Zhang, W.C. Hepatic HuR protects against the pathogenesis of non-alcoholic fatty liver disease by targeting PTEN. Cell Death Dis. 2021, 12, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, P.; Gargani, S.; Palladini, A.; Chatzimike, M.; Grzybek, M.; Peitzsch, M.; Papanastasiou, A.D.; Pyrina, I.; Ntafis, V.; Gercken, B.; et al. The RNA binding protein human antigen R is a gatekeeper of liver homeostasis. Hepatology 2022, 75, 881–897. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.Y.; Tai, Y.-L.; Way, G.; Zeng, J.; Zhao, D.; Su, L.Y.; Jiang, X.X.; Jackson, K.G.; Wang, X.; Gurley, E.C.; et al. RNA binding protein HuR protects against NAFLD by suppressing long noncoding RNA H19 expression. Cell Biosci. 2022, 12, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Hartley, R.S. HuR contributes to cyclin E1 deregulation in MCF-7 breast cancer cells. Cancer Res. 2006, 66, 7948–7956. [Google Scholar] [CrossRef] [PubMed]
- Linker, K.; Pautz, A.; Fechir, M.; Hubrich, T.; Greeve, J.; Kleinert, H. Involvement of KSRP in the post-transcriptional regulation of human iNOS expression-complex interplay of KSRP with TTP and HuR. Nucleic Acids Res. 2005, 33, 4813–4827. [Google Scholar] [CrossRef] [PubMed]
- Dean, J.L.E.; Wait, R.; Mahtani, K.R.; Sully, G.; Clark, A.R.; Saklatvala, J. The 3′ untranslated region of tumor necrosis factor alpha mRNA is a target of the mRNA-stabilizing factor HuR. Mol. Cell. Biol. 2001, 21, 721–730. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, J.; Viggiano, T.R.; McGill, D.B.; Ott, B.J. Nonalcoholic steatohepatitis: Mayo Clinic experiences with a hitherto unnamed disease. Mayo Clin. Proc. 1980, 55, 434–438. [Google Scholar] [CrossRef] [PubMed]
- Day, C.P.; James, O.F.W. Steatohepatitis: A tale of two “hits”? Gastroenterology 1998, 114, 842–845. [Google Scholar] [CrossRef] [PubMed]
- Buzzetti, E.; Pinzani, M.; Tsochatzis, E.A. The multiple-hit pathogenesis of non-alcoholic fatty liver disease (NAFLD). Metab.-Clin. Exp. 2016, 65, 1038–1048. [Google Scholar] [CrossRef] [PubMed]
- Yeh, H.-Y.; Lin, S.-W.; Shen, H.-C.; Li, T.-H.; Tsai, H.-C.; Yang, Y.-Y.; Lin, H.-C.; Hou, M.-C. Complexity of Metabolic dysfunction-associated steatotic liver disease (MASLD): State of art review. J. Chin. Med. Assoc. 2025. [Google Scholar] [CrossRef] [PubMed]
- Heeren, J.; Scheja, L. Metabolic-associated fatty liver disease and lipoprotein metabolism. Mol. Metab. 2021, 50, 101238. [Google Scholar] [CrossRef] [PubMed]
- Donnelly, K.L.; Smith, C.I.; Schwarzenberg, S.J.; Jessurun, J.; Boldt, M.D.; Parks, E.J. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J. Clin. Investig. 2005, 115, 1343–1351. [Google Scholar] [CrossRef] [PubMed]
- Marchesini, G.; Brizi, M.; Morselli-Labate, A.M.; Bianchi, G.; Bugianesi, E.; McCullough, A.J.; Forlani, G.; Melchionda, N. Association of nonalcoholic fatty liver disease with insulin resistance. Am. J. Med. 1999, 107, 450–455. [Google Scholar] [CrossRef] [PubMed]
- Arab, J.P.; Arrese, M.; Trauner, M. Recent Insights into the Pathogenesis of Nonalcoholic Fatty Liver Disease. Annu. Rev. Pathol. 2018, 13, 321–350. [Google Scholar] [CrossRef] [PubMed]
- Feldstein, A.E.; Canbay, A.; Angulo, P.; Taniai, M.; Burgart, L.J.; Lindor, K.D.; Gores, G.J. Hepatocyte apoptosis and Fas expression are prominent features of human nonalcoholic steatohepatitis. Gastroenterology 2003, 125, 437–443. [Google Scholar] [CrossRef] [PubMed]
- Caballero, F.; Fernández, A.; De Lacy, A.M.; Fernández-Checa, J.C.; Caballería, J.; García-Ruiz, C. Enhanced free cholesterol, SREBP-2 and StAR expression in human NASH. J. Hepatol. 2009, 50, 789–796. [Google Scholar] [CrossRef] [PubMed]
- Marí, M.; Caballero, F.; Colell, A.; Morales, A.; Caballeria, J.; Fernandez, A.; Enrich, C.; Fernandez-Checa, J.C.; García-Ruiz, C. Mitochondrial free cholesterol loading sensitizes to TNF- and Fas-mediated steatohepatitis. Cell Metab. 2006, 4, 185–198. [Google Scholar] [CrossRef] [PubMed]
- Fromenty, B.; Roden, M. Mitochondrial alterations in fatty liver diseases. J. Hepatol. 2023, 78, 415–429. [Google Scholar] [CrossRef] [PubMed]
- Angulo, P.; Machado, M.V.; Diehl, A.M. Fibrosis in Nonalcoholic Fatty Liver Disease: Mechanisms and Clinical Implications. Semin. Liver Dis. 2015, 35, 132–145. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.J.; Zong, C.; Jiang, M.Y.; Hu, H.; Cheng, X.L.; Ni, J.H.; Yi, X.; Jiang, B.; Tian, F.; Chang, M.-W.; et al. Hepatic HuR modulates lipid homeostasis in response to high-fat diet. Nat. Commun. 2020, 11, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Cho, E.E.L.; Ang, C.Z.; Quek, J.; Fu, C.E.; Lim, L.K.E.; Heng, Z.E.Q.; Tan, D.J.H.; Lim, W.H.; Yong, J.N.; Zeng, R.B.C.; et al. Global prevalence of non-alcoholic fatty liver disease in type 2 diabetes mellitus: An updated systematic review and meta-analysis. Gut 2023, 72, 2138–2148. [Google Scholar] [CrossRef] [PubMed]
- Titchenell, P.M.; Lazar, M.A.; Birnbaum, M.J. Unraveling the Regulation of Hepatic Metabolism by Insulin. Trends Endocrinol. Metab. 2017, 28, 497–505. [Google Scholar] [CrossRef] [PubMed]
- Lomonaco, R.; Ortiz-Lopez, C.; Orsak, B.; Webb, A.; Hardies, J.; Darland, C.; Finch, J.; Gastaldelli, A.; Harrison, S.; Tio, F.; et al. Effect of adipose tissue insulin resistance on metabolic parameters and liver histology in obese patients with nonalcoholic fatty liver disease. Hepatology 2012, 55, 1389–1397. [Google Scholar] [CrossRef] [PubMed]
- Petersen, K.F.; Dufour, S.; Savage, D.B.; Bilz, S.; Solomon, G.; Yonemitsu, S.; Cline, G.W.; Befroy, D.; Zemany, L.; Kahn, B.B.; et al. The role of skeletal muscle insulin resistance in the pathogenesis of the metabolic syndrome. Proc. Natl. Acad. Sci. USA 2007, 104, 12587–12594. [Google Scholar] [CrossRef] [PubMed]
- Wong, J.T.; Kim, P.T.W.; Peacock, J.W.; Yau, T.Y.; Mui, A.L.-F.; Chung, S.W.; Sossi, V.; Doudet, D.; Green, D.; Ruth, T.J.; et al. Pten (phosphatase and tensin homologue gene) haploinsufficiency promotes insulin hypersensitivity. Diabetologia 2007, 50, 395–403. [Google Scholar] [CrossRef] [PubMed]
- Stiles, B.; Wang, Y.; Stahl, A.; Bassilian, S.; Lee, W.P.; Kim, Y.-J.; Sherwin, R.; Devaskar, S.; Lesche, R.; Magnuson, M.A.; et al. Liver-specific deletion of negative regulator Pten results in fatty liver and insulin hypersensitivity. Proc. Natl. Acad. Sci. USA 2004, 101, 2082–2087. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.H.; Ginsberg, H.N. Increased very low density lipoprotein (VLDL) secretion, hepatic steatosis, and insulin resistance. Trends Endocrinol. Metab. 2011, 22, 353–363. [Google Scholar] [CrossRef] [PubMed]
- Adiels, M.; Taskinen, M.-R.; Packard, C.; Caslake, M.J.; Soro-Paavonen, A.; Westerbacka, J.; Vehkavaara, S.; Häkkinen, A.; Olofsson, S.-O.; Yki-Järvinen, H.; et al. Overproduction of large VLDL particles is driven by increased liver fat content in man. Diabetologia 2006, 49, 755–765. [Google Scholar] [CrossRef] [PubMed]
- Eaton, S.; Bartlett, K.B.; Pourfarzam, M. Mammalian mitochondrial β-oxidation. Biochem. J. 1996, 320, 345–357. [Google Scholar] [CrossRef] [PubMed]
- Kawano, Y.; Cohen, D.E. Mechanisms of hepatic triglyceride accumulation in non-alcoholic fatty liver disease. J. Gastroenterol. 2013, 48, 434–441. [Google Scholar] [CrossRef] [PubMed]
- Marais, A.D. Apolipoprotein E in lipoprotein metabolism, health and cardiovascular disease. Pathology 2019, 51, 165–176. [Google Scholar] [CrossRef] [PubMed]
- Welty, F.K. Hypobetalipoproteinemia and abetalipoproteinemia: Liver disease and cardiovascular disease. Curr. Opin. Lipidol. 2020, 31, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Vacca, M.; Kamzolas, I.; Harder, L.M.; Oakley, F.; Trautwein, C.; Hatting, M.; Ross, T.; Bernardo, B.; Oldenburger, A.; Hjuler, S.T.; et al. An unbiased ranking of murine dietary models based on their proximity to human metabolic dysfunction-associated steatotic liver disease (MASLD). Nat. Metab. 2024, 6, 1178–1196. [Google Scholar] [CrossRef] [PubMed]
- Horn, C.L.; Morales, A.L.; Savard, C.; Farrell, G.C.; Ioannou, G.N. Role of Cholesterol-Associated Steatohepatitis in the Development of NASH. Hepatol. Commun. 2022, 6, 12–35. [Google Scholar] [CrossRef] [PubMed]
- Sato, R. Sterol metabolism and SREBP activation. Arch. Biochem. Biophys. 2010, 501, 177–181. [Google Scholar] [CrossRef] [PubMed]
- Rodwell, V.W.; Bender, D.; Botham, K.M.; Kennelly, P.J.; Weil, P.A. Harper’s Illustrated Biochemistry, 31st ed.; Rodwell, V.W., Bender, D., Botham, K.M., Kennelly, P.J., Weil, P.A., Eds.; McGraw-Hill Education: New York, NY, USA, 2018. [Google Scholar]
- Musso, G.; Gambino, R.; Cassader, M. Cholesterol metabolism and the pathogenesis of non-alcoholic steatohepatitis. Prog. Lipid Res. 2013, 52, 175–191. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Chen, Y.X.; Tang, R.K.; Chen, Y.; Li, Q.; Gong, J.P.; Huang, A.L.; Varghese, Z.; Moorhead, J.F.; Ruan, X.Z. Inflammatory stress exacerbates hepatic cholesterol accumulation via increasing cholesterol uptake and de novo synthesis. J. Gastroenterol. Hepatol. 2011, 26, 875–883. [Google Scholar] [CrossRef] [PubMed]
- van Rooyen, D.M.; Larter, C.Z.; Haigh, W.G.; Yeh, M.M.; Ioannou, G.; Kuver, R.; Lee, S.P.; Teoh, N.C.; Farrell, G.C. Hepatic Free Cholesterol Accumulates in Obese, Diabetic Mice and Causes Nonalcoholic Steatohepatitis. Gastroenterology 2011, 141, 1393–1403.e5. [Google Scholar] [CrossRef] [PubMed]
- Ikonen, E. Cellular cholesterol trafficking and compartmentalization. Nat. Rev. Mol. Cell Biol. 2008, 9, 125–138. [Google Scholar] [CrossRef] [PubMed]
- Puri, P.; Daita, K.; Joyce, A.; Mirshahi, F.; Santhekadur, P.K.; Cazanave, S.; Luketic, V.A.; Siddiqui, M.S.; Boyett, S.; Min, H.-K.; et al. The presence and severity of nonalcoholic steatohepatitis is associated with specific changes in circulating bile acids. Hepatology 2018, 67, 534–548. [Google Scholar] [CrossRef] [PubMed]
- Grzych, G.; Chávez-Talavera, O.; Descat, A.; Thuillier, D.; Verrijken, A.; Kouach, M.; Legry, V.; Verkindt, H.; Raverdy, V.; Legendre, B.; et al. NASH-related increases in plasma bile acid levels depend on insulin resistance. JHEP Rep. 2021, 3, 100222. [Google Scholar] [CrossRef] [PubMed]
- Choudhuri, S.; Klaassen, C.D. Molecular Regulation of Bile Acid Homeostasis. Drug Metab. Dispos. 2022, 50, 425–455. [Google Scholar] [CrossRef] [PubMed]
- Chávez-Talavera, O.; Haas, J.; Grzych, G.; Tailleux, A.; Staels, B. Bile acid alterations in nonalcoholic fatty liver disease, obesity, insulin resistance and type 2 diabetes: What do the human studies tell? Curr. Opin. Lipidol. 2019, 30, 244–254. [Google Scholar] [CrossRef] [PubMed]
- Jiao, N.; Baker, S.S.; Chapa-Rodriguez, A.; Liu, W.S.; Nugent, C.A.; Tsompana, M.; Mastrandrea, L.; Buck, M.J.; Baker, R.D.; Genco, R.J.; et al. Suppressed hepatic bile acid signalling despite elevated production of primary and secondary bile acids in NAFLD. Gut 2018, 67, 1881–1891. [Google Scholar] [CrossRef] [PubMed]
- Schreuder, T.C.M.A.; Marsman, H.A.; Lenicek, M.; van Werven, J.R.; Nederveen, A.J.; Jansen, P.L.M.; Schaap, F.G. The hepatic response to FGF19 is impaired in patients with nonalcoholic fatty liver disease and insulin resistance. Am. J. Physiol.-Gastrointest. Liver Physiol. 2010, 298, G440–G445. [Google Scholar] [CrossRef] [PubMed]
- Lake, A.D.; Novak, P.; Shipkova, P.; Aranibar, N.; Robertson, D.; Reily, M.D.; Lu, Z.Q.; Lehman-McKeeman, L.D.; Cherrington, N.J. Decreased hepatotoxic bile acid composition and altered synthesis in progressive human nonalcoholic fatty liver disease. Toxicol. Appl. Pharmacol. 2013, 268, 132–140. [Google Scholar] [CrossRef] [PubMed]
- Klaassen, C.D.; Cui, J.Y. Review: Mechanisms of How the Intestinal Microbiota Alters the Effects of Drugs and Bile Acids. Drug Metab. Dispos. 2015, 43, 1505–1521. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.; Guo, Y.; Klaassen, C.D. Effect of Gender and Various Diets on Bile Acid Profile and Related Genes in Mice. Drug Metab. Dispos. 2021, 49, 62–71. [Google Scholar] [CrossRef] [PubMed]
- Ikegami, T.; Hyogo, H.; Honda, A.; Miyazaki, T.; Tokushige, K.; Hashimoto, E.; Inui, K.; Matsuzaki, Y.; Tazuma, S. Increased serum liver X receptor ligand oxysterols in patients with non-alcoholic fatty liver disease. J. Gastroenterol. 2012, 47, 1257–1266. [Google Scholar] [CrossRef] [PubMed]
- Raselli, T.; Hearn, T.; Wyss, A.; Atrott, K.; Peter, A.; Frey-Wagner, I.; Spalinger, M.R.; Maggio, E.M.; Sailer, A.W.; Schmitt, J.; et al. Elevated oxysterol levels in human and mouse livers reflect nonalcoholic steatohepatitis. J. Lipid Res. 2019, 60, 1270–1283. [Google Scholar] [CrossRef] [PubMed]
- Pandak, W.M.; Kakiyama, G. The acidic pathway of bile acid synthesis: Not just an alternative pathway. Liver Res. 2019, 3, 88–98. [Google Scholar] [CrossRef] [PubMed]
- Li, T.G.; Chiang, J.Y.L. Bile Acid Signaling in Metabolic Disease and Drug Therapy. Pharmacol. Rev. 2014, 66, 948–983. [Google Scholar] [CrossRef] [PubMed]
- Koliaki, C.; Szendroedi, J.; Kaul, K.; Jelenik, T.; Nowotny, P.; Jankowiak, F.; Herder, C.; Carstensen, M.; Krausch, M.; Knoefel, W.T.; et al. Adaptation of Hepatic Mitochondrial Function in Humans with Non-Alcoholic Fatty Liver Is Lost in Steatohepatitis. Cell Metab. 2015, 21, 739–746. [Google Scholar] [CrossRef] [PubMed]
- Moore, M.P.; Cunningham, R.P.; Meers, G.M.; Johnson, S.A.; Wheeler, A.A.; Ganga, R.R.; Spencer, N.M.; Pitt, J.B.; Diaz-Arias, A.; Swi, A.I.A.; et al. Compromised hepatic mitochondrial fatty acid oxidation and reduced markers of mitochondrial turnover in human NAFLD. Hepatology 2022, 76, 1452–1465. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Carreras, M.; Del Hoyo, P.; Martín, M.A.; Rubio, J.C.; Martín, A.; Castellano, G.; Colina, F.; Arenas, J.; Solis-Herruzo, J.A. Defective hepatic mitochondrial respiratory chain in patients with nonalcoholic steatohepatitis. Hepatology 2003, 38, 999–1007. [Google Scholar] [CrossRef] [PubMed]
- Adam-Vizi, V.; Chinopoulos, C. Bioenergetics and the formation of mitochondrial reactive oxygen species. Trends Pharmacol. Sci. 2006, 27, 639–645. [Google Scholar] [CrossRef] [PubMed]
- Loboda, A.; Damulewicz, M.; Pyza, E.; Jozkowicz, A.; Dulak, J. Role of Nrf2/HO-1 system in development, oxidative stress response and diseases: An evolutionarily conserved mechanism. Cell. Mol. Life Sci. 2016, 73, 3221–3247. [Google Scholar] [CrossRef] [PubMed]
- Ferrigno, A.; Campagnoli, L.I.M.; Barbieri, A.; Marchesi, N.; Pascale, A.; Croce, A.C.; Vairetti, M.; Di Pasqua, L.G. MCD Diet Modulates HuR and Oxidative Stress-Related HuR Targets in Rats. Int. J. Mol. Sci. 2023, 24, 9808. [Google Scholar] [CrossRef] [PubMed]
- Radosavljevic, T.; Brankovic, M.; Samardzic, J.; Djuretić, J.; Vukicevic, D.; Vucevic, D.; Jakovljevic, V. Altered Mitochondrial Function in MASLD: Key Features and Promising Therapeutic Approaches. Antioxidants 2024, 13, 906. [Google Scholar] [CrossRef] [PubMed]
- Green, A.; Hossain, T.; Eckmann, D.M. Mitochondrial dynamics involves molecular and mechanical events in motility, fusion and fission. Front. Cell Dev. Biol. 2022, 10, 1010232. [Google Scholar] [CrossRef] [PubMed]
- Legaki, A.-I.; Moustakas, I.I.; Sikorska, M.; Papadopoulos, G.; Velliou, R.-I.; Chatzigeorgiou, A. Hepatocyte Mitochondrial Dynamics and Bioenergetics in Obesity-Related Non-Alcoholic Fatty Liver Disease. Curr. Obes. Rep. 2022, 11, 126–143. [Google Scholar] [CrossRef] [PubMed]
- Steffen, J.; Ngo, J.; Wang, S.-P.; Williams, K.; Kramer, H.F.; Ho, G.; Rodriguez, C.; Yekkala, K.; Amuzie, C.; Bialecki, R.; et al. The mitochondrial fission protein Drp1 in liver is required to mitigate NASH and prevents the activation of the mitochondrial ISR. Mol. Metab. 2022, 64, 101566. [Google Scholar] [CrossRef] [PubMed]
- Undamatla, R.; Fagunloye, O.G.; Chen, J.; Edmunds, L.R.; Murali, A.; Mills, A.; Xie, B.; Pangburn, M.M.; Sipula, I.; Gibson, G.; et al. Reduced mitophagy is an early feature of NAFLD and liver-specific PARKIN knockout hastens the onset of steatosis, inflammation and fibrosis. Sci. Rep. 2023, 13, 7575. [Google Scholar] [CrossRef] [PubMed]
- Zhou, T.; Chang, L.; Luo, Y.; Zhou, Y.; Zhang, J. Mst1 inhibition attenuates non-alcoholic fatty liver disease via reversing Parkin-related mitophagy. Redox Biol. 2019, 21, 101120. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.W.; McKeen, T.; Zhang, J.H.; Ding, W.-X. Role and Mechanisms of Mitophagy in Liver Diseases. Cells 2020, 9, 837. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Komatsu, M. Autophagy: Renovation of Cells and Tissues. Cell 2011, 147, 728–741. [Google Scholar] [CrossRef] [PubMed]
- Filali-Mouncef, Y.; Hunter, C.; Roccio, F.; Zagkou, S.; Dupont, N.; Primard, C.; Proikas-Cezanne, T.; Reggiori, F. The ménage à trois of autophagy, lipid droplets and liver disease. Autophagy 2022, 18, 50–72. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.H.; Palanisamy, K.; Sun, K.T.; Li, X.; Wang, Y.M.; Lin, F.Y.; Chen, K.B.; Wang, I.K.; Yu, T.M.; Li, C.Y. Human antigen R regulates hypoxia-induced mitophagy in renal tubular cells through PARKIN/BNIP3L expressions. J. Cell. Mol. Med. 2021, 25, 2691–2702. [Google Scholar] [CrossRef] [PubMed]
- Ji, E.; Kim, C.; Kang, H.; Ahn, S.; Jung, M.; Hong, Y.; Tak, H.; Lee, S.; Kim, W.; Lee, E.K. Correction for Ji et al., “RNA Binding Protein HuR Promotes Autophagosome Formation by Regulating Expression of Autophagy-Related Proteins 5, 12, and 16 in Human Hepatocellular Carcinoma Cells”. Mol. Cell. Biol. 2021, 41, e00643-20. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.L.; Chen, J.C.; Jiang, H.; Li, N.; Li, X.J.; Liu, Y.H.; Zong, C. Hepatocyte-Specific HuR Protects Against Acetaminophen-Induced Liver Injury in Mice. J. Cell. Mol. Med. 2024, 28, e70246. [Google Scholar] [CrossRef] [PubMed]
- Riedl, S.J.; Shi, Y. Molecular mechanisms of caspase regulation during apoptosis. Nat. Rev. Mol. Cell Biol. 2004, 5, 897–907. [Google Scholar] [CrossRef]
- Ye, K.; Chen, Z.; Xu, Y. The double-edged functions of necroptosis. Cell Death Dis. 2023, 14, 163. [Google Scholar] [CrossRef] [PubMed]
- Broz, P. Pyroptosis: Molecular mechanisms and roles in disease. Cell Res. 2025, 35, 334–344. [Google Scholar] [CrossRef] [PubMed]
- Stockwell, B.R. Ferroptosis turns 10: Emerging mechanisms, physiological functions, and therapeutic applications. Cell 2022, 185, 2401–2421. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.L.; Wan, S.R.; An, Y.; Wu, Q.; Xing, Y.H.; Deng, C.H.; Zhang, P.P.; Long, Y.; Xu, B.T.; Jiang, Z.Z. Targeting cell death in NAFLD: Mechanisms and targeted therapies. Cell Death Discov. 2024, 10, 399. [Google Scholar] [CrossRef] [PubMed]
- Arab, J.P.; Hernández-Rocha, C.; Morales, C.; Vargas, J.I.; Solís, N.; Pizarro, M.; Robles, C.; Sandoval, D.; Ponthus, S.; Benítez, C.; et al. Serum cytokeratin-18 fragment levels as noninvasive marker of nonalcoholic steatohepatitis in the chilean population. Gastroenterol. Y Hepatol. 2017, 40, 388–394. [Google Scholar] [CrossRef] [PubMed]
- Simion, V.; Zhou, H.Y.; Haemmig, S.; Pierce, J.B.; Mendes, S.; Tesmenitsky, Y.; Pérez-Cremades, D.; Lee, J.F.; Chen, A.F.; Ronda, N.; et al. A macrophage-specific lncRNA regulates apoptosis and atherosclerosis by tethering HuR in the nucleus. Nat. Commun. 2020, 11, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Abdelmohsen, K.; Gorospe, M. Posttranscriptional regulation of cancer traits by HuR. Wiley Interdiscip. Rev.-Rna 2010, 1, 214–229. [Google Scholar] [CrossRef] [PubMed]
- Abdelmohsen, K.; Lal, A.; Kim, H.H.; Gorospe, M. Posttranscriptional orchestration of an anti-apoptotic program by HuR. Cell Cycle 2007, 6, 1288–1292. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, S.H.; Kohli, R.; Gores, G.J. Mechanisms of Lipotoxicity in NAFLD and Clinical Implications. J. Pediatr. Gastroenterol. Nutr. 2011, 53, 131–140. [Google Scholar] [CrossRef] [PubMed]
- Alkhouri, N.; Carter-Kent, C.; Feldstein, A.E. Apoptosis in nonalcoholic fatty liver disease: Diagnostic and therapeutic implications. Expert Rev. Gastroenterol. Hepatol. 2011, 5, 201–212. [Google Scholar] [CrossRef] [PubMed]
- Malhi, H.; Barreyro, F.J.; Isomoto, H.; Bronk, S.F.; Gores, G.J. Free fatty acids sensitise hepatocytes to TRAIL mediated cytotoxicity. Gut 2007, 56, 1124–1131. [Google Scholar] [CrossRef] [PubMed]
- Malhi, H.; Bronk, S.F.; Werneburg, N.W.; Gores, G.J. Free fatty acids induce JNK-dependent hepatocyte lipoapoptosis. J. Biol. Chem. 2006, 281, 12093–12101. [Google Scholar] [CrossRef] [PubMed]
- Gancheva, S.; Kahl, S.; Pesta, D.; Mastrototaro, L.; Dewidar, B.; Strassburger, K.; Sabah, E.; Esposito, I.; Weiβ, J.; Sarabhai, T.; et al. Impaired Hepatic Mitochondrial Capacity in Nonalcoholic Steatohepatitis Associated With Type 2 Diabetes. Diabetes Care 2022, 45, 928–937. [Google Scholar] [CrossRef] [PubMed]
- Lin, Z.; Song, J.; Gao, Y.; Huang, S.; Dou, R.; Zhong, P.; Huang, G.; Han, L.; Zheng, J.; Zhang, X.; et al. Hypoxia-induced HIF-1α/lncRNA-PMAN inhibits ferroptosis by promoting the cytoplasmic translocation of ELAVL1 in peritoneal dissemination from gastric cancer. Redox Biol. 2022, 52, 102312. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.; Kim, S.H.; Armaly, A.M.; Aubé, J.; Xu, L.; Wu, X. RNA-binding protein HuR inhibition induces multiple programmed cell death in breast and prostate cancer. Cell Commun. Signal 2024, 22, 580. [Google Scholar] [CrossRef] [PubMed]
- Vachliotis, I.D.; Polyzos, S.A. The Role of Tumor Necrosis Factor-Alpha in the Pathogenesis and Treatment of Nonalcoholic Fatty Liver Disease. Curr. Obes. Rep. 2023, 12, 191–206. [Google Scholar] [CrossRef] [PubMed]
- Ertunc, M.E.; Hotamisligil, G.S. Lipid signaling and lipotoxicity in metaflammation: Indications for metabolic disease pathogenesis and treatment. J. Lipid Res. 2016, 57, 2099–2114. [Google Scholar] [CrossRef] [PubMed]
- Beutler, B.; Cerami, A. The biology of cachectin/TNF—A primary mediator of the host response. Annu. Rev. Immunol. 1989, 7, 625–655. [Google Scholar] [CrossRef] [PubMed]
- Sedlyarov, V.; Fallmann, J.; Ebner, F.; Huemer, J.; Sneezum, L.; Ivin, M.; Kreiner, K.; Tanzer, A.; Vogl, C.; Hofacker, I.; et al. Tristetraprolin binding site atlas in the macrophage transcriptome reveals a switch for inflammation resolution. Mol. Syst. Biol. 2016, 12, 868. [Google Scholar] [CrossRef] [PubMed]
- Tiedje, C.; Ronkina, N.; Tehrani, M.; Dhamija, S.; Laass, K.; Holtmann, H.; Kotlyarov, A.; Gaestel, M. The p38/MK2-Driven Exchange between Tristetraprolin and HuR Regulates AU-Rich Element-Dependent Translation. PLoS Genet. 2012, 8, e1002977. [Google Scholar] [CrossRef] [PubMed]
- Abdelsam, S.S.; Ghanem, S.K.; Zahid, M.A.; Abunada, H.H.; Bader, L.; Raïq, H.; Khan, A.; Parray, A.; Djouhri, L.; Agouni, A. Human antigen R: Exploring its inflammatory response impact and significance in cardiometabolic disorders. J. Cell Physiol. 2024, 239, e31229. [Google Scholar] [CrossRef] [PubMed]
- Ouhara, K.; Munenaga, S.; Kajiya, M.; Takeda, K.; Matsuda, S.; Sato, Y.; Hamamoto, Y.; Iwata, T.; Yamasaki, S.; Akutagawa, K.; et al. The induced RNA-binding protein, HuR, targets 3′-UTR region of IL-6 mRNA and enhances its stabilization in periodontitis. Clin. Exp. Immunol. 2018, 192, 325–336. [Google Scholar] [CrossRef] [PubMed]
- Yiakouvaki, A.; Dimitriou, M.; Karakasiliotis, I.; Eftychi, C.; Theocharis, S.; Kontoyiannis, D.L. Myeloid cell expression of the RNA-binding protein HuR protects mice from pathologic inflammation and colorectal carcinogenesis. J. Clin. Investig. 2012, 122, 48–61. [Google Scholar] [CrossRef] [PubMed]
- Rhee, W.J.; Ni, C.W.; Zheng, Z.L.; Chang, K.; Jo, H.; Bao, G. HuR regulates the expression of stress-sensitive genes and mediates inflammatory response in human umbilical vein endothelial cells. Proc. Natl. Acad. Sci. USA 2010, 107, 6858–6863. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.S.; Ishmael, F.T.; Fang, X.; Myers, A.; Cheadle, C.; Huang, S.K.; Atasoy, U.; Gorospe, M.; Stellato, C. Chemokine Transcripts as Targets of the RNA-Binding Protein HuR in Human Airway Epithelium. J. Immunol. 2011, 186, 2482–2494. [Google Scholar] [CrossRef] [PubMed]
- de Bruin, R.G.; Rabelink, T.J.; van Zonneveld, A.J.; van der Veer, E.P. Emerging roles for RNA-binding proteins as effectors and regulators of cardiovascular disease. Eur. Heart J. 2017, 38, 1380–1388D. [Google Scholar] [CrossRef] [PubMed]
- Barreau, C.; Paillard, L.; Osborne, H.B. AU-rich elements and associated factors: Are there unifying principles? Nucleic Acids Res. 2005, 33, 7138–7150. [Google Scholar] [CrossRef] [PubMed]
- Sobolewski, C.; Dubuquoy, L.; Legrand, N. MicroRNAs, Tristetraprolin Family Members and HuR: A Complex Interplay Controlling Cancer-Related Processes. Cancers 2022, 14, 3516. [Google Scholar] [CrossRef] [PubMed]
- Brooks, S.A.; Blackshear, P.J. Tristetraprolin (TTP): Interactions with mRNA and proteins, and current thoughts on mechanisms of action. Biochim. Et. Biophys. Acta-Gene Regul. Mech. 2013, 1829, 666–679. [Google Scholar] [CrossRef] [PubMed]
- Taylor, G.A.; Carballo, E.; Lee, D.M.; Lai, W.S.; Thompson, M.J.; Patel, D.D.; I Schenkman, D.; Gilkeson, G.S.; Broxmeyer, H.E.; Haynes, B.F.; et al. A pathogenetic role for TNF alpha In the syndrome of cachexia, arthritis, and autoimmunity resulting from tristetraprolin (TTP) deficiency. Immunity 1996, 4, 445–454. [Google Scholar] [CrossRef] [PubMed]
- Sawada, K.; Chung, H.K.; Softic, S.; Moreno-Fernandez, M.E.; Divanovic, S. The bidirectional immune crosstalk in metabolic dysfunction-associated steatotic liver disease. Cell Metab. 2023, 35, 1852–1871. [Google Scholar] [CrossRef] [PubMed]
- Syn, W.K.; Oo, Y.H.; Pereira, T.A.; Karaca, G.F.; Jung, Y.M.; Omenetti, A.; Witek, R.P.; Choi, S.S.; Guy, C.D.; Fearing, C.M.; et al. Accumulation of Natural Killer T Cells in Progressive Nonalcoholic Fatty Liver Disease. Hepatology 2010, 51, 1998–2007. [Google Scholar] [CrossRef] [PubMed]
- Papadaki, O.; Milatos, S.; Grammenoudi, S.; Mukherjee, N.; Keene, J.D.; Kontoyiannis, D.L. Control of Thymic T Cell Maturation, Deletion and Egress by the RNA-Binding Protein HuR. J. Immunol. 2009, 182, 6779–6788. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Martindale, J.L.; Abdelmohsen, K.; Kumar, G.; Fortina, P.M.; Gorospe, M.; Rostami, A.; Yu, S.G. RNA-Binding Protein HuR Promotes Th17 Cell Differentiation and Can Be Targeted to Reduce Autoimmune Neuroinflammation. J. Immunol. 2020, 204, 2076–2087. [Google Scholar] [CrossRef] [PubMed]
- Angulo, P.; Kleiner, D.E.; Dam-Larsen, S.; Adams, L.A.; Björnsson, E.S.; Charatcharoenwitthaya, P.; Mills, P.R.; Keach, J.C.; Lafferty, H.D.; Stahler, A.; et al. Liver Fibrosis, but No Other Histologic Features, Is Associated With Long-term Outcomes of Patients With Nonalcoholic Fatty Liver Disease. Gastroenterology 2015, 149, 389–397. [Google Scholar] [CrossRef] [PubMed]
- Vilar-Gomez, E.; Calzadilla-Bertot, L.; Wong, V.W.S.; Castellanos, M.; Fuente, R.A.-D.L.; Metwally, M.; Eslam, M.; Gonzalez-Fabian, L.; Sanz, M.A.Q.; Conde-Martín, A.F.; et al. Fibrosis Severity as a Determinant of Cause-Specific Mortality in Patients With Advanced Nonalcoholic Fatty Liver Disease: A Multi-National Cohort Study. Gastroenterology 2018, 155, 443–457. [Google Scholar] [CrossRef] [PubMed]
- Schwabe, R.F.; Tabas, I.; Pajvani, U.B. Mechanisms of Fibrosis Development in Nonalcoholic Steatohepatitis. Gastroenterology 2020, 158, 1913–1928. [Google Scholar] [CrossRef] [PubMed]
- Gandhi, C.R. Hepatic stellate cell activation and pro-fibrogenic signals. J. Hepatol. 2017, 67, 1104–1105. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.Y.; Kim, K.; Wang, X.B.; Bartolome, A.; Salomao, M.; Dongiovanni, P.; Meroni, M.; Graham, M.J.; Yates, K.P.; Diehl, A.M.; et al. Hepatocyte Notch activation induces liver fibrosis in nonalcoholic steatohepatitis. Sci. Transl. Med. 2018, 10, eaat0344. [Google Scholar] [CrossRef] [PubMed]
- Li, X.J.Y.; Liu, R.P.; Yang, J.; Sun, L.X.; Zhang, L.Y.; Jiang, Z.Z.; Puri, P.; Gurley, E.C.; Lai, G.H.; Tang, Y.P.; et al. The Role of Long Noncoding RNA H19 in Gender Disparity of Cholestatic Liver Injury in Multidrug Resistance 2 Gene Knockout Mice. Hepatology 2017, 66, 869–884. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.P.; Li, X.J.Y.; Zhu, W.W.; Wang, Y.Y.; Zhao, D.; Wang, X.; Gurley, E.C.; Liang, G.; Chen, W.D.; Lai, G.H.; et al. Cholangiocyte-Derived Exosomal Long Noncoding RNA H19 Promotes Hepatic Stellate Cell Activation and Cholestatic Liver Fibrosis. Hepatology 2019, 70, 1317–1335. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.L.; Yao, Z.; Wang, L.; Ding, H.; Shao, J.J.; Chen, A.P.; Zhang, F.; Zheng, S.Z. Activation of ferritinophagy is required for the RNA-binding protein ELAVL1/HuR to regulate ferroptosis in hepatic stellate cells. Autophagy 2018, 14, 2083–2103. [Google Scholar] [CrossRef] [PubMed]
- Polyzos, S.A.; Kountouras, J.; Mantzoros, C.S. Obesity and nonalcoholic fatty liver disease: From pathophysiology to therapeutics. Metabolism 2019, 92, 82–97. [Google Scholar] [CrossRef] [PubMed]
- Blüher, M. Obesity: Global epidemiology and pathogenesis. Nat. Rev. Endocrinol. 2019, 15, 288–298. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.P.; Wu, W.Y.; Ma, C.F.; Du, C.Y.; Huang, Y.R.; Xu, H.X.; Li, C.C.; Cheng, X.F.; Hao, R.J.; Xu, Y.J. RNA-Binding Proteins in the Regulation of Adipogenesis and Adipose Function. Cells 2022, 11, 2357. [Google Scholar] [CrossRef] [PubMed]
- Hwang, J.S.; Lee, W.J.; Hur, J.; Lee, H.G.; Kim, E.; Lee, G.H.; Choi, M.J.; Lim, D.S.; Paek, K.S.; Seo, H.G. Rosiglitazone-dependent dissociation of HuR from PPAR-γ regulates adiponectin expression at the posttranscriptional level. FASEB J. 2019, 33, 7707–7720. [Google Scholar] [CrossRef] [PubMed]
- Guarnieri, A.R.; Anthony, S.R.; Gozdiff, A.; Green, L.C.; Fleifil, S.M.; Slone, S.; Nieman, M.L.; Alam, P.; Benoit, J.B.; Owens, A.P.; et al. Adipocyte-specific deletion of HuR induces spontaneous cardiac hypertrophy and fibrosis. Am. J. Physiol.-Heart Circ. Physiol. 2021, 321, H228–H241. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.; Han, J.; Wang, H.Y.; Meng, Q.Y.; Chen, L.L.; Liu, Y.G.; Feng, Y.; Wu, G.H. Cachexia-related long noncoding RNA, CAAlnc1, suppresses adipogenesis by blocking the binding of HuR to adipogenic transcription factor mRNAs. Int. J. Cancer 2019, 145, 1809–1821. [Google Scholar] [CrossRef] [PubMed]
- Vanni, E.; Marengo, A.; Mezzabotta, L.; Bugianesi, E. Systemic Complications of Nonalcoholic Fatty Liver Disease: When the Liver Is Not an Innocent Bystander. Semin. Liver Dis. 2015, 35, 236–249. [Google Scholar] [CrossRef] [PubMed]
- Targher, G.; Corey, K.E.; Byrne, C.D.; Roden, M. The complex link between NAFLD and type 2 diabetes mellitus-mechanisms and treatments. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 599–612. [Google Scholar] [CrossRef] [PubMed]
- Targher, G.; Day, C.P.; Bonora, E. Risk of Cardiovascular Disease in Patients with Nonalcoholic Fatty Liver Disease. N. Engl. J. Med. 2010, 363, 1341–1350. [Google Scholar] [CrossRef] [PubMed]
- Parker, E.D.; Lin, J.N.C.; Mahoney, T.; Ume, N.; Yang, G.C.; Gabbay, R.A.; ElSayed, N.A.; Bannuru, R.R. Economic Costs of Diabetes in the US in 2022. Diabetes Care 2024, 47, 26–43. [Google Scholar] [CrossRef] [PubMed]
- Forbes, J.M.; Cooper, M.E. Mechanisms of Diabetic Complications. Physiol. Rev. 2013, 93, 137–188. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.; Zhang, Z.G.; Zheng, C.; Wintergerst, K.A.; Keller, B.B.; Cai, L. Mechanisms of diabetic cardiomyopathy and potential therapeutic strategies: Preclinical and clinical evidence. Nat. Rev. Cardiol. 2020, 17, 585–607. [Google Scholar] [CrossRef] [PubMed]
- Green, L.C.; Anthony, S.R.; Slone, S.; Lanzillotta, L.; Nieman, M.L.; Wu, X.Q.; Robbins, N.; Jones, S.M.; Roy, S.; Owens, A.P.; et al. Human antigen R as a therapeutic target in pathological cardiac hypertrophy. J. Clin. Investig. 2019, 4, e121541. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.; Jiang, M.Y.; Cao, Y.P.; Zhang, Z.J.; Jiang, B.; Tian, F.; Feng, J.; Dou, Y.L.; Gorospe, M.; Zheng, M.; et al. HuR regulates phospholamban expression in isoproterenol-induced cardiac remodelling. Cardiovasc. Res. 2019, 116, 944–955. [Google Scholar] [CrossRef] [PubMed]
- Slone, S.; Anthony, S.R.; Green, L.C.; Parkins, S.; Acharya, P.; Kasprovic, D.A.; Reynolds, K.; Jaggers, R.M.; Nieman, M.L.; Alam, P.; et al. HuR inhibition reduces post-ischemic cardiac remodeling by dampening myocyte-dependent inflammatory gene expression and the innate immune response. FASEB J. 2025, 39, e70433. [Google Scholar] [CrossRef] [PubMed]
- Green, L.C.; Slone, S.; Anthony, S.R.; Guarnieri, A.R.; Parkins, S.; Shearer, S.M.; Nieman, M.L.; Roy, S.; Aube, J.; Wu, X.Q.; et al. HuR-dependent expression of Wisp1 is necessary for TGFβ-induced cardiac myofibroblast activity. J. Mol. Cell. Cardiol. 2023, 174, 38–46. [Google Scholar] [CrossRef] [PubMed]
- Govindappa, P.K.; Patil, M.; Garikipati, V.N.S.; Verma, S.K.; Saheera, S.; Narasimhan, G.; Zhu, W.Q.; Kishore, R.; Zhang, J.Y.; Krishnamurthy, P. Targeting exosome-associated human antigen R attenuates fibrosis and inflammation in diabetic heart. FASEB J. 2020, 34, 2238–2251. [Google Scholar] [CrossRef] [PubMed]
- Patil, M.; Singh, S.; Dubey, P.K.; Tousif, S.; Umbarkar, P.; Zhang, Q.K.; Lal, H.; Sewell-Loftin, M.K.; Umeshappa, C.S.; Ghebre, Y.T.; et al. Fibroblast-Specific Depletion of Human Antigen R Alleviates Myocardial Fibrosis Induced by Cardiac Stress. Jacc-Basic. Transl. Sci. 2024, 9, 754–770. [Google Scholar] [CrossRef] [PubMed]
- Krishnamurthy, P.; Lambers, E.; Verma, S.; Thorne, T.; Qin, G.J.; Losordo, D.W.; Kishore, R. Myocardial knockdown of mRNA-stabilizing protein HuR attenuates post-MI inflammatory response and left ventricular dysfunction in IL-10-null mice. FASEB J. 2010, 24, 2484–2494. [Google Scholar] [CrossRef] [PubMed]
- Jeyabal, P.; Thandavarayan, R.A.; Joladarashi, D.; Babu, S.S.; Krishnamurthy, S.; Bhimaraj, A.; Youker, K.A.; Kishore, R.; Krishnamurthy, P. MicroRNA-9 inhibits hyperglycemia-induced pyroptosis in human ventricular cardiomyocytes by targeting ELAVL1. Biochem. Biophys. Res. Commun. 2016, 471, 423–429. [Google Scholar] [CrossRef] [PubMed]
- Lachiondo-Ortega, S.; Delgado, T.C.; Baños-Jaime, B.; Velázquez-Cruz, A.; Díaz-Moreno, I.; Martinez-Chantar, M.L. Hu Antigen R (HuR) Protein Structure, Function and Regulation in Hepatobiliary Tumors. Cancers 2022, 14, 2666. [Google Scholar] [CrossRef] [PubMed]
- Vyas, K.; Patel, M.M. Insights on drug and gene delivery systems in liver fibrosis. Asian J. Pharm. Sci. 2023, 18, 100779. [Google Scholar] [CrossRef] [PubMed]
- Majumder, M.; Chakraborty, P.; Mohan, S.; Mehrotra, S.; Palanisamy, V. HuR as a molecular target for cancer therapeutics and immune-related disorders. Adv. Drug Deliv. Rev. 2022, 188, 114442. [Google Scholar] [CrossRef] [PubMed]
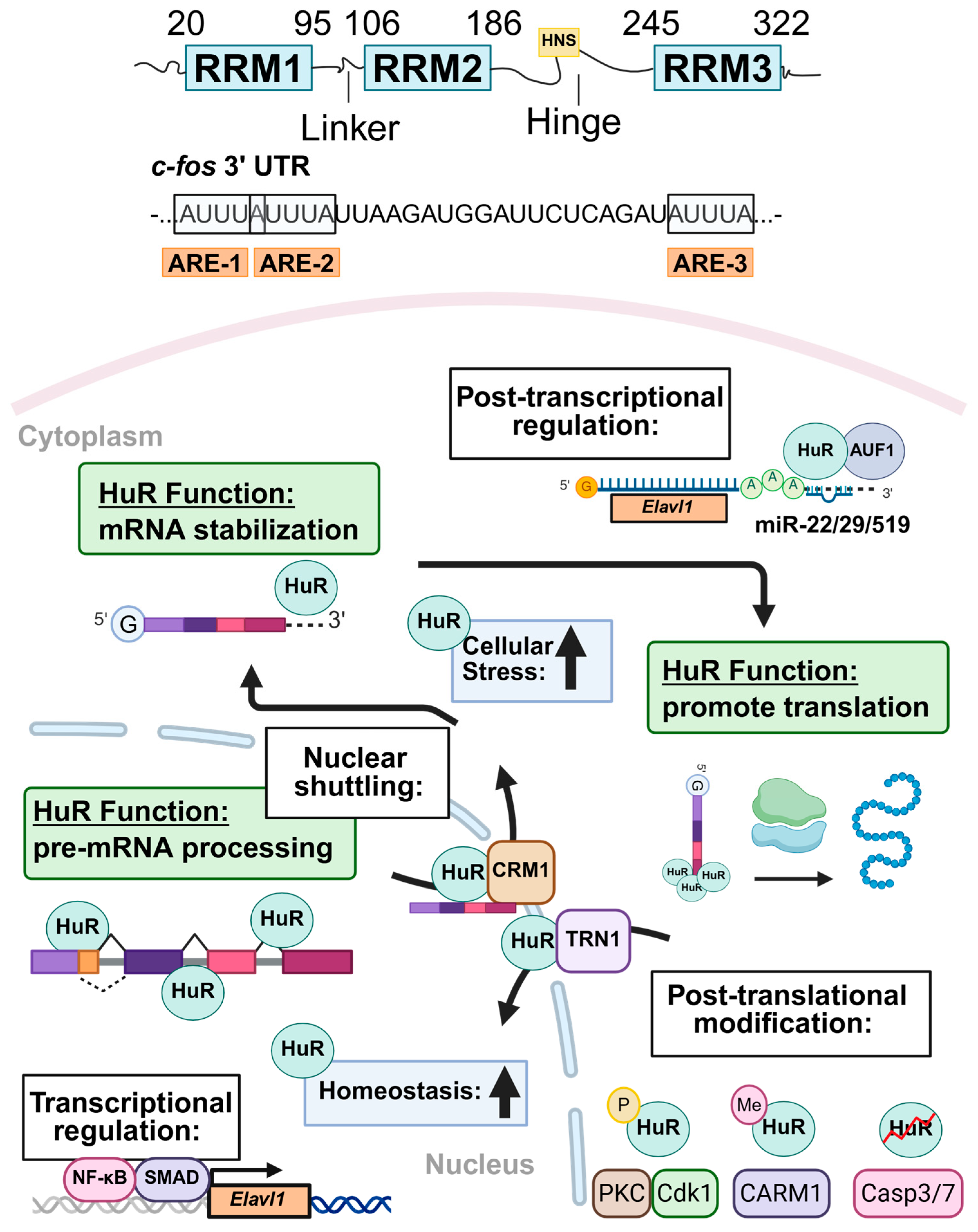
| Regulator | Context | Effect on HuR | Ref. |
|---|---|---|---|
| Transcriptional | |||
| NFκB | Cell survival/proliferation in MKN74 cells | Long gene variant expression | [67] |
| SMAD | ATP depletion in LLC-PK1 cells | Short gene variant expression | [68] |
| Post-transcriptional | |||
| HuR | Human diploid fibroblasts HeLa cells | Cytoplasmic localization | [36,69] |
| AUF1 | HeLa cells | mRNA destabilization | [69,71] |
| miR-22 | SW480 colorectal cancer cells | mRNA destabilization | [72] |
| miR-29 | Acute myeloid leukemia cells | mRNA destabilization | [73] |
| miR-519 | Human carcinoma cells | Impaired translation | [74] |
| Post-translational | |||
| Phosphorylation | |||
| CDK1 | Irradiated HeLa cells | Nuclear localization | [55] |
| CHK2 | H2O2-treated HeLa cells | Altered mRNA binding affinity | [74,77] |
| MAPK p38 | Irradiated HaCaT human keratinocytes | Cytoplasmic localization | [39,79] |
| PKC-α | ATP-treated human mesangial cells | Cytoplasmic localization | [80] |
| PKC-δ | HCV-infected Huh7.5 cells | Cytoplasmic localization | [63] |
| Methylation | |||
| CARM1 | Human diploid fibroblasts | Altered mRNA binding affinity | [35] |
| Protein cleavage | |||
| CASP3/7 | Staurosporine-treated HeLa cells | Cytoplasmic localization; pro-apoptotic | [33,34] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Eppler, N.; Jones, E.; Ahamed, F.; Zhang, Y. Multifaceted Human Antigen R (HuR): A Key Player in Liver Metabolism and MASLD. Livers 2025, 5, 33. https://doi.org/10.3390/livers5030033
Eppler N, Jones E, Ahamed F, Zhang Y. Multifaceted Human Antigen R (HuR): A Key Player in Liver Metabolism and MASLD. Livers. 2025; 5(3):33. https://doi.org/10.3390/livers5030033
Chicago/Turabian StyleEppler, Natalie, Elizabeth Jones, Forkan Ahamed, and Yuxia Zhang. 2025. "Multifaceted Human Antigen R (HuR): A Key Player in Liver Metabolism and MASLD" Livers 5, no. 3: 33. https://doi.org/10.3390/livers5030033
APA StyleEppler, N., Jones, E., Ahamed, F., & Zhang, Y. (2025). Multifaceted Human Antigen R (HuR): A Key Player in Liver Metabolism and MASLD. Livers, 5(3), 33. https://doi.org/10.3390/livers5030033