1. Introduction
Conventional plastics are synthetic materials made from petroleum-based polymers, which are durable and very slow to degrade, leading to depleted resources and massive waste [
1]. By 2019, the global production of petrochemical plastics amounted to almost 359 million tons per year [
2]. The degradation of these plastics is very problematic since large amounts of CO
2 and many other toxic compounds are emitted during their incineration process [
3,
4]. To minimise the environmental impact of petrochemical-based plastic packaging, new sustainable materials, such as biodegradable and compostable bioplastics, have been developed. In this sense, poly(lactic acid) (PLA) is a thermoplastic, compostable, and biocompatible polymer derived from renewable resources [
5]. PLA is nowadays seen as one of the most promising compostable polymers for commercial use as a plastic substitute [
6].
Biopolymer-based materials have some limitations, including low water and chemical resistance, low heat resistance, and brittleness [
7]. Several alternatives and approaches have been proposed to improve the performance of these biomaterials. These strategies, including the addition of fillers, compatibilisers, plasticisers, polymer functionalisation, or biodegradable multilayer systems, have been successfully investigated [
8,
9]. The use of cellulose fibres from lignocellulosic waste, as reinforcing agents, to improve the properties of biomaterials has also been widely explored [
10,
11,
12]. Their incorporation as fillers in polymeric films offers a potential improvement in the functional properties of the material, while contributing to the sustainability, compostability, and biodegradability of the material [
13]. Nevertheless, the process of obtaining cellulose fibres from biomass usually implies the use of a great amount of chemicals to isolate cellulose from the lignocellulosic complex, and more sustainable and greener processes are necessary to exploit cellulose from a large number of cellulose-rich biomass residues. In this sense, Camarena-Bononad et al. [
14] obtained cellulose fibres from
Posidonia oceanica (PO) waste (24–25 g/100 g PO) by a greener method, using subcritical water extraction (SWE) and subsequent bleaching process with hydrogen peroxide (H
2O
2) or with sodium chlorite (NaClO
2). Therefore, PO waste can be considered as a potential source of cellulose, while incorporating cellulose extracted from PO into different polymer matrices could revalorise cellulose-rich waste, which incurs important costs when removed from beaches and disposed of in landfills, with significant losses of organic matter. The use of subcritical water extraction (SWE) represents a green method to remove an important fraction of non-cellulosic components, while making the lignocellulosic substrate more accessible to other purification agents. This is due to the high extractive power of water under subcritical conditions, when the properties of water (dielectric constant, diffusion, surface tension, …) change selectively with the temperature, improving the solubility of less-polar compounds to the level of organic solvents [
14].
One possible strategy for using cellulose fibres as fillers in food packaging material is to develop biocomposites. Due to its polysaccharide nature, cellulose is compatible with chitosan, a biodegradable cationic, non-toxic, and biocompatible polysaccharide with antimicrobial activity and excellent film-forming properties [
15,
16]. A chitosan–cellulose mixture yielded composites with interesting properties by casting techniques [
17]. In cellulose–chitosan blends, the cellulose would act as a reinforcement material for chitosan, providing mechanical strength, while chitosan, due to its peculiar properties, could provide composite films with antimicrobial activity and protection of bioactive molecules [
18,
19].
Different studies analysed the incorporation of cellulose fibres of different structures (such as nanofibres, microfibres, or nanocrystals) into the PLA polymeric matrix, obtaining composite materials with enhanced functional performance, particularly better oxygen barrier properties and mechanical strength [
20,
21]. This approach has also been previously used with PO cellulose fibres with different levels of purification to develop PLA composite films [
22]. However, no studies have been found for PLA–cellulose laminates (bilayer films) that could also have suitable properties for food packaging purposes.
The aim of this study was to explore the capacity of PO cellulosic fibres obtained by subcritical water extraction at 170 °C, and bleaching with hydrogen peroxide or sodium chlorite, to develop different materials for food packaging: (a) cellulosic films, (b) chitosan–cellulose biocomposites, and (c) cellulose–PLA laminates. These different materials were characterised by their relevant functional properties for packaging applications, such as mechanical performance and barrier capacity to water vapour and oxygen. Likewise, the optical properties, thermal stability, and microstructure of the materials were also analysed.
2. Materials and Methods
2.1. Chemicals
Medium molecular weight chitosan, with a deacetylation degree of ≥75% and a viscosity of 200–800 cps (at 1% w/w in 1% w/w acetic acid solution), glucose, arabinose, sodium chlorite (NaClO2), sodium hydroxide (NaOH), and sodium acetate were supplied by Sigma-Aldrich (St. Louis, MO, USA). Amorphous PLA 4060D, density 1.24 g.cm−3, with an average molecular weight of 106,226 D and 40% low molecular weight fraction (275D) was obtained from Natureworks (Plymouth, MN, USA). Phosphorus pentoxide (P2O5, 98.2%), magnesium nitrate (Mg(NO3)2), hydrogen peroxide (H2O2, 30%), sulphuric acid (H2SO4, 98%), sodium carbonate (Na2CO3, 99.5%), acetic acid (CH3COOH), and ethanol (C2H6O, 98%) were purchased from Panreac Quimica S.L.U (Castellar del Vallés, Barcelona, Spain). D(+)-xylose was obtained from Merck KGaA (Darmstadt, Germany).
2.2. Obtaining Cellulose Fibres from Posidonia oceanica
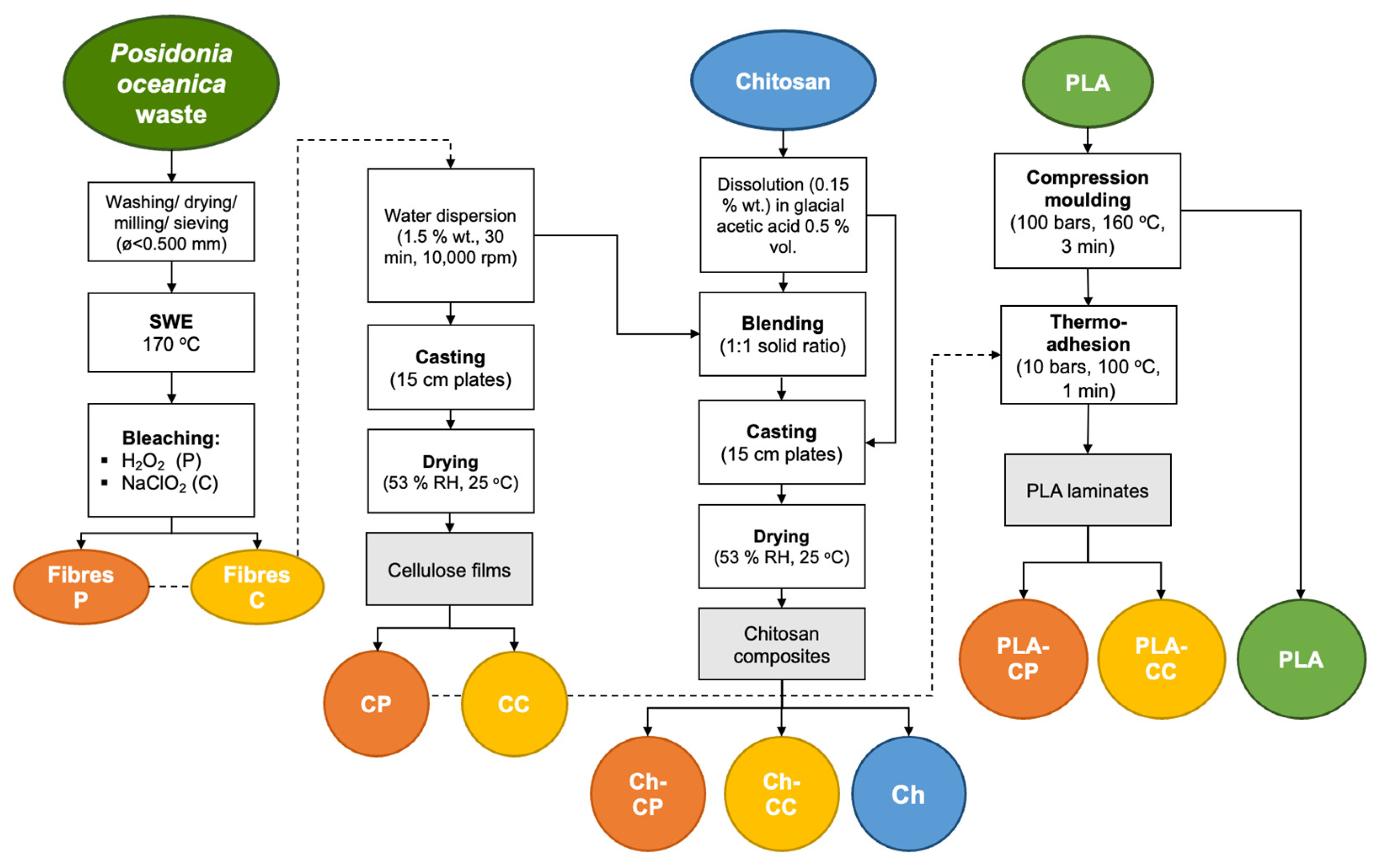
PO waste was provided by a landfill located in Denia (Alicante, Spain) and fibres were obtained as described by Camarena-Bononad et al. [
14]. As shown in
Figure 1, the PO leaves were sorted, washed, dried (50 °C for 72 h), milled (Model SM300 stainless, Retsch GmbH, Haan, Germany), and sieved to obtain particles less than 0.50 mm. Subsequently, the sieved PO leaves were subjected to subcritical water extraction (SWE) at 170 °C and 9.5 bar, 150 rpm for 30 min, using a Pressure Reactor (Model 1-T-A-P-CE, 5 L capacity, Amar Equipment PVT. Ltd., Mumbai, India), with a PO–distilled water ratio of 1:15 (
w/
v). Afterward, the insoluble residue was submitted to different bleaching processes: (a) treatment with hydrogen peroxide (H
2O
2) at 8% (v/v), pH 12, 4 cycles of 1 h, 40 °C, and (b) treatment with sodium chlorite (NaClO
2), under reflux heating, 5 cycles of 4 h, at pH 4.5. The bleached fibres were dried in a vacuum oven (S. P. Selecta, s. a., Barcelona, Spain) at 60 °C for 3 days, ground (Model SM300 stainless, Retsch GmbH, Haan, Germany), sieved (Ø < 0.125 mm), and stored in a desiccator with P
2O
5 until use.
The cellulose, hemicellulose, and lignin contents of the different cellulose fibres were determined following the NREL/TP-510-42618-2008 method [
23], as described in a previous study [
14]. Briefly, the fibre samples were submitted to acid hydrolysis with H
2SO
4 in a two-step process, and acid-insoluble Klason lignin was determined gravimetrically from the insoluble fraction. The cellulose and hemicellulose contents were determined from the monosaccharide analysis (glucose, xylose, and arabinose) in the soluble fraction, by using HPLC equipment (Agilent Technologies, model 1120 Compact LC, Waldbronn, Germany), with RezexTM RCM-Monosaccharide Ca
2+ column (150 × 7.8 mm) and an evaporative light scattering detector (ELSD Agilent Technologies 1200 Series, Waldbronn, Germany). Milli-Q water was used as eluent in isocratic mode, at a 0.4 mL·min
−1 flow rate. The cellulose content was obtained from the glucose concentration, and the hemicellulose content was determined from the sum of the xylose and arabinose concentrations. In both cases, the correction factors related to the water molecular weight were considered [
23]. So, this method overestimates the cellulose content, since glucose is also present in hemicelluloses.
Likewise, the fibre whiteness index (WI) was also analysed as described in
Section 2.6 for the films.
2.3. Obtaining Cellulose Films from PO Fibres
Cellulose films were obtained by casting the PO fibre dispersion in distilled water (0.015 g/mL), previously homogenised with Ultra-Turrax (model IKA T25 digital, IKA-Werke GmbH & Co., Staufen, Germany) for 30 min at 10,000 rpm (
Figure 1). Each film (0.6 g cellulose) was obtained by drying the water dispersion poured onto Teflon plates (15 cm in diameter) for 2 days in a chamber with controlled humidity (RH) and temperature conditions (53% RH, 25 °C). Two kinds of cellulosic films were obtained: CP from fibres bleached with hydrogen peroxide, and CC from fibres bleached with sodium chlorite. The films were stored in a desiccator with Mg(NO
3)
2 (53% RH) or P
2O
5 (0% RH) until further use.
2.4. Obtaining Chitosan–Cellulose Composites
Chitosan–cellulose composites were prepared, using a 1:1 ratio of the polymers, by solvent casting due to the non-thermoplastic nature of chitosan. For this purpose, chitosan (1.5% wt.) was dispersed in glacial acetic acid solution (0.5% v/v), maintained at 50 °C for 16 h under continuous stirring, and filtered (
Figure 1). Cellulose fibres (1.5% wt.) were dispersed in distilled water and homogenised using Ultra-Turrax for 30 min at 10,000 rpm. The chitosan and cellulose dispersions were mixed, and the final solution was stirred for 20 min. Then, 0.6 g of cellulose–chitosan blend was poured onto casting Teflon plates (15 cm in diameter) and dried at 53% RH, 25 °C, for 2 days. Composite films were obtained with peroxide-bleached fibres (Ch-CP) and chlorite-bleached fibres (Ch-CC). Chitosan control films (Ch) were also obtained from the pure chitosan solution applying the same procedure. All films were conditioned in desiccators with Mg(NO
3)
2 (53% RH) or P
2O
5 (0% RH) until their characterisation.
2.5. Obtaining PLA–Cellulose Laminates
Amorphous PLA was selected based on its better heat-sealing properties as compared to semicrystalline PLA. The films were thermoprocessed, as is usual on an industrial scale, due to the thermoplastic nature of PLA. First, the PLA films (2 g) were obtained by compression moulding of the previously dried PLA pellets (60 °C, under vacuum, for 24 h) using a hydraulic press (Model LP20, Labtech Engineering Company Ltd., Samut Prakan, Thailand) (
Figure 1). To this end, preheating at 160 °C for 3 min, compression at 100 bar at 160 °C for 3 min, and final cooling to 70 °C were applied.
PLA-CC- and PLA-CP-laminated films were obtained by the thermo-adhesion of a PLA film with the CP or CC films. This process was performed by placing the films between Teflon sheets in the preheated hydraulic press for 1 min at 100 °C, followed by compression at 10 bar and 100 °C for 1 min, and cooling to 70 °C. All films were conditioned in desiccators with Mg(NO3)2 (53% RH) or P2O5 (0% RH) until their characterisation.
2.6. Optical Properties of the Films
The optical properties of the films were analysed using a spectrocolorimeter (CM-3600d, Minolta Co., Tokyo, Japan), obtaining the reflection spectra (R) of the films between 420 and 700 nm on white (R
g) and black (R
0) backgrounds. The Kubelka–Munk multiple scattering theory [
24] was used to estimate the internal transmittance (T
i) and infinite reflectance (R
∞) (Equations (1)–(4)). Likewise, through the R
∞ spectra, the
CIEL*
a*
b* colour coordinates were obtained, considering the illuminant D65 and observer 10°. Then, chroma (
Cab*), hue angle (
hab*), whiteness index (WI), and total colour difference (∆
E*) with respect to the control films were calculated using Equations (5)–(8). Each sample was measured in triplicate.
where
;
;
; and
,
,
are the colour coordinates of the control films (Ch and PLA).
2.7. Mechanical and Barrier Properties of the Films
The tensile test was carried out according to ASTM D882-12 [
25], using a universal testing machine (Stable Micro Systems, TA.XT plus, Stable Micro Systems, Godalming, UK). Elongation at break (E%), tensile strength at break (TS), and elastic modulus (EM) were determined from the stress vs. strain curves. The film samples (25 × 100 mm), conditioned at 53% RH, were placed between two grips initially separated by 50 mm and stretched at a crosshead speed of 50 mm·min
−1. For each formulation, eight samples were evaluated.
The tear test proposed by Yamauchi and Tanaka [
26] for paper was used to determine the tear index of the films. Pre-conditioned samples (53% RH for 1 week) were cut and placed between two grips initially separated by 50 mm, and stretched at a crosshead speed of 500 mm·min
−1 using a universal testing machine (Stable Micro Systems, TA.XT plus, Stable Micro Systems, Godalming, UK). The test was performed in 6 samples for each formulation. From the obtained force–distance curves, the tear index was obtained using Equation (9).
The oxygen permeability (OP) of the conditioned films at 53% RH was determined according to ASTM D3985-05 [
27] using an Oxygen Permeation Analyzer (Model 8101e, Systech Illinois, McHenry, IL, USA) at 25 °C and 53% of RH. The oxygen transmission rate (OTR) was determined every 15 min until equilibrium was raised, with an exposure film area of 50 cm
2. The OP of the films was obtained by considering film thickness, the OTR, and the difference in partial pressure of oxygen between the two sides of the films (~1 atm). For each formulation, the measurements were taken in duplicate.
The water vapour permeability (WVP) of the films conditioned at 53% RH was determined according to the ASTM E96/E96M [
28] methodology. Films were cut (Ø = 35 mm), placed, and sealed in Payne permeability cups filled with 5 mL of distilled water (100% RH). Then, the cups were placed into desiccators containing an over-saturated solution of Mg(NO
3)
2 (53% RH) at 25 °C and weighed periodically, using an analytical balance (ME36S, Sartorius, ±0.00001 g, Fisher Scientific, Hampton, NH, USA). The measurements were taken in triplicate every 2 h for 42 h. The WVP was determined from the slope of the weight loss vs. time curves, considering the water vapour pressure gradient and the film thickness.
2.8. Microstructural Analysis
The cross-section of the films was observed using a High-resolution Field Emission Scanning Electron Microscope (HRFESEM, GeminiSEM 500, Zeiss, Oberkochen, Germany). The samples were previously conditioned at 0% RH for 1 week. Before the observation, films were immersed in liquid nitrogen and cryo-fractured and then coated with platinum for 60 s using an EM MED020 sputter coater (Leica BioSystems, Barcelona, Spain). Images were obtained at an accelerating voltage of 1.50 kV and 600×, 800×, and 2000× magnification.
2.9. Thermal Properties of the Films
The thermal stability of the films conditioned at 0% RH was determined in duplicate by thermogravimetric analysis (TGA) using a thermogravimetric analyser (TGA 1 Stare System analyser, Mettler-Toledo, Greifensee, Switzerland). Samples of about 3–4 mg were heated from 25 to 900 °C at 10 °C·min−1, under a constant nitrogen flow of 10 mL·min−1. STARe evaluation software (version V12.00a, Mettler-Toledo, Inc., Greifensee, Switzerland) was used to obtain the TGA curves and their first derivative curves (DTGA).
2.10. Statistical Analysis
The experimental data were submitted to analysis of variance (ANOVA) at a 95% confidence level using Statgraphics Centurion XIX (version 19-X64). Differences between the formulations were determined by the Fisher test, using the least significant difference of 5% (α = 0.05).
3. Results and Discussion
3.1. Composition and Whiteness of the Cellulose Fibres
Table 1 presents the composition of the fibres, in terms of structural components and ashes; the effect of the different bleaching treatments on the whiteness index of the cellulose fibres and their composition is also shown, as are the obtained yields in the process (fibre mass with respect to the initial biomass solids). The obtained values were similar to those reported for these fibres in a previous study [
14].
Fibres beached with sodium chlorite had a higher whiteness index (WI) than those bleached with hydrogen peroxide, coherently with the greater cellulose purification and higher removal of coloured compounds such as lignin. However, bleaching yields were very similar in both cases, suggesting that chlorite treatment was more effective in removing coloured lignin while peroxide removed a higher proportion of colourless compounds. Concerning the ash content of the fibres, a lower content was observed for the chlorite treatment (≈0.5%), obtaining values similar to those of Tarchoun et al. [
29] and Camarena-Bononad et al. [
14].
3.2. Optical Properties
Figure 2 shows the internal transmittance (T
i) spectra of the different films and their visual appearance, which were affected by the colour of the incorporated fibres, the type of polymer, and the assembly of components (cellulosic films, chitosan–cellulose composites, or cellulose–PLA laminates). The cellulosic films (CC and CP) were very opaque with very low T
i values, CC films being the ones that showed the lowest transmittance values and, therefore, the highest opacity. This can be attributed to the greater cohesion of the fibres bleached with sodium chlorite, or deduced from their slightly lower thickness with the same solid content. The higher selective absorption at a low wavelength in CP film can be attributed to the higher presence of coloured compounds of the lignin fraction that absorb in this wavelength range.
PLA lamination greatly increased the transparency of the films, increasing the Ti values, which were markedly lower than those of PLA control film. In PLA laminates, the polymer partially fills the pores of the cellulose film during the thermocompression assembly, which leads to an increase in the internal transmittance of the laminated films as compared to pure cellulose films. The selective absorption at low wavelength was also observed in PLA-CP laminates, coherently with the lower WI of the fibres.
Chitosan-based films were more transparent than PLA laminates and cellulosic films. The incorporation of the cellulose fibres in the chitosan films reduced their transparency, although these films were still more transparent than the PLA laminates. The dispersed fibres in the chitosan matrix provoked the expected light scattering effect, increasing the opacity of the film and decreasing the T
i values with respect to the neat chitosan film, as observed in previous studies [
30]. The different WI of the fibres was also reflected in the T
i spectra, which shows a higher absorbance (lower T
i) at low wavelengths, especially in Ch-CP films where fibres contained more coloured compounds.
The colour coordinates of the films resulted from the optical effects commented on above, the light scattering effects provoked by dispersed fibres, and selective light absorption of the fibre-coloured compounds.
Table 2 shows the values of lightness (
L*), chroma (
Cab*), and hue (
hab*), as well as the whiteness index (WI) and the total colour difference (∆
E*) of the cellulose-containing films with respect to the corresponding control film.
For cellulosic films, CC films were lighter, more yellowish, and less saturated in colour than the CP films, coherently with the higher WI index and cellulose content of the CC fibres. In the composites/laminates, the colour was significantly influenced by cellulose arrangement in the assembly. Fibres embedded in the chitosan matrix led to lighter and more saturated colour, coherently with the light scattering effect of the dispersed cellulose. The greater ratio of coloured compounds in CP fibres resulted in lower L* values and higher Cab* values than those obtained for Ch-CC films. As concerns the PLA laminates, fibres provoked similar effects to those observed in chitosan composites, but with higher lightness, associated with prevalent light reflexion due to the high opacity of the cellulose layer.
3.3. Mechanical and Barrier Properties
The tensile and barrier properties of the chitosan composites and PLA laminates are shown in
Table 3. These properties could not be determined in pure cellulose films (CC and CP) due to their low mechanical strength (in the detection limit of the equipment) and porous structure that allows for the free flow of gases. The fibres, either dispersed in the chitosan composites or laminated with the PLA films, decreased the elastic modulus (EM) and tensile strength (TS) with respect to the control films of both pure polymers, without relevant effects on their stretchability. The effect was more pronounced for peroxide-bleached fibres (CP films) than for chlorite-bleached fibres (CC films).
Previous studies in cellulose nanocomposites reported an increase in elastic modulus and tensile strength with a reduction in extensibility due to the generation of stress concentrations and increased stiffness, depending on the fibre aspect ratio [
31,
32]. Nevertheless, the increase in the tensile strength did not effectively occur in microfibres due to their lower aspect ratio [
22]. This reinforcing effect was not observed in the obtained chitosan composites with dispersed fibres, which can be attributed to the high fibre content (50% wt.), which reduces the cohesive forces of the polymer matrix, weakening the polymer network responsible for mechanical strength.
In PLA-laminated films, the fibres had limited interaction with the polymer matrix, although the cellulose film adhered to the PLA has very low strength, contributing to lowering the overall strength of the bilayer assembly. Nevertheless, the laminates presented very close EM and TS values to those of pure PLA film, mainly those obtained with CC, whose structure was more cohesive and less porous. This is attributable to the higher cellulose purity of chlorite-bleached fibres, which plays an important role in the mechanical properties of cellulosic films [
33]. Likewise, the higher concentration of non-cellulosic components in CP fibres, such as lignin, could lead to their migration towards the PLA matrix, affecting the interaction forces of PLA chains and, therefore, the matrix toughness. Likewise, the bound water of cellulose fibres could also migrate into the PLA film during the thermo-adhesion step, partially promoting the hydrolysis of the PLA chains, thus weakening the film resistance.
The composite and the laminate films were also mechanically characterised by their tearing resistance (tear index in
Table 3), which determines the perpendicular force to the plane of the paper required to tear a sheet of paper. This index reflected the influence of factors related to the cellulose fibres, including length, density, grammage, flexibility, orientation, cellulose–hemicellulose ratio, and degree of compaction and bonding of the microfibrils [
34,
35]. Pure PLA films exhibited greater tear index values than the chitosan films (7.5 vs. 1.8 Nm
2·kg
−1), while cellulose fibres highly reduced this index in both chitosan composites and, especially, in PLA laminates, as occurred with the tensile strength. For both, composites and laminates, those formulated with the CC fibres (with a cellulose–hemicellulose ratio of 32) showed higher tear index values than those obtained with the CP fibres with a cellulose–hemicellulose ratio of ~11. This agrees with previous studies that reported that the increase in the cellulose–hemicellulose ratio led to an increase in the tear index [
34,
36]. Other authors also observed higher resistance to tearing for paper sheets from cellulose nanofibrils from non-wood sources when these contained less lignin [
37].
The oxygen permeability (OP) and water vapour permeability (WVP) values of the different films are shown in
Table 3. The neat chitosan films showed high WVP, but low OP values, consistent with the hydrophilic nature of the polymer. These values were in the range determined by other authors [
38,
39]. As expected, the incorporation of the fibres significantly reduced the oxygen permeability (~1400–1800%) with no noticeable effect on WVP. This is attributable to the increased tortuosity factor for mass transport introduced by the fibres in the matrix, which reduces the transport rate when the permeant gas is poorly soluble in the dispersed phase. Water molecules have a high affinity for cellulose, and therefore, the dispersed fibres did not significantly slow down their transport through the matrix. Nevertheless, this effect was noticeable for oxygen molecules, with lower chemical affinity with the fibres. It is remarkable that the peroxide-bleached fibre had a higher oxygen barrier effect than the chlorite-bleached fibre, probably due to its higher lignin content, which could have an antioxidant and scavenger effect on oxygen molecules, contributing to limiting their transport [
40].
PLA films had a high water vapour barrier and limited oxygen barrier [
41]. The obtained values for the pure amorphous PLA films were in the range observed by other authors [
42,
43]. In PLA laminates, the adhesion of cellulosic films generally increased the WVP values, particularly for the CP fibres, while CP and CC fibres had an opposite effect on the oxygen barrier capacity. The CC layer promoted the OP decrease in the laminates whereas the CP layer reduced the oxygen barrier capacity. This could be related to the different structure of the PLA–cellulose interface. The chlorite-bleached cellulose layer (CC) had a more densely packed structure with lower porosity, which would affect the PLA penetration into the cellulose pores near the interface of the laminate during thermo-adhesion, giving rise to an interfacial layer richer in cellulose than the CP layer. Therefore, the interface structure could play a crucial role in the barrier capacity of this kind of laminate. The filling of the cellulose matrix pores with melted polymer could create an interfacial layer with increased tortuosity, potentially enhancing the barrier properties of the laminate, depending on the final structure formed. Further studies are required to validate this hypothesis.
3.4. Thermal Properties
The thermal stability of the different samples can be seen in
Figure 3, which includes the weight loss curves (TGA) and the derived curves (DTGA) for the composite and the laminated films, compared to the cellulose films (CC and CP) and the control films (PLA and Ch). As shown in
Figure 3, the cellulose films and chitosan composites presented a first weight loss step up to around 100 °C, associated with the loss of bound water (~4%), as described by other authors [
44]. In contrast, PLA films and PLA laminates did not show this water loss step (
Figure 3b), due to the absence of bound water. In all cellulose-containing films, the weight loss associated with the thermo-degradation of lignocellulosic components of the fibres—cellulose (275–450 °C), the residual hemicellulose (150–250 °C), and lignin (370–650 °C)—was also observed, at the typical temperatures [
45]. As shown in the DGTA curves, CP film had a broader and less intense peak at 289 °C, while the CC film exhibited a narrower and more pronounced peak at around 325 °C, coherently with the higher purity of cellulose in CC film. In PLA control films, the typical temperature peak (maximum degradation rate) around 350 °C was also observed, as previously reported [
42,
46,
47]. However, in the laminates, PLA degradation overlapped with the degradation peaks of cellulose and residual lignin present in the cellulose fibres. Only small effects derived from the interaction between components were observed for the laminates. In particular, the PLA-CP laminates exhibited slightly lower T
on and T
p values than those observed for PLA and PLA-CC films, which may be attributable to the migration of components between the layers during the thermo-adhesion step. The higher lignin concentration in the CP fibres may have led to greater migration of this component to the PLA layer, significantly affecting both the cohesion of the matrix and the hydrolysis of the PLA chains. In fact, the presence of phenolic acids in the lignin structure could provoke partial hydrolysis of the PLA chains, making it less thermally stable.
As shown in
Figure 3c, chitosan presented the maximum degradation rate peak at around 313 °C, with a second peak associated with the degradation of secondary products, as described by other authors [
48,
49]. This temperature peak was close to that of cellulose degradation, leading to a partial overlapping of both peaks in the composite films. Likewise, the second degradation step of the secondary products partially overlapped with the decomposition of residual lignin in the fibres. Oca-Vásquez et al. [
50] also reported non-noticeable changes in the thermal degradation profile of chitosan composite films with cellulose nanofibres from agave bagasse.
Therefore, the TGA curves show that the thermal stability of the different components in the materials was not significantly affected by the blending effect in composites or laminates. Nonetheless, the presence of non-cellulosic components, such as lignin, may have a slightly negative impact on the thermal stability of PLA in laminates.
3.5. Microstructural Properties of the Films
Figure 4 shows the micrographs of the cross-section of the PLA and chitosan (Ch) control films, the cellulose films (CC and CP), the chitosan–cellulose composites (Ch-CC and Ch-CP), and the PLA-laminated films (PLA-CC and PLA-CP). The selected magnifications highlight the most relevant structural elements in each case. The cross-section of the cellulose films (CC and CP) shows the aggregation of fibres in the film, generating high porous structures, which reveals weak cohesive forces between the fibres with low compactness of the structured films. During film drying, the water-soluble compounds of the fibres, such as hemicellulose, contribute to forming the film matrix with the embedded cellulose fibrils. This is especially noticeable in the CP films which exhibited a higher proportion of this continuous matrix, which was consistent with their lower cellulose purity.
The chitosan–cellulose composites showed cellulose particles of different sizes embedded in the continuous chitosan matrix, with good interfacial adhesion. Other authors reported good compatibility between chitosan and cellulosic fractions [
50,
51]. In general, fewer dispersed cellulose particles and larger aggregates were observed in the Ch-CP films than in the Ch-CC films. This suggests the better dispersion of the CC particles in the aqueous chitosan dispersion and films. This structural difference is coherent with a higher tortuosity factor for mass transport through the film, which could explain the greater reduction in water vapour permeability of the chlorite-bleached fibres in the chitosan composites, as well as the better mechanical performance of the Ch-CC composites.
HRFESEM images of PLA–cellulose laminates show the good thermo-adhesion of both layers, reflecting the typical homogeneous structure of the PLA layer [
42] and the aggregated fibres in the cellulose sheet. The thickness of the cellulose layer in laminates, as deduced from the micrographs, was reduced by about 50% with respect to the initial cellulose films during the thermocompression. This can be explained by the promotion of fibre aggregation by the thermal effect, in part due to the elimination of bound water. Likewise, the molten PLA could penetrate to a certain depth into the cellulose layer, masking the fibre structure in the micrographs, and giving rise to very good adhesion between layers (
Figure 5). Likewise, an irregular interface can be observed for the laminates, with variable thicknesses of the cellulose layer. The latter can be attributed to the lack of fluidity of the solid cellulose fibres during thermo-adhesion whereas amorphous PLA flows by thermal softening against the cellulose layer with heterogenous surface solid density.
4. Conclusions
Cellulose films obtained with fibres isolated from Posidonia oceanica waste, by subcritical water extraction and bleaching with hydrogen peroxide or sodium chlorite, exhibited different whiteness depending on the degree of delignification produced by the bleaching process. Treatment with sodium chlorite was more effective for fibre delignification and whitening. These cellulose films showed very low mechanical strength and porous structure, especially when hydrogen peroxide was used as the bleaching agent. Chitosan–cellulose composites showed lower elastic modulus, tensile strength, and tear index than pure chitosan films, this decrease being more significant in the composites with cellulose bleached with hydrogen peroxide. These fibres had a similar, but attenuated, effect when laminated with PLA layers. Cellulose fibres improved the oxygen barrier capacity of the chitosan composites, but in PLA laminates, this effect was only observed for fibres treated with sodium chlorite. In no case did cellulose improve the water vapour barrier capacity of the films compared to pure polymers. The thermal stability of the different film components was not notably altered by the blending effect, thus reflecting the absence of significant interactions between the fibres and the polymer. Further studies are needed to improve the functionality of cellulose fibres from PO waste for their incorporation as fillers or laminates in biodegradable food packaging materials. Nevertheless, PO waste can be harnessed as a renewable source of cellulose, with a high potential for developing packaging materials, mitigating the costs of beach collection as well as the polluting effects of accumulated waste.








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}