4.1. Pectins in Nasal Delivery: The Case of PecFent®
Pectins (MW~100,000 Da) are heterogeneous polysaccharides comprised of a backbone of galacturonic acid units linked by α-1,4 bonds with a neutral sugar such as galactose, xylose, rhamnose, or arabinose either in the backbone or as side chains [
61]. The pectin excipient is commonly utilized in the food industry and medicinal products as a gelling agent and is Generally Regarded as Safe (GRAS) by the FDA. In Europe, it is classified as a food additive (E440). In the medical field, pectin is traditionally employed in managing diarrhea, constipation, and in obesity treatment. Additionally, pectin is included in certain pharmaceutical products such as Orabase
® paste and Stomahesive
®, which are used for treating oromucosal lesions. There are two basic types of pectin, classified by the degree of esterification (DE) on its galacturonic residues and consequent mechanism of gelation: high methoxyl pectins (HM), which have a DE higher than 50%, and low methoxyl pectins (LM), where the DE is up to 50% [
62,
63]. High methoxyl pectin undergoes thermoreversible gelation at low pH and high solid contents, often facilitated by the addition of sucrose. Conversely, low methoxyl pectin undergoes an in situ gelling process where ionic interactions occur between functional groups of pectin chains and cations, such as calcium ions. These interactions, including hydrogen bonds with carboxylic groups, are pivotal in biological secretions, resulting in the formation of a structured three-dimensional network within the gel. Consequently, when the liquid solution of pectin encounters mucosal secretions, a gel forms, effectively entrapping the active compound within the pectin matrix [
37]. Owing to its properties to form hydrogels in the presence of divalent cations, naturally occurring in nasal fluids, this natural polysaccharide is increasingly used as excipient for nasal drug delivery systems.
The use of pectin is common in drug delivery systems, especially in orally administrated formulations, to provide gastro-retention and colon targeting. However, in situ gelling systems have emerged as a novel approach in the intranasal delivery of therapeutics, and significant advances have been made in the development of innovative formulations. Currently, there are two drug delivery systems based on LM Aloe pectin in development: GelSite
® and GelVac
®. GelSite
® is an in situ gelling liquid formulation evaluated as a depot injection, whereas GelVac
® is a powder formulation capable of transforming into a gel upon contact with body fluids at the administration site. GelVac
® has been explored for the nasal delivery of an inactivated H5N1 influenza vaccine. This approach is particularly intriguing for vaccine delivery as it can delay mucociliary clearance, thereby prolonging exposure of Norwalk virus-like-particle antigens to immune effector sites. Furthermore, the dry powder formulation provides greater microbial and chemical stability compared to current formulations. This characteristic facilitates mass production and vaccination efforts in both developed and developing countries [
64]. The in situ gelling of GelVac
® was assessed in animal models, and the data obtained demonstrated that this formulation induces robust systemic and mucosal immunity. As a result, GelVac
® has received approval for human testing by the FDA, and clinical studies were announced but their results have not been published [
24].
Currently, the sole liquid formulation of pectin engineered to gel upon application to mucosal surfaces is PecSys
®. This recent delivery system relies on low methoxyl pectin gel, specifically designed for application to mucosal surfaces like the nasal cavity. PecSys
® does not function as an absorption enhancer system; instead, it modulates the pharmacokinetic profile of drugs that are readily absorbed in the nasal cavity, such as lipophilic drugs [
61]. In their investigation of permeation modulation in HT29-MTX cell monolayers using a well-absorbed drug model, Hagesaether and colleagues examined the capacity of various pectins and other polymers to enhance drug permeation across cellular epithelia. Their study affirmed that low methoxyl pectin reduced the permeability of drugs across epithelial barriers [
60].
Probably the most important case of a pectin-based formulation for intranasal delivery is PecFent, a PecSys
®-based formulation, approved by FDA and marked as nasal spray for pain cancer management (Lazanda, Archimedes, Bedminster, NJ, USA). This product, based on an LM pectin co-formulated with the opioid analgesic fentanyl, had successfully completed Phase 3 clinical studies and was the first product using a mucoadhesive approach approved for marketing in Europe in 2009 and in the USA in 2011. Currently, PecFent is commercially available in two dosages: 100 µg or 400 µg for each actuation. It may be administered at singular doses of 100 µg or 400 µg (one actuation vaporized in one nostril) or double doses of 200 µg or 800 µg (one actuation vaporized per nostril) [
65]. After being administered, the active compound, fentanyl, diffuses from the pectin gel and is absorbed according to an absorption profile suitable for the treatment of breakthrough cancer pain (BTCP). BTCP, according to a study presented at ESMO 2018, has an estimated prevalence of 59% and is present in 39% of outpatient cases and 80% of hospitalized patients [
66]. The occurrence of these pain episodes is unpredictable in about 50% of cases and is characterized by a rapid onset of about a minute. Their maximum intensity is reached within three minutes, and their average length is 20–30 min. Painful accesses end after about 30–60 min. These episodes occur with a frequency of 1.5–6 times a day and most are of moderate or severe intensity. The treatment of these episodes justifies the adaptation of pharmacokinetics and pharmacodynamics of the analgesics to the characteristics of the symptom. To better perform their action, in fact, analgesics must have a rapid onset of therapeutic effect and a duration of action that coincides with the episode’s time course. The maximum concentration (C
max) of the analgesic should occur early (T
max), and its half-life must be sufficiently long to cover the duration of the painful event. However, it should also be relatively short to prevent the drug from accumulating during intervals between doses, thereby limiting the risk of harmful effects.
Many studies investigated the pharmacokinetic of opioids, demonstrating that fentanyl and its derivatives have the highest bioavailability rate and rapid onset of action after nasal administration. Fisher et al. led a Phase I, randomized, single dose, open-label, clinical pharmacokinetic study involving 18 opioid-naïve healthy adult volunteers aged between 18 and 50 years [
67]. In this study, clinical tolerance and relevant pharmacokinetic characteristics for BTCP treatment (C
max, T
max and systemic bioavailability) of different fentanyl citrate formulations were compared. In detail, three gel-polysaccharide formulations for nasal spray application were evaluated, including gels of fentanyl pectin (FPNS/PecFent), fentanyl chitosan (FChNS), and fentanyl chitosan-poloxamer 188 (FChPNS). The oral solid formulation of fentanyl citrate currently marketed (OTFC, Actiq, TEVA Pharma B.V., Utrecht, The Netherlands) was employed for comparison purposes. Each of the three gels contained 10 µg of fentanyl in 100 mL, while the tablets had a dosage of 200 µg. The gel nasal sprays provided superior bioavailability of the drug. The relative bioavailability compared to the oral transmucosal fentanyl were 132.4% for FPNS, 154.1% for FChNS, and 122.3% for FChPNS. Fentanyl nasal sprays also provided a significantly reduced half-life ranging from 10 to 20 min, against 90 min of OTFC. Nasal formulations have shown not only a higher bioavailability than the compared oral formulation (
p < 0.05), reducing T
max significantly from 1.5 h for OTFC to 0.33 h for pectin formulation, but PecFent also demonstrates the most favorable tolerability profile (
Figure 3). In fact, PecFent can be perceived by most patients within the first five minutes following dosing, with noticeable effects often experienced within 10 min [
67].
In another study, researchers compared the pharmacokinetic profile and bioavailability of the PecFent 100, 200, 400, and 800 µg doses with 200 µg OTFC. Fentanyl PecFent showed a higher C
max (2.3-fold higher compared to OTFC) and a shorter T
max values with median ranging from 15 to 20 min post-dose, in a dose-independent way. Moreover, mean- relative bioavailability of FPNS ranged from 103 to 163%. PecFent achieves intranasal fentanyl release with an absorption profile suited for specific pain treatment, according to the authors, characterized by quick release (shortened T
max), controlled plasma concentrations corresponding to pain duration, and consistent dosing without fluctuations (reducing risk of swallowing compared to aqueous fentanyl without pectin) [
68].
A recent study investigated the development of pectin-based, ion-responsive in situ gelling formulations for nasal sprayable fluticasone propionate (FP) as a localized treatment for chronic rhinosinusitis. The approach integrated fluticasone propionate nanocrystals into an in situ gelling nanosuspension composed of pectin and sodium hyaluronate as key polymers. The nanocrystals (133.0 ± 0.8 nm) significantly enhanced solubility and diffusion, while pectin improved gel consistency and mucoadhesion. Formulation testing revealed robust gelation properties, with a rapid transition to a viscoelastic gel upon contact with nasal fluids—critical for mucosal retention. The combination of pectin and sodium hyaluronate enhanced mucoadhesion through interactions with mucus glycoproteins, while maintaining excellent biocompatibility and high cell viability (~96.7%). The formulation’s zero-shear viscosity (9.3–13.4 mPa·s) ensured optimal sprayability alongside prolonged mucosal retention. The droplet size distribution complied with EMA and FDA standards, while the narrow spray cone angle (21.3°) and optimized deposition profiles facilitated precise drug delivery to the middle turbinate. Deposition studies using a 3D nasal cavity model demonstrated that 51.8% of the administered dose localized to the middle meatus, a critical site for effectively treating chronic rhinosinusitis. The nanosuspension technology achieved an 11.8-fold increase in FP solubility and a rapid in vitro release (~85% within 8 h), enhancing therapeutic efficacy and highlighting the potential of pectin-based formulations for local treatments [
69].
Pectin is also explored as an excipient in other formulation approaches for nasal delivery, such as nasal powders. Indeed, in a 2021 study, pectin was used in combination with hypromellose (HPMC) to create microspheres by spray drying for the nasal delivery of dexamethasone sodium phosphate. The pectin/HPMC combination provided several benefits including a modulation of the swelling of the microspheres in contact with nasal fluid due to pectin crosslinking with calcium ions present in the nasal environment. The controlled swelling behavior also helped to prolong drug release from the microparticles, with a complete release of dexamethasone in 90 min after an initial rapid release (50% of drug released in 30 min). Pectin improved the mucoadhesive properties of the formulation, allowing better adhesion to and interaction with the porcine nasal mucosa used as the model (work of adhesion over 10 times greater than the pure drug). Finally, the microspheres improved the permeation of dexamethasone across a Calu-3 cells monolayer, used as a model of the nasal epithelium, by 1.7-fold. Overall, the combination of pectin and HPMC created an optimized polymer matrix that provided appropriate drug release profiles for nasal administration in view of the envisioned nose-to-brain delivery of the glucocorticoid to control neuroinflammation processes in patients with severe COVID-19 [
70].
4.2. Chitosan and Derivatives
Chitosan (Chit) is a natural cationic polysaccharide made up of randomly ordered units of glucosamine (2-amino-2-deoxy-β-D-glycopyranose, GlcN) and N-acetylglucosamine (2-acetamide-2-deoxy-β-D-glucopyranose, GlcNAc). The two monomers differ with respect to the C2-substituent in the sugar ring, which is either an amino or an acetamide group [
71]. Chitosan can be derived by the partial deacetylation of chitin, a material found in abundance in shells of crustaceans, but it is also found in some microorganisms like yeast and fungi [
36]. A wide range of chitosan polymers is available depending on the molecular weights (50 kDa–2000 kDa) and degrees of deacetylation (40–98%), all of which may affect physical-chemical properties of chitosan polymers, i.e., solubility, biocompatibility, mucoadhesive behavior and degradation, properties that influence the effectiveness of chitosan in pharmaceutical formulations.
At acid pH, chitosan is positively charged and its apparent pKa (6.1–7.3) is connected to the degree of deacetylation. The solubility and conformation of chitosan is also dependent on ionic strength, in fact, with increasing ionic strength, the solubility decreases and the folding conformation increases. At alkaline and neutral pH values, chitosan is insoluble but able to form salts with inorganic and organic acids (hydrochloric acid, lactic acid, acetic acid, and glutamic acid). In solution, chitosan salts present protonated amino groups and, overall, the polysaccharide results are positively charged [
72]. Exploiting its positive charge, chitosan can strongly bind to negatively charged materials such as the epithelial surface and mucus layers. In fact, among the various components forming the mucus, there are mucins which contain a significant proportion of sialic acid. At physiological pH, sialic acid is negatively charged and thus can interact with the positively charged amino functions of chitosan. This strong electrostatic interaction is responsible for the bioadhesive properties of this polysaccharide. Chitosan-based bioadhesive systems are able to slow down the MCC process and, so far, enhance the period of contact between the formulation and the nasal mucosa [
73].
In addition to its mucoadhesive effect, chitosan is believed to alter the permeability of the epithelial membrane by transiently opening intercellular tight junctions. This allows hydrophilic molecules to cross the epithelium via the paracellular route. Tight junctions are composed of complex transmembrane proteins (occludin, claudin, and junctional adhesion molecule or JAM), cytoplasmic proteins (ZO-1, ZO-2, ZO-3), and various associated proteins. NH
2 and -COOH termini of occludin are in the cytoplasm, with the COOH terminus anchored to the N-termini of scaffolding proteins ZO-1, ZO-2, and ZO-3, which, in turn, are linked to the F-actin of the cytoskeleton [
74]. The formation of the junction itself and probably also its function is apparently regulated by the phosphorylation of the tight junctional proteins.
In a study by Smith and colleagues on cultured CaCo2 intestinal epithelial cells, chitosan was shown to affect tight junctions. The disruption and opening of tight junctions led to increased permeability, causing a dose-dependent reduction in trans-epithelial electrical resistance (TEER) of up to 83% and increased the permeability of model drug compounds by up to 4.5%. The increase in paracellular permeability was attributed to chitosan’s action as a Protein Kinase C (PKC) inhibitor, which transiently opens tight junctions [
75]. Moreover, a correlation between the mucoadhesive effect and the increased paracellular transport of drugs was reported when using chitosan’s with higher MW and a lower degree of acetylation, resulting in higher positive charge [
53].
However, Bruinsmann and co-workers, studying the effect of chitosan molecular weight on the pharmaceutical properties of polymeric nanocapsules produced using poly(ε-caprolactone), capric/caprylic triglyceride, and sorbitan monostearate, and coated with chitosan, found that high molecular weight chitosan coated nanocapsules promoted lower permeation of the model drug simvastatin across both in vitro and ex vivo models of the nasal mucosa in comparison to low molecular weight chitosan coating. Nevertheless, both nanoformulations enhanced the simvastatin permeation compared to the free drug (
Figure 4). The results were explained considering both the lipophilic nature of the drug, less likely to exploit paracellular transport across the mucosal barrier and the slower simvastatin release observed using high molecular weight chitosan for the nanocapsule coating [
76].
The same group in a follow-up in vivo study demonstrated that simvastatin-loaded lipid core nanocapsules significantly enhanced the amount of drug in rat brain tissue after intranasal administration compared with free simvastatin and that the drug induced a significant decrease in tumor growth and malignancy in glioma-bearing rats without relevant systemic toxicity [
77].
The ability of chitosan to enhance nasal drug absorption was also assessed with calcitonin. Salmon calcitonin was formulated with chitosan, or its glutamate salt, and the resulting formulations were compared with hydroxypropyl- and dimethyl-β-cyclodextrins, which are known permeation enhancers, as will be discussed below. In particular, after nasal administration to rats, the 1% chitosan formulation at pH 4 exhibited a comparable enhancing activity with respect to 5% dimethyl-β-cyclodextrins with a less irritating effect [
78].
To enhance chitosan’s properties, various derivatives have been developed, notable examples include trimethylated, PEGylated, carboxylated, and thiolated chitosans. N-trimethyl chitosan chloride (TMC), a partially quaternized derivative, was synthesized to improve chitosan’s solubility at neutral and alkaline pH levels. This is particularly important in conditions like rhinitis, where the nasal fluid pH shifts from slightly acidic to neutral, rendering chitosan insoluble and limiting its use. Unlike chitosan, TMC remains highly soluble in water across a wide pH and concentration range [
53]. PEGylated chitosans have also shown an increased solubility at basic pH values compared to the unmodified polymer. Moreover, Casettari et al. demonstrated that mPEG-g-chitosan also had a reduced cytotoxicity [
79]. Thiolated chitosan demonstrated superior mucoadhesive and permeation-enhancing properties compared to unmodified chitosan. The thiol groups enable thiolated chitosan to form disulfide bonds with mucus glycoproteins, as well as inter- and intramolecular disulfide bonds, enhancing its mucoadhesion. Additionally, the thiol groups can inhibit protein tyrosine phosphatase, an enzyme involved in regulating tight junctions, thereby significantly improving chitosan’s permeation-enhancing effects [
80].
Chitosan is generally considered non-toxic, well-tolerated by humans, and biologically compatible. It is approved as a dietary food ingredient in Japan, Italy, and Finland, and is recognized as a Generally Recognized as Safe (GRAS) material. Numerous preclinical and clinical studies, including those by Archimedes and other groups, have investigated intranasal administration of chitosan. Data showed that chitosan was generally well tolerated, with no adverse effects or mild symptoms such as erythema, itchy nose, rhinorrhea, and sore throat. The non-irritating nature of chitosan and its low local and systemic toxicity are well documented in the literature. This is supported by a filled DMF (Type IV) with FDA and EMA approval, by a monograph in the European Pharmacopeia 6.0/EP 1774 (2008) for chitosan chloride, and, more recently, mention in the USP 34/National Formulary 29 (2011) [
71].
Due to its low toxicity, high tolerance, mucoadhesive properties, biocompatibility, ease of administration, low cost, and ability to stimulate a broad antibody response, chitosan has been widely explored as an adjuvant for nasal mucosa vaccines [
73]. Indeed, chitosan’s ability to slow mucociliary transport enhances antigen contact and uptake by nasal lymphoid tissue. Also, the transient increase in paracellular transport can boost the immune response. Animal studies on influenza, pertussis, and diphtheria vaccines demonstrated that nasal administration of chitosan-antigen vaccines induced serum IgG responses comparable to parenteral administration, while significantly increasing secretory IgA levels [
72]. The adjuvant activity of chitosan in the nose is the result of a combination of molecular weight, degree of deacetylation, particle size and solubility, as demonstrated by Scherließ et al., by comparing three commercialized chitosans with different molecular weight and degree of deacetylation [
81].
Parra and colleagues developed a norovirus virus-like particle (VLP) vaccine, formulated as a spray-dried powder composed of chitosan (ChiSys
®, Archimedes Development Ltd., Bedminster, NJ, USA) and norovirus VLP antigen with monophosphoryl lipid (MPL) as an immune enhancer. The norovirus vaccine originated distinct immune responses in rabbits after intranasal vaccination [
82].
Intranasal vaccination with p36/LACK-DNA combined with chitosan microparticles (LACK-DNA/CMC) provided enhanced and long-lasting immunity against visceral leishmaniasis. Mice vaccinated with LACK-DNA/CMC showed significant reductions in liver and spleen parasite burdens at 1 week and 3 months compared to control groups, with chitosan microparticles outperforming LACK-DNA alone at later challenges (6 months after vaccination). Splenocytes from these mice exhibited robust lymphoproliferative responses and increased IFN-γ production, crucial for protective immunity, while controlling IL-10 and TNF-α levels associated with disease progression. The nasal administered CMC vaccine induced no systemic toxicity, demonstrated by stable ALT, AST, and creatinine levels. Association of chitosan with LACK DNA on previous immunized mice caused a significant swelling (up to 6 months) of the hypersensitivity responses (DTH) after skin challenge with antigen. The successful use of chitosan-DNA association given intranasally enhanced antigen uptake by antigen-presenting cells (APCs), prolonged antigen release, and improved immune response via mucosal epithelial permeation [
36].
An in vivo immunogenicity study on 6 to 8-week-old female BALB mice evaluated chitosan for controlled nasal delivery of the Hepatitis B virus surface antigen (HBsAg). HBsAg was successfully formulated in N,N,N-trimethyl chitosan nanoparticles (N-TMC NPs), leveraging the mucoadhesive properties of N-TMC to enhance adherence and prolong residence time on the nasal mucosa. The study compared nasal delivery of HBsAg-loaded N-TMC NPs with free antigen and a co-administered N-TMC solution with soluble antigen. Forty-three days after a single nasal administration, the IgG immune response with N-TMC NPs (10.46 ± 0.47 mIU/mL) was significantly stronger than with free antigen (0.79 ± 0.21 mIU/mL) and N-TMC antigen solution (1.39 ± 0.37 mIU/mL) (
p < 0.05). This indicates that N-TMC NPs are potent immunostimulants for nasal vaccines, making them particularly attractive for vaccine delivery [
83]. Immunological results showed that the IgA titters induced by HBsAg-loaded N-TMC nanoparticles (NPs) were significantly higher and more stable than those from alum-adsorbed HBsAg standards. After three immunizations, only the N-TMC NPs induced IgA titers in all mice (48.03 ± 5.24 mIU/mL) compared to the Alum-adsorbed HBsAg standard (8.02 ± 1.26 mIU/mL). Results may suggest that N-TMC nanoparticles could also be promising for preventing or treating allergic rhinitis, asthma, and chronic obstructive pulmonary disease.
Recently, Altay Benetti and co-workers proposed a novel chitosan-lipid formulation loaded with mRNA for the immunization against SARS-CoV-2. The formulations developed preserved mRNA integrity after lyophilization and resulted in a satisfactory expression of the Spike protein receptor-binding domain in various human cell lines. Chitosan inclusion in the formulation played a pivotal role not only providing mucoadhesion but triggering specific local immune response in animal studies. However, the formulation tested did not induce systemic antibody response [
84].
The capacity of chitosan to act as an adjuvant was also demonstrated in clinical trials in which the polysaccharide was combined with mutant diphtheria toxin CMR
197 to study the induction of protective levels of antitoxin antibodies. A powder formulation, in which chitosan glutamate (7 mg) was added to 50 µg of CMR
197, was used as treatment via two intranasal administrations (days 0 and 28) in 20 healthy volunteers and then compared to intranasal and intramuscular (IM) administrations of chitosan-free formulations. After the second nasal immunization, systemic protective immunity against
Corynebacterium diphtheriae toxin in terms of neutralizing antibody response was significantly higher (peak 20 IU/mL) for the chitosan-containing formulation than control groups (below 10 IU/mL). Interestingly, local antitoxin sIgA in nasal-lavage fluid was increased significantly for the chitosan group (>10-fold) compared to the intranasal delivery of the formulation without chitosan, but this increase was detected only in the nostril that received powder formulation [
85].
Additionally, chitosan is an attractive excipient for formulations intended to deliver drugs from the nasal cavity to the CNS. Chitosan can provide bioadhesion and absorption enhancement, which is crucial for brain-targeting drugs that require increased residence time and membrane penetration in specific nasal regions [
86]. Illum and collaborators have studied chitosan for delivering various pharmacological agents including low-molecular-weight polar drugs, such as morphine and other compounds, for migraine treatment. Products for treating migraine and cancer pain have reached Phase II clinical trials [
87,
88]. A study compared different chitosan-based formulations, either in solution or lyophilized, to a solution of morphine after nasal application in a sheep model. Bioavailability was determined by comparing C
max values to those from an intravenous morphine infusion (C
max = 2593 nM). Both chitosan formulations showed increased nasal absorption compared to the morphine hydrochloride solution, which had just 10% bioavailability (C
max = 151 nM). The morphine-chitosan solution exhibited rapid absorption, with a T
max of around 15 min and a C
max of 657 nM, resulting in a bioavailability of 23%. The best results were observed with the freeze-dried morphine-chitosan microparticles which achieved a rapid plasma peak at 8 min and a bioavailability of nearly 60% [
87].
The same morphine-chitosan formulations were also tested in a three-way crossover designee in human volunteers by comparing the chitosan-morphine solution, morphine-chitosan freeze-dried microparticles powder intranasally administered, and morphine sulfate administered by intravenous infusion, used as control. The nasal solution and powder formulations employing chitosan resulted in substantially identical morphine plasma profiles with rapid and high peak plasma concentrations, which were both similar to the profile obtained for IV. T
max for the nasal powder formulation was slightly longer (21 min) compared to the solution formulation (15 min). Moreover, the results obtained in humans showed a bioavailability of nearly 60%, similar to the sheep model, with low levels of morphine metabolites [
87]. These results support the hypothesis of a direct absorption of morphine from the nose into the systemic circulation, bypassing the first-pass metabolism, the major factor responsible for metabolite production and prevention of morphine brain targeting.
Successively, the morphine–chitosan solution formulation was tested in 14 cancer patients experiencing BCTP, demonstrating a rapid onset of pain relief within 5 min, progressively improving and peaking at 45 min [
89]. The chitosan–morphine solution was generally well tolerated by patients who could self-administrate morphine noninvasively, obtaining a plasma profile comparable to that of intravenous administration. A morphine–chitosan product for nasal administration, Rylomine
®, is under development as an alternative to parenteral morphine. Rylomine
® has reached Phase III clinical trials and studies have demonstrated its effectiveness in the treatment of breakthrough pain. Rylomine
® showed statistically significant pain relief when compared with the placebo in addition to an onset time comparable to parenteral morphine in timing and degree of analgesia [
90].
Illum and collaborators have also exploited chitosan for delivering peptide and protein drugs, including parathyroid hormone, calcitonin, and insulin. They were pioneers in demonstrating that chitosan significantly enhances the absorption of both small molecular weight polar drugs and larger peptides and proteins across the nasal membrane [
71]. Indeed, in recent years, numerous studies have evaluated chitosan as an absorption enhancer for nasal insulin delivery. Insulin, due to its hydrophilicity and large molecular size (insulin monomer diameter: 26.8 Å; nasal epithelial tight junction diameter: 3.9 to 8.4 Å), faces poor absorption challenges. Factors such as low nasal mucosa permeability, rapid clearance, and proteolytic enzymes further hinder nasal insulin absorption, resulting in bioavailability typically below 1%. Nevertheless, nasal insulin delivery offers potential advantages over parenteral administration, including patient self-medication due to easy access and the ability to achieve pharmacokinetic profiles resembling natural pulsatile insulin secretion in healthy individuals [
91]. Currently, chitosan and its derivatives represent the most interesting absorption enhancers for intranasal insulin delivery, and as such, they are extensively studied. In an in vitro study, Schipper et al. reported that the absorption enhancement of hydrophilic peptide hormones like insulin are connected to the structural properties of chitosan, such as molecular weight and degree of acetylation. They observed that a low degree of acetylation and/or a high molecular weight appears to be necessary for chitosan to increase the epithelial permeability [
92].
Illum et al. described the use of a chitosan glutamate solution at 0.5%
w/
v for the enhancement of insulin transport across sheep nasal mucosa. Systemic absorption of insulin was indirectly monitored by measuring arterial blood glucose levels. Results indicated a decrease in plasma glucose levels by 43% from the baseline value within 90 min after a single nasal administration. Correspondingly, insulin levels rapidly increased, with AUC values rising 7.2-fold compared to nasal instillation of insulin solution without chitosan. Subsequently, a phase 1 clinical trial demonstrated that the bioavailability of the chitosan–insulin solution was 10% higher compared to subcutaneous injection [
93].
In another study conducted by Aspdend and colleagues, the ability of various chitosans to enhance nasal absorption of peptides was evaluated using a rat model. For this study, five chitosans with different molecular weights and degrees of deacetylation have been selected. All chitosan formulations produced clinically relevant levels of insulin in the blood and considerable glucose reduction compared to plain insulin administered nasally to the rats (4 IU/kg), i.e., without absorption promoters. Moreover, chitosan was shown to have little or no adverse effect on the rat nasal membrane, as indicated by histological evaluation [
94]. Subsequently, three different formulations of chitosan (solution, nanoparticles, and powder) were compared for insulin nasal administration in sheep. Here, the insulin-chitosan nanoparticles showed a nearly 3-fold lower bioavailability compared to the solution formulation. The bioavailability of the powder formulation, instead, was found to be 5-fold higher. Increased powder bioavailability was attributed to a stronger resistance to nasal clearance. The powder formulation, in fact, showed a half-life of 115 min while for chitosan-insulin solution it was about 43 min [
95]. Also, an in vivo study performed by Mao et al. investigated nasal administration of insulin in rats. They found no added benefits in terms of improved absorption when using chitosan nanoparticles relative to a chitosan solution [
96]. Conversely, contrasting these results, a Spanish research group conducted by Maria Jose Alonso found that insulin administered in chitosan nanoparticles (300–400 nm) induced a more pronounced lowering of plasma glucose levels compared to a chitosan solution [
97]. The conflicting results among these studies could be attributed to variations in the type of chitosan used, its molecular weight, degree of acetylation, and other properties that influence the characteristics of chitosan-based formulations.
Krauland et al. instead evaluated the effect of thiolated chitosan, a chitosan derivative, for the nasal delivery of insulin. The authors compared microparticles of thiolated chitosan-insulin with microparticles of chitosan-insulin combined with glutathione. After nasal administration to rats as dry particles, the thiolated chitosan formulation showed nearly double the insulin bioavailability (6.9 ± 1.5%). However, this increase was not considered very distinctive. Nevertheless, the improved absorption was attributed to the higher mucoadhesive properties of the thiolated chitosan formulation [
98].
A research group from Leiden University, led by Junginger, was among the pioneers in investigating the use of another quaternized derivative of chitosan, N-trimethyl-chitosan (TMC), for nasal delivery of insulin. As previously underlined, TMC is freely soluble over a wider pH range and exhibits absorption-enhancing effects even in neutral and basic pH environments. One study evaluated the use of chitosan HCl TMC derivatives with high (61.2%) and low (12.3%) degrees of quaternization, at two concentrations (0.25% and 0.5%
w/
v), for enhancing nasal insulin delivery in rats. At acidic pH (4.4), all polymers increased insulin absorption, but only the 61.2% quaternized TMC enhanced absorption at physiological pH (7.4) [
99].
Currently, TMC is under investigation for developing spray-dried polymer-coated liposomes. Liposomes offer several advantages for the nasal delivery of proteins, such as protecting entrapped proteins, disrupting mucosal membranes, and increasing their residence time on the negatively charged mucosal surfaces. However, their susceptibility to mucociliary clearance limits liposomes application. To address this, researchers explored coating liposomes with mucoadhesive polymers like chitosan, TMC, or alginate. To benefit from liposome properties and the mucoadhesion of polymers, researchers used this approach to produce a spray-dried formulation for nasal antigen delivery. Using bovine serum albumin (BSA) as a model antigen, spray-dried BSA-loaded liposomes were obtained with high encapsulation efficiency, protein protection, mucoadhesive properties, and nasal mucosal penetration. They compared uncoated and polymer-coated liposome formulations with their corresponding spray-dried liposome powders. The findings revealed that TMC-coated liposome powders exhibited superior mucoadhesive strength, higher glass transition temperature (Tg), and encapsulation efficiency compared to those coated with alginate and chitosan. Furthermore, TMC-coated liposomes showed promising nasal mucosal tissue penetration when administered as dispersions, with improved mucoadhesion and drug loading observed in dry powder form. Overall, TMC-coated liposomes represent a promising approach for nasal vaccine delivery [
100].
Another interesting approach for the nasal administration of peptides involves using chitosan in thermosensitive gel forms. When applied as drops or a spray into the nose at 35–37 °C, these formulations form a gel that reduces nasal mucociliary clearance rate and enables sustained drug release [
71]. In vivo experiments on rats conducted by Wu et al. exploited an insulin-hydrogel system based on N-[(2-hydroxy-3-trimethylammonium) propyl] chitosan (HTCC) and PEG. After nasal administration, a decrease in the blood glucose concentration of about 50% of the initial levels was observed for approximately 4–5 h [
101]. Moreover, sustained nasal drug release based on thermosensitive gels composed of TMC, PEG and glycerophosphate, proposed by Nazar et al., allowed a modulated release of insulin up to 20 h. In vivo results confirmed that insulin-extended release conferred a greatly prolonged hyperglycemic effect, improving pharmacological efficiency [
102].
Based on the studies presented, bioadhesive microsphere delivery systems or water-insoluble powders incorporating chitosan or its derivatives appear to be the most promising for nasal absorption of insulin. Nanoparticles also show potential, although it remains unclear whether their benefits over chitosan solutions stem from drug encapsulation and protection against enzymatic activity and/or prolonged residence time in the nasal cavity [
95,
103].
At present, no product using chitosan derivatives for enhanced nasal drug absorption has reached the market. However, the ability to formulate products with neutral or basic pH values makes chitosan and its derivatives a highly promising approach, driving interest in developing novel products with these polysaccharides.
4.3. Hyaluronic Acid
Hyaluronic acid (HA) is a naturally occurring polysaccharide which consists of a linear chain of monomers of D-glucuronic acid and N-acetyl-glucosamine linked by alternating β-1,3 and-β-1,4 glycosidic bonds. HA is an abundant constituent of the extracellular matrix of connective tissue, synovial fluid, embryonic mesenchyme, vitreous humor, skin, and several other tissues and organs of the human body. Commercially produced HA is obtained either from animal sources or from bacteria through fermentation or direct isolation [
104]. Hyaluronan (the polyanion form of hyaluronic acid) is also present on the airway surfaces and is an important component of the normal airway secretions. It plays an important role in the physiological homeostasis of the respiratory apparatus, especially at the upper airway level. In the nasal mucosa, hyaluronan plays a role regulating vasomotor tone and gland secretion, contributing also to mucosal host defense. In fact, HA can stimulate the ciliary clearance of foreign bodies, while also retaining enzymes crucial for maintaining homeostasis on the apical surface [
105]. Apart from its mechanical functions in the human body, HA also plays a role in mediating physiological functions through interactions with binding proteins and cell surface receptors. These functions include morphogenesis, regeneration, wound healing, and tumor invasion [
106]. Hyaluronan may also be an important regulator of inflammatory response. The signaling function of HA is closely tied to its molecular weight. Indeed, during inflammation, high molecular weight HA is enzymatically broken down by reactive oxygen species (ROS) induced by allergens in the airway epithelium. Low molecular weight fragments resulting from this catabolism (<300 kDa) deliver signals about tissue damage and inflammation and mobilize immune cells. These fragments stimulate cell proliferation, initiate inflammatory pathways, and enhance ciliary beat frequency (CBF), thus helping in removing mucous deposits and irritants, including pathogens, through the RHAMM receptor (receptor for hyaluronic acid-mediated motility) or by binding to CD44, a receptor involved in inflammation signaling, found on various cell types including leukocytes, chondrocytes, fibroblasts, endothelial, and epithelial cells. On the other hand, high molecular weight HA acts as proliferation inhibitor, suppresses immune responses, preventing excessive exacerbations of inflammation, and does not affect CBF [
107].
Hyaluronan, which is a highly hygroscopic macromolecule, has been reported to have a much greater capacity to bind water compared to other polysaccharides like alginate, carrageenan, and guar gum. In solution, HA can form a scaffold, and it is often found in association with several protein cores, such as aggrecans, which are composed of very large proteoglycan aggregates. The most relevant property of proteoglycans is their ability to bind water molecules, resulting in hydration to such an extent that a gel-like system is formed [
105]. The networks of proteoglycan-HA aggregates shift the Newtonian region to lower shear rates and the forming gels have an increased dynamic viscoelasticity compared to HA-HA networks [
104]. Viscoelastic properties of HA are also influenced by its molecular weight and pH of the aqueous solution. Indeed, it has been observed that the higher the molecular weight and concentration of HA in the aqueous solution, the greater the viscoelasticity exhibited by the solution [
108]. Regarding pH conditions, hyaluronan has a pKa value around 3.0. Changes in pH influence the ionization of HA chains, altering intermolecular interactions between HA molecules and thereby modifying the rheological properties of the compound. Gibbs et al., in fact, compared viscoelastic properties of HA solution at different pH and found a higher viscoelasticity at pH 2.5 compared to pH values of 1.5 or 7.0. Moreover, the charges on HA chains are sensitive to specific ionic conditions. For instance, the addition of different ions (Ca
2+, Mn
2+, Na
+, K
+, and Mg
2+) alters the viscoelasticity of HA solutions due to the distinct effects that these ions have on the flexibility of HA chains [
109].
Rheological properties are related to mucoadhesive properties of HA, as demonstrated by Saettone et al. and Durrani et al.: the degree of mucoadhesion exhibited by HA was both pH and molecular weight dependent and increasing the molecular weight of HA from 134,000 to 4 million Da or decreasing pH from neutral to acidic was found to promote adhesion [
110,
111].
The viscoelastic nature of hyaluronan, along with its excellent mucoadhesive capacity, high biocompatibility, low immunogenicity, and overall high level of safety, has led to its use in various cosmetic, medical, and pharmaceutical applications including the treatment arthritis [
112]. Formulations of sodium hyaluronate (SH) have also been developed for topical administration as coadjutant treatment in clinical cases of acute and chronic pathologies in the upper aerodigestive tract (UADT).
Hyaluronic acid has also been shown to have beneficial effects in experimental models of chronic respiratory diseases. Indeed, thanks to its water-retaining properties, HA can humidify and protect the respiratory airways against injury, providing anti-inflammatory properties. Sodium hyaluronate has been suggested for use in patients suffering from Empty Nose Syndrome (ENS) along with atrophic rhinitis [
113,
114]. This clinical condition is characterized by excessive widening of the nasal meatus and symptoms include chronic dryness of the nose, dyspnea sensation, headache, and depressive states. It has been demonstrated that hyaluronate, unlike other materials, exhibits superior water absorption, providing an additional advantage by enhancing the moisturizing of the mucous membrane. Moreover, in a recent study performed by Gavina and collaborators, nebulized HA has been shown to be effective in controlling inflammation in vivo in mice cystic fibrosis (CF) airways and in vitro in human airway epithelial cells, thus highlighting its potential as anti-inflammatory in CF therapy [
112]. In fact, SH is used in conjunction with tobramycin for patients with CF, in addition to treating recurrent upper respiratory tract infections in the pediatric population. Di Cicco et al. reported the effectiveness of SH combined with tobramycin for treating bacterial rhinosinusitis in CF patients. They tested the tolerability and efficacy of a nasal spray formulation containing 0.2% sodium hyaluronate and 3% tobramycin compared to a control formulation containing 0.2% sodium hyaluronate alone [
115]. This randomized double-blind study found that the nasal spray formulation containing tobramycin and HA was well-tolerated and more effective than the one with HA alone. The combined spray not only reduced symptoms but also restored the health of the nasal mucosa. The authors suggested that the symptomatic relief is likely due to HA’s ability to form a softening and protective film over the nasal mucosa, which acts as a barrier and prevents dehydration. HA also facilitates the regeneration and hydration of the mucous layer, improving its viscoelasticity. Additionally, it helps remove mucous deposits and irritants, including pathogens, by binding to RHAMM and stimulating ciliary beating. The enhanced therapeutic benefit from combining tobramycin with HA may be due to tobramycin’s ability to reduce bacterial load, thus slowing the enzymatic degradation of HA caused by bacterial hyaluronidase. This prolongs HA’s activity and enhances its water-retaining capacity. However, it is important to highlight that the study’s limitation is that it is not placebo-controlled, so the specific effects of the tobramycin-HA combination cannot be fully assessed since HA was administered to both groups.
Sodium hyaluronate has been widely investigated as a drug delivery agent for ophthalmic, pulmonary, parenteral, nasal, and dermal routes [
116]. Sodium hyaluronate has been exploited as a component of vehicles for the nasal delivery of small molecular drugs and peptides to increase their bioavailability as a result of the polymer bioadhesion and penetration enhancement [
117]. Morimoto et al. demonstrated that the use of HA solution as a vehicle for intranasal administration in rats increased absorption of vasopressin and a vasopressin analog. Following nasal administration, bioavailability of vasopressin and 1-deamino-8-D-arginine vasopressin were increased more than 2-fold and 1.6-fold, respectively, after formulation with HA. The authors correlated the bioavailability increasing effect with the molecular weight and concentration of the polymer. It emerged that high molecular weight HA fractions (>300 kDa) determined an increase in bioavailability, while low molecular weight HA (55 kDa) seems to possess no effect [
118].
Other researchers investigated the possibility to increase mucoadhesive properties of a formulation incorporating HA into microparticulate delivery systems such as mucoadhesive microspheres and nanoparticles. In an interesting study by the Ben Forbes group at King’s College, the authors compared the bioavailability of different mucoadhesive microspheres prepared using chitosan hydroglutamate (CH), hyaluronic acid, and a combination of both polymers for nasally delivering a hydrophilic model drug, gentamicin [
119,
120]. Gentamicin was also administered nasally to rabbits as a solution and powder (a physical mixture of gentamicin and lactose), intravenously (IV) and intramuscularly (IM). A poor bioavailability of gentamicin administered as a nasal solution (1.1%) and dry powder (2.1%) was observed compared with IV. Instead, the nasal administration of gentamycin in rabbits using HA-based microspheres significantly increased bioavailability up to 23.3% (relative to intravenous dosing) compared to unformulated drug, even if it was not as high as that using chitosan-based particles (31.4%). Interestingly, the bioavailability of gentamicin in the HA/CH formulation (42.9%) was found to be significantly greater than the HA and CH microparticulate formulations alone, even if chitosan-loading gentamicin nanoparticles presented the highest C
max and shortest T
max (
Figure 5).
However, the combination of HA and CH in the same system allowed a high and prolonged serum concentration of gentamicin, increasing bioavailability. This was likely due to a synergistic effect between the two polymers, namely from HA’s mucoadhesive properties, which extend the formulation’s residence time in the nasal cavity, and CH’s penetration-enhancing effect. This synergistic combination of two polymers for nasal delivery appears to be a promising approach for achieving higher bioavailability and prolonged plasma release profiles of drugs, making it an interesting strategy for the controlled delivery of various medications.
It is important to denote that the flexible usage of polysaccharides as a delivery platform for many drugs contributes to the great interest in the study and application of such materials. For example, drugs like fexofenadine hydrochloride (HCl), which normally is only available as an oral formulation, have been introduced into HA-based microspheres containing PEG 6000 and/or sodium taurocholate (NaTC) [
121]: the pharmacokinetic in vivo study showed an enhanced AUC and C
max after nasal administration of HA microspheres compared to the corresponding fexofenadine-HCl solution.
In addition, for its excellent mucoadhesive properties, hyaluronic acid has been investigated in particulate form in the development of subunit mucosal vaccine delivery. In fact, most of the vaccines under development today are subunit vaccines, based on highly purified and well-characterized antigens derived from the respective pathogens against which one wants to protect. However, the higher safety profile has a counterpart, a lower immunogenicity compared to inactivated or attenuated pathogens. To improve the immunogenicity of the antigens and determine adequate protective immune responses, those vaccines have been formulated with adjuvants, i.e., delivery systems and/or immune potentiators [
29]. Co-formulation of adjuvants and antigens in micro and nanoparticles was found to increase effectiveness in eliciting immune responses compared to the plain solution, probably due to the improved protection of the antigen against degradation. Moreover, particles have also been shown to be better taken up by the first immunological line, i.e., the antigen presenting cells (APCs), and they may prolong the residence time of the antigen at the site of action [
30,
122].
A study investigated poly(L-lysine)-
b-polylactide (PLys
+-b-PLLA) HA-coated micelles as a promising platform for nasal vaccination, focusing on their preparation, cellular uptake, and immunological responses. PLys
+-b-PLLA micelles were successfully synthesized and coated with hyaluronic acid to encapsulate OVA (antigen) and CpG-DNA (adjuvant). HA-micelles exhibited a size of ~157 nm with a negative zeta potential (−21 mV), confirming successful coating. Cellular uptake studies demonstrated a significantly enhanced delivery of OVA and CpG-DNA into bone marrow-derived dendritic cells (BMDCs) using HA-coated micelles, leading to significant cytokine expression (INF-g and IL-4) and MHC class II activation compared to control micelles. In vivo, the nasal administration of HA-micelles induced robust OVA-specific IgG and IgA antibody responses, surpassing the performance of dextran-coated micelles (positive control) and a solution-based delivery. The authors attributed the enhanced mucosal immune activation of HA-coated micelles to the specific uptake of HA by mucosal epithelial cells with HA receptors, highlighting its potential for effective nasal vaccine delivery systems [
123].
Following the strategy of combining the synergistic effects of HA with another polysaccharide, Verheul et al. prepared covalently stabilized nanoparticles composed of thiolated hyaluronic acid (HA-SH) and thiolated trimethyl chitosan (TMC-SH), two oppositely charged partially thiolated polymers, to enhance antigen immunogenicity in vaccine formulations. Ovalbumin (OVA) was used as a model antigen. Nanoparticles were produced in two different ways: using the polymers in their thiolated and non-thiolated forms. The intermolecular disulfide bonds of the thiolated polyelectrolyte complexes increased the stability of the TMC-S-S-HA nanoparticles compared to their non-thiolated counterparts (TMC/HA particles), which are held together only by electrostatic interactions. In vivo tests on mice showed that OVA-loaded TMC-S-S-HA particles induced superior immunogenicity compared to non-stabilized particles, indicated by higher IgG titers. Furthermore, results imply that the stabilized TMC-S-S-HA nanoparticles form a highly versatile and promising vaccine carrier system, offering options for further modifications. The thiol groups on the surface of TMC-S-S-HA particles are available for several chemical modifications, such as PEGylation [
124].
Polyelectrolyte complexes based on hyaluronic acid combination with chitosan have also been explored to produce novel mucoadhesive nasal inserts aiming to investigate their possible application for peptide and protein delivery. This technology exploits the capacity of chitosan to form a three-dimensional network when it is cross-linked or complexed with an oppositely charged polyelectrolyte, such as HA. The drug can be incorporated in this network and its release controlled. The physicochemical properties of polyelectrolyte complexes have been widely characterized, but just a few works present their application in the field of drug delivery.
The group of Barbara Luppi from the University of Bologna, for instance, evaluated the release behavior of two peptide drugs with different ionic natures and molecular weights: vancomycin, a glycopeptide with low MW, and insulin, a protein with high MW, from mucoadhesive nasal inserts based on chitosan/hyaluronate polyelectrolyte complexes [
125]. The researchers prepared polyelectrolyte complexes at various pH levels and molar ratios, selecting polymer concentration (5.0 mM) and pH range (2.0–5.0) based on the solubility, solution viscosity, and pKa values of chitosan and hyaluronic acid. These conditions allowed modulation of the swelling behavior and release of the drugs at the administration site. In vitro results showed that the presence of insulin in the nasal inserts increased water uptake, while vancomycin reduced it. At pH 7.4, an excess of free negative charges was observed in all polyelectrolyte complexes, further increased by the presence of insulin (pI 5.4), but not by vancomycin (pI 7.2). This excess of negative charges led to an increased water uptake in insulin-loaded inserts. However, this did not significantly influence drug release. Instead, the molecular weight of the drugs was the key factor affecting their availability [
78].
Mucoadhesive properties of sodium hyaluronate have also been exploited to produce nasal spray formulations containing micro- or nanosized particles. Meloxicam, a non-steroidal anti-inflammatory drug (NSAID), can be nasally administered for analgesic effect. Nasal formulations were prepared using a pre-dispersion of micronized or nanonized MEL [
126]. To prepare the intranasal formulations, a low concentration of HA was added to a pre-dispersion of the drug in the presence of polyvinyl alcohol (PVA) as a stabilizing agent. MEL particle reduction to a nano-sized range increased saturation solubility, dissolution rates, and surface adhesiveness. In addition, HA ensured a longer residence time and uniform distribution of nano-MEL spray across artificial membranes and nasal mucosa improving drug diffusion and increasing blood concentration of MEL. The HA-PVA nano-MEL dispersion system also enhanced drug-controlled release. PVA-coated particles reduced the degradation rate of sodium hyaluronate and prevented its rapid dissolution in biological fluids.
Similarly, hyaluronic acid (MW 1400 kDa) was selected as mucoadhesive agent for the development of a nanosuspension of the poorly soluble (BCS class II) anti-allergic drug loratadine [
127]. Loratadine nanocrystals (311 nm) combined with sodium hyaluronate enhanced bioadhesion properties, produced a faster dissolution rate, and ultimately an increased bioavailability after intranasal administration in comparison to oral delivery (5.54-fold increase).
Despite its wide application in several drug delivery systems, hyaluronate has not been explored much yet in terms of nose-to-brain drug delivery. Horvàt and colleagues worked in the development of a formulation based on sodium hyaluronate in combination with Cremophor RH 40. Cremophor is a non-ionic surfactant which acts as an absorption enhancer to increase hydrophilic compounds delivery to the brain via the olfactory route [
44]. To evaluate the combined effectiveness of a viscosity and bioadhesion-enhancing polymer with an absorption-enhancing surfactant, a fluorescein isothiocyanate-labeled 4 kDa dextran (FD-4) was incorporated into the formulation. Formulations were administered nasally to Wistar rats, and the nose-to-brain transport of FD-4 was determined through fluorescent spectrophotometry in brain areas. The highest concentrations of FD-4 were highlighted in the olfactory bulbs, frontal and parietal cortex regions. Significantly higher FD-4 level was also measured in hippocampus, cerebellum, midbrain, and pons.
These results are in accordance with data collected by Thorne et al., who detected a similar brain distribution of insulin-like growth factors after nasal administration using an analogous formulation. Also, a morphological examination of the olfactory system revealed that the vehicle containing sodium hyaluronate and Cremophor RH 40 did not induce tissue damage, epithelial or subepithelial toxicity, nor ciliotoxicity. This confirms that the combination of the mucoadhesive properties of hyaluronic derivatives with penetration enhancers could be exploited to deliver drugs molecules, peptides, or fragments to the nervous system exploiting the nose-to-brain route [
128].
In an interesting recent application, hyaluronic acid was conjugated to FG loop peptide (FGL), a neural cell adhesion molecule-mimetic peptide with remarkable properties as anti-depressant and neuroprotective agent in neurodegeneration and ischemic models [
129]. The conjugate was synthesized via the coupling reaction of an intermediate HA–aldehyde to the amine group of FGL and showed an increased protection against degradation induced by incubation with trypsin. Moreover, the hyaluronate-peptide conjugate demonstrated higher binding to human nasal RPMI2650 cells. In vivo bioimaging experiments in rats evidenced a more efficient brain delivery after intranasal administration for low molecular weight hyaluronate conjugates (10 kDa) in comparison to high molecular weight (100 kDa). Finally, the therapeutic effect of the low MW hyaluronate conjugate was evaluated in an animal model of hypoxic–ischemic encephalopathy, showing, after nasal administration (5 µg of FGL peptide in 10 µL), promising results: a significant reduction in the infarction area, of neuronal damage, of the expression of inflammatory factors (IL-1b, TNFa and IL-6), as well as better performance in neurobehavioral tests 28 days after the injury.
Taken together the presented works suggest that hyaluronate might be the key to successful development of efficient nose-to-brain delivery systems for the non-invasive treatment of brain diseases.
4.4. Cyclodextrins
The use of cyclodextrins for the nasal delivery of drugs has been recently reviewed by Rassu et al., to whom the reader is referred [
130].
Cyclodextrins (CDs) are cyclic ring-shaped oligosaccharides composed of at least six D-(+)-glucopyranose units linked into a macrocycle by α-1,4 glycosidic bonds. CDs possess a torus-like shape (toroid structure), with a hydrophilic outer surface, due to the presence of hydroxyl groups and a hydrophobic internal cavity, in which skeletal carbons with hydrogen atoms and oxygen bridges are located [
131]. In nature, three main CDs exist: α, β and γ, composed of six, seven, and eight D-glucopyranose residues, respectively, differing between the internal ring size and physicochemical properties. They contain 18 (α-CD), 21 (β-CD), or 24 (γ-CD) hydroxyl groups that can be chemically modified to improve CDs physicochemical properties. Natural cyclodextrins present a relatively low solubility, both in water and organic solvents, probably due to the relatively strong binding of the cyclodextrins molecules in the crystalline state [
132]. This limits their use in pharmaceutical formulations. For this reason, many types of CDs derivatives have been developed, e.g., hydrophilic (methylated, hydroxyalkylated, and branched), lipophilic (ethylated), or ionic (sulphated and phosphated). Among these molecules, methylated and hydroxyalkylated β-CDs have especially been used to overcome the lack of water solubility of the natural CDs. Controlling the degree of substitution of these derivatives is crucial for balancing their water solubility and complexing capability [
133].
The unique nature of cyclodextrins’ structure confers to these polysaccharides’ particular importance in pharmaceutical fields. In fact, CDs can trap inside their hydrophobic cavity many lipophilic molecules or their portions, thus creating inclusion complexes. The formation of inclusion compounds can modify the physical and chemical properties of the guest molecule, especially in terms of apparent water solubility, dissolution rate, bioavailability, and stability of drugs. Each cyclodextrin has its peculiar ability to form inclusion complexes with specific guest drugs, as this depends on a proper fit of the guest molecule into CDs hydrophobic cavity [
133,
134]. Not all molecules, in fact, can form stable complexes: for example, highly water-soluble substances generally cannot be included. The size and geometry of the guest molecule are important features as well [
135]. If the molecule has adequate properties, the bond between the guest molecule and the internal region of cyclodextrin (host), is non-covalent and regulated by a dynamic equilibrium process of association/dissociation where free guest molecules in aqueous solution are in equilibrium with molecules in the complex [
136].
The drug release from the CD complex is mainly governed by a simple drug-dissociation, after the dilution of the complex in fluids. However, topical applications, such as ocular, nasal, rectal, or dermal, offer minimal or no dilution chances: in this case the release of the drug from CD complex is explained by drug uptake by the tissue. If the drug has physicochemical properties that enable it to penetrate biological membranes, the tissue acts as a sink, causing dissociation of the drug complex. Only the free form of the drug, which is in equilibrium with the complexed form in solution, can penetrate lipophilic barriers such as mucosal epithelia or stratified cell layers [
131].
Several factors affecting the dissociation equilibrium of the complex can be adjusted to optimize the bioavailability of poorly water-soluble drugs from cyclodextrin complexes. For instance, maximum absorption enhancement is achieved by using just enough cyclodextrin to solubilize all the drug in solution. Adding more cyclodextrins reduces the free fraction of the drug, thereby decreasing its bioavailability. Moreover, pharmaceutical excipients contained in the formulation and endogenous substances existing at the absorption site can influence the speed of absorption. Indeed, endogenous and exogenous small lipophilic substances can compete with the drug for the cyclodextrin cavity. This displacement of the guest drug from the cavity is responsible for accelerating drug release. In conclusion, high dissolution rates and the relative stability of the complexes (Ki > Kc, where Ki is the stability constant of the complex of the competing agent with the cyclodextrin and Kc is the stability constant of the complex of the drug with the cyclodextrin), favor the free drug, making it readily available for absorption [
136,
137].
Cyclodextrins act as solubilizing agents and absorption promoters also by solubilizing specific lipids from biological membranes through a rapid and reversible formation of inclusion complexes, thereby increasing membrane permeability. To realize the potential of cyclodextrins in pharmaceutical formulations, their biological profiles, including in vivo fate and toxic effects, have been investigated in numerous papers, by analyzing, for example, changes in nasal morphology in vivo, effects on ciliary beat frequency in vitro, in vivo release of marker compounds, erythrocyte hemolysis test in vitro, and in vitro cytotoxicity. The acute histological effects of cyclodextrins on the nasal epithelium was investigated in rats by the group of Illum and collaborators using light microscopy [
102]. After a single nasal administration of 2% randomly methylated β-cyclodextrin (RM-β-CD) or 2% dimethyl-β-cyclodextrin (DM-β-CD), no significant variations were observed in the appearance of the apical cell membranes and the cilia. These results were similar to the data obtained after nasal administration of the physiological saline in which the absorption enhancers were dissolved [
87]. In a study performed in an in vivo rat model, after administration of 2% RM-β-CD and 2% DM-β-CD, larger amounts of relased protein and cholesterol were noticed compared to the control (physiological saline). However, the amounts released were significantly lower than those observed for absorption enhancers such as 1% sodium taurodihydrofusidate, 1% laureth-9 and 1% L-a-lysophosphatidylcholine currently used in nasal formulations. Moreover, differently from these substances, no intracellular release of the enzyme acid phosphatase, which is responsible for severe nasal membrane damage, was detected after nasal administration of 2% and 5% MB-β-CD [
87,
105]. The toxicity data of CDs have been finally resumed by EMA, including CDs in the short list of excipients that are compatible and tolerated by the nasal mucosa and the airways. In particular, less than 10% of HP-β-CD or RM-β-CD and less than 1.5% β-CD are regarded as safe (EMA, Background review for cyclodextrins used as excipients EMA/CHMP/333892/2013). The safety of these substances mainly derives from their low water solubility, preventing their systemic absorption and paving the way for their application in commercial formulations. Indeed, CDs have been used to improve nasal delivery of lipophilic drugs that normally are poorly soluble in water. However, CDs have been employed also for large hydrophilic drugs like peptides and proteins, which typically exhibit insufficient nasal absorption due to their size and hydrophilicity. Using several types of cyclodextrins as absorption enhancers, very efficient nasal drug absorption has been reported [
138].
In a paper by Prakapenka et al., different types of CDs (randomly methylated β-CD, 2HP-β-CD, β-CD, and γ-CD) were compared for their ability to deliver 17-β-estradiol to the brain via nasal route in a rat model of surgical menopause. The 2HP-β-CD significantly increased the uptake of the drug in dorsal hippocampus [
139]. Exploiting CDs properties, the group of Merkus and collaborators studied the nasal administration of the lipophilic steroid hormone estradiol in animals and humans. Nasal-administered estradiol complexed with dimethyl-β-cyclodextrin (DM-β-CD) to rats and rabbits showed high levels of bioavailability up to 94.6% and 67.2%, respectively. In addition, estradiol and DM-β-CD complexes were also administered nasally in oophorectomized women in a pilot study, achieving a rapid absorption of the compound (average T
max approximately within 24 min). Moreover, after a 6-month human trial, no side effects were detected [
140].
A nasal spray product containing 17-β-estradiol solubilized in DM-β-CD (Aerodiol
®, Les Laboratoires Servier, Suresnes, France) was found effective in the treatment of menopause symptoms. Several clinical studies compared the pharmacokinetics, toxicity, and efficacy of the nasal formulation with the current administered oral and transdermal hormone replacement therapies. Nasal administration of Aerodiol
® to menopausal women provided the same bioavailability (AUC > 1000 pg·h/mL of 2 mg and 300 μg doses, oral and IN administered, respectively) and efficacy (estrogenisation increased up to 85%) of orally and transdermally administered formulations. Aerodiol
® was shown to provide pulses estrogen therapy (T
max = 10–30 min, decreasing after 12 h post-treatment), with similar effectiveness at lower dosages (300 μg/day) compared to those observed after oral administration (2 mg/day). Moreover, the good tolerability and reduced side effects of Aerodiol
® offer a safe, compliant, and highly effective treatment to relieve menopausal symptoms. A similar nasal formulation of progesterone complexed with dimethyl-β-cyclodextrin has been tested in human volunteers. Results showed values of progesterone blood levels comparable to those obtained after intravenous administration, once again suggesting that cyclodextrins not only increase hormones solubility but also have an absorption enhancement effect on the nasal mucosa [
141,
142].
In another study, researchers explored the nasal administration of melatonin in combination with β-CD. Melatonin, a lipophilic hormone known for slow oral absorption, commonly presents low and variable bioavailability: in a trial involving eight volunteers, the formulation with β-CD showed remarkably rapid and efficient nasal absorption. Peak levels of melatonin were found to be 50 times higher than those achieved through oral administration [
143].
The combination of lipophilic-drug-cyclodextrins has been advantageously applied also to nasal administration of granisetron (GNR). Granisetron is a selective 5-HT3 receptor antagonists used as antiemetic to treat nausea and vomiting following chemotherapy. Since the dose of GRN is very low (1 or 2 mg) and rapid onset of action is required, the nasal administration of GRN represents an interesting alternative to oral and intravenous administration. However, GNR has a relatively low aqueous solubility, and an increase in its solubility is crucial for the absorption through the nasal membrane. So, Hyun-Jong Cho et al. studied the possibility to use microparticles (MPs) of GRN in combination with hydroxypropyl-β-cyclodextrin (HP-β-CD) and the mucoadhesive polymer sodium carboxymethylcellulose (CMC-Na). To predict the in vivo performance of the GNR-loaded microparticles, in vitro drug release was performed in PBS pH 6.4 for 120 min and compared with the release profile of GNR powder. Studies revealed that the presence of HP-β-CD determined a significantly higher drug release rate compared to the pure GNR (95% against 50%, respectively). Moreover, it was found that the presence of HP-β-CD combined with CMC-Na greatly enhanced the in vitro permeation of the MPs through a human nasal epithelium cells monolayer grown in air-liquid-interface: for the GNP powder 10% of permeation was observed within studied time, while freeze-dried microparticles increased GNP permeation up to 50%) [
144].
Luppi and co-workers evaluated the potential of cyclodextrins to improve nasal delivery of nanosystems loaded with tacrine, a potent centrally active reversible cholinesterase inhibitor licensed for the treatment of Alzheimer’s disease. Bovine serum albumin nanoparticles carrying β-CD and two hydrophilic derivatives (HP-β-CD and sulphobutylether-β-cyclodextrin, SBE-β-CD) were tested to establish if and how the presence of these cyclic oligosaccharides could modify drug release, mucoadhesion, and permeation through nasal mucosa. The presence of the native and derivatives β-CD determined a significant decrease in particle size from 220 nm to 135 nm. This reduction is likely due to the β-CDs masking the hydrophobic regions of albumin molecules, suppressing their interactions and enhancing protein unfolding. Consequently, during albumin coacervation in the presence of the various β-CDs, the extensive protein unfolding favored the formation of smaller nanoparticles. In addition, the presence of β-CD and HP-β-CD in the nanoparticle network did not affect drug loading. Conversely, the presence of SBE-β-CD caused an increase in the drug loading, almost duplicating the drug content (TnP %L = 14.7 ± 0.9 vs. TnP SBE-β-CD %L = 22.0 ± 0.05). Ex vivo permeation studies using sheep nasal mucosa revealed that the presence of different β-CDs enhanced drug permeation and nanoparticle mucoadhesion compared to albumin nanoparticles alone, particularly with SBE-β-CD. The improved absorption of tacrine from nanoparticles in the presence of β-CDs was attributed to the cyclodextrins’ ability to interact with the lipophilic components of biological membranes, altering their permeability. Additionally, HP-β-CD and SBE-β-CD, which have high solubility (>600 mg/mL and >500 mg/mL, respectively), significantly increased the nanoparticles’ ability to hydrate, improving nanoparticles mucoadhesion and drug permeation [
145].
Another molecule considered for the treatment of Alzheimer’s disease is curcumin: its complexation with HP-β-CDs was proven superior to its encapsulation in nanoparticles prepared with chitosan and PLGA for intranasal delivery. Both formulations improved the solubility and the stability of the active molecule with respect to the solution, but, in vitro, cellular uptake by SHSY-5Y and BV-2 cell lines was increased when HP-β-CD complex was used. Moreover, when curcumin-CDs complexes were administered intranasally at 2 mg/kg to C57BL/6 mice, the C
max in plasma was almost twice with respect to curcumin solution or in nanoparticles (9.71 ng/mL vs. 5.1 and 4.02 ng/mL, respectively) and, in the brain, they provided the higher AUC, supporting a prevalent delivery to the brain rather than systemic [
146].
SBE-β-CD was also used to help the dissolution in saline of allopregnanolone, an antiseizure drug, for intranasal administration, to overcome its poor bioavailability. A dose of 16 mg/kg was administered to NIH Swiss male mice resulting in high levels of drug in the olfactory bulb (C
max 24 times higher than the rest of the brain) and a very fast nose-to-brain delivery (<30 s) with fewer side effects with respect to injection [
147].
Classically, CDs are widely used as solubilizers of hydrophobic drugs; however, it has been shown that CDs can also offer interesting possibilities for regulating the delivery of hydrophilic drugs. Cyclodextrins, in fact, may serve as novel hydrophobic carriers to control the release of water-soluble drugs, such as peptide and protein drugs, in various routes of administration, including nasal delivery [
148]. Most peptides and proteins cannot achieve sufficient bioavailability on their own. Cyclodextrins can enhance their availability mainly by preventing their enzymatic degradation and by increasing membrane permeability.
The potential of cyclodextrins as nasal absorption enhancers for peptides and proteins has been demonstrated for luteinizing hormone-releasing hormone agonists, adrenocorticotropic hormone analog, calcitonin, granulocyte colony-stimulating factor, insulin-like growth factor-1, and glucagon.
In particular, glucagon is included in a nasal commercial product by Eli Lilly, Baqsimi
TM. The product is intended for the treatment of severe hyperglycemic reactions in emergency situations as an alternative to the parenteral formulation that requires reconstitution from a lyophilized form. The formulation contains 10%
w/
w glucagon, β-CD and dodecylphosphocoline. Safety was assessed by Reno et al. in rats at two doses (0.1 and 0.2 mg/day). In the clinical trial performed on the intranasal product (IN, 3 mg) by comparison with the intramuscular (IM, 1 mg), no significant differences were observed in terms of T
max, but the C
max increased by almost 60% (6130 pg/mL IN vs. 35,750 pg/mL IM), confirming the non-inferiority of the product [
149].
Merkus and co-workers explored cyclodextrins to increase nasal absorption of insulin from different pharmaceutical forms tested in two animal models. In the first study, the authors found that a solution of insulin and 5% of dimethyl-β-cyclodextrin (DM-β-CD) did not enhance insulin absorption after nasal instillation in rabbits [
150]. However, this finding contradicted previous observations where the same formulation significantly improved insulin absorption in rats [
151]. The intranasal bioavailability of insulin from DM-β-CD, in fact, was found to be 100% in rats, although any insulin was detected in rabbits or humans. Still, when administered as a powder form to rabbits and humans, insulin absorption increased up to 13% in rabbits and about 5% in diabetes mellitus patients. Despite these findings, researchers concluded that the nasal bioavailability of insulin remains too low, and its association with cyclodextrins is not currently a viable alternative to subcutaneous insulin injections.
In a paper by Nonaka et al., the pituitary adenylate cyclase-activating polypeptide (PACAP) was formulated with CDs for intranasal delivery to the brain. The authors evidenced that the presence of CDs, in particular β-CD, generally increased the uptake of the peptide in the brain. Moreover, different types of CDs produced variation in the regional distribution of PACAP, with α-CD increasing the accumulation in olfactory bulb, while HP-β-CD promoted uptake by thalamus [
152].
Cyclodextrins can also be used in association with other excipients, as in the case of HP-β-CD and chitosan aspartate, to help the polar drug buspirone HCl cross the blood–brain barrier and avoid the extensive first-pass effect after oral administration. The drug was formulated in a microemulsion and administered intranasally, showing that both excipients contributed to a 4.3 folds increase in C
max in the brain with respect to the intravenous administration [
153].
4.5. Alginate
Alginate (AL) polymers are a family of linear unbranched polysaccharides which contain varying amounts of β-D-mannuronic acid and α-L-guluronic acid residues linked by β-1,4-glycosidic bonds. Along the polysaccharidic chain, there are homopolymeric regions of β-D-mannuronic acid blocks and α-L-guluronic acid blocks interdispersed with regions of alternating structure. Therefore, the composition and sequence of the residues may vary widely. Alginate is a naturally occurring biopolymer extracted from different brown algae (kelp), where it exists as an insoluble Ca
2+ cross-linked gel. The organism and tissue from which the alginates are isolated influences significantly the composition, the extent of the chain sequence and the molecular weight, thus determining their functional and physical properties. Alginates with a content of α-L-guluronic acid greater than 70% are called “high G” and may form a gel characterized by high porosity, high mechanical strength and stability, but also rigidity and brittleness. Conversely, alginates with a low content of α-L-guluronic and low molecular weight form a more elastic gel and undergo a faster degradation releasing embedded molecules at much faster rate [
154].
For its safety and non-immunogenic properties, alginate has been successfully used in the food and beverage industry as colloidal stabilizer, thickening, and gelling agent. Moreover, due to its high biocompatibility and gel formation ability, it is also widely used as a polymer matrix for the entrapment and/or delivery of many biological agents [
155].
Alginates, with their carboxylic end groups, are classified as anionic water-soluble polysaccharides and possess, among other features, a bioadhesive property which could serve as a potential advantage in mucosal drug delivery. So, due to their ability to form hydrogen bonds with mucin-like glycoproteins through carboxyl–hydroxyl interactions, alginates have been widely investigated as potential delivery vehicles to enhance effectiveness and bioavailability of drugs applied to nasal mucosa [
156].
This polymer has been exploited to develop a solid insert designed to hydrate rapidly in the nasal cavity into a bioadhesive gel. This in situ gelling formulation presents several advantages. It combines the advantages of solid single unit dosage form, such as high stability and dosing accuracy, with those of gel preparations, which avoid foreign body sensation and allow for increased residence time, thereby extending drug release. Moreover, due to the dissolution of the gel and mucociliary clearance towards the nasopharynx, there is no need for mechanical removal of the insert after drug release. Nasal inserts are typically prepared by freeze-drying aqueous solutions containing polymeric carriers and drugs. This production method ensures the formation of highly porous polymeric sponges that incorporate the drug [
157].
Based on the properties of this innovative nasal dosage form, Bertram et al., in particular, evaluated the bioadhesion potential, wetting time, water uptake, drug release, mechanical properties and the physical state of different polymers, including alginate, and the drug within the insert. Different water-soluble polymers were analyzed after freeze-drying from a 2%
w/
w aqueous solution, both visually and by X-ray diffraction, to characterize the appearance of the product and the physical state of the polymer. It emerged that all the amorphous polymers tested, including Na-alginate, remained amorphous after freeze-drying, and formed the desired sponge-like structure suitable for manufacturing nasal inserts. In addition, the inserts must be hard enough to be easily removed from their packaging and to be placed intact into the nasal cavity, but at the same time they have to be elastic enough to recover their sponge-like structure after compression. It was found that inserts with a larger, more layered structure as Na-alginate had a lower elasticity compared with inserts with a more pronounced network structure, as PVP 90. Also, when evaluating the potential of bioadhesion, sodium alginate inserts presented a low bioadhesion potential, justified by their rather low molecular weight and therefore low solution viscosity. Moreover, in vitro test simulating nasal conditions showed that inserts of low viscosity polymers, like sodium alginate, hydrated more rapidly but dissolved just as quickly. In fact, the complete disappearance of the insert occurred after 4 h, against the 8 h or more for polymers with high molecular weight (carrageenan, chitosan, Carbopol, xanthan gum). So, due to the rapid dissolution and almost instantaneous displacement of the gel matrix, inserts prepared from Na-alginate released the drug (oxymetazoline HCl) more rapidly [
158].
In another study, Farid et al. formulated in situ gelling mucoadhesive inserts with different polymers, for systemic drug delivery of the model drug salbutamol sulfate through the nasal route. Salbutamol sulfate (SS) was chosen as a drug model which undergoes strong first hepatic metabolism and consequently presents poor bioavailability, despite it is well-absorbed when orally administered. Each formulation was prepared with 1.4%
w/
w SS in inserts of 10 mm diameter and a different mucoadhesive polymer, namely high molecular weight alginate (AL), medium molecular weight chitosan (Chi), sodium Na-CMC or HPMC. From this study emerged the observation that water and moisture uptake were superior from inserts made of Na-CMC and alginate than HPMC or Chi as well as for in vitro mucoadhesion and drug release. The authors attributed these results to the ionic nature of AL which provides high tendency towards water uptake. Also in this study, mucoadhesion properties were tested by displacement vertical method. After 9 h of observation, no insert, except for HPMC, showed downwards movement. According to these data, despite the negative charge of alginate polymer, a good mucoadhesion performance was observed and may be related to its good balance between available hydrogen bonding sites and the open expanded conformation. As for drug release, Na-CMC and AL inserts exhibited better prolongation of drug release than the Chit insert. The drug release profile from alginate inserts exhibited a delayed release pattern with a percentage of dissolution efficiency (%DE) value of 55.16. In fact, during dissolution, AL absorbs a significant amount of water to hydrate, swells and forms a stable hydrogel upon exposure to the divalent cations Ca
2+ present in simulated nasal fluid, thus immobilizing the incorporated drug in the polymer matrix. Therefore, the in situ gel-forming insert works as a reservoir which release drug from the matrix depending on the pore size of the Ca-alginate gel [
159].
Besides nasal inserts, alginate gels could also be used to encapsulate other delivery systems including microspheres and liposomes. Tafaghodi et al. explored the possibility to use alginates, poly-lactic-co gliycolic acid (PLGA) or Sephadex (dextran) to prepare microspheres potentially applicable as nasal drug and antigen delivery systems. Therefore, they evaluated the nasal clearance characteristics of those three kinds of microspheres using gamma-scintigraphy in a human study. Lactose powder was used as negative control. All of microspheres studied in this work were polyanionic and, in general, showed good mucoadhesive potential. Results obtained from this study showed that the control lactose powder was cleared rapidly (half-life of nasopharynx clearance was 1.5 h), while the bioadhesive delivery systems were retained within the nasal cavity for extended periods of time (half-lives of nasopharynx clearance were >2.5 h). Moreover, among microspheres studied, the least clearance rate from nasopharynx region was shown by alginate microspheres: after 4 h, 45.0% of alginate microspheres (similar to PLGA microspheres) were cleared from nasopharynx, while, at the same time, 63.1% and 74.5% of Sephadex microspheres and lactose powders were cleared, respectively [
156].
In association with other polymers like chitosan or TMC, alginates have also been described for the development of spray dried coated liposomes. In a recent study, Chen et al. used those polymers to produce spray-dried coated mucoadhesive liposomes for nasal delivery of antigens, in which bovine serum albumin was used as a model. From this study emerged the observation that coating liposomes with such mucoadhesive polymers offers enhanced penetration over uncoated liposomes through the nasal mucosa when delivered as a dry powder, which can result in increased drug bioavailability. Concerning the mucoadhesion of spray-dried uncoated and polymer-coated liposome powders, it was found that chitosan and TMC-coated liposomes exhibited a significantly higher mucoadhesive strength than alginate-coated or uncoated liposomes. This was most likely ascribed to the ability of the positive charge of chitosan and TMC to interact with the sialic groups on mucin. However, evaluating important parameters of manufacturing process, it was noticed that alginate-coated liposomes retained the smallest size and rehydration after coating [
100].
For a long time, nasal administration has been investigated as an attractive route for administration of cardiovascular drugs such as propranolol, nifedipine, nitroglycerin, metoprolol tartrate and carvedilol [
160]. These last two have been widely investigated for nasal administration associated with mucoadhesive agents, including sodium alginate. To enhance the systemic delivery of metoprolol tartrate and obtain improved therapeutic efficacy in the treatment of hypertension and angina pectoris, nasal bioadhesive microspheres composed of sodium alginate have been developed. The microspheres were prepared by emulsification followed by cross-linking with calcium chloride: the salt solution was mixed with the aqueous phase of alginate-containing drug, forming a gel instantaneously and entrapping the drug in the resultant three-dimensional lattice. In vivo studies showed that alginate microspheres improved therapeutic efficiency of the drug and provided a sustained and controlled delivery of metoprolol tartrate when compared with intravenous and oral routes [
161].
Carvedilol, a non-selective β-adrenergic antagonist, has also been investigated for nasal administration in the treatment of hypertension and stable angina pectoris. In fact, although well absorbed after oral administration, carvedilol suffers a significant first-pass metabolism, thus determining an absolute bioavailability of only 25%. An in vivo study conducted by Patti and co-workers evaluated the nasal application on rabbits of an alginate microspheres formulation encapsulating carvedilol. Concerning the bioavailability of the formulation, microsphere systems presented values greater than 65%, with respect to intravenous administration of the drug. The mucociliary transport rate was evaluated using the non-invasive gamma scintigraphy method and showed that alginate microspheres formulation was retained in the nasal cavity for an extended period compared to the control lactose powder. In fact, the latter was cleared rapidly (half-life of nasal clearance was less than 1 h) whereas the mucoadhesive delivery system was retained within the nasal cavity for longer time (half-life of nasal clearance were more than 2.5 h). Moreover, after 4 h, 61.55% of alginate microspheres were cleared from the nasal cavity compared to 87.36% of the lactose powder. These results remark the high mucoadhesive strength of this particulate systems which is very interesting considering that the normal half-life of nasal clearance in man is about 20 min. Thus, alginate polysaccharide associated with controlled release micro/nano-systems provides an improvement of drugs bioavailability, prolonging their therapeutic effect [
162].
While chitosan is widely acknowledged for its immunoadjuvant capabilities, other research indicates that surface-coating it with alginate can amplify these effects. For instance, authors investigated alginate-coating effect on chitosan (CHI) and trimethylchitosan (TMC) nanovaccines for enhancing immune response in a PR8 influenza vaccine study involving nasal immunization in BALB/c mice. Alginate-coating increased nanoparticles size (>100 nm) and decreased zeta potential, indicating effective presence of the polysaccharide in the coating layer. PR8-CHT-ALG nanoparticles showed lower efficacy compared to the non-coated nanoparticles due to particles agglomeration, reducing uptake by antigen-presenting cells. Conversely, immunization studies in BALB/c mice demonstrated that alginate-coated PR8-TMC-ALG nanoparticles significantly induced a Th1-based immune response compared to non-coated particles, evidenced by a higher IgG2a/IgG1 ratio, critical for combating viral infections. These findings validate the potential of employing alginate-coating as efficient mucosal adjuvant to generate a superior immune response when combining selected polysaccharides on vaccines preparations against similar pathogens [
163].




 ,
, 


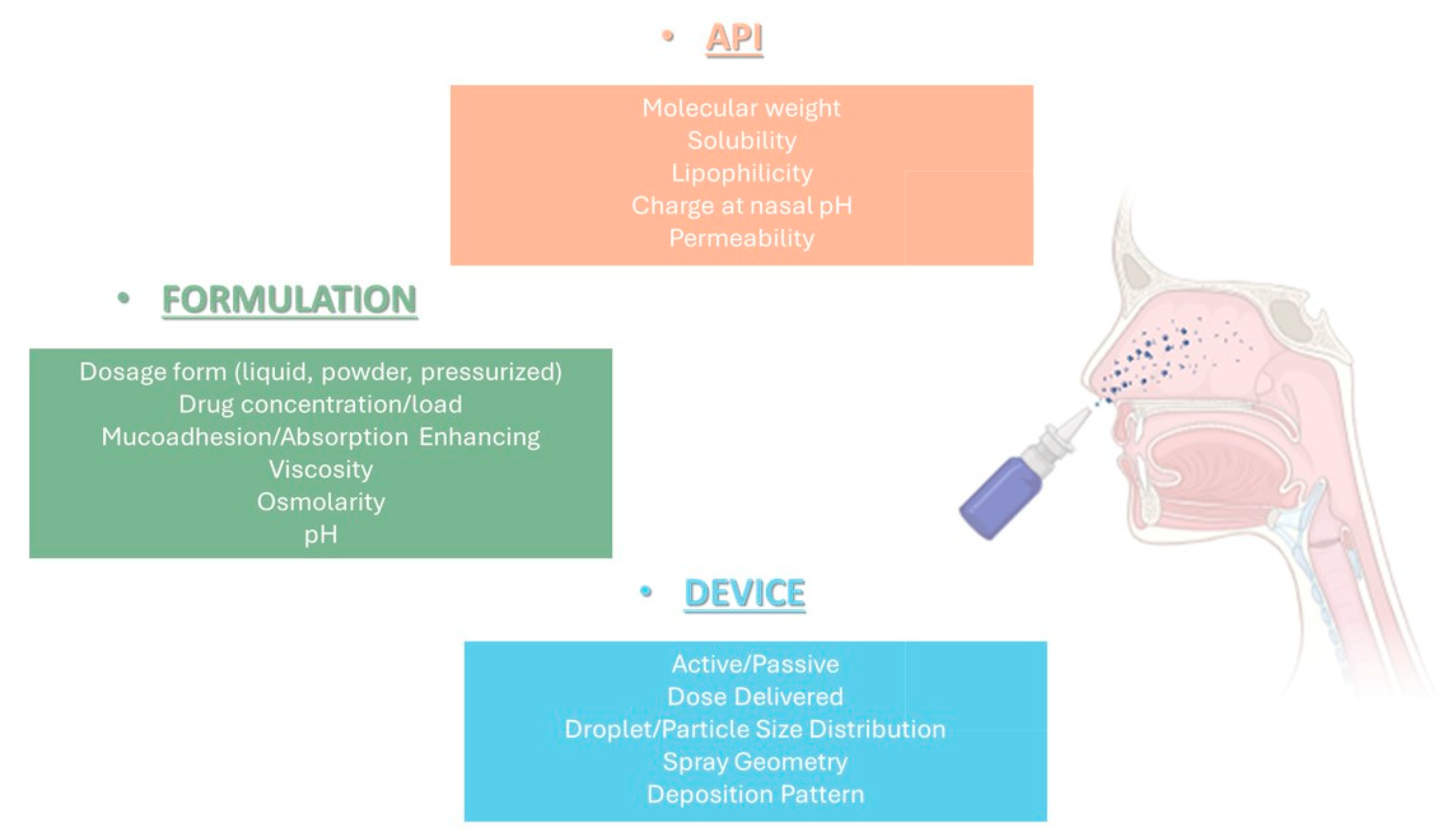
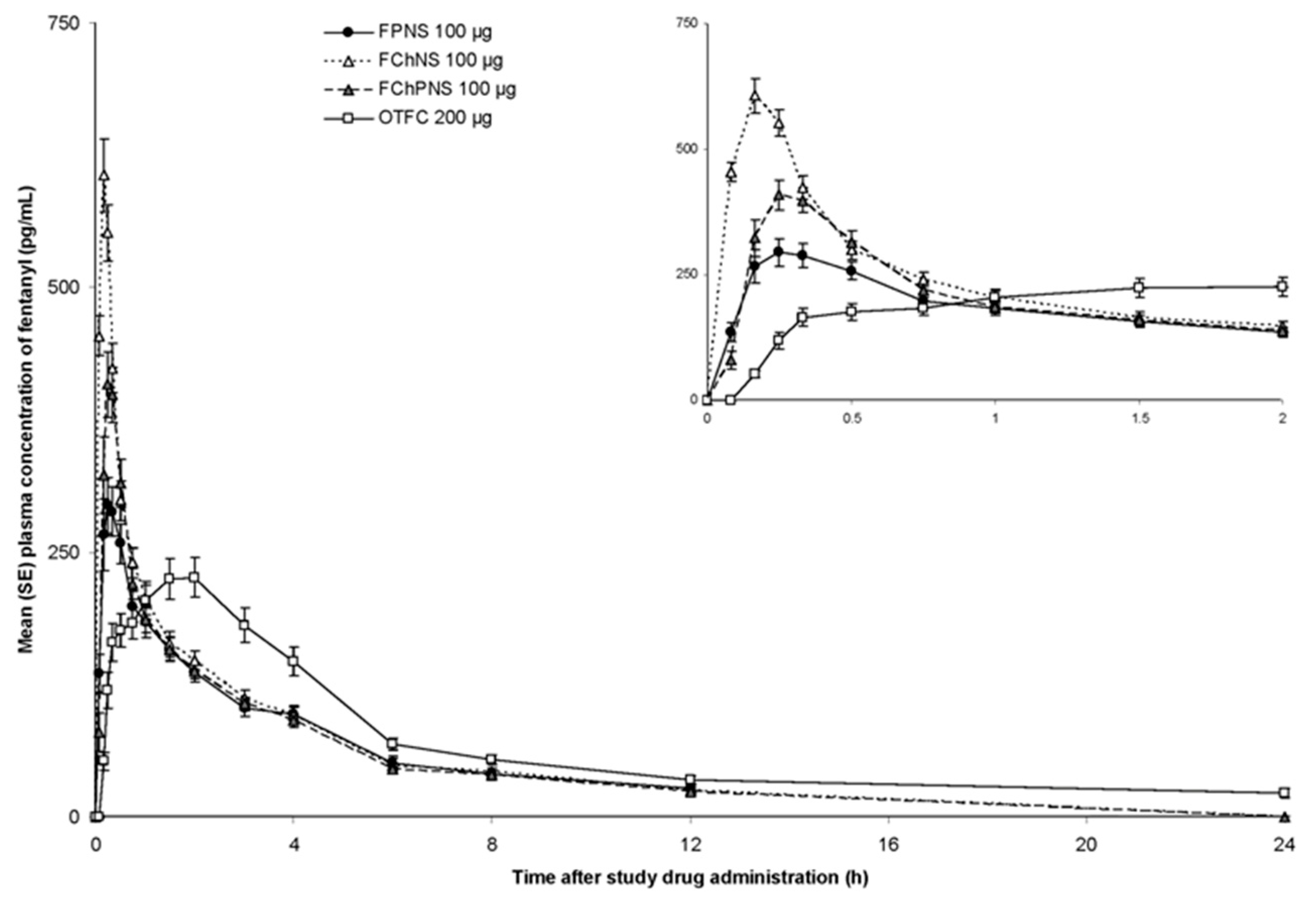
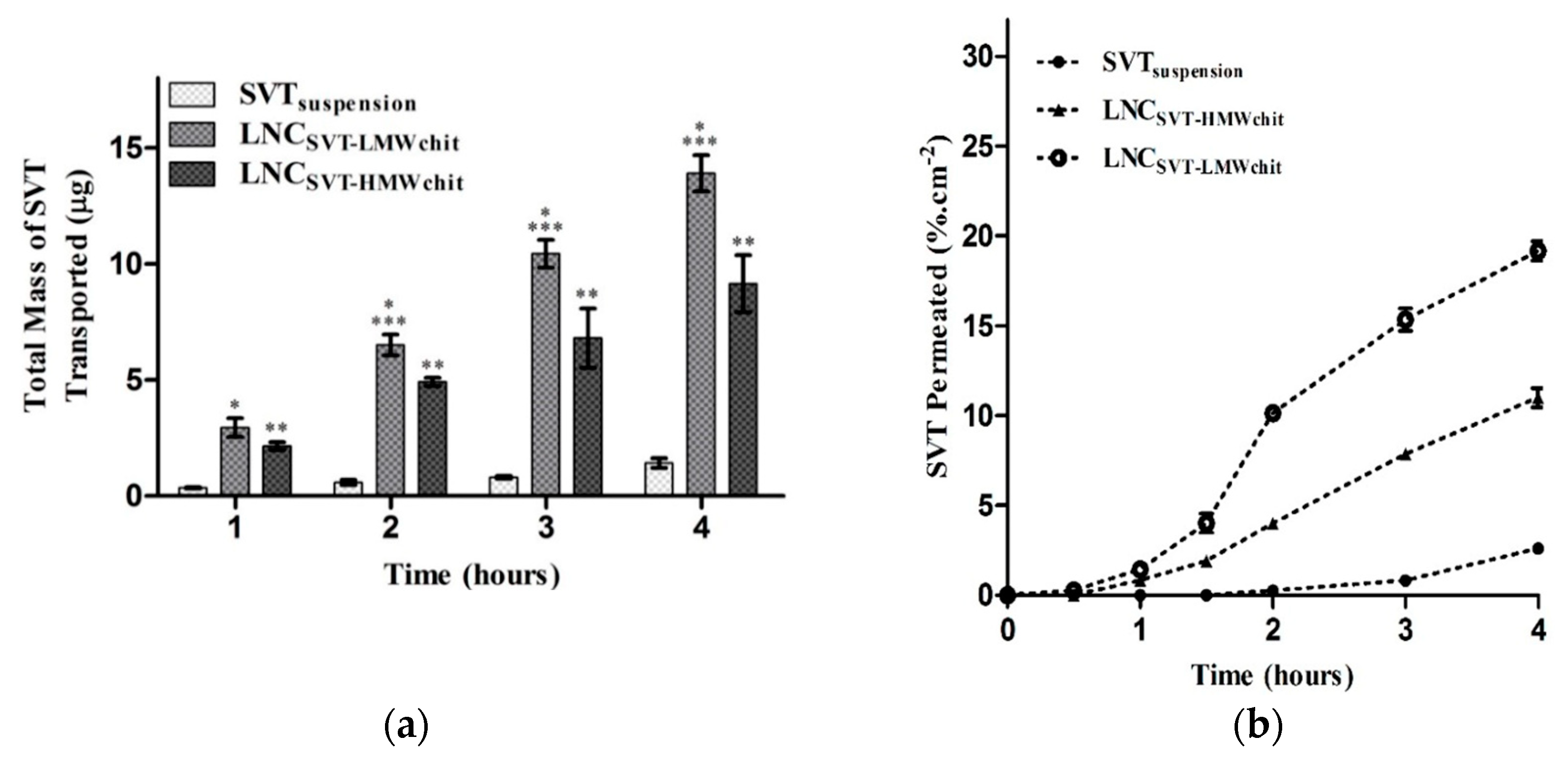
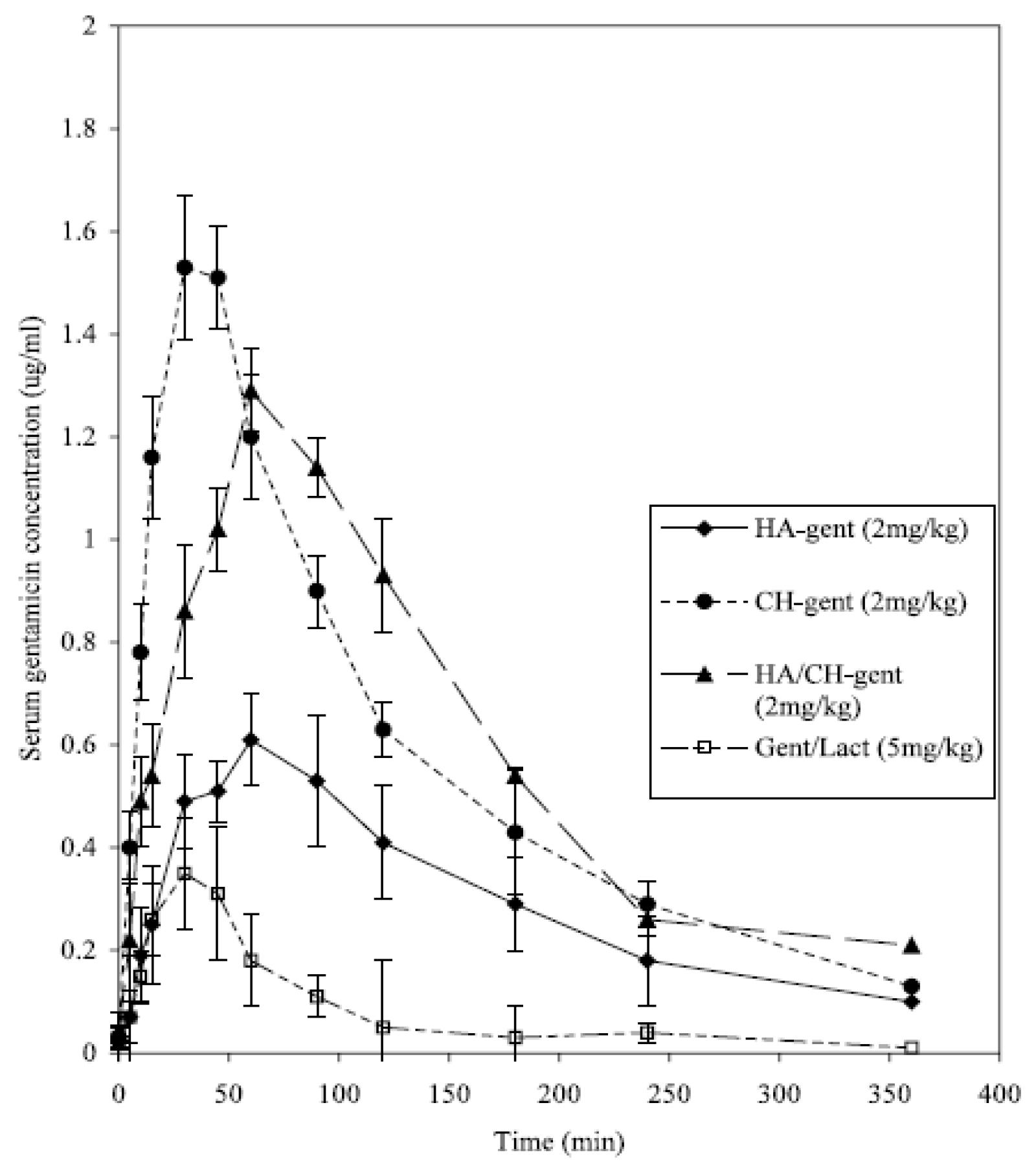

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}