1. Introduction
Hydrogels are composed of a three-dimensional network of polymer chains dispersed in water. These polymers have a high affinity for water due to their hydrophilic nature, allowing them to absorb and retain large amounts of water while maintaining their structural integrity. Hydrogels are obtained by crosslinking of synthetic- or biobased polymers under aqueous conditions through different mechanisms, including covalent bonding, supramolecular self-assembling, and physical interaction [
1,
2,
3]. Due to their high water content, hydrogels are intensively studied in biomedical areas such as wound healing, tissue engineering, regenerative medicine, and drug delivery [
4,
5,
6]. Further fields of applications for hydrogels in areas such as energy storage and conversion, environmental technologies, and agriculture have emerged recently [
7,
8,
9].
Polysaccharide-based materials provide many advantages over materials obtained from synthetic, fossil-based polymers. By nature’s design, they are typically biocompatible, biodegradable, and/or bioactive, as well as sustainable and renewable [
10]. Polysaccharide-based hydrogels can be obtained from different plant-, microbial-, or animal-derived polysaccharides such as cellulose, dextran, chitosan, and hyaluronic acid [
11,
12,
13,
14]. In most cases, artificial crosslinking through physical interaction (electrostatic interaction, hydrogen bonding, hydrophobic/hydrophilic aggregation) or covalent bonding is required to initiate gelation. Several polysaccharides from algae, such as alginate, carrageenan, ulvan, and agarose, are often studied in the context of developing hydrogel materials [
15]. They inherently form hydrogels in aqueous media and are readily available as sustainable biomass. Among these, agarose is of particular interest because it readily forms hydrogels simply by dissolution of the polymer in hot aqueous media followed by cooling. Moreover, agarose is a non-charged polymer, although it can frequently contain minor traces of the sulfated polysaccharide agaropectin.
Hydrogels derived from agarose have been studied extensively for various biomedical applications [
16,
17,
18,
19]. The materials are biocompatible but do not stimulate cell adhesion [
20]. Thus, different methods have been studied to add functionalities to agarose hydrogels and tune their physical, chemical, and biological properties. A feasible approach is to blend agarose gels with other polysaccharides (e.g., chitosan, hyaluronic acid) or inorganic materials (e.g., hydroxyapatite, silicates) [
20,
21,
22,
23]. Therein, agarose acts as the gel-forming matrix, and long-term compatibility with the functional additives must be ensured. Well-defined chemical modification in the hydrogel state is difficult to realize due to the combination of aqueous reaction media (prevents the use of many efficient derivatization reagents) and heterogeneous reaction course (even functionalization of the surface/bulk material is strongly hindered by diffusion). An alternative approach for obtaining functional agarose hydrogels is the chemical modification of the polysaccharide by a polymer analogous reaction prior to the gel formation.
Agarose is composed of a disaccharide repeating unit with alternating 1,3-linked β-D-galactose and 1,4-linked 3,6-anhydro-α-L-galactose. The mechanism that was proposed for the thermoreversible gelation of agarose involves the self-assembling of agarose molecules into double helixes that further aggregate upon cooling to form crosslinking points and finally a three-dimensional gel network [
15,
16]. In principle, chemical modification of hydroxy groups in the polymer backbone is feasible by conventional methods such as etherification, esterification, oxidation, and nucleophilic displacement reactions. However, the innate ability to form gels is lost if the molecular structure is modified extensively. Introducing ionic or hydrophobic groups will improve water solubility and disturb the supramolecular assembling into double helixes. Introducing hydrophobic moieties will ultimately result in water-insoluble agarose derivatives. It can be postulated that the ability to form hydrogels is retained if the overall degree of substitution (DS) is relatively small. Furthermore, meticulous control over the distribution of functionalities within the repeating unit and along the polymer chain is essential to ensure that the supramolecule self-assembling is not disrupted. Such a level of control over the molecular structure of the polysaccharide requires efficient synthesis strategies and is best achieved under well-controlled homogeneous reaction conditions.
The goal of this work was to introduce new functionalities by chemical modification of agarose in such a way that the native ability to form hydrogels by thermoreversible self-assembling is not impaired. By this approach, it would ultimately be possible to create novel agarose-based gels that are tailored, e.g., for interaction with specific cells or the uptake and release of specific molecules such as drugs or pollutants. In this context, three different routes for chemical derivatization of agarose under homogeneous reaction conditions were studied: (i) sulfation to introduce anionic sulfate groups, (ii) tosylation followed by nucleophilic displacement reaction to introduce reactive azido groups, and (iii) preparation of aryl carbonates followed by conversion into functional carbamates for introducing tertiary amino and quaternary ammonium groups. Furthermore, the impact of chemical modification of the agarose backbone on the ability to form hydrogels as well as on the mechanical and biological properties of the final hydrogels was studied. Agarose gels with anionic (e.g., sulfate) or cationic (amino or ammonium) groups are desired for drug delivery purposes because the charges can tune the uptake and release kinetics. It is also possible to further modify the surface and/or bulk of the charged gels by adsorption of oppositely charged polymers such as bioactive aminoglycans. Agarose gels with azido groups are very versatile and can potentially be used for many different applications. Depending on the specific needs, the very efficient and selective azide–alkyne cycloaddition can be used to introduce sensitive molecules such as dyes, drugs, antibodies, and other biomolecules in a highly predictable and effective manner.
3. Results and Discussion
The objective of this work was to introduce new functionalities into the backbone of agarose to tune the physical, chemical, and potentially biological properties of the biopolymer and the hydrogels obtainable therefrom. It was hypothesized that the native ability to form hydrogels by thermoreversible self-assembling in aqueous media is retained if the functionalities are hydrophilic (i.e., the water solubility is not imparted) and if the degree of substitution (DS) is low (<0.5). Agarose is soluble in different dipolar aprotic solvents like
N,
N-dimethylacetetamide (DMA),
N,
N-dimethylformamide (DMF), and 1,3-dimethyl-2-imidazolidinon (DMI), as well as in certain ionic liquids, including 1-butyl-3-methylimidazolium chloride (BMIMCl) [
26]. Thus, chemical derivatization of agarose under homogeneous reaction conditions was studied (
Figure 1). Anionic agarose sulfates (ASs), agarose tosylates (ATOSs), and reactive azido agarose derivatives (AZs), as well as agarose phenyl carbonates (APhCs), which are useful to synthesize cationic agarose carbamate (AC) derivatives, were of particular interest in this work.
3.1. Synthesis of Agarose Sulfates
Heterogeneous sulfation of agarose in
N,
N-dimethylformamide (DMF) has been reported previously with the intention to obtain anticoagulant compounds [
27]. The agarose was swollen and dispersed in the reaction medium and treated with chlorosulfonic acid. The products were described as insoluble below 40 °C with no information on their gel-forming ability. Moreover, homogeneous methods for acetosulfation (parallel acetylation and sulfation followed by hydrolysis of acetyl moieties) and heterogeneous methods for sulfation using ammonium sulfamate were reported [
28,
29]. The focus of these previous studies was to achieve high DS values, i.e., water-soluble AS derivatives. The intention of this work was to obtain AS with a reasonable amount of sulfate moieties that are still able to form gels in aqueous media. Thus, sulfation under homogeneous reaction conditions was performed to achieve an even distribution of ionic moieties along the polymer chain. DMA without and with LiCl was evaluated as the reaction medium (
Table 1). Different amounts of SO
3 pyridine were employed with the intention of obtaining products with a reasonable DS that still dissolve in hot water and form gels upon cooling.
At a molar ratio of three equivalents per repeating unit, a product with a high DS
sulfate of 1.28 was obtained (
AS 1) in DMA. Decreasing the amount of reagent to two equivalents resulted in a lower DS
sulfate of 0.66 (
AS 2). Both AS derivatives were fully water-soluble even at lower temperatures and did not form hydrogels. Further decreasing the molar ratio to one equivalent finally yielded a sulfated product (
AS 3) with a reasonable DS
sulfate of 0.26 that still was able to form hydrogels by dissolution in hot water followed by cooling to room temperature. It has been demonstrated that the addition of LiCl can enhance the dissolution of agarose in dipolar aprotic solvents by lowering the temperature needed for obtaining optically clear solutions [
26]. Likewise, the presence of LiCl can influence the regioselectivity. Sulfation of agarose in DMA/LiCl yielded similar products. The DS values were lower when compared to the reactions in the absence of LiCl. The reason for this finding is not clear yet. Nevertheless, the AS derivatives prepared in DMA/LiCl showed similar solubility properties. At low DS values of 0.27 (
AS 5) and 0.14 (
AS 6), the samples formed gels similarly to agarose. However, the sample with a DS of 0.42 (
AS 4) was fully water soluble and showed no gelation, which can be attributed to the higher number of anionic groups that promote water solubility.
All AS derivatives dissolved in DMSO, which enabled NMR spectroscopic characterization (
Figure 2). For the sample
AS 1 with the highest DS obtained in this study (1.28), it was found that the peak around 60 ppm, which corresponds to an unmodified G-6 position, disappeared completely. Instead, a new peak around 64 ppm appeared that can be attributed to a conversion of the primary hydroxy group into a sulfate group. The peak shift is in accordance with studies on the sulfation of other polysaccharides such as cellulose [
25]. In addition to the shift in the G-6-related peak, a new peak of around 72.5 ppm emerged in all spectra. The peak can be assigned to a carbon atom in a G-5 position that is adjacent to a sulfated G-6 position. A similar shift has also been observed for ATOS derivatives with a tosylate moiety in position G-6 [
26]. The relative intensity of the peaks related to sulfation at G-6 correlates well with the overall DS. Thus, it can be stated that the homogeneous sulfation in DMA is regioselective, and significant conversion of the other hydroxy moieties can be neglected at DS values below one. For sample
AS 1, however, a splitting of the LA-1-related peak was observed. This is an indication that at DS above one, the secondary hydroxy groups in the adjacent position LA-2 were partly converted into sulfate groups as well. It has been reported for the homogeneous tosylation of agarose in dipolar aprotic solvents that different regioselectivities are achieved if the reactions are performed in the absence (only G-6) or presence of LiCl (G-6 and LA-2) [
26]. This shift in regioselectivity was not observed for the sulfation. The spectra of ASs prepared in DMA/LiCl strongly resembled those obtained from DMA.
It can be summarized that sulfation of agarose under homogeneous conditions is feasible and regioselective for the position G-6 (below DS of 1) followed by LA-2 (above DS of 1). AS derivatives with a DS below 0.3 exhibited behavior akin to agarose, forming gels in aqueous media. At higher DS values above 0.4, the AS derivatives became fully water-soluble at all temperatures. It can be speculated that introducing anionic moieties increases intermolecular repulsion, thus hindering self-assembling into double-helix structures. The gel-forming AS 6 was selected for further studies.
3.2. Synthesis of Azido Agarose and Functional Agarose Carbamates
AZA derivatives were obtained by a two-step homogeneous synthesis that involved the synthesis and subsequent conversion of ATOSs into the final product (
Figure 1). First, agarose was converted into ATOSs by homogeneous conversion with
p-toluenesulfonyl chloride (
Table 2), as reported previously [
26]. By adjusting the amount of reagent of reagent, the DS
tosyl was tuned from 0.13 (
ATOS 1) to 0.82 (
ATOS 2), which was in the range desired for this work. The tosyl moiety can be replaced by several nucleophiles through a nucleophilic displacement reaction. The TSOA derivatives were converted with sodium azide in order to obtain AZA derivatives (
Table 3). The products no longer contain sulfur but rather a certain amount of nitrogen. The corresponding DS
azide values correlated well with the staring DS
tosyl, i.e., a quantitative nucleophilic displacement occurred in all cases.
AZA 4 with the highest DS
azide of 0.75 was soluble in DMSO but insoluble in water, which was expected considering that the azido group is somewhat hydrophobic. The AZA with lower DS values were soluble in hot water.
AZA 2 with a DS of 0.32 remained soluble upon cooling while
AZA 1 with a lower DS of 0.13 and
AZA 3 with a higher DS of 0.45 formed gels. As reported before, the reason for this unexpected behavior is not clear yet [
26].
Polysaccharide tosylates can also be used to introduce amino groups into the polymer backbone, either directly (by conversion with diamines) or indirectly (by reduction of azido derivatives) [
30,
31]. An alternative and more versatile approach for the modular synthesis of functional polysaccharide derivatives uses polysaccharide aryl carbonates [
32,
33]. The carbonates are prepared with a defined molecular structure and the subsequent conversion with primary and secondary amines can yield a broad variety of functional polysaccharide carbamates. Among others, it is possible to introduce tertiary amino and quaternary ammonium groups by this approach [
24,
34]. Thus, the synthesis of APhC derivatives was studied with the intention of obtaining low DS values (<0.3) for the final AC derivatives (
Table 2). The ionic liquid BMIMCl in combination with the co-solvent pyridine was chosen as the reaction medium. The homogeneous conversion of agarose with up to two equivalents of phenyl chloroformate yielded products with the desired carbonate groups. The corresponding
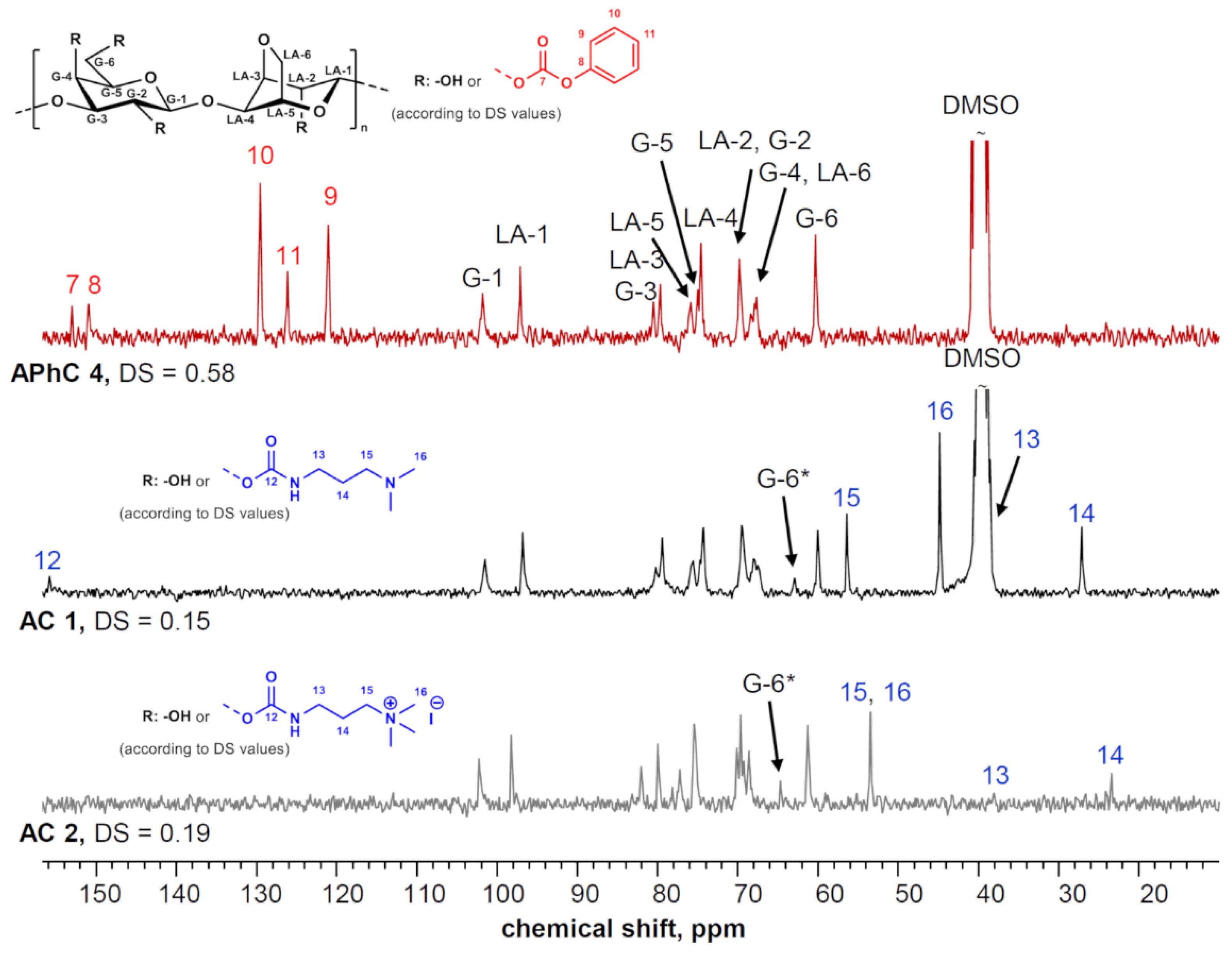
13C NMR spectra clearly showed the presence of the aromatic moiety as well as the carbonyl moieties of the newly introduced carbonate linkage (
Figure 3). DS
carbonate values of up to 0.58 (
APhC 4) were obtained. The reactivity of agarose in this particular derivatization reaction was lower compared to other polysaccharides such as xylan and cellulose that yield higher DS values under similar conditions [
32,
35]. The reaction seems to be rather sensitive to the molecular structure of the particular polysaccharide. Preliminary tests have shown that also starch might show a lower reactivity as well. Nevertheless, the APhC derivatives obtained were suitable for further functionalization into ACs.
A goal of this work was to introduce substituents that are either permanently cationic (quaternary ammonium groups) or can be protonated in aqueous systems (tertiary amino groups) to induce a cationic charge (
Table 3).
APhC 4 with the highest DS
carbonate obtained in this study was converted with two different functional amines that either carried a tertiary amin (
N1,
N1-dimethylpropane-1,3-diamine) or the corresponding quaternary ammonium group (3-amino-
N,
N,
N-trimethylpropan-1-aminium iodide). NMR spectroscopic characterization of both products obtained (
AC 1 and
AC 2) showed a complete removal of the aromatic phenyl ring (
Figure 3). At the same time, peaks appeared that could be assigned to aliphatic carbon atoms in the newly introduced carbamate group and to the carbonyl moiety. The DS values of
AC 1 (0.15, tertiary amino group) and
AC 2 (0.19, quaternary ammonium group) were lower than the starting DS
carbonate, which indicates a certain degree of hydrolysis during the conversion that has been observed previously [
24]. Both ACs dissolved in hot water and formed gels upon cooling. Thus, both compounds were suitable for the subsequent experiments on hydrogels.
3.3. Preparation and Characterization of Agarose-Based Gels
It could be demonstrated in this study that additional anionic, cationic, and reactive functionalities can be introduced into the agarose backbone by homogeneous synthesis in such a way that the native gel-forming ability is retained. Thus, it was studied how the different chemical modifications affect the properties of the final gels. Representative AS, AC, and AZA samples were selected that showed gelation behavior in preliminary tests. Cylindrical gel specimens were prepared by dissolving agarose in boiling water with mass concentrations between 1 and 3%, pouring the warm solution into molds, and cooling the solutions to room temperature. The same facile procedure could be used for the gel-forming agarose derivatives. In addition, mixed gels containing agarose and an agarose derivative were prepared in some cases. In all cases discussed below, complete gelation of the initially transparent aqueous solutions occurred meaning that no residual liquid solution remained. Furthermore, all gel specimens were stable enough to be handled manually without breaking.
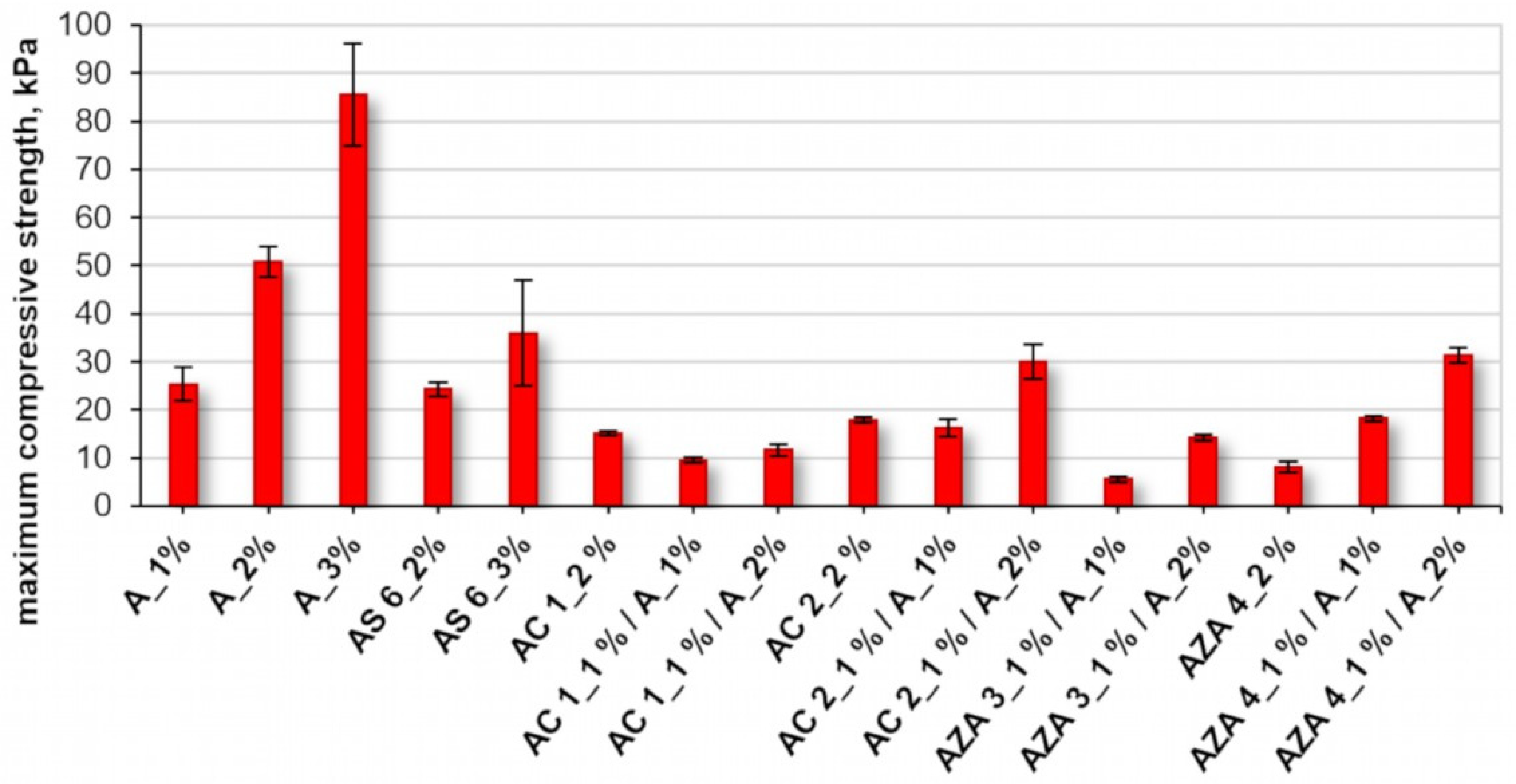
The mechanical stability of the different hydrogel samples was evaluated by compression tests. The agarose-based gels displayed an elastic behavior as described previously [
22]. They were compressible to a certain maximum compressive stress. Upon further increasing strain, macroscopic cracks appeared, and the gel specimens were destroyed. For agarose gels, the maximum compressive strength observed before breaking increased with the mass concentration of agarose from 25.4 kPa (1%) to 50.8 kPa (2%) and finally 85.6 kPa (3%;
Figure 4).
The increase in mechanical stability can be explained by the increasing density of the three-dimensional polymer network. In comparison, hydrogels obtained from chemically modified agarose derivatives showed the same elastic properties but lower overall mechanical stability. As an example, AS 6 hydrogels with a 2% mass concentration showed a similar maximum compression strength (24.3 kPa) as an agarose gel with a 1% mass concentration. The two AC derivative yield gels broke at maximum compressive strengths of 15.2kPa (AC 1) and 17.9 kPa (AC 2). The lowest mechanical stability was observed for gels prepared from AZ 4. Nevertheless, it should be noted that all gel specimens derived from agarose derivatives were stable enough for manual handling and further experiments.
Chemical modification of agarose could yield fully water-soluble derivatives. Most notably,
AZA 3 with a DS
azide of 0.25 did not form hydrogels on its own. However, gelation could be induced by mixing the water-soluble derivative with agarose. Thus, it was possible to obtain stable mixed hydrogels. Adding agarose to gel-forming derivatives is a possibility to further alter the mechanical properties. For the cationic
AC 2 and the reactive
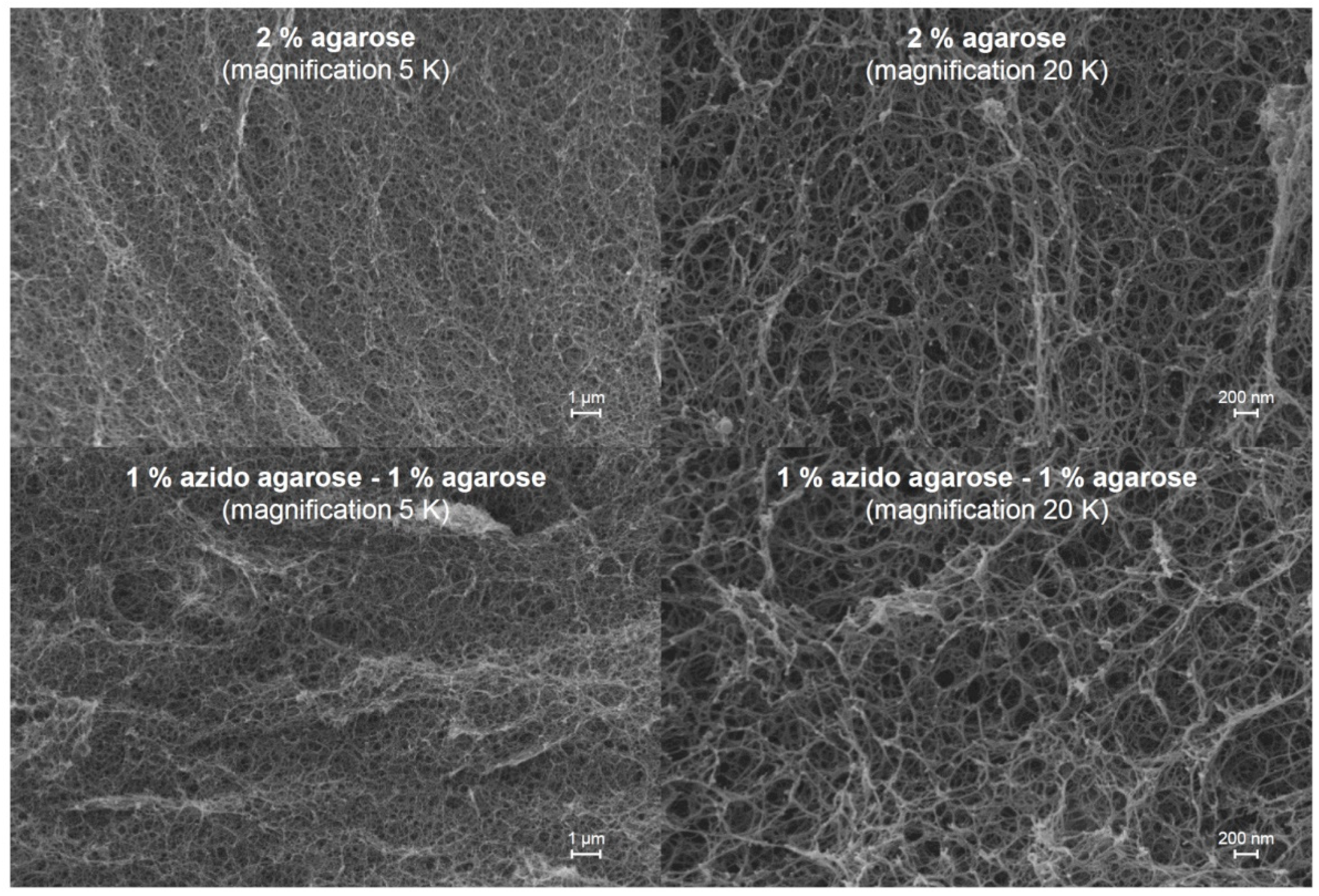
AZA 4, it was observed that the mixed hydrogels with agarose featured higher mechanical stability compared to the gels prepared from the agarose derivatives alone when the overall polymer mass concentration was 2% in both cases. Thus, blending agarose gels with agarose derivatives (gel-forming or fully water-soluble) is a feasible approach to obtain functional hydrogel materials. It can be speculated that agarose derivatives with low DS of additional functionalities still contain larger segments of the unaltered agarose backbone. Hence, the polymers can still interact with each other or with unmodified agarose molecules to self-assemble into double helixes and form hydrogels. Scanning electron microscopy (SEM) experiments were performed on hydrogel materials (
Figure 5). To preserve the native structure of the polymer network, water was exchanged stepwise to ethanol and liquid CO
2, which was finally removed under mild supercritical conditions. The SEM images of the gels obtained from agarose alone, as well as from a mixture of agarose and
AZA 4, show very similar structures of elongated nanosized fibrils with no significant differences. No phase separation, aggregation, or defect structures were visible, which indicates that both polymers were well integrated within the three-dimensional network.
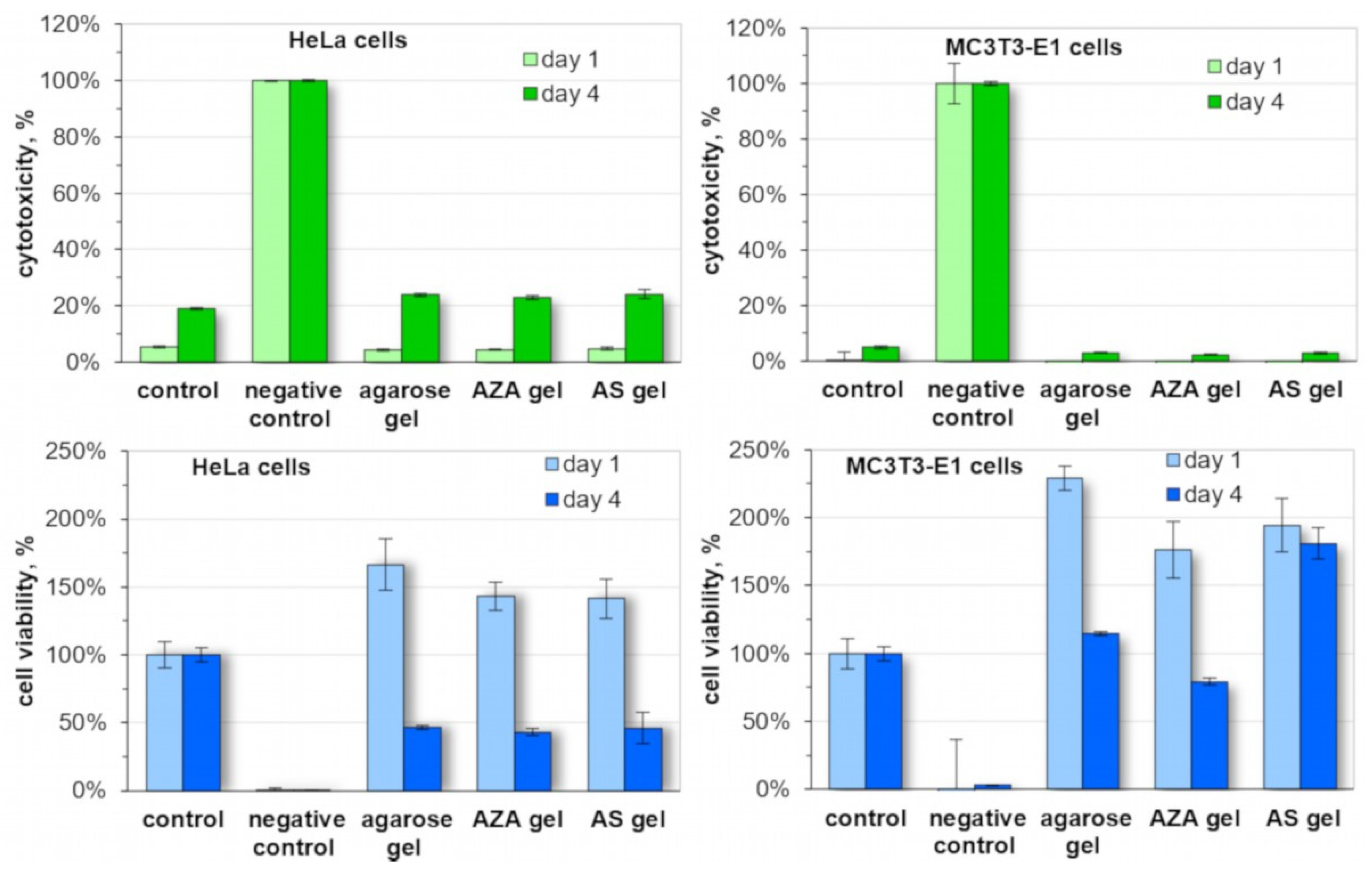
The biocompatibility of selected hydrogels was studied by colonization tests using cell lines of different origins (epithelial, osseous;
Figure 6). Agarose gels and gels prepared from AS and AZA showed no cytotoxic effect. The results after one and four days were similar to the positive control in all cases. It should be noted that the increased cytotoxicity after 4 days is the result of an accumulation of cell decomposition products in the medium, which would not occur under physiological conditions. Cultivation of the different cells on the surface of the gels had no negative impact on the cell viability either, when compared to the positive control. Interestingly, the cell viability slightly increased after one day, especially for the osseous cell line (MC3T3-E1). It decreased after four days in the case of epithelial cells (HeLa), which could be an indication that cells are detaching from the surface due to the lack of stronger bio-interaction. For osseous cell lines, this decrease was less pronounced. Interestingly, the AS hydrogel showed no decrease in cell viability after four days. This could be an indication that the anionic sulfate moieties can induce a beneficial interaction of the gel surface with these cells, which will be further studied in future experiments. Overall, it can be summarized that the gels derived from agarose and functional agarose derivatives are biocompatible and thus suited for biomedical applications.
4. Conclusions
In this study, various agarose derivatives were prepared, including reactive azido agarose (AZA), cationic agarose carbamates (ACs), and anionic agarose sulfates (ASs). The molecular structure and degrees of substitution (DS) of these derivatives were successfully tuned through homogeneous synthesis. It was demonstrated that the innate ability of agarose to form hydrogels is sensitive to alterations within the polymer repeating unit. However, functional agarose derivatives with reasonable DS values were obtained that still allowed the facile preparation of functional hydrogels. Furthermore, the impact of different chemical derivatizations on the mechanical and biological properties of the hydrogels was investigated. The defined chemical modification of agarose explored in this study offers a versatile approach to creating functional hydrogel materials for advanced applications. Analogous to recent findings, anionic and cationic agarose gels could also be utilized in a layer-by-layer approach for tailored sustained release, particularly of hydrophilic drugs. Moreover, these compounds are potentially bioactive (antimicrobial, antiviral, anticoagulant) based on previous studies on similar ionic derivatives derived from other polysaccharides such as cellulose. Thus, incorporating these agarose derivatives can yield hydrogels with interesting bioactive properties. The findings also suggest that AS-based gels may exhibit altered interactions with cells and tissues. Hydrogels derived from AZA are amenable to further modification through well-defined click-chemistry reactions, which are currently under investigation in a comprehensive study.


 ,
, 





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}