
A Review of Cellulose Coarse-Grained Models and Their Applications



Abstract
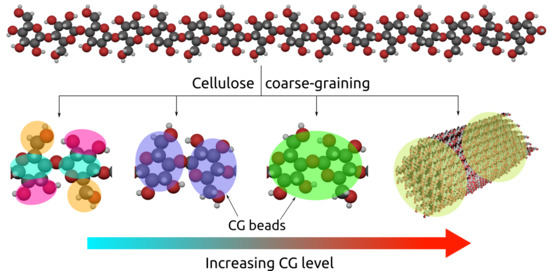
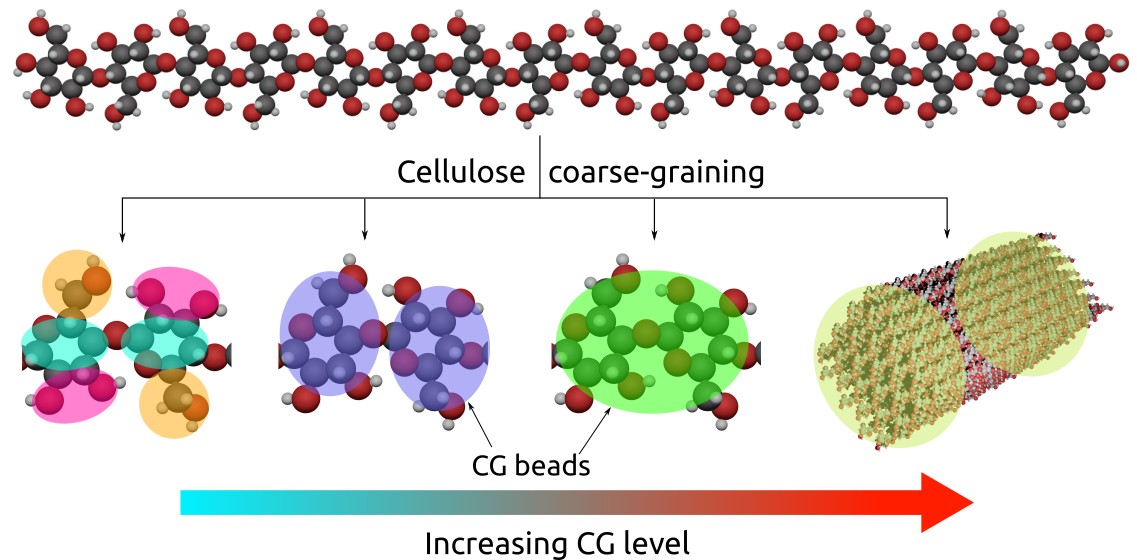{kind=link}
{kind=link}
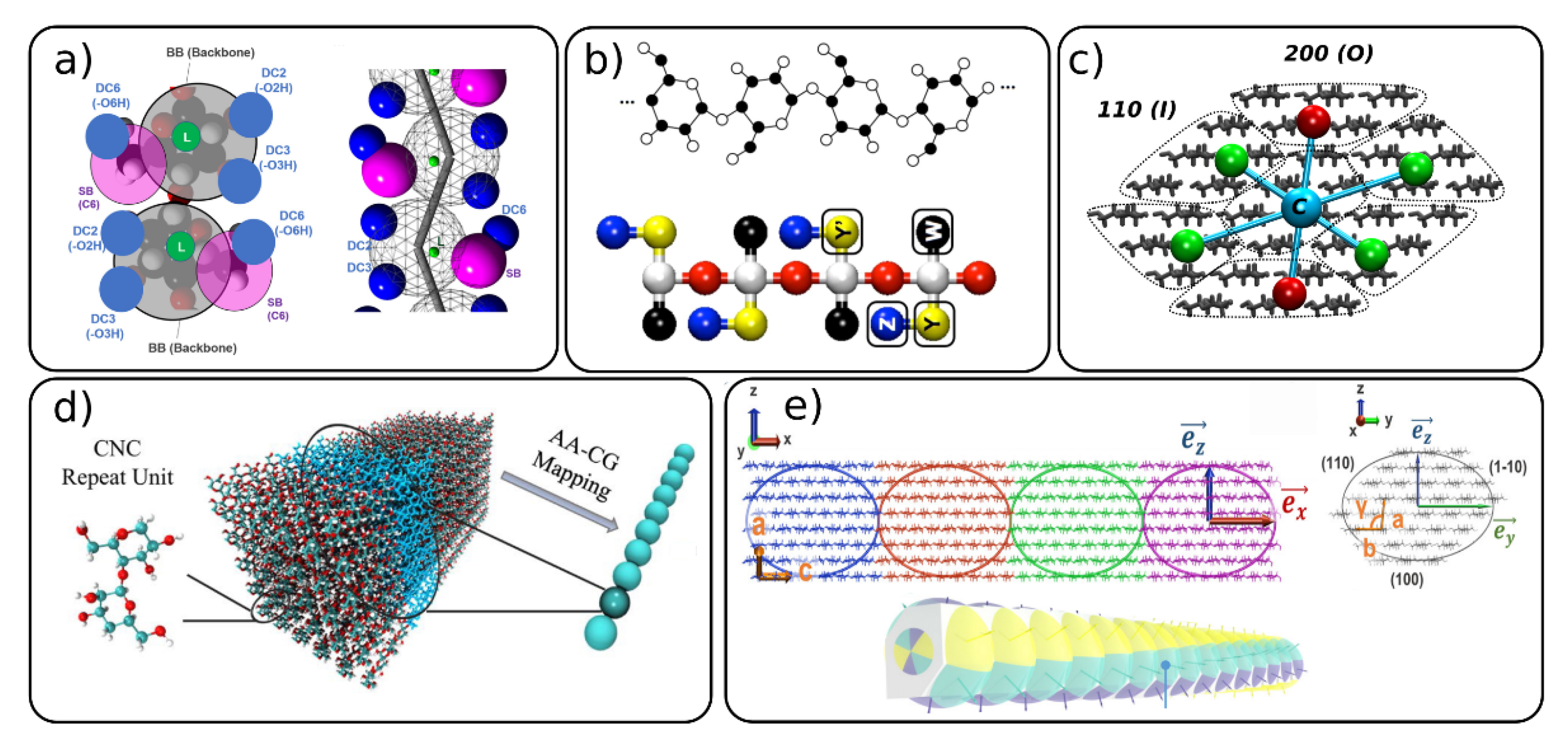{kind=link}
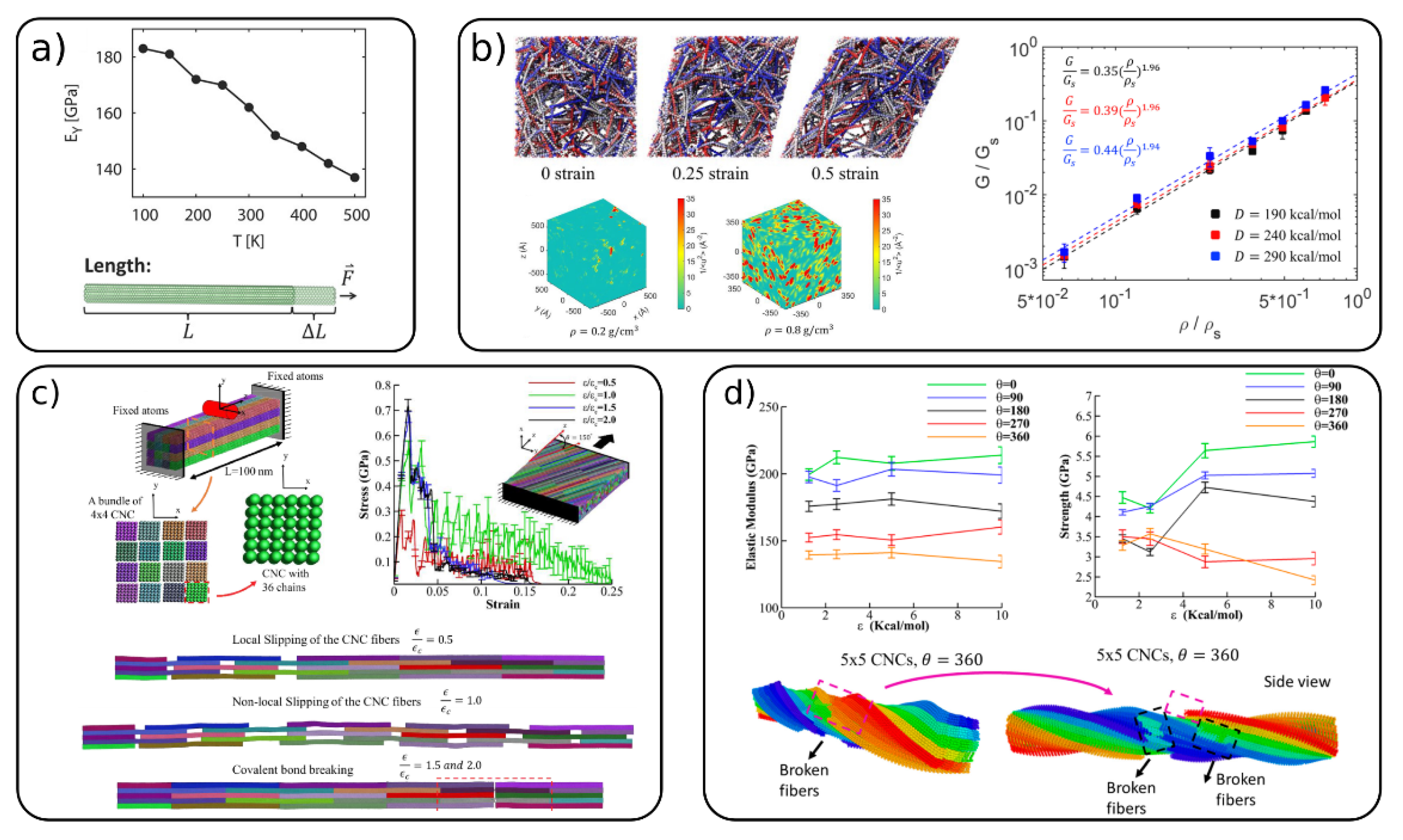{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Coarse-Grained Modelling
3. Coarse-Grained Models of Carbohydrates
4. Applications of Cellulose CG Models
4.1. Mechanical Properties
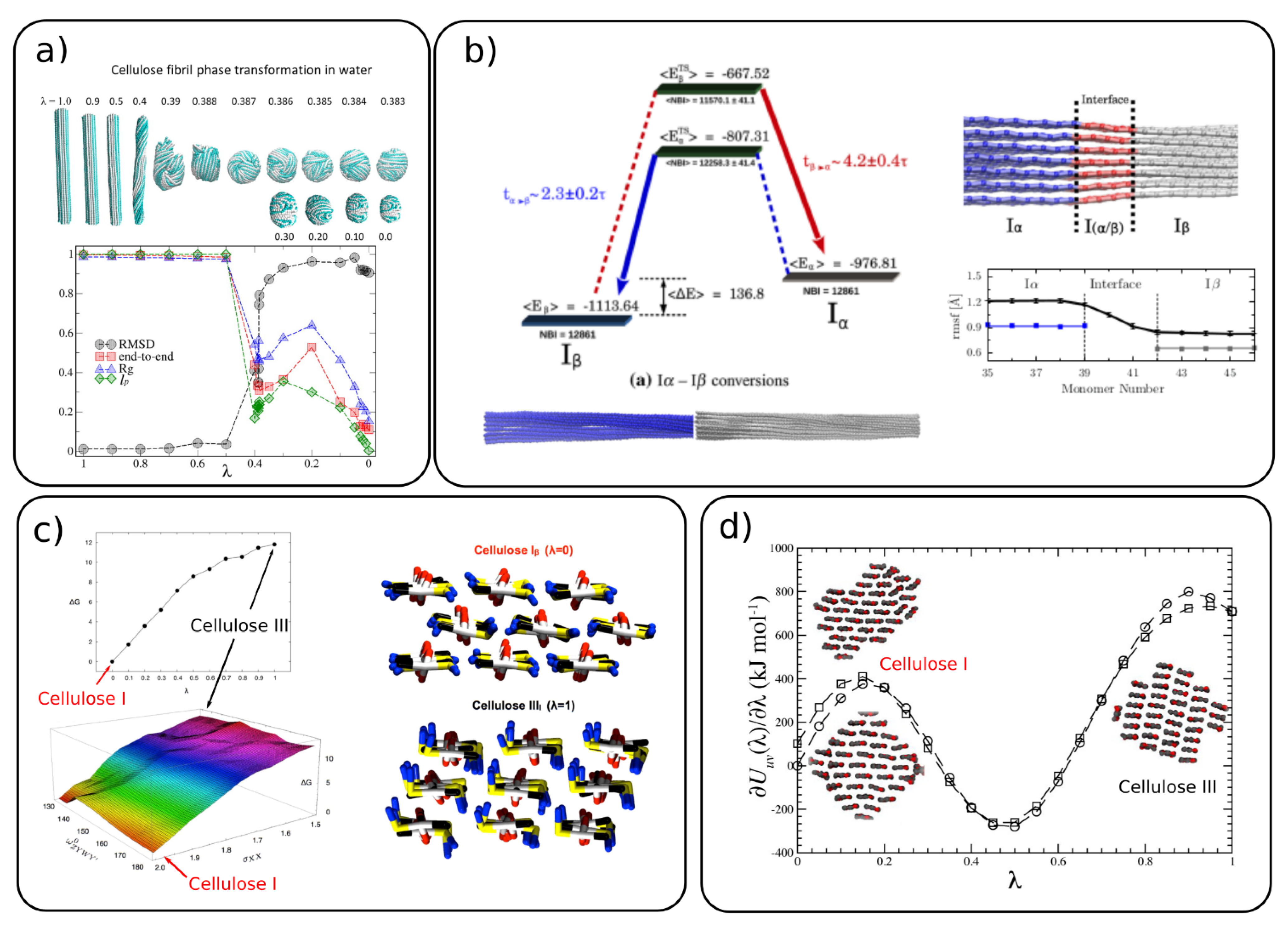
4.2. Conversion between Different Cellulose Allomorphs
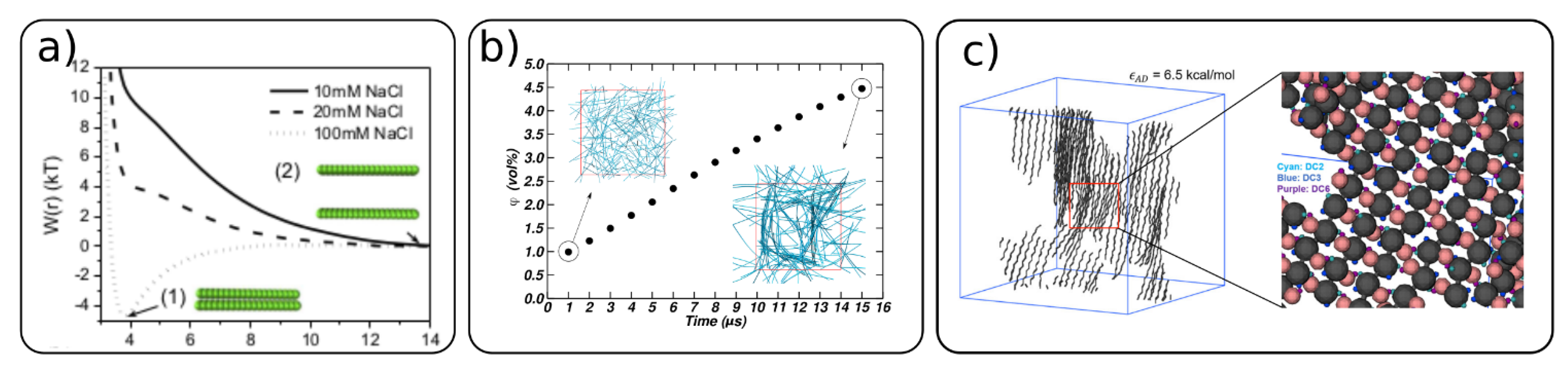
4.3. Cellulose-Cellulose Interactions and Self-Assembly of Cellulose Network
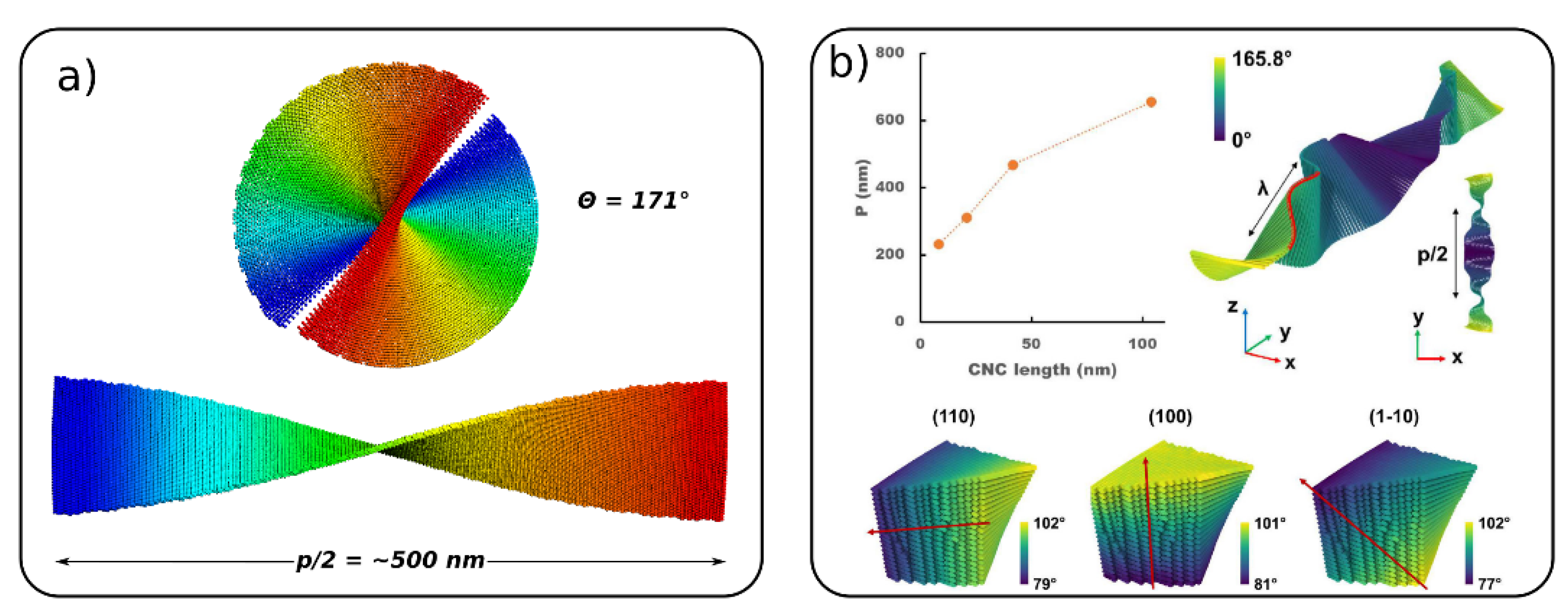
4.4. Chiral Nematic Phases
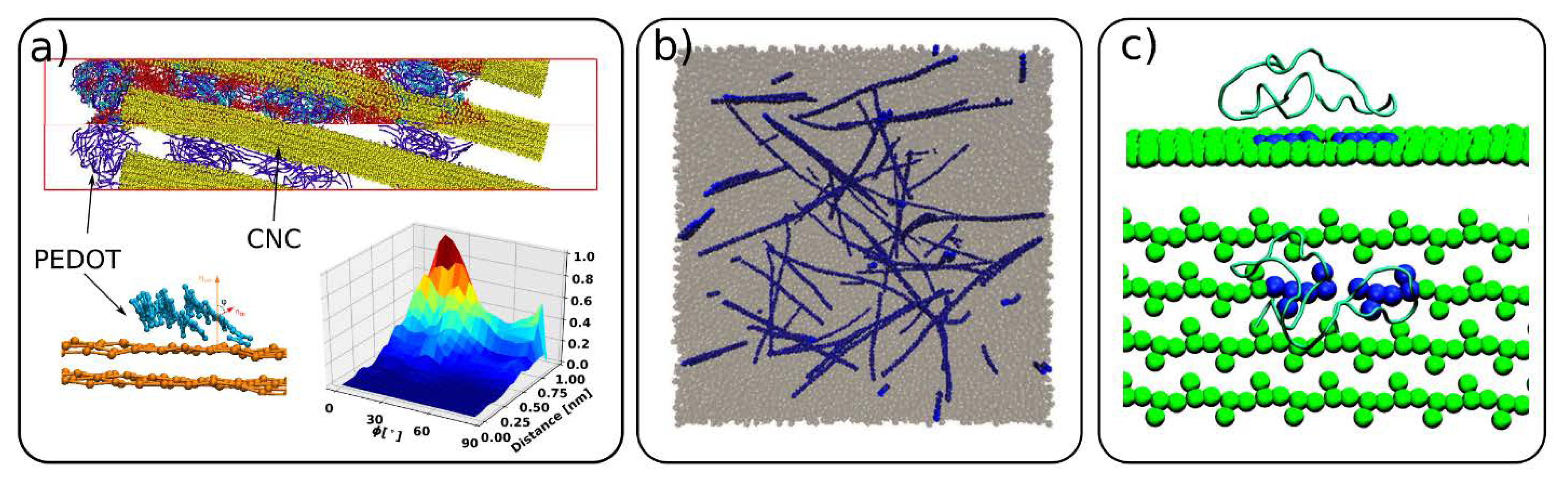
4.5. Cellulose Composite Materials and Interactions with Other Molecules
5. Conclusions and Outlook
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Klemm, D.; Heublein, B.; Fink, H.-P.; Bohn, A. Cellulose: Fascinating Biopolymer and Sustainable Raw Material. Angew. Chem. Int. Ed. 2005, 44, 3358–3393. [Google Scholar] [CrossRef]
- Eyley, S.; Thielemans, W. Surface Modification of Cellulose Nanocrystals. Nanoscale 2014, 6, 7764. [Google Scholar] [CrossRef] [PubMed]
- Dufresne, A. Nanocellulose: A New Ageless Bionanomaterial. Mater. Today 2013, 30, 1704285. [Google Scholar] [CrossRef]
- Jarvis, M.C. Structure of Native Cellulose Microfibrils, the Starting Point for Nanocellulose Manufacture. Philos. Trans. R. Soc. A Math. Phys. Eng. Sci. 2018, 376, 20170045. [Google Scholar] [CrossRef]
- Ding, S.-Y.; Zhao, S.; Zeng, Y. Size, Shape, and Arrangement of Native Cellulose Fibrils in Maize Cell Walls. Cellulose 2014, 2, 1863–1871. [Google Scholar] [CrossRef]
- Nixon, B.T.; Mansouri, K.; Singh, A.; Du, J.; Davis, J.K.; Lee, J.-G.; Slabaugh, E.; Vandavasi, V.G.; O’Neill, H.; Roberts, E.M.; et al. Comparative Structural and Computational Analysis Supports Eighteen Cellulose Synthases in the Plant Cellulose Synthesis Complex. Sci. Rep. 2016, 6, 28696. [Google Scholar] [CrossRef] [PubMed]
- Ling, S.; Kaplan, D.L.; Buehler, M.J. Nanofibrils in Nature and Materials Engineering. Nat. Rev. Mater. 2018, 3, 1–15. [Google Scholar] [CrossRef]
- Trache, D.; Hussin, M.H.; Haafiz, M.K.M.; Thakur, V.K. Recent Progress in Cellulose Nanocrystals: Sources and Production. Nanoscale 2017, 9, 1763–1786. [Google Scholar] [CrossRef]
- Moon, R.J.; Martini, A.; Nairn, J.; Simonsenf, J.; Youngblood, J. Cellulose Nanomaterials Review: Structure, Properties and Nanocomposites. Chem. Soc. Rev. 2011, 40, 3941–3994. [Google Scholar] [CrossRef]
- Shankaran, D.R. Chapter 14—Cellulose Nanocrystals for Health Care Applications. In Applications of Nanomaterials; Mohan Bhagyaraj, S., Oluwafemi, O.S., Kalarikkal, N., Thomas, S., Eds.; Micro and Nano Technologies; Woodhead Publishing: Cambridge, UK, 2018; pp. 415–459. ISBN 978-0-08-101971-9. [Google Scholar]
- Fan, J.; Zhang, S.; Li, F.; Shi, J. Cellulose-Based Sensors for Metal Ions Detection. Cellulose 2020, 27, 5477–5507. [Google Scholar] [CrossRef]
- Fan, J.; Zhang, S.; Li, F.; Yang, Y.; Du, M. Recent Advances in Cellulose-Based Membranes for Their Sensing Applications. Cellulose 2020, 27, 9157–9179. [Google Scholar] [CrossRef]
- Chen, W.; Yu, H.; Lee, S.-Y.; Wei, T.; Li, J.; Fan, Z. Nanocellulose: A Promising Nanomaterial for Advanced Electrochemical Energy Storage. Chem. Soc. Rev. 2018, 47, 2837. [Google Scholar] [CrossRef]
- Zhang, L.; Han, J.; Wang, H.; Car, R.; Weinan, E. DeePCG: Constructing Coarse-Grained Models via Deep Neural Networks. J. Chem. Phys. 2018, 149, 034101. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, M.; Xu, C.; White, A.D. Encoding and Selecting Coarse-Grain Mapping Operators with Hierarchical Graphs. J. Chem. Phys. 2018, 149, 134106. [Google Scholar] [CrossRef]
- Li, Z.; Wellawatte, G.P.; Chakraborty, M.; Gandhi, H.A.; Xu, C.; White, A.D. Graph Neural Network Based Coarse-Grained Mapping Prediction. Chem. Sci. 2020, 11, 9524–9531. [Google Scholar] [CrossRef]
- Lyubartsev, A.P.; Laaksonen, A. Calculation of Effective Interaction Potentials from Radial Distribution Functions: A Reverse Monte Carlo Approach. Phys. Rev. E 1995, 52, 3730–3737. [Google Scholar] [CrossRef]
- Lyubartsev, A.P.; Naômé, A.; Vercauteren, D.P.; Laaksonen, A. Systematic Hierarchical Coarse-Graining with the Inverse Monte Carlo Method. J. Chem. Phys. 2015, 143, 243120. [Google Scholar] [CrossRef]
- Shell, M.S. The Relative Entropy Is Fundamental to Multiscale and Inverse Thermodynamic Problems. J. Chem. Phys. 2008, 129, 144108. [Google Scholar] [CrossRef] [PubMed]
- Izvekov, S.; Voth, G.A. A Multiscale Coarse-Graining Method for Biomolecular Systems. J. Phys. Chem. B 2005, 109, 2469–2473. [Google Scholar] [CrossRef]
- Izvekov, S.; Voth, G.A. Multiscale Coarse Graining of Liquid-State Systems. J. Chem. Phys. 2005, 123, 134105. [Google Scholar] [CrossRef]
- Moritsugu, K.; Smith, J.C. Coarse-Grained Biomolecular Simulation with REACH: Realistic Extension Algorithm via Covariance Hessian. Biophys. J. 2007, 93, 3460–3469. [Google Scholar] [CrossRef]
- Noid, W.G. Perspective: Coarse-Grained Models for Biomolecular Systems. J. Chem. Phys. 2013, 139, 090901. [Google Scholar] [CrossRef]
- Brini, E.; Algaer, E.A.; Ganguly, P.; Li, C.; Rodríguez-Ropero, F.; van der Vegt, N.F.A. Systematic Coarse-Graining Methods for Soft Matter Simulations—A Review. Soft Matter 2013, 9, 2108–2119. [Google Scholar] [CrossRef]
- Peter, C.; Kremer, K. Multiscale Simulation of Soft Matter Systems—from the Atomistic to the Coarse-Grained Level and Back. Soft Matter 2009, 5, 4357. [Google Scholar] [CrossRef]
- Marrink, S.J.; Risselada, H.J.; Yefimov, S.; Tieleman, D.P.; de Vries, A.H. The MARTINI Force Field: Coarse Grained Model for Biomolecular Simulations. J. Phys. Chem. B. 2007, 111, 7812–7824. [Google Scholar] [CrossRef] [PubMed]
- López, C.A.; Rzepiela, A.J.; de Vries, A.H.; Dijkhuizen, L.; Hünenberger, P.H.; Marrink, S.J. Martini Coarse-Grained Force Field: Extension to Carbohydrates. J. Chem. Theory Comput. 2009, 5, 3195–3210. [Google Scholar] [CrossRef] [PubMed]
- Wohlert, J.; Berglund, L.A. A Coarse-Grained Model for Molecular Dynamics Simulations of Native Cellulose. J. Chem. Theory Comput. 2011, 7, 753–760. [Google Scholar] [CrossRef]
- López, C.A.; Bellesia, G.; Redondo, A.; Langan, P.; Chundawat, S.P.S.; Dale, B.E.; Marrink, S.J.; Gnanakaran, S. MARTINI Coarse-Grained Model for Crystalline Cellulose Microfibers. J. Phys. Chem. B 2015, 119, 465–473. [Google Scholar] [CrossRef] [PubMed]
- Shivgan, A.T.; Marzinek, J.K.; Huber, R.G.; Krah, A.; Henchman, R.H.; Matsudaira, P.; Verma, C.S.; Bond, P.J. Extending the Martini Coarse-Grained Force Field to N -Glycans. J. Chem. Inf. Model. 2020, 60, 3864–3883. [Google Scholar] [CrossRef]
- Molinero, V.; Goddard, W.A. M3B: A Coarse Grain Force Field for Molecular Simulations of Malto-Oligosaccharides and Their Water Mixtures. J. Phys. Chem. B 2004, 108, 1414–1427. [Google Scholar] [CrossRef]
- Molinero, V.; Çaǧın, T.; Goddard, W.A. Mechanisms of Nonexponential Relaxation in Supercooled Glucose Solutions: The Role of Water Facilitation. J. Phys. Chem. A 2004, 108, 3699–3712. [Google Scholar] [CrossRef]
- Queyroy, S.; Neyertz, S.; Brown, D.; Müller-Plathe, F. Preparing Relaxed Systems of Amorphous Polymers by Multiscale Simulation: Application to Cellulose. Macromolecules 2004, 37, 7338–7350. [Google Scholar] [CrossRef]
- Molinero, V.; Goddard, W.A. Microscopic Mechanism of Water Diffusion in Glucose Glasses. Phys. Rev. Lett. 2005, 95, 045701. [Google Scholar] [CrossRef]
- Liu, P.; Izvekov, S.; Voth, G.A. Multiscale Coarse-Graining of Monosaccharides. J. Phys. Chem. B 2007, 111, 11566–11575. [Google Scholar] [CrossRef] [PubMed]
- Bu, L.; Beckham, G.T.; Crowley, M.F.; Chang, C.H.; Matthews, J.F.; Bomble, Y.J.; Adney, W.S.; Himmel, M.E.; Nimlos, M.R. The Energy Landscape for the Interaction of the Family 1 Carbohydrate-Binding Module and the Cellulose Surface Is Altered by Hydrolyzed Glycosidic Bonds. J. Phys. Chem. B 2009, 113, 10994–11002. [Google Scholar] [CrossRef]
- Srinivas, G.; Cheng, X.; Smith, J.C. A Solvent-Free Coarse Grain Model for Crystalline and Amorphous Cellulose Fibrils. J. Chem. Theory Comput. 2011, 7, 2539–2548. [Google Scholar] [CrossRef]
- Hynninen, A.-P.; Matthews, J.F.; Beckham, G.T.; Crowley, M.F.; Nimlos, M.R. Coarse-Grain Model for Glucose, Cellobiose, and Cellotetraose in Water. J. Chem. Theory Comput. 2011, 7, 2137–2150. [Google Scholar] [CrossRef]
- Markutsya, S.; Kholod, Y.A.; Devarajan, A.; Windus, T.L.; Gordon, M.S.; Lamm, M.H. A Coarse-Grained Model for β-d-Glucose Based on Force Matching. Chem. Acc. 2012, 131, 1162. [Google Scholar] [CrossRef]
- Glass, D.C.; Moritsugu, K.; Cheng, X.; Smith, J.C. REACH Coarse-Grained Simulation of a Cellulose Fiber. Biomacromolecules 2012, 13, 2634–2644. [Google Scholar] [CrossRef]
- Bellesia, G.; Chundawat, S.P.S.; Langan, P.; Redondo, A.; Dale, B.E.; Gnanakaran, S. Coarse-Grained Model for the Interconversion between Native and Liquid Ammonia-Treated Crystalline Cellulose. J. Phys. Chem. B 2012, 116, 8031–8037. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Zhan, H.; Wang, H.; Ju, Y. Secondary Structure Analysis of Native Cellulose by Molecular Dynamics Simulations with Coarse Grained Model. Chin. J. Chem. Phys. 2012, 25, 191–198. [Google Scholar] [CrossRef]
- Markutsya, S.; Devarajan, A.; Baluyut, J.Y.; Windus, T.L.; Gordon, M.S.; Lamm, M.H. Evaluation of Coarse-Grained Mapping Schemes for Polysaccharide Chains in Cellulose. J. Chem. Phys. 2013, 138, 214108. [Google Scholar] [CrossRef]
- Srinivas, G.; Cheng, X.; Smith, J.C. Coarse-Grain Model for Natural Cellulose Fibrils in Explicit Water. J. Phys. Chem. B 2014, 118, 3026–3034. [Google Scholar] [CrossRef]
- Rusu, V.H.; Baron, R.; Lins, R.D. PITOMBA: Parameter Interface for Oligosaccharide Molecules Based on Atoms. J. Chem. Theory Comput. 2014, 10, 5068–5080. [Google Scholar] [CrossRef]
- Li, L.; Pérré, P.; Frank, X.; Mazeau, K. A Coarse-Grain Force-Field for Xylan and Its Interaction with Cellulose. Carbohydr. Polym. 2015, 127, 438–450. [Google Scholar] [CrossRef] [PubMed]
- Poma, A.B.; Chwastyk, M.; Cieplak, M. Polysaccharide-Protein Complexes in a Coarse-Grained Model. J. Phys. Chem. B 2015, 119, 12028–12041. [Google Scholar] [CrossRef]
- Fan, B.; Maranas, J.K. Coarse-Grained Simulation of Cellulose Iβ with Application to Long Fibrils. Cellulose 2015, 22, 31–44. [Google Scholar] [CrossRef]
- Phan-Xuan, T.; Thuresson, A.; Skepö, M.; Labrador, A.; Bordes, R.; Matic, A. Aggregation Behavior of Aqueous Cellulose Nanocrystals: The Effect of Inorganic Salts. Cellulose 2016, 23, 3653–3663. [Google Scholar] [CrossRef]
- Poma, A.B.; Chwastyk, M.; Cieplak, M. Coarse-Grained Model of the Native Cellulose I-Alpha and the Transformation Pathways to the I-Beta Allomorph. Cellulose 2016, 23, 1573–1591. [Google Scholar] [CrossRef]
- Poma, A.; Chwastyk, M.; Cieplak, M. Elastic Moduli of Biological Fibers in a Coarse-Grained Model: Crystalline Cellulose and Beta-Amyloids. Phys. Chem. Chem. Phys. 2017, 19, 28195–28206. [Google Scholar] [CrossRef]
- Ramezani, M.G.; Golchinfar, B. Mechanical Properties of Cellulose Nanocrystal (CNC) Bundles: Coarse-Grained Molecular Dynamic Simulation. J. Compos. Sci. 2019, 3, 57. [Google Scholar] [CrossRef]
- Shishehbor, M.; Zavattieri, P.D. Effects of Interface Properties on the Mechanical Properties of Bio-Inspired Cellulose Nanocrystal (CNC)-Based Materials. J. Mech. Phys. Solids 2019, 124, 871–896. [Google Scholar] [CrossRef]
- Mehandzhiyski, A.Y.; Rolland, N.; Garg, M.; Wohlert, J.; Linares, M.; Zozoulenko, I. A Novel Supra Coarse-Grained Model for Cellulose. Cellulose 2020, 27, 4221–4234. [Google Scholar] [CrossRef]
- Rolland, N.; Mehandzhiyski, A.Y.; Garg, M.; Linares, M.; Zozoulenko, I.V. New Patchy Particle Model with Anisotropic Patches for Molecular Dynamics Simulations: Application to a Coarse-Grained Model of Cellulose Nanocrystal. J. Chem. Theory Comput. 2020, 16, 3699–3711. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Beltran-Villegas, D.J.; Jayaraman, A. Development of a New Coarse-Grained Model to Simulate Assembly of Cellulose Chains Due to Hydrogen Bonding. J. Chem. Theory Comput. 2020, 16, 4599–4614. [Google Scholar] [CrossRef]
- Reyes, G.; Aguayo, M.G.; Fernández Pérez, A.; Pääkkönen, T.; Gacitúa, W.; Rojas, O.J. Dissolution and Hydrolysis of Bleached Kraft Pulp Using Ionic Liquids. Polymers 2019, 11, 673. [Google Scholar] [CrossRef] [PubMed]
- Dong, H.; Sliozberg, Y.R.; Snyder, J.F.; Steele, J.; Chantawansri, T.L.; Orlicki, J.A.; Walck, S.D.; Reiner, R.S.; Rudie, A.W. Highly Transparent and Toughened Poly(Methyl Methacrylate) Nanocomposite Films Containing Networks of Cellulose Nanofibrils. Acs Appl. Mater. Interfaces 2015, 7, 25464–25472. [Google Scholar] [CrossRef] [PubMed]
- Ray, U.; Pang, Z.; Li, T. Mechanics of Cellulose Nanopaper Using a Scalable Coarse-Grained Modeling Scheme. Cellulose 2021. [Google Scholar] [CrossRef]
- Li, Z.; Xia, W. Coarse-Grained Modeling of Nanocellulose Network towards Understanding the Mechanical Performance. Extrem. Mech. Lett. 2020, 40, 100942. [Google Scholar] [CrossRef]
- Qin, X.; Feng, S.; Meng, Z.; Keten, S. Optimizing the Mechanical Properties of Cellulose Nanopaper through Surface Energy and Critical Length Scale Considerations. Cellulose 2017, 24, 3289–3299. [Google Scholar] [CrossRef]
- Beltran-Villegas, D.J.; Intriago, D.; Kim, K.H.C.; Behabtu, N.; Londono, J.D.; Jayaraman, A. Coarse-Grained Molecular Dynamics Simulations of α-1,3-Glucan. Soft Matter 2019, 15, 4669–4681. [Google Scholar] [CrossRef]
- Kern, N.; Frenkel, D. Fluid–Fluid Coexistence in Colloidal Systems with Short-Ranged Strongly Directional Attraction. J. Chem. Phys. 2003, 118, 9882–9889. [Google Scholar] [CrossRef]
- Shishehbor, M.; Dri, F.L.; Moon, R.J.; Zavattieri, P.D. A Continuum-Based Structural Modeling Approach for Cellulose Nanocrystals (CNCs). J. Mech. Phys. Solids 2018, 111, 308–332. [Google Scholar] [CrossRef]
- Garg, M.; Linares, M.; Zozoulenko, I. Theoretical Rationalization of Self-Assembly of Cellulose Nanocrystals: Effect of Surface Modifications and Counterions. Biomacromolecules 2020, 21, 3069–3080. [Google Scholar] [CrossRef]
- Benselfelt, T.; Nordenström, M.; Hamedi, M.M.; W\aagberg, L. Ion-Induced Assemblies of Highly Anisotropic Nanoparticles Are Governed by Ion-Ion Correlation and Specific Ion Effects. Nanoscale 2019, 11, 3514–3520. [Google Scholar] [CrossRef] [PubMed]
- Parker, R.M.; Guidetti, G.; Williams, C.A.; Zhao, T.; Narkevicius, A.; Vignolini, S.; Frka-Petesic, B. The Self-Assembly of Cellulose Nanocrystals: Hierarchical Design of Visual Appearance. Adv. Mater. 2018, 30, 1704477. [Google Scholar] [CrossRef] [PubMed]
- Mehandzhiyski, A.Y.; Zozoulenko, I. Computational Microscopy of PEDOT:PSS/Cellulose Composite Paper. ACS Appl. Energy Mater. 2019, 2, 3568–3577. [Google Scholar] [CrossRef]
- Peterson, A.; Mehandzhiyski, A.Y.; Svenningsson, L.; Ziolkowska, A.; Kádár, R.; Lund, A.; Sandblad, L.; Evenäs, L.; Lo Re, G.; Zozoulenko, I.; et al. A Combined Theoretical and Experimental Study of the Polymer Matrix Mediated Stress Transfer in a Cellulose Nanocomposite. Macromolecules 2021. [Google Scholar] [CrossRef]
- Belaineh, D.; Andreasen, J.W.; Palisaitis, J.; Malti, S.; Håkansson, K.; Wågberg, L.; Crispin, X.; Engquist, I.; Berggren, M. Controlling the Organization of PEDOT: PSS on Cellulose Structures. Acs Appl. Polym. Mater. 2019, 1, 2342–2351. [Google Scholar] [CrossRef]
- Modarresi, M.; Franco-Gonzalez, J.F.; Zozoulenko, I. Morphology and Ion Diffusion in PEDOT:Tos. A Coarse Grained Molecular Dynamics Simulation. Phys. Chem. Chem. Phys. 2018, 20, 17188. [Google Scholar] [CrossRef]
- Holm, M.V.C.; Smiatek, J. Coarse-Grained Simulations of Polyelectrolyte Complexes: MARTINI Models for Poly(Styrene Sulfonate) and Poly(Diallyldimethylammonium). J. Chem. Phys. 2015, 143, 243151. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mehandzhiyski, A.Y.; Zozoulenko, I. A Review of Cellulose Coarse-Grained Models and Their Applications. Polysaccharides 2021, 2, 257-270. https://doi.org/10.3390/polysaccharides2020018
Mehandzhiyski AY, Zozoulenko I. A Review of Cellulose Coarse-Grained Models and Their Applications. Polysaccharides. 2021; 2(2):257-270. https://doi.org/10.3390/polysaccharides2020018
Chicago/Turabian StyleMehandzhiyski, Aleksandar Y., and Igor Zozoulenko. 2021. "A Review of Cellulose Coarse-Grained Models and Their Applications" Polysaccharides 2, no. 2: 257-270. https://doi.org/10.3390/polysaccharides2020018
APA StyleMehandzhiyski, A. Y., & Zozoulenko, I. (2021). A Review of Cellulose Coarse-Grained Models and Their Applications. Polysaccharides, 2(2), 257-270. https://doi.org/10.3390/polysaccharides2020018