1. Introduction
Raman spectroscopy is extensively utilized in pharmaceutical analysis, enabling precise identification of chemical compounds, quality control, and the development of Active Pharmaceutical Ingredients (APIs). Recent advances in Raman-based analytics have significantly expanded their role in pharmaceutical applications.
Recent advances and emerging trends in Raman spectroscopy are significantly reshaping pharmaceutical analysis and quality control, particularly through their impact on real-time and on-site applications. Raman spectroscopy is being increasingly recognized by regulatory bodies such as the European and US Pharmacopoeias as a valid analytical process. However, it still lacks specific inclusion in individual substance monographs, which has limited its broader clinical adoption [
1]. Raman spectroscopy offers several advantages, including non-destructive and label-free analysis, compatibility with various physical states (solids, liquids, gases), and minimal sample preparation. These features make it a powerful tool for detecting counterfeit drugs, supporting the development of personalized medicines, and enabling continuous pharmaceutical manufacturing processes.
Spontaneous Raman spectroscopy is effective for the identification of raw material and the monitoring of physical forms such as polymorphs [
2]. UV and deep-UV Raman techniques enhance sensitivity to specific molecular vibrations, making them suitable for biological macromolecule analysis [
3]. Surface-enhanced Raman spectroscopy (SERS) increases signal intensity by using metallic nanostructures, thus enabling the detection of trace analytes [
4]. Coherent Raman methods like CARS and SRS allow for rapid, label-free molecular imaging, useful for in vivo studies and understanding drug distribution [
5].
Technological innovations have significantly expanded the capabilities of Raman spectroscopy. Raman microscopes now facilitate hyperspectral 3D imaging. Immersion fiber optic probes facilitate in-line monitoring during manufacturing. Handheld devices are increasingly being used for field analysis and verification of raw materials. Spatial Offset Raman Spectroscopy (SORS) enables analysis through opaque packaging, while Fiber-Enhanced Raman Spectroscopy (FERS) allows for sensitive detection of compounds in gases and liquids [
6]. Machine learning and chemometric techniques enable classification and quantitative analysis of complex datasets. User-friendly software tools such as RAMANMETRIX help non-expert users perform sophisticated spectral analyses. However, the standardization and reproducibility of data processing workflows remain challenges for broader industrial and clinical adoption [
7]. Also, Raman spectroscopy has been used for the quality control of 3D-printed medications, virus detection via SERS-based microdevices, and monitoring of drug resistance in pathogens [
8].
Raman spectroscopy has become a versatile analytical tool in pharmaceutical science, with techniques such as dispersive Raman, FT-Raman, SERS, and resonance Raman being widely applied for quality control, counterfeit detection, and drug formulation analysis [
9,
10]. Recent advances have demonstrated the integration of Raman data with machine learning (ML) and deep learning (DL) methods to enhance classification, quantification, and process monitoring tasks. For example, deep learning-based SERS strategies have been used for identifying protein binding sites [
11], while CNNs have been employed for real-time quality control in tablet manufacturing [
12].
Several studies have demonstrated the efficacy of ML algorithms—including SVMs, Random Forest, XGBoost, and artificial neural networks—for spectral classification and drug dissolution prediction [
13,
14,
15]. Additionally, applications range from particle identification in injectable drug production [
16] to the detection of drug residues on latent fingermarks [
17]. ML-enhanced Raman spectroscopy has also proven effective for colonic drug delivery optimization [
18], falsified medicine detection [
19], and CHO cell culture monitoring [
20]. Furthermore, collaborative platforms have enabled large-scale model development for monoclonal antibody quantification [
21]. These works collectively underscore the increasing synergy between Raman spectroscopy and ML in advancing pharmaceutical research and quality assurance.
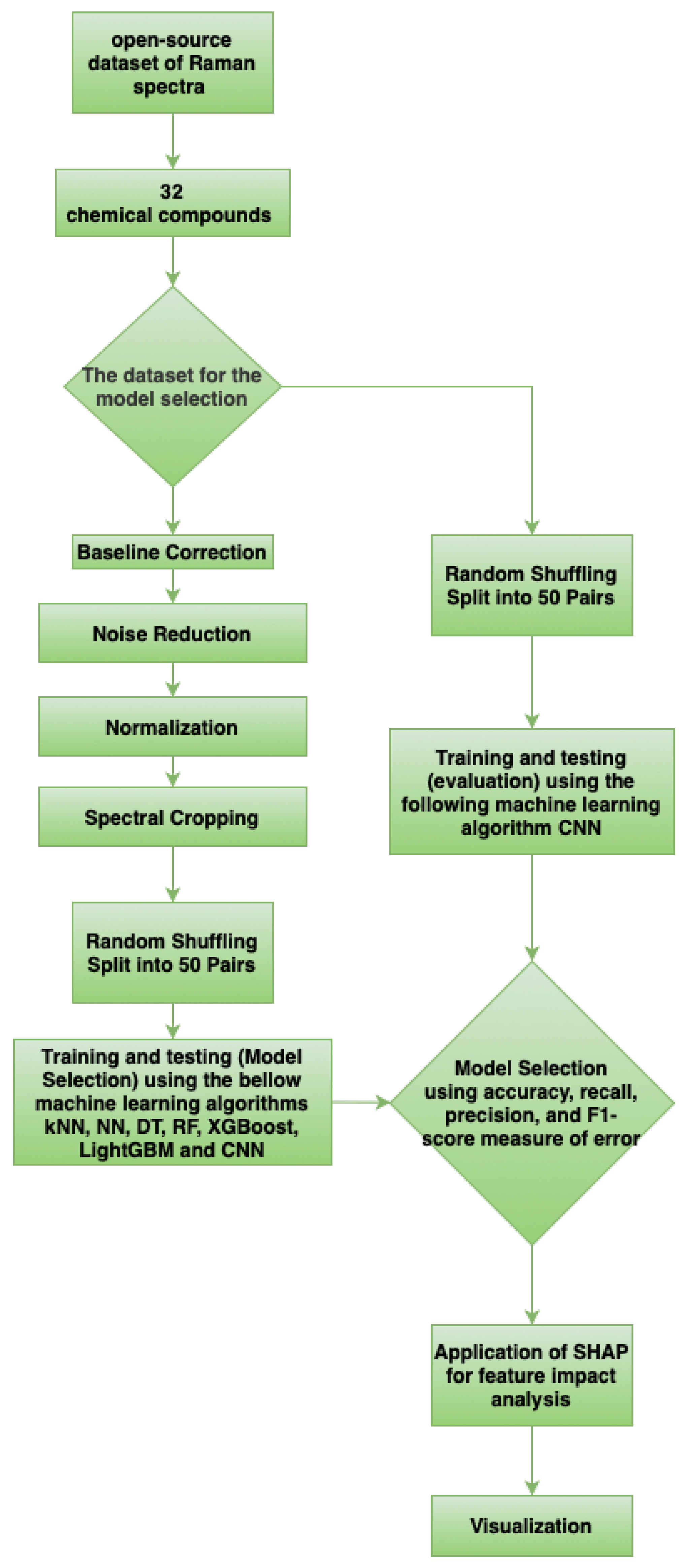
In this study, we present a systematic comparison of multiple machine learning algorithms for the classification of pharmaceutical compounds based on Raman spectral data. The evaluated models include both traditional approaches—such as Support Vector Machines (SVMs), Random Forests, and k-Nearest Neighbors (k-NN)—as well as a deep learning architecture in the form of a one-dimensional Convolutional Neural Network (1D CNN). In addition to assessing classification performance, we incorporate SHAP-based explainability techniques to interpret the decision-making process of the models. This integration not only enhances transparency but also offers deeper chemical insight into the spectral regions that are most influential for compound differentiation.
4. Discussion
Raman spectroscopy is evolving into a robust analytical tool that spans pharmaceutical research, manufacturing, and clinical diagnostics. With ongoing advancements in instrumentation and interdisciplinary collaboration, it is poised to become an integral part of pharmaceutical quality control. Regulatory integration and user-friendly technology will be critical for its widespread acceptance in hospitals and pharmacies.
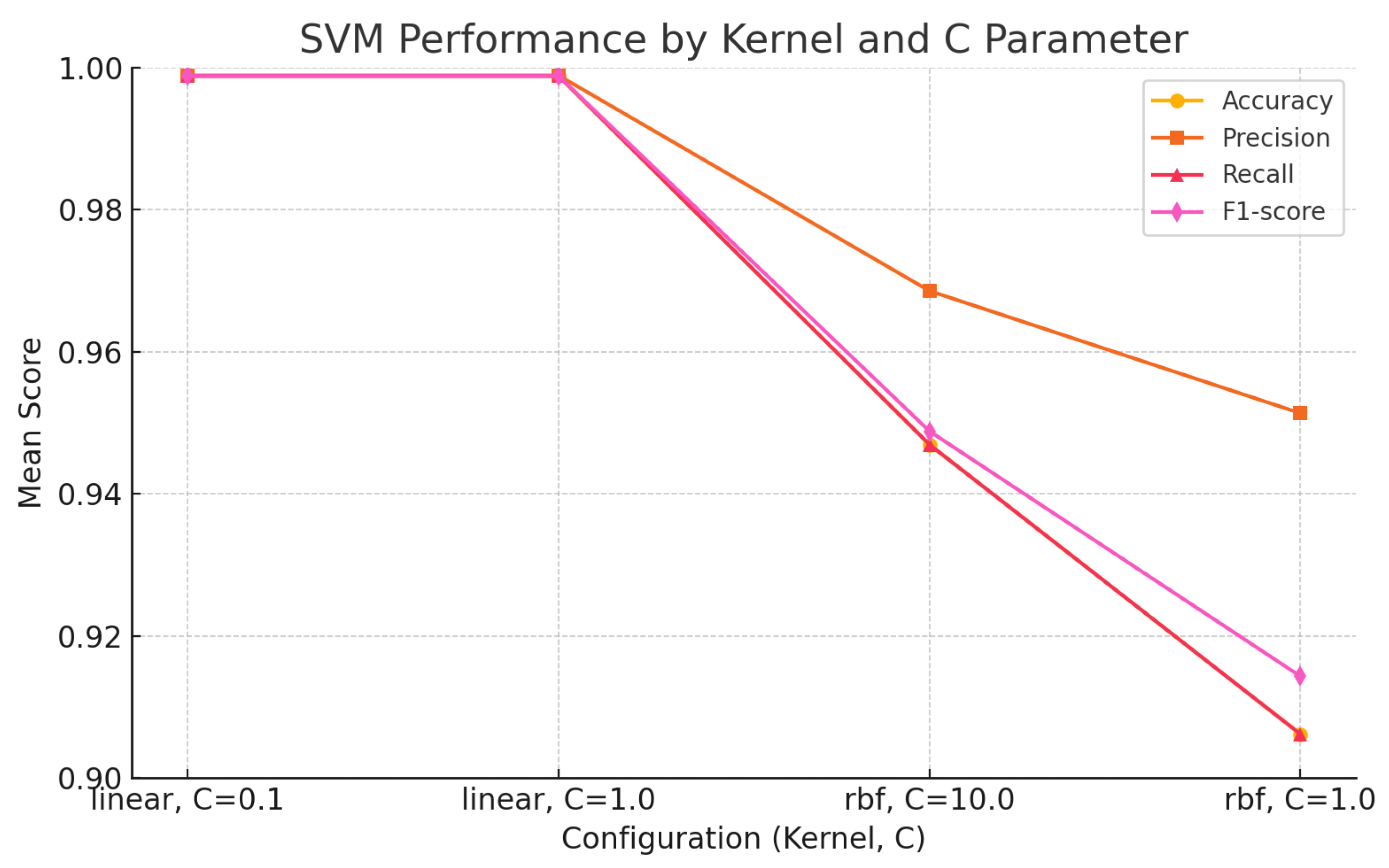
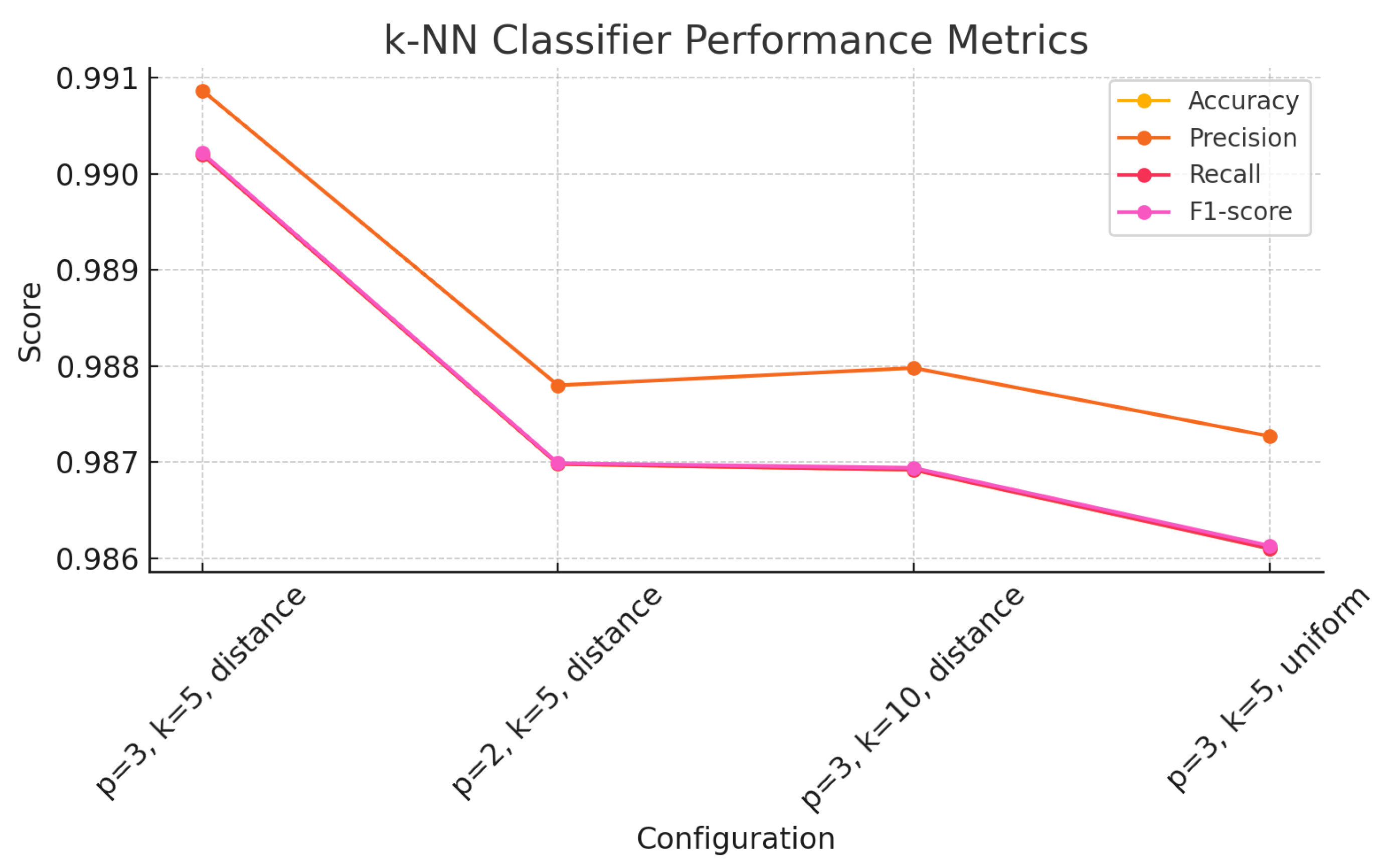
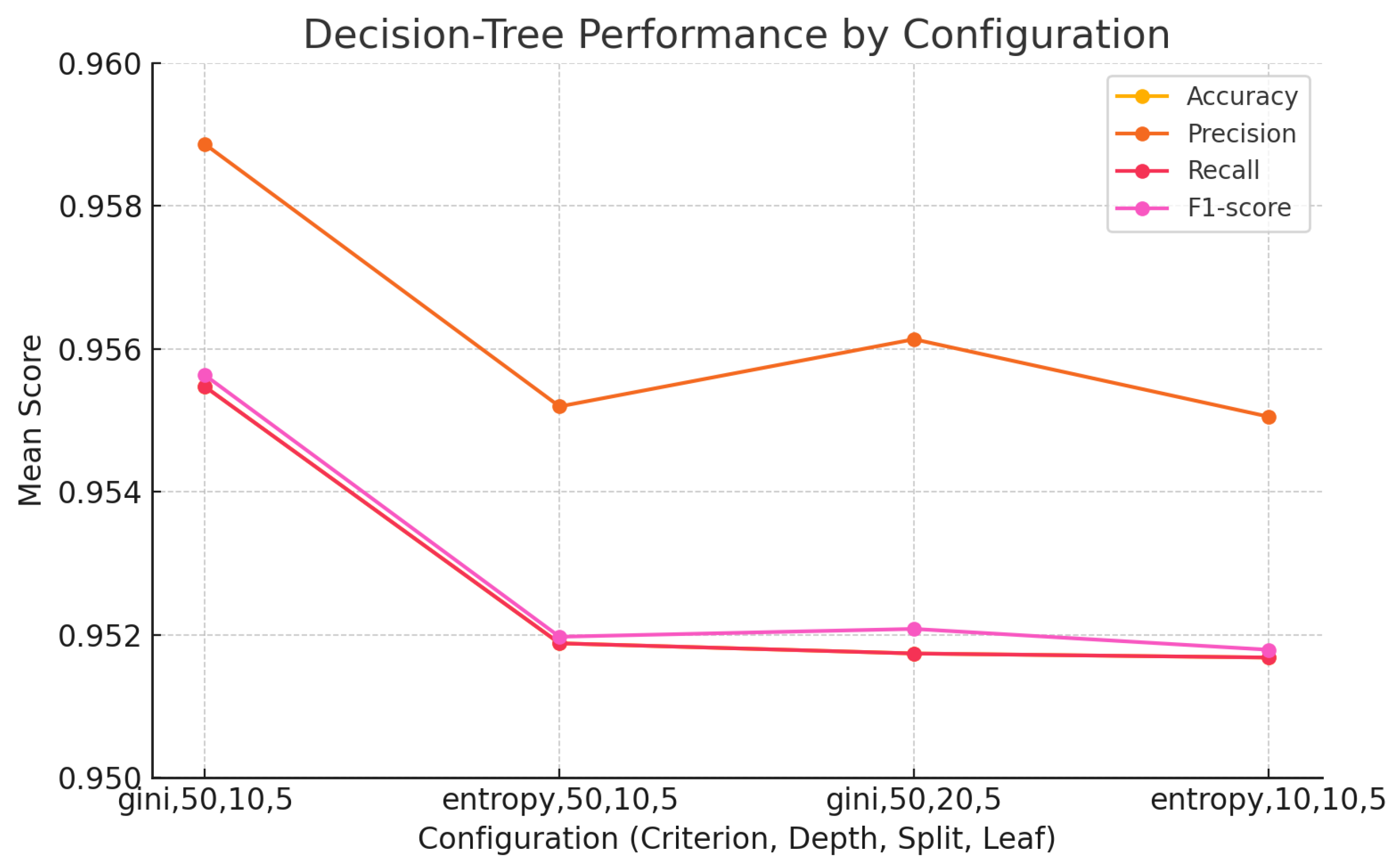
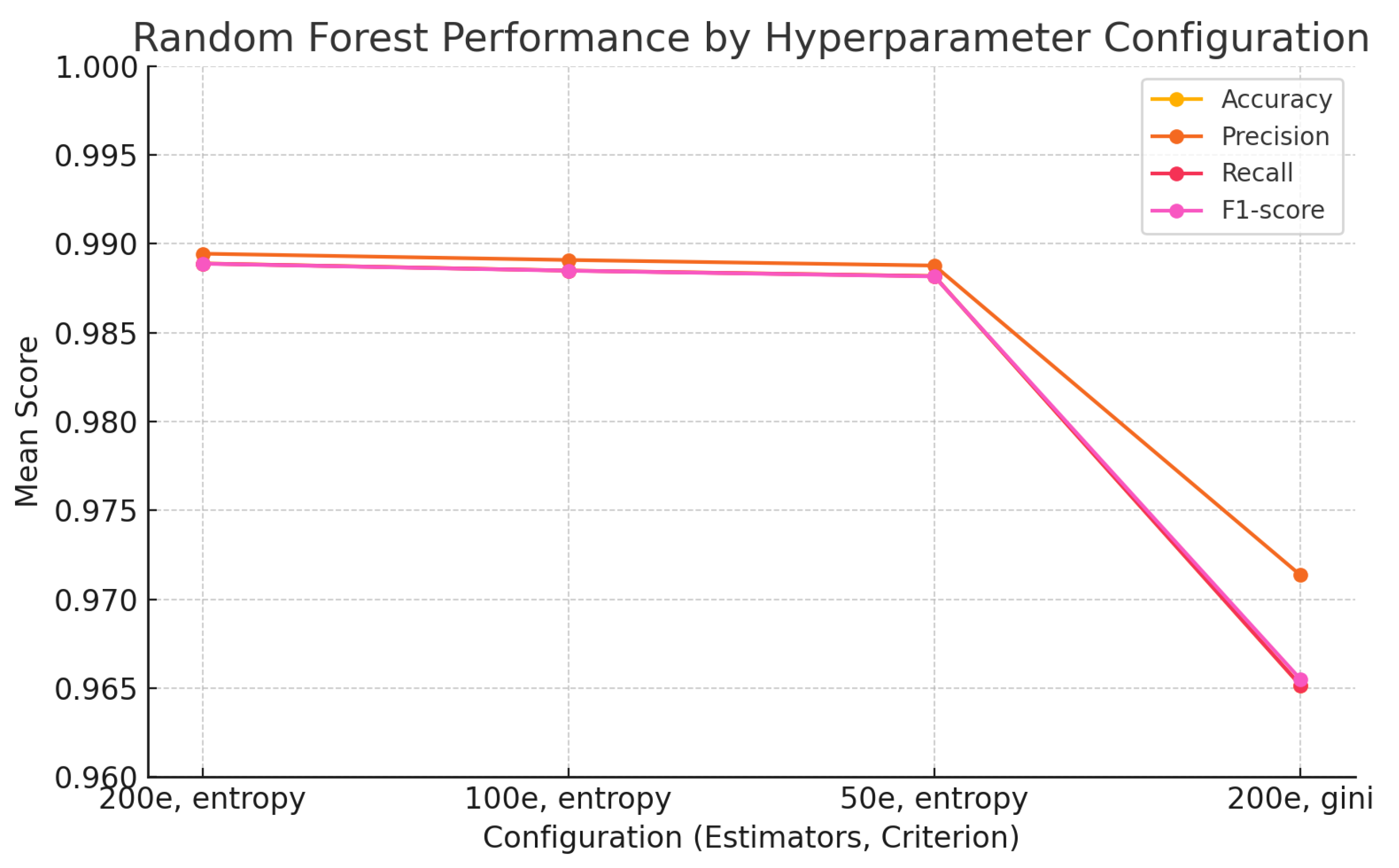
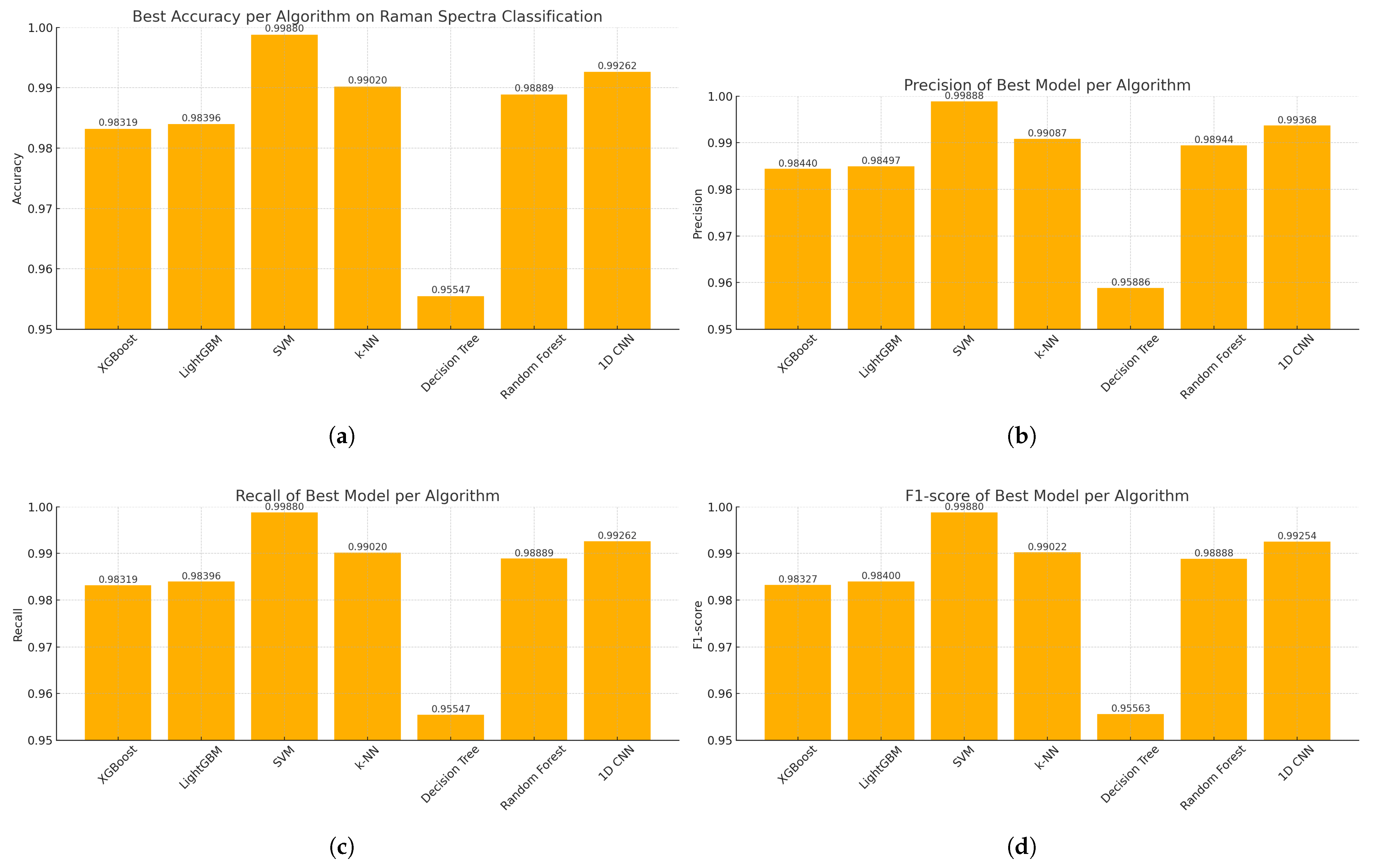
The findings of this study underscore the growing relevance of explainable machine learning techniques in the context of pharmaceutical spectroscopy. Among the evaluated models, the Support Vector Machine (SVM) with a linear kernel achieved the highest classification accuracy (99.88%), highlighting its effectiveness in handling high-dimensional spectral data when class boundaries are well defined. Meanwhile, the 1D Convolutional Neural Network (CNN) also demonstrated strong performance (accuracy of 99.26%) and offered unique advantages in terms of interpretability through SHAP-based explanations.
Notably, the SHAP heatmap generated for the CNN revealed localized spectral regions—particularly in the 300–350 cm−1, 600–920 cm−1 and 1050 cm−1 ranges—that were consistently influential across multiple classes. These regions correspond to known Raman-active vibrational modes, indicating that the CNN model was able to autonomously learn chemically meaningful features directly from raw spectral inputs. In contrast, the SVM’s SHAP visualization exhibited a more diffuse pattern of feature importance, suggesting that while the model performed well, it distributed decision relevance more broadly across the spectrum without clear localization.
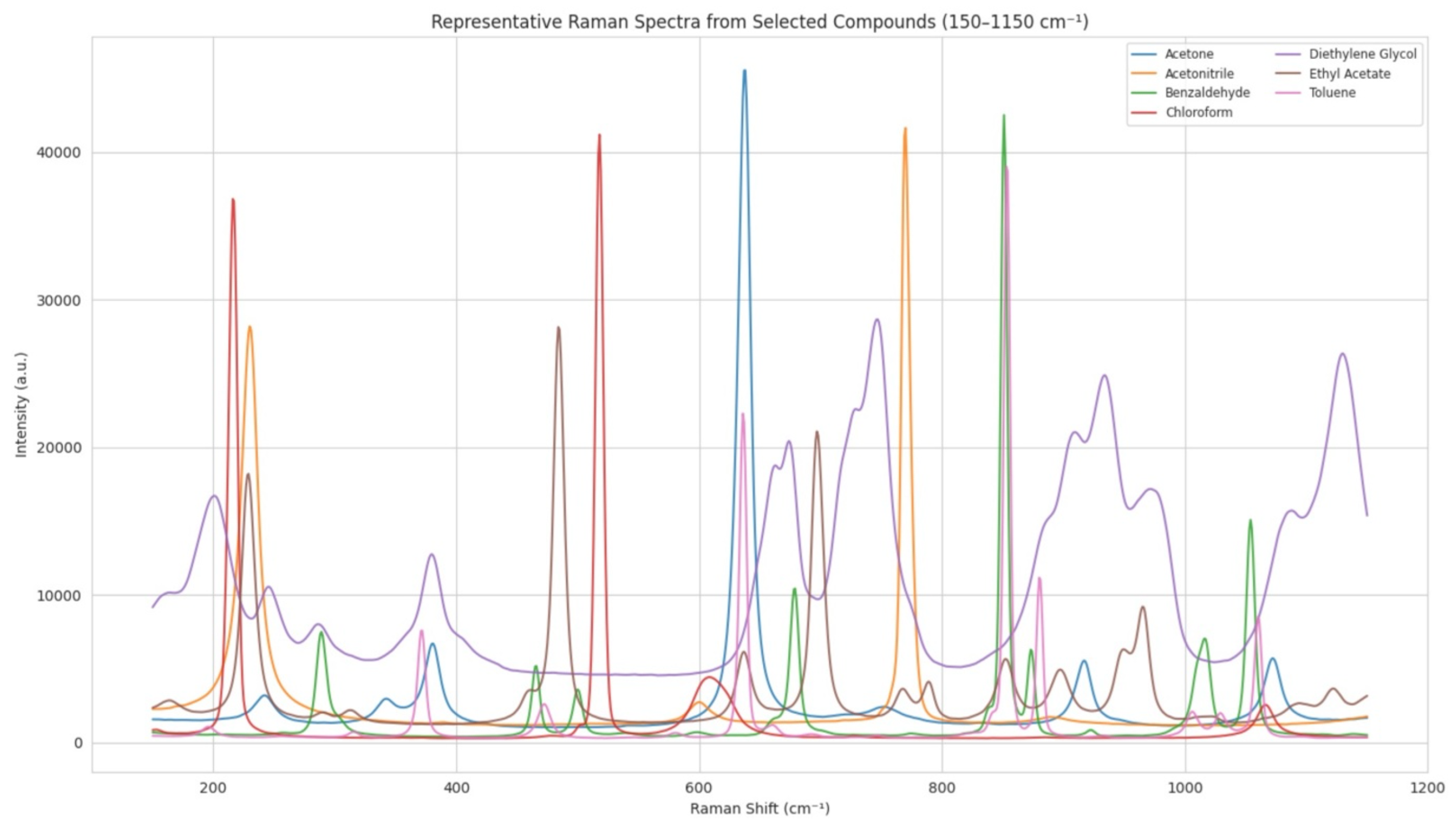
The SHAP analysis revealed that the machine learning model places significant importance on Raman shift regions that correspond to chemically meaningful vibrational modes. As detailed in
Table 10, many of the most influential spectral regions align with characteristic functional group vibrations well documented in the Raman spectroscopy literature. For instance, the 790–920 cm
−1 region, which showed high SHAP values, corresponds to out-of-plane C–H bending in mono- and disubstituted aromatic rings—functional motifs frequently found in pharmacologically active compounds such as toluene derivatives. Similarly, the 1050–1150 cm
−1 region, associated with C–O and C–N stretching vibrations, is indicative of ether, ester, and amine functionalities, which are prevalent in a wide range of bioactive molecules and excipients. These findings not only affirm that the model is capturing structurally and pharmacologically relevant spectral features but also enhance the interpretability and mechanistic plausibility of the classification results [
28,
29].
This analysis revealed important spectral regions used by the model to distinguish compounds. While some of these regions correspond to well-known vibrational modes (e.g., C–O or ring deformation bands), others do not coincide with visually dominant peaks. This suggests that the model may exploit subtle, low-amplitude patterns, such as weak shoulders or overlapping bands, highlighting the potential of explainable AI to uncover latent spectral information beyond conventional inspection.
This distinction in interpretability has practical implications. In pharmaceutical quality control scenarios, where understanding which spectral regions contribute to a prediction is critical, CNN-based models may provide not only high accuracy but also actionable insights. Such transparency supports regulatory compliance, enhances trust in automated systems, and facilitates the identification of anomalies or adulterations in real-world settings.
This study demonstrates that integrating explainability tools such as SHAP with high-performing machine learning models enhances both the transparency and utility of Raman spectroscopy in pharmaceutical analysis. Future work may explore extending this framework to other spectroscopic modalities, implementing real-time classification pipelines, and expanding the chemical space to include complex formulations and biological matrices.
In future work, we plan to extend our benchmarking to include classical chemometric techniques such as Principal Component Analysis (PCA) and Linear Discriminant Analysis (LDA), which are standard tools in spectroscopy. This would allow for a direct comparison between traditional statistical approaches and machine learning models in terms of both classification performance and interpretability.
Also, we plan to extend the SHAP-based interpretation of our models by systematically analyzing whether the most influential spectral regions correspond to known vibrational modes associated with specific chemical structures. While this was not feasible in the present study due to the large number of classes (32 compounds), such analysis could be performed in focused studies with fewer, well-characterized compounds. This would enhance the interpretability of ML-based spectral classification and potentially provide new insights into structure–spectrum relationships in Raman spectroscopy.






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}