
The Nature of Lignin and Implications for Its Technical Use as a Source for Biogenic Aromatics—A Review


Abstract
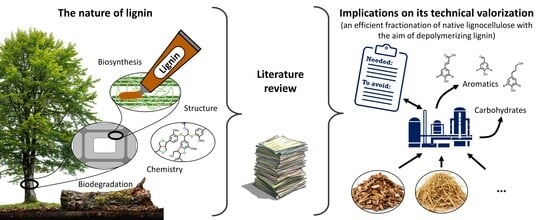
1. Introduction
2. Biosynthesis of Lignocellulose
- Apparently there is a clear order in the synthesis of lignocellulose: first, a matrix of carbohydrate polymers is built up, into which lignin is then incorporated and where it acts as a “curing agent” [16]. It seems logical to proceed in the opposite direction when degrading the material and remove the lignin first.
- Regions with highest lignin concentrations are more difficult to access as they are found in the compound middle lamella in between two cells, thus surrounded by secondary cell walls. Lignified plant cells are typically hollow on the inside and can therefore be accessed from the inside. However, the high mobility of lignin monomers within the cell wall indicates a certain diffusibility of the cell walls, which is examined further in Section 4.1.
- Unlike the formation of other biopolymers like cellulose, lignin polymerization is not the result of a direct enzymatic action, but a chemical process. Accordingly, a reverse, selective enzymatic cleavage of lignin bonds does not appear to be the obvious solution for depolymerization. The natural biodegradation of lignin is further explored in Section 5.
- The lack of regulation leads to a high degree of heterogeneity and poses a challenge for lignin depolymerization. The insight that the formation of β-O-4 ether bonds is favored may prove helpful for the development of depolymerization approaches.
3. Structure of Lignocellulose

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Classification | Examples | Lignin Content in % [73,74] | Monolignol Shares in % [43,74,75,76] | Typical Unconventional Monomers [29] | Bond Frequencies in % 2 | |
|---|---|---|---|---|---|---|
| Gymnosperms | ↦ Softwoods | Spruce, Pine | 27–31 | H: <5 G: >95 S: →0 | Dihydroconiferyl alcohol, guaiacylpropane-1,3-diol | β-O-4: ~60 [30,31,32,33,34,35,36,37,38,39,40,41,42,43,44,45,46,47,48,49,50] 2 PC: ~10 Res: ~5 DBDOX: ~10 Sp: 1–2 BP: ~1 |
| Angiosperms | ↦Dicots 1 ↦Hardwoods 1 | Poplar, birch, Beech | 19–25 | H: <5 G: 20–50 S: 45–75 | Acylation with p-hydroxybenzoates and acetates | β-O-4: ~80 [60,61,62,63,64,65,66,67,68,69,70,71,72,73,74,75] 2 PC: ~5 Res: ~5 DBDOX: ~3 Sp: 1–2 BP: 1–2 |
| ↦Monocots 1 ↦Grasses 1 | Straw, switch-grass, corn stalk, Miscanthus | 6–23 | H: <5 G: 23–80 S: 20–75 | Acylation with p-coumarates and acetate; ferulate-polysaccharide-esters; tricin | β-O-4: ~75 [30,31,32,33,34,35,36,37,38,39,40,41,42,43,44,45,46,47,48,49,50,51,52,53,54,55,56,57,58,59,60] 2 PC: <10 THF: ~5 DBDOX: ~10 Sp: 1–2 BP: 1–2 |
- Lignin represents a group of phenolic macromolecules with an extremely heterogeneous composition. It consists of a large number of monomers that are connected by very different types of bonds, and furthermore, lignin concentration, composition and structure vary within the cell wall. This means that, firstly, there is no single type of bond that can be used as a point of attack to cleave all monomers from one another. Secondly, this heterogeneity naturally imposes restrictions on the maximally achievable product selectivity.
- The majority of the bonds are ether bonds, in particular β-O-4 bonds. These bonds are therefore the most suitable effective target for selective cleavage. Thus, a focus will be set on the chemistry of these ether bonds. The maximally achievable monomer yield by cleaving the ether bonds can then be estimated by the square of the ether bond content [118].
- In order to release lignin from the cell wall, not only the bonds between lignin monomers have to be considered but also the bonds between the lignin molecules and the carbohydrates. These appear to be cleavable mainly through the cleavage of carbon–oxygen bonds (ester, ether and glycosidic bonds). However, there are still many unknowns regarding the covalent linkages between lignin and carbohydrates.
- Lignin cross-links primarily with hemicellulose, thus filling the space between aggregates of cellulose microfibrils and also filling existing pores in the carbohydrate matrix. The removal of lignin could thus lead to a considerably higher porosity and hence diffusibility of the cell wall.
4. Chemistry of Lignocellulose and Lignin
4.1. Solubility
- Lignin appears to be rather alkali-soluble than acid-soluble in aqueous media.
- Lignin has an intermediate solubility with both rather polar and rather non-polar moieties. As evidenced by different approaches, mixtures of water with an organic solvent appear to be optimal for solubilizing native lignin and thereof derived structures that are not further functionalized.
- Smaller lignin fragments tend to be more soluble in a broader range of solvents.
- Besides a matching solubility, a high moisture content is essential for a good diffusion of small molecules through the secondary cell wall.
4.2. Bond Stabilities
- Lignin bonds directly involving the aromatic ring (4-O-5, β-1, 5-5, β-5) are clearly more stable than bonds involving only aliphatic carbon or oxygen (α-O, α-β, β-O). Based on the calculated bond dissociation enthalpies (BDEs) regarding homolytic bond dissociation, the β-O-4 structure and the α-β bond in phenylcoumaran structures appear to be the simplest target for complete cleavage of two monomers.
- The oxidation and, to a lesser extent, the methoxylation of aliphatic OH groups apparently decrease the stabilities of the β-O-4 bond.
- The lignin–carbohydrate complexes (LCCs) predominating according to the current state of knowledge contain bonds not directly involving the aromatic ring; consequently, their stability seems to be in a similar range to the stability of the β-O-4 bond.
- Side groups, especially the α-OH group, are less stable than the inter-unit bonds, so lignin modifications typically occur at much milder conditions than depolymerization reactions.
| Bond Structure | Specific Bonds and Their BDE in kJ/mol | |
|---|---|---|
| Lignin bonds | β-O-4 ether [149,150] | β-O ≈ 268–301, α-β ≈ 314–322 |
| Biphenyl ether [149] | 4-O/O-5 ≈ 326–347 | |
| Phenylcoumaran [152] | β-5′ ≈ 393–422, α-O ≈ 188–226, α-β ≈ 238–293 | |
| Resinol [155] | β-β′ ≈ 339–343, α-β ≈ 272–280, β-γ ≈ 330–339, α-O ≈ 284, γ-O ≈ 330–334 | |
| DBDOX [149,150,155] | 5′-5″ ≈ 468–497, α-O ≈ 176–192, β-O ≈ 238 | |
| Spirodienone [156] | α-β ≈ 380, α′-β′ ≈ 301, α-O ≈ 343, α′-O ≈ 376, 1-α′ ≈ 192, 1-β′ ≈ 213, β-O ≈ 301 | |
| LCCs | α-ether 1 [160] | α-O ≈ 319-340, O-S5 ≈ 274-291 |
| α-ester 1 [160] | α-O ≈ 327, O-S5 ≈ 413 | |
| Phenyl glycoside 1 [160] | 4-O ≈ 415-427, O-S1 ≈ 243–280 |
4.3. Effect of Thermo-Chemical Treatments
4.3.1. Effect of Heat
- When only heated, lignin might already change from a glassy to a rubbery state at temperatures below 100 °C. Significant depolymerization of native lignin in the lignocellulose complex, however, starts to occur only at temperatures around 350 °C. Repolymerization reactions are already possible at lower temperatures than 250 °C and might lead to the formation of new, stable bonds to aromatic nuclei.
- Lignin apparently has a higher mobility than the carbohydrate polymers.
4.3.2. Effect of Solvents
- In combination with the application of heat, solvents clearly have an important effect on lignin reactions in various ways.
- The important tasks are the stabilization and transport of released lignin fragments, and the implementation of these tasks is essential for which reactions occur next. Thereby, the suitability of a solvent depends on the solubility and thus on the characteristics of lignin fragments released by a process.
- Solvents can act as reactants themselves and donate hydrogen or attach by nucleophilic attack and thereby influence cleavage reactions but also condensation reactions.
4.3.3. Effect of Acidic Environments
- Ether bonds within lignin as well as lignin–carbohydrate complexes (LCCs) are cleavable under acidic conditions with benzylic carbocations as typical intermediates. However, lignin fragments are only removed when they are sufficiently soluble in the solvent used. Repolymerization reactions, e.g., through the formation of reactive carbocations, can easily occur under acidic conditions.
- Besides these reactions on lignin, carbohydrates (especially hemicellulose) are significantly hydrolyzed under acidic conditions.
4.3.4. Effect of Alkaline Environments
- Under alkaline conditions, ether bonds can be cleaved with quinone methide structures as typical intermediates and released fragments are typically soluble, so a delignification can be achieved. However, different intermediates are prone to repolymerization reactions.
- Among the lignin–carbohydrate complexes (LCCs), ester bonds in particular are easily cleavable by alkali, leading to the high solubilization of ester-bound fragments, whereas other LCCs appear to be more challenging to cleave.
- Alkaline treatment removes lignin and hemicellulose and leaves cellulose more or less intact.
4.3.5. Effect of Reductive Environments
- As essential effect, reductive environments stabilize reactive fragments released during lignin fragmentation and depolymerization and thereby reduce repolymerization reactions and increase monomer yields.
- It is uncertain, and depends on the process conditions and lignin structure, how and to what extent reductive environments furthermore induce ether bond cleavage by hydrogenolysis reactions. For such reactions, the existence of an oxygen functionality at the α-carbon appears to be essential, under the reaction conditions considered.
4.3.6. Effect of Oxidative Environments
- Oxidative cleavage apparently rather occurs at carbon–carbon bonds than at ether bonds. Thus, a larger lignin fraction might be depolymerizable by oxidative methods.
- There seems to be an increased risk of side and repolymerization reactions under oxidative conditions.
5. Biodegradation of Lignocellulose
- Nature’s response to the complex lignin structure is an oxidative and nonspecific lignin degradation mediated by an extracellular system [267]. Similarly to its role during lignin synthesis, enzymatic action is rather indirect during lignin degradation, which is instead largely based on radical formation as well. There is no recycling to the original lignin monomers, which would require selective, reductive cleavage. Instead, further oxidation is the main pathway for lignin degradation in nature, leading to either degraded lignin (brown-rot) or a variety of lignin fragments, which are biologically funneled and incorporated in metabolism (white-rot). Can these natural mechanisms be utilized or copied?
- ◦
- Copying this mechanism in vitro would presumably require a complex set of enzymes, co-factors and auxiliary molecules and finally end up in a complex, heterogeneous mixture of possibly condensed lignin fragments. This process appears to be much more complex than the in vitro enzymatic cleavage of cellulose, which requires a small subset of enzymes selectively leading to the original glucose monomers, which nevertheless already poses some challenges in terms of costs and reaction rates [307].
- ◦
- Adapting the use of redox-active molecules for lignin degradation from brown-rot fungi, but generating these chemically is a nature-mimicking strategy currently being investigated [308,309,310,311]. However, condensation reactions in the lignin can be expected due to the non-selective, radical-based procedure.
- ◦
- Applying lignin-degrading microorganisms is generally regarded as one possibility of biological pre-treatment of lignocellulose. However, biological degradation is a slow process and microorganisms will degrade both lignin and carbohydrates so that pre-treatment comes along with a loss in both [312].
- ◦
- Applying genetically modified microorganisms to conduct lignolysis as well as biological funneling to specific reactive intermediates could be used to avoid the expense of enzyme production and achieve a high selectivity (even single products) [280]. However, the challenges of slow reaction rates remain and the challenges of handling fermentations with genetically modified organisms are added.
- Biological processes typically have the advantage of high selectivity and specificity, but these do not appear to be provided in the case of lignin biodegradation. Instead, there are some challenges and disadvantages for biodegradation that do not exist or could be overcome in thermo-chemical processes (among them are the insolubility, high molecule sizes, possible toxicity of degradation products, the high carbon-to-nitrogen ratio of lignocellulose and the slow apparent reaction rates in the case of biodegradation). What remains are the challenges of heterogeneity and bond stability, and the disadvantage of reactivity is even more severe in the case of thermo-chemical processes.
6. Discussion of the Derived Implications
- 1.
- The primary focus should be on the gentle removal of lignin and possibly hemicellulose, enabling the utilization of all fractions;
- 2.
- Lignin–carbohydrate complexes (LCCs) and β-O-4 ether bonds should be cleaved;
- 3.
- Liberated lignin fragments must be soluble;
- 4.
- Reactive lignin fragments should be actively stabilized.
7. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Weng, J.-K.; Chapple, C. The origin and evolution of lignin biosynthesis. New Phytol. 2010, 187, 273–285. [Google Scholar] [CrossRef]
- Lowry, B.; Lee, D.; Hébant, C. The origin of land plants: A new look at an old problem. Taxon 1980, 29, 183–197. [Google Scholar] [CrossRef]
- Renault, H.; Alber, A.; Horst, N.A.; Basilio Lopes, A.; Fich, E.A.; Kriegshauser, L.; Wiedemann, G.; Ullmann, P.; Herrgott, L.; Erhardt, M.; et al. A phenol-enriched cuticle is ancestral to lignin evolution in land plants. Nat. Commun. 2017, 8, 14713. [Google Scholar] [CrossRef] [PubMed]
- Raven, J.A. Physiological correlates of the morphology of early vascular plants. Bot. J. Linn. Soc. 1984, 88, 105–126. [Google Scholar] [CrossRef]
- Guerriero, G.; Hausman, J.-F.; Strauss, J.; Ertan, H.; Siddiqui, K.S. Lignocellulosic biomass: Biosynthesis, degradation, and industrial utilization. Eng. Life Sci. 2016, 16, 1–16. [Google Scholar] [CrossRef]
- Meents, M.J.; Watanabe, Y.; Samuels, A.L. The cell biology of secondary cell wall biosynthesis. Ann. Bot. 2018, 121, 1107–1125. [Google Scholar] [CrossRef]
- Kadereit, J.W.; Körner, C.; Kost, B.; Sonnewald, U. Strasburger—Lehrbuch der Pflanzenwissenschaften; Springer: Berlin/Heidelberg, Germany, 2014; ISBN 978-3-642-54435-4. [Google Scholar]
- Ludmila, H.; Michal, J.; Andrea, Š.; Aleš, H. Lignin, potential products and their market value. Wood Res. 2015, 60, 973–986. [Google Scholar]
- Diercks, R.; Arndt, J.-D.; Freyer, S.; Geier, R.; Machhammer, O.; Schwartze, J.; Volland, M. Raw Material Changes in the Chemical Industry. Chem. Eng. Technol. 2008, 31, 631–637. [Google Scholar] [CrossRef]
- Vishtal, A.; Kraslawski, A. Challenges in industrial applications of technical lignins. BioResources 2011, 6, 3547–3568. [Google Scholar] [CrossRef]
- Park, J.; Riaz, A.; Insyani, R.; Kim, J. Understanding the relationship between the structure and depolymerization behavior of lignin. Fuel 2018, 217, 202–210. [Google Scholar] [CrossRef]
- Phongpreecha, T.; Hool, N.C.; Stoklosa, R.J.; Klett, A.S.; Foster, C.E.; Bhalla, A.; Holmes, D.; Thies, M.C.; Hodge, D.B. Predicting lignin depolymerization yields from quantifiable properties using fractionated biorefinery lignins. Green Chem. 2017, 19, 5131–5143. [Google Scholar] [CrossRef]
- Abu-Omar, M.M.; Barta, K.; Beckham, G.T.; Luterbacher, J.S.; Ralph, J.; Rinaldi, R.; Román-Leshkov, Y.; Samec, J.S.M.; Sels, B.F.; Wang, F. Guidelines for performing lignin-first biorefining. Energy Environ. Sci. 2021, 14, 262–292. [Google Scholar] [CrossRef]
- Donaldson, L.A. Lignification and lignin topochemistry—An ultrastructural view. Phytochemistry 2001, 57, 859–873. [Google Scholar] [CrossRef] [PubMed]
- Terashima, N.; Yoshida, M.; Hafrén, J.; Fukushima, K.; Westermark, U. Proposed supramolecular structure of lignin in softwood tracheid compound middle lamella regions. Holzforschung 2012, 66, 907–915. [Google Scholar] [CrossRef]
- Henriksson, G. What are the biological functions of lignin and its complexation with carbohydrates? Nord. Pulp Pap. Res. J. 2017, 32, 527–541. [Google Scholar] [CrossRef]
- Saka, S.; Thomas, R.J.; Gratzl, J.S.; Abson, D. Topochemistry of delignification in Douglas-fir wood with soda, soda-anthraquinone and karft pulping as determined by SEM-EDXA. Wood Sci. Technol. 1982, 16, 139–153. [Google Scholar] [CrossRef]
- Ji, Z.; Ma, J.-F.; Zhang, Z.-H.; Xu, F.; Sun, R.-C. Distribution of lignin and cellulose in compression wood tracheids of Pinus yunnanensis determined by fluorescence microscopy and confocal Raman microscopy. Ind. Crops Prod. 2013, 47, 212–217. [Google Scholar] [CrossRef]
- Bollhöner, B.; Zhang, B.; Stael, S.; Denancé, N.; Overmyer, K.; Goffner, D.; van Breusegem, F.; Tuominen, H. Post mortem function of AtMC9 in xylem vessel elements. New Phytol. 2013, 200, 498–510. [Google Scholar] [CrossRef]
- Cornelis, S.; Hazak, O. Understanding the root xylem plasticity for designing resilient crops. Plant Cell Environ. 2022, 45, 664–676. [Google Scholar] [CrossRef]
- Pesquet, E.; Zhang, B.; Gorzsás, A.; Puhakainen, T.; Serk, H.; Escamez, S.; Barbier, O.; Gerber, L.; Courtois-Moreau, C.; Alatalo, E.; et al. Non-cell-autonomous postmortem lignification of tracheary elements in Zinnia elegans. Plant Cell 2013, 25, 1314–1328. [Google Scholar] [CrossRef]
- Smith, R.A.; Schuetz, M.; Roach, M.; Mansfield, S.D.; Ellis, B.; Samuels, L. Neighboring parenchyma cells contribute to Arabidopsis xylem lignification, while lignification of interfascicular fibers is cell autonomous. Plant Cell 2013, 25, 3988–3999. [Google Scholar] [CrossRef] [PubMed]
- Yao, T.; Feng, K.; Xie, M.; Barros, J.; Tschaplinski, T.J.; Tuskan, G.A.; Muchero, W.; Chen, J.-G. Phylogenetic Occurrence of the Phenylpropanoid Pathway and Lignin Biosynthesis in Plants. Front. Plant Sci. 2021, 12, 704697. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.-J.; Miao, Y.-C.; Zhang, K.-W. Sequestration and transport of lignin monomeric precursors. Molecules 2011, 16, 710–727. [Google Scholar] [CrossRef] [PubMed]
- Chapelle, A.; Morreel, K.; Vanholme, R.; Le-Bris, P.; Morin, H.; Lapierre, C.; Boerjan, W.; Jouanin, L.; Demont-Caulet, N. Impact of the absence of stem-specific β-glucosidases on lignin and monolignols. Plant Physiol. 2012, 160, 1204–1217. [Google Scholar] [CrossRef]
- Terashima, N.; Ko, C.; Matsushita, Y.; Westermark, U. Monolignol glucosides as intermediate compounds in lignin biosynthesis. Revisiting the cell wall lignification and new 13 C-tracer experiments with Ginkgo biloba and Magnolialiliiflora. Holzforschung 2016, 70, 801–810. [Google Scholar] [CrossRef]
- Le Roy, J.; Huss, B.; Creach, A.; Hawkins, S.; Neutelings, G. Glycosylation Is a Major Regulator of Phenylpropanoid Availability and Biological Activity in Plants. Front. Plant Sci. 2016, 7, 735. [Google Scholar] [CrossRef]
- Chandrakanth, N.N.; Zhang, C.; Freeman, J.; de Souza, W.R.; Bartley, L.E.; Mitchell, R.A.C. Modification of plant cell walls with hydroxycinnamic acids by BAHD acyltransferases. Front. Plant Sci. 2022, 13, 1088879. [Google Scholar] [CrossRef]
- Del Río, J.C.; Rencoret, J.; Gutiérrez, A.; Kim, H.; Ralph, J. Unconventional lignin monomers—Extension of the lignin paradigm. In Lignin and Hydroxycinnamic Acids: Biosynthesis and the Buildup of the Cell Wall; Sibout, R., Ed.; Elsevier: Amsterdam, The Netherlands, 2022; pp. 1–39. ISBN 9780323912204. [Google Scholar]
- Karlen, S.D.; Zhang, C.; Peck, M.L.; Smith, R.A.; Padmakshan, D.; Helmich, K.E.; Free, H.C.A.; Lee, S.; Smith, B.G.; Lu, F.; et al. Monolignol ferulate conjugates are naturally incorporated into plant lignins. Sci. Adv. 2016, 2, e1600393. [Google Scholar] [CrossRef]
- Vanholme, R.; Demedts, B.; Morreel, K.; Ralph, J.; Boerjan, W. Lignin biosynthesis and structure. Plant Physiol. 2010, 153, 895–905. [Google Scholar] [CrossRef]
- Ralph, J.; Schatz, P.F.; Lu, F.; Kim, H.; Akiyama, T.; Nelsen, S.F. Quinone Methides in Lignification. In Quinone Methides; Rokita, S.E., Ed.; Wiley: Hoboken, NJ, USA, 2009; pp. 385–420. ISBN 9780470192245. [Google Scholar]
- Freudenberg, K. Entstehung des Holzes und des Lignins. Holz Als Roh-Und Werkst. 1960, 18, 282–287. [Google Scholar] [CrossRef]
- Ralph, J.; Lundquist, K.; Brunow, G.; Lu, F.; Kim, H.; Schatz, P.F.; Marita, J.M.; Hatfield, R.D.; Ralph, S.A.; Christensen, J.H.; et al. Lignins: Natural polymers from oxidative coupling of 4-hydroxyphenyl- propanoids. Phytochem. Rev. 2004, 3, 29–60. [Google Scholar] [CrossRef]
- Balk, M.; Sofia, P.; Neffe, A.T.; Tirelli, N. Lignin, the Lignification Process, and Advanced, Lignin-Based Materials. Int. J. Mol. Sci. 2023, 24, 11668. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Koda, K.; Yoshinaga, A.; Takabe, K.; Shimomura, M.; Hirai, Y.; Tamai, Y.; Uraki, Y. Dehydrogenative polymerization of coniferyl alcohol in artificial polysaccharides matrices: Effects of xylan on the polymerization. J. Agric. Food Chem. 2015, 63, 4613–4620. [Google Scholar] [CrossRef] [PubMed]
- Navarro-Santos, P.; Rodriguez-Olalde, N.E.; Gallo, M.; Vargas, R.; Garza, J.; López-Albarrán, P. On the initial stages of lignin polymerization through spin-polarized density functional theory. Chem. Phys. Lett. 2019, 730, 289–296. [Google Scholar] [CrossRef]
- Akiyama, T.; Magara, K.; Meshitsuka, G.; Lundquist, K.; Matsumoto, Y. Absolute Configuration of β- and α-Asymmetric Carbons within β-O-4-Structures in Hardwood Lignin. J. Wood Chem. Technol. 2015, 35, 8–16. [Google Scholar] [CrossRef]
- Ralph, J.; Peng, J.; Lu, F.; Hatfield, R.D.; Helm, R.F. Are lignins optically active? J. Agric. Food Chem. 1999, 47, 2991–2996. [Google Scholar] [CrossRef]
- Oliveira, D.M.; Cesarino, I. Finding my way: The role of dirigent proteins in lignin assembly. Mol. Plant 2024, 17, 230–232. [Google Scholar] [CrossRef]
- Gao, Y.-Q.; Huang, J.-Q.; Reyt, G.; Song, T.; Love, A.; Tiemessen, D.; Xue, P.-Y.; Wu, W.-K.; George, M.W.; Chen, X.-Y.; et al. A dirigent protein complex directs lignin polymerization and assembly of the root diffusion barrier. Science 2023, 382, 464–471. [Google Scholar] [CrossRef]
- Sederoff, R.R.; MacKay, J.J.; Ralph, J.; Hatfield, R.D. Unexpected variation in lignin. Curr. Opin. Plant Biol. 1999, 2, 145–152. [Google Scholar] [CrossRef]
- Ralph, J.; Lapierre, C.; Boerjan, W. Lignin structure and its engineering. Curr. Opin. Biotechnol. 2019, 56, 240–249. [Google Scholar] [CrossRef]
- Sangha, A.K.; Parks, J.M.; Standaert, R.F.; Ziebell, A.; Davis, M.; Smith, J.C. Radical coupling reactions in lignin synthesis: A density functional theory study. J. Phys. Chem. B 2012, 116, 4760–4768. [Google Scholar] [CrossRef]
- Maia, R.A.; Ventorim, G.; Batagin-Neto, A. Reactivity of lignin subunits: The influence of dehydrogenation and formation of dimeric structures. J. Mol. Model. 2019, 25, 228. [Google Scholar] [CrossRef]
- Sánchez-González, Á.; Martín-Martínez, F.J.; Dobado, J.A. The role of weak interactions in lignin polymerization. J. Mol. Model. 2017, 23, 80. [Google Scholar] [CrossRef]
- Miyagawa, Y.; Tobimatsu, Y.; Lam, P.Y.; Mizukami, T.; Sakurai, S.; Kamitakahara, H.; Takano, T. Possible mechanisms for the generation of phenyl glycoside-type lignin-carbohydrate linkages in lignification with monolignol glucosides. Plant J. 2020, 104, 156–170. [Google Scholar] [CrossRef]
- Yi Chou, E.; Schuetz, M.; Hoffmann, N.; Watanabe, Y.; Sibout, R.; Samuels, A.L. Distribution, mobility, and anchoring of lignin-related oxidative enzymes in Arabidopsis secondary cell walls. J. Exp. Bot. 2018, 69, 1849–1859. [Google Scholar] [CrossRef]
- Tobimatsu, Y.; Schuetz, M. Lignin polymerization: How do plants manage the chemistry so well? Curr. Opin. Biotechnol. 2019, 56, 75–81. [Google Scholar] [CrossRef] [PubMed]
- Obst, J.R.; Laaducci, L.L. The Syringyl Content of Softwood Lignin. J. Wood Chem. Technol. 1986, 6, 311–327. [Google Scholar] [CrossRef]
- Kim, H.; Padmakshan, D.; Li, Y.; Rencoret, J.; Hatfield, R.D.; Ralph, J. Characterization and Elimination of Undesirable Protein Residues in Plant Cell Wall Materials for Enhancing Lignin Analysis by Solution-State Nuclear Magnetic Resonance Spectroscopy. Biomacromolecules 2017, 18, 4184–4195. [Google Scholar] [CrossRef] [PubMed]
- Rinaldi, R.; Jastrzebski, R.; Clough, M.T.; Ralph, J.; Kennema, M.; Bruijnincx, P.C.A.; Weckhuysen, B.M. Paving the Way for Lignin Valorisation: Recent Advances in Bioengineering, Biorefining and Catalysis. Angew. Chem. Int. Ed. Engl. 2016, 55, 8164–8215. [Google Scholar] [CrossRef]
- Beck, S.; Mushrif, S.H. Novel lignin polymerization pathway leading to branching in the structure. Org. Biomol. Chem. 2023, 21, 4596–4600. [Google Scholar] [CrossRef]
- Zhu, X.; Sipilä, J.; Potthast, A.; Rosenau, T.; Balakshin, M. Exploring Alkyl-O-Alkyl Ether Structures in Softwood Milled Wood Lignins. J. Agric. Food Chem. 2023, 71, 580–591. [Google Scholar] [CrossRef]
- Capanema, E.A.; Balakshin, M.Y.; Kadla, J.F. A comprehensive approach for quantitative lignin characterization by NMR spectroscopy. J. Agric. Food Chem. 2004, 52, 1850–1860. [Google Scholar] [CrossRef]
- Balakshin, M.; Capanema, E.A.; Zhu, X.; Sulaeva, I.; Potthast, A.; Rosenau, T.; Rojas, O.J. Spruce milled wood lignin: Linear, branched or cross-linked? Green Chem. 2020, 22, 3985–4001. [Google Scholar] [CrossRef]
- Yue, F.; Lu, F.; Ralph, S.; Ralph, J. Identification of 4-O-5-Units in Softwood Lignins via Definitive Lignin Models and NMR. Biomacromolecules 2016, 17, 1909–1920. [Google Scholar] [CrossRef] [PubMed]
- Adler, E. Lignin chemistry—Past, present and future. Wood Sci. Technol. 1977, 11, 169–218. [Google Scholar] [CrossRef]
- Kishimoto, T.; Hiyama, A.; Yamashita, A.; Takano, T.; Tobimatsu, Y.; Urabe, D. Existence of Syringyl α-Carbonyl-Type Tetrahydrofuran β–β Structure in Hardwood Lignins. ACS Sustain. Chem. Eng. 2022, 10, 12394–12401. [Google Scholar] [CrossRef]
- Lu, F.; Ralph, J. Novel tetrahydrofuran structures derived from beta-beta-coupling reactions involving sinapyl acetate in Kenaf lignins. Org. Biomol. Chem. 2008, 6, 3681–3694. [Google Scholar] [CrossRef]
- Karhunen, P.; Rummakko, P.; Sipilä, J.; Brunow, G.; Kilpeläinen, I. Dibenzodioxocins; a novel type of linkage in softwood lignins. Tetrahedron Lett. 1995, 36, 169–170. [Google Scholar] [CrossRef]
- Zhang, L.; Gellerstedt, G.; Ralph, J.; Lu, F. NMR Studies on the Occurrence of Spirodienone Structures in Lignins. J. Wood Chem. Technol. 2006, 26, 65–79. [Google Scholar] [CrossRef]
- Ralph, J. Hydroxycinnamates in lignification. Phytochem. Rev. 2010, 9, 65–83. [Google Scholar] [CrossRef]
- Lan, W.; Rencoret, J.; Lu, F.; Karlen, S.D.; Smith, B.G.; Harris, P.J.; Del Río, J.C.; Ralph, J. Tricin-lignins: Occurrence and quantitation of tricin in relation to phylogeny. Plant J. 2016, 88, 1046–1057. [Google Scholar] [CrossRef]
- Souto, F.; Calado, V. Mystifications and misconceptions of lignin: Revisiting understandings. Green Chem. 2022, 24, 8172–8192. [Google Scholar] [CrossRef]
- Crestini, C.; Melone, F.; Sette, M.; Saladino, R. Milled wood lignin: A linear oligomer. Biomacromolecules 2011, 12, 3928–3935. [Google Scholar] [CrossRef] [PubMed]
- Lai, Y.-Z.; Guo, X.-P. Variation of the phenolic hydroxyl group content in wood lignins. Wood Sci. Technol. 1991, 25, 467–472. [Google Scholar] [CrossRef]
- Sapouna, I.; Lawoko, M. Deciphering lignin heterogeneity in ball milled softwood: Unravelling the synergy between the supramolecular cell wall structure and molecular events. Green Chem. 2021, 23, 3348–3364. [Google Scholar] [CrossRef]
- Ruwoldt, J. A Critical Review of the Physicochemical Properties of Lignosulfonates: Chemical Structure and Behavior in Aqueous Solution, at Surfaces and Interfaces. Surfaces 2020, 3, 622–648. [Google Scholar] [CrossRef]
- Tolbert, A.; Akinosho, H.; Khunsupat, R.; Naskar, A.K.; Ragauskas, A.J. Characterization and analysis of the molecular weight of lignin for biorefining studies. Biofuels Bioprod. Bioref. 2014, 8, 836–856. [Google Scholar] [CrossRef]
- Micic, M.; Jeremic, M.; Radotic, K.; Mavers, M.; Leblanc, R.M. Visualization of artificial lignin supramolecular structures. Scanning 2000, 22, 288–294. [Google Scholar] [CrossRef]
- Kishimoto, T.; Uraki, Y.; Ubukata, M. Chemical synthesis of beta-O-4 type artificial lignin. Org. Biomol. Chem. 2006, 4, 1343–1347. [Google Scholar] [CrossRef]
- Monono, E.M.; Nyren, P.E.; Berti, M.T.; Pryor, S.W. Variability in biomass yield, chemical composition, and ethanol potential of individual and mixed herbaceous biomass species grown in North Dakota. Ind. Crops Prod. 2013, 41, 331–339. [Google Scholar] [CrossRef]
- Gellerstedt, G.; Henriksson, G. Lignins: Major Sources, Structure and Properties. In Monomers, Polymers and Composites from Renewable Resources; Reprinted; Belgacem, M.N., Gandini, A., Eds.; Elsevier: Amsterdam, The Netherlands, 2008; pp. 201–224. ISBN 978-0-08-045316-3. [Google Scholar]
- Eloy, N.B.; Voorend, W.; Lan, W.; Saleme, M.d.L.S.; Cesarino, I.; Vanholme, R.; Smith, R.A.; Goeminne, G.; Pallidis, A.; Morreel, K.; et al. Silencing CHALCONE SYNTHASE in Maize Impedes the Incorporation of Tricin into Lignin and Increases Lignin Content. Plant Physiol. 2017, 173, 998–1016. [Google Scholar] [CrossRef]
- Wang, Z.; Zhu, X.; Deuss, P.J. The effect of ball milling on birch, pine, reed, walnut shell enzymatic hydrolysis recalcitrance and the structure of the isolated residual enzyme lignin. Ind. Crops Prod. 2021, 167, 113493. [Google Scholar] [CrossRef]
- Lapierre, C. Determining Lignin Structure by Chemical Degradations. In Lignin and Lignans: Advances in Chemistry; Heitner, C., Dimmel, D., Schmidt, J.A., Eds.; CRC Press: Boca Raton, FL, USA, 2010; pp. 11–48. ISBN 9781574444865. [Google Scholar]
- Schutyser, W.; Renders, T.; van den Bosch, S.; Koelewijn, S.-F.; Beckham, G.T.; Sels, B.F. Chemicals from lignin: An interplay of lignocellulose fractionation, depolymerisation, and upgrading. Chem. Soc. Rev. 2018, 47, 852–908. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.H. Pretreatment of Lignocellulosic Biomass. In Bioprocessing Technologies in Biorefinery for Sustainable Production of Fuels, Chemicals, and Polymers; Yang, S.-T., El-Enshasy, H.A., Thongchul, N., Eds.; Wiley: Hoboken, NJ, USA, 2013; pp. 91–110. ISBN 9780470541951. [Google Scholar]
- Wohlert, M.; Benselfelt, T.; Wågberg, L.; Furó, I.; Berglund, L.A.; Wohlert, J. Cellulose and the role of hydrogen bonds: Not in charge of everything. Cellulose 2022, 29, 1–23. [Google Scholar] [CrossRef]
- Cosgrove, D.J. Re-constructing our models of cellulose and primary cell wall assembly. Curr. Opin. Plant Biol. 2014, 22, 122–131. [Google Scholar] [CrossRef]
- Berglund, J.; Angles d’Ortoli, T.; Vilaplana, F.; Widmalm, G.; Bergenstråhle-Wohlert, M.; Lawoko, M.; Henriksson, G.; Lindström, M.; Wohlert, J. A molecular dynamics study of the effect of glycosidic linkage type in the hemicellulose backbone on the molecular chain flexibility. Plant J. 2016, 88, 56–70. [Google Scholar] [CrossRef]
- Scheller, H.V.; Ulvskov, P. Hemicelluloses. Annu. Rev. Plant Biol. 2010, 61, 263–289. [Google Scholar] [CrossRef]
- Addison, B.; Bu, L.; Bharadwaj, V.; Crowley, M.F.; Harman-Ware, A.E.; Crowley, M.F.; Bomble, Y.J.; Ciesielski, P.N. Atomistic, macromolecular model of the Populus secondary cell wall informed by solid-state NMR. Sci. Adv. 2024, 10, eadi7965. [Google Scholar] [CrossRef]
- Bromley, J.R.; Busse-Wicher, M.; Tryfona, T.; Mortimer, J.C.; Zhang, Z.; Brown, D.M.; Dupree, P. GUX1 and GUX2 glucuronyltransferases decorate distinct domains of glucuronoxylan with different substitution patterns. Plant J. 2013, 74, 423–434. [Google Scholar] [CrossRef]
- Kirui, A.; Zhao, W.; Deligey, F.; Yang, H.; Kang, X.; Mentink-Vigier, F.; Wang, T. Carbohydrate-aromatic interface and molecular architecture of lignocellulose. Nat. Commun. 2022, 13, 538. [Google Scholar] [CrossRef]
- Donaldson, L. Cellulose microfibril aggregates and their size variation with cell wall type. Wood Sci. Technol. 2007, 41, 443–460. [Google Scholar] [CrossRef]
- Kang, X.; Kirui, A.; Dickwella Widanage, M.C.; Mentink-Vigier, F.; Cosgrove, D.J.; Wang, T. Lignin-polysaccharide interactions in plant secondary cell walls revealed by solid-state NMR. Nat. Commun. 2019, 10, 347. [Google Scholar] [CrossRef]
- Vermaas, J.V.; Crowley, M.F.; Beckham, G.T. A Quantitative Molecular Atlas for Interactions Between Lignin and Cellulose. ACS Sustain. Chem. Eng. 2019, 7, 19570–19583. [Google Scholar] [CrossRef]
- Silveira, R.L.; Stoyanov, S.R.; Gusarov, S.; Skaf, M.S.; Kovalenko, A. Supramolecular Interactions in Secondary Plant Cell Walls: Effect of Lignin Chemical Composition Revealed with the Molecular Theory of Solvation. J. Phys. Chem. Lett. 2015, 6, 206–211. [Google Scholar] [CrossRef] [PubMed]
- Erdmann, J. Ueber die Concretionen in den Birnen. Justus Liebigs Ann. Der Chem. 1866, 139, 1–19. [Google Scholar] [CrossRef]
- Freudenberg, K.; Harkin, J.M. Modelle für die Bindung des Lignins an die Kohlenhydrate. Chem. Ber. 1960, 93, 2814–2819. [Google Scholar] [CrossRef]
- Min, D.; Yang, C.; Chiang, V.; Jameel, H.; Chang, H. The influence of lignin–carbohydrate complexes on the cellulase-mediated saccharification II: Transgenic hybrid poplars (Populus nigra L. and Populus maximowiczii A.). Fuel 2014, 116, 56–62. [Google Scholar] [CrossRef]
- Beck, S.; Choi, P.; Mushrif, S.H. Physico-chemical interactions within lignocellulosic biomass and their importance in developing solvent based deconstruction methods. React. Chem. Eng. 2022, 7, 2471–2487. [Google Scholar] [CrossRef]
- Cresswell, R.; Dupree, R.; Brown, S.P.; Pereira, C.S.; Skaf, M.S.; Sorieul, M.; Dupree, P.; Hill, S. Importance of Water in Maintaining Softwood Secondary Cell Wall Nanostructure. Biomacromolecules 2021, 22, 4669–4680. [Google Scholar] [CrossRef]
- Gao, Y.; Lipton, A.S.; Wittmer, Y.; Murray, D.T.; Mortimer, J.C. A grass-specific cellulose-xylan interaction dominates in sorghum secondary cell walls. Nat. Commun. 2020, 11, 6081. [Google Scholar] [CrossRef]
- Giummarella, N.; Pu, Y.; Ragauskas, A.J.; Lawoko, M. A critical review on the analysis of lignin carbohydrate bonds. Green Chem. 2019, 21, 1573–1595. [Google Scholar] [CrossRef]
- Beck, S.; Choi, P.; Mushrif, S.H. Origins of covalent linkages within the lignin-carbohydrate network of biomass. Phys. Chem. Chem. Phys. 2022, 24, 20480–20490. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, H.; Kamiya, A.; Nagata, T.; Katahira, M.; Watanabe, T. Direct evidence for α ether linkage between lignin and carbohydrates in wood cell walls. Sci. Rep. 2018, 8, 6538. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Helm, R.F. Synthesis and Rearrangement Reactions of Ester-Linked Lignin-Carbohydrate Model Compounds. J. Agric. Food Chem. 1995, 43, 2098–2103. [Google Scholar] [CrossRef]
- Giummarella, N.; Balakshin, M.; Koutaniemi, S.; Kärkönen, A.; Lawoko, M. Nativity of lignin carbohydrate bonds substantiated by biomimetic synthesis. J. Exp. Bot. 2019, 70, 5591–5601. [Google Scholar] [CrossRef]
- Yuan, T.-Q.; Sun, S.-N.; Xu, F.; Sun, R.-C. Characterization of lignin structures and lignin-carbohydrate complex (LCC) linkages by quantitative 13C and 2D HSQC NMR spectroscopy. J. Agric. Food Chem. 2011, 59, 10604–10614. [Google Scholar] [CrossRef]
- Ralph, J.; Grabber, J.H.; Hatfield, R.D. Lignin-ferulate cross-links in grasses: Active incorporation of ferulate polysaccharide esters into ryegrass lignins. Carbohydr. Res. 1995, 275, 167–178. [Google Scholar] [CrossRef]
- Buranov, A.U.; Mazza, G. Lignin in straw of herbaceous crops. Ind. Crops Prod. 2008, 28, 237–259. [Google Scholar] [CrossRef]
- Tarasov, D.; Leitch, M.; Fatehi, P. Lignin-carbohydrate complexes: Properties, applications, analyses, and methods of extraction: A review. Biotechnol. Biofuels 2018, 11, 269. [Google Scholar] [CrossRef]
- Jin, Z.; Katsumata, K.S.; Lam, T.B.T.; Iiyama, K. Covalent linkages between cellulose and lignin in cell walls of coniferous and nonconiferous woods. Biopolymers 2006, 83, 103–110. [Google Scholar] [CrossRef]
- Aminzadeh, S.; Zhang, L.; Henriksson, G. A possible explanation for the structural inhomogeneity of lignin in LCC networks. Wood Sci. Technol. 2017, 51, 1365–1376. [Google Scholar] [CrossRef]
- Fengel, D.; Wegener, G. Wood: Chemistry, Ultrastructure, Reactions; De Gruyter: Berlin, Germany, 1989; ISBN 9783110839654. [Google Scholar]
- Thornburg, N.E.; Pecha, M.B.; Brandner, D.G.; Reed, M.L.; Vermaas, J.V.; Michener, W.E.; Katahira, R.; Vinzant, T.B.; Foust, T.D.; Donohoe, B.S.; et al. Mesoscale Reaction-Diffusion Phenomena Governing Lignin-First Biomass Fractionation. ChemSusChem 2020, 13, 4495–4509. [Google Scholar] [CrossRef] [PubMed]
- Davison, B.H.; Parks, J.; Davis, M.F.; Donohoe, B.S. Plant Cell Walls: Basics of Structure, Chemistry, Accessibility and the Influence on Conversion. In Aqueous Pretreatment of Plant Biomass for Biological and Chemical Conversion to Fuels and Chemicals; Wyman, C.E., Ed.; Wiley: Hoboken, NJ, USA, 2013; pp. 23–38. ISBN 9780470972021. [Google Scholar]
- Fahlén, J.; Salmén, L. Cross-sectional structure of the secondary wall of wood fibers as affected by processing. J. Mater. Sci. 2003, 38, 119–126. [Google Scholar] [CrossRef]
- Cao, M.; Ren, W.; Zhu, J.; Wang, H.; Guo, J.; Zhang, X.; Yu, Y. Cell wall pore structures of bamboo evaluated using gas adsorption methods. Holzforschung 2022, 76, 754–764. [Google Scholar] [CrossRef]
- Kellogg, R.M.; Wangaard, F.F. Variation in The Cell-Wall Density of Wood. WFS 1969, 3, 180–204. [Google Scholar]
- Gao, X.; Zhuang, S.; Jin, J.; Cao, P. Bound Water Content and Pore Size Distribution in Swollen Cell Walls Determined by NMR Technology. BioResources 2015, 10, 8208–8224. [Google Scholar] [CrossRef]
- Nopens, M.; Sazama, U.; König, S.; Kaschuro, S.; Krause, A.; Fröba, M. Determination of mesopores in the wood cell wall at dry and wet state. Sci. Rep. 2020, 10, 9543. [Google Scholar] [CrossRef]
- Li, X.; Zhao, Z. Time domain-NMR studies of average pore size of wood cell walls during drying and moisture adsorption. Wood Sci. Technol. 2020, 54, 1241–1251. [Google Scholar] [CrossRef]
- Donaldson, L.A.; Cairns, M.; Hill, S.J. Comparison of Micropore Distribution in Cell Walls of Softwood and Hardwood Xylem. Plant Physiol. 2018, 178, 1142–1153. [Google Scholar] [CrossRef]
- Yan, N.; Zhao, C.; Dyson, P.J.; Wang, C.; Liu, L.; Kou, Y. Selective degradation of wood lignin over noble-metal catalysts in a two-step process. ChemSusChem 2008, 1, 626–629. [Google Scholar] [CrossRef]
- Huang, J.; Fu, S.; Gan, L. Structure and Characteristics of Lignin. In Lignin Chemistry and Applications; Huang, J., Fu, S., Gan, L., Eds.; Elsevier: Amsterdam, The Netherlands, 2019; pp. 25–50. ISBN 9780128139417. [Google Scholar]
- Glasser, W.G. About Making Lignin Great Again-Some Lessons from the Past. Front. Chem. 2019, 7, 565. [Google Scholar] [CrossRef]
- Pu, Y.; Hu, F.; Huang, F.; Ragauskas, A.J. Lignin Structural Alterations in Thermochemical Pretreatments with Limited Delignification. Bioenerg. Res. 2015, 8, 992–1003. [Google Scholar] [CrossRef]
- Raikwar, D.; van Aelst, K.; Vangeel, T.; Corderi, S.; van Aelst, J.; van den Bosch, S.; Servaes, K.; Vanbroekhoven, K.; Elst, K.; Sels, B.F. Elucidating the effect of the physicochemical properties of organosolv lignins on its solubility and reductive catalytic depolymerization. Chem. Eng. J. 2023, 461, 141999. [Google Scholar] [CrossRef]
- Schuerch, C. The Solvent Properties of Liquids and Their Relation to the Solubility, Swelling, Isolation and Fractionation of Lignin. J. Am. Chem. Soc. 1952, 74, 5061–5067. [Google Scholar] [CrossRef]
- Horvath, A.L. Solubility of Structurally Complicated Materials: I. Wood. J. Phys. Chem. Ref. Data 2006, 35, 77–92. [Google Scholar] [CrossRef]
- Vermaas, J.V.; Crowley, M.F.; Beckham, G.T. Molecular Lignin Solubility and Structure in Organic Solvents. ACS Sustain. Chem. Eng. 2020, 8, 17839–17850. [Google Scholar] [CrossRef]
- Smith, M.D.; Mostofian, B.; Cheng, X.; Petridis, L.; Cai, C.M.; Wyman, C.E.; Smith, J.C. Cosolvent pretreatment in cellulosic biofuel production: Effect of tetrahydrofuran-water on lignin structure and dynamics. Green Chem. 2016, 18, 1268–1277. [Google Scholar] [CrossRef]
- Pingali, S.V.; Smith, M.D.; Liu, S.-H.; Rawal, T.B.; Pu, Y.; Shah, R.; Evans, B.R.; Urban, V.S.; Davison, B.H.; Cai, C.M.; et al. Deconstruction of biomass enabled by local demixing of cosolvents at cellulose and lignin surfaces. Proc. Natl. Acad. Sci. USA 2020, 117, 16776–16781. [Google Scholar] [CrossRef] [PubMed]
- Renders, T.; van den Bosch, S.; Vangeel, T.; Ennaert, T.; Koelewijn, S.-F.; van den Bossche, G.; Courtin, C.M.; Schutyser, W.; Sels, B.F. Synergetic Effects of Alcohol/Water Mixing on the Catalytic Reductive Fractionation of Poplar Wood. ACS Sustain. Chem. Eng. 2016, 4, 6894–6904. [Google Scholar] [CrossRef]
- Jahan, N.; Huda, M.M.; Tran, Q.X.; Rai, N. Effect of Solvent Quality on Structure and Dynamics of Lignin in Solution. J. Phys. Chem. B 2022, 126, 5752–5764. [Google Scholar] [CrossRef]
- Wang, J.; Qian, Y.; Li, L.; Qiu, X. Atomic Force Microscopy and Molecular Dynamics Simulations for Study of Lignin Solution Self-Assembly Mechanisms in Organic-Aqueous Solvent Mixtures. ChemSusChem 2020, 13, 4420–4427. [Google Scholar] [CrossRef]
- Zhao, W.; Xiao, L.-P.; Song, G.; Sun, R.-C.; He, L.; Singh, S.; Simmons, B.A.; Cheng, G. From lignin subunits to aggregates: Insights into lignin solubilization. Green Chem. 2017, 19, 3272–3281. [Google Scholar] [CrossRef]
- Hansen, C.M. Solubility Parameters—An introduction. Chapter 1. In Hansen Solubility Parameters: A User’s Handbook, 2nd ed.; Hansen, C.M., Ed.; CRC Press: Boca Raton, FL, USA, 2007; pp. 1–26. ISBN 0-8493-7248-8. [Google Scholar]
- Mohan, M.; Huang, K.; Pidatala, V.R.; Simmons, B.A.; Singh, S.; Sale, K.L.; Gladden, J.M. Prediction of solubility parameters of lignin and ionic liquids using multi-resolution simulation approaches. Green Chem. 2022, 24, 1165–1176. [Google Scholar] [CrossRef]
- Thielemans, W.; Wool, R.P. Lignin esters for use in unsaturated thermosets: Lignin modification and solubility modeling. Biomacromolecules 2005, 6, 1895–1905. [Google Scholar] [CrossRef] [PubMed]
- Lê, H.Q.; Zaitseva, A.; Pokki, J.-P.; Ståhl, M.; Alopaeus, V.; Sixta, H. Solubility of Organosolv Lignin in γ-Valerolactone/Water Binary Mixtures. ChemSusChem 2016, 9, 2939–2947. [Google Scholar] [CrossRef]
- Ni, Y.; Hu, Q. Alcell® lignin solubility in ethanol–water mixtures. J. Appl. Polym. Sci. 1995, 57, 1441–1446. [Google Scholar] [CrossRef]
- Wang, Q.; Chen, K.; Li, J.; Yang, G.; Liu, S.; Xu, J. The solubility of lignin from bagasse in a 1,4-butanediol/water system. BioResources 2011, 6, 3034–3043. [Google Scholar] [CrossRef]
- Quesada-Medina, J.; López-Cremades, F.J.; Olivares-Carrillo, P. Organosolv extraction of lignin from hydrolyzed almond shells and application of the delta-value theory. Bioresour. Technol. 2010, 101, 8252–8260. [Google Scholar] [CrossRef]
- Ma, Q.; Yu, C.; Zhou, Y.; Hu, D.; Chen, J.; Zhang, X. A review on the calculation and application of lignin Hansen solubility parameters. Int. J. Biol. Macromol. 2024, 256, 128506. [Google Scholar] [CrossRef]
- Hansen, C.M.; Björkman, A. The Ultrastructure of Wood from a Solubility Parameter Point of View. Holzforschung 1998, 52, 335–344. [Google Scholar] [CrossRef]
- Jakes, J.E.; Hunt, C.G.; Zelinka, S.L.; Ciesielski, P.N.; Plaza, N.Z. Effects of Moisture on Diffusion in Unmodified Wood Cell Walls: A Phenomenological Polymer Science Approach. Forests 2019, 10, 1084. [Google Scholar] [CrossRef]
- Stamm, A.J. SHRINKING and SWELLING of WOOD. Ind. Eng. Chem. 1935, 27, 401–406. [Google Scholar] [CrossRef]
- Bossu, J.; Le Moigne, N.; Corn, S.; Trens, P.; Di Renzo, F. Sorption of water–ethanol mixtures by poplar wood: Swelling and viscoelastic behaviour. Wood Sci. Technol. 2018, 52, 987–1008. [Google Scholar] [CrossRef]
- Ong, R.G.; Chundawat, S.P.S.; Hodge, D.B.; Keskar, S.; Dale, B.E. Linking Plant Biology and Pretreatment: Understanding the Structure and Organization of the Plant Cell Wall and Interactions with Cellulosic Biofuel Production. In Plants and BioEnergy; McCann, M.C., Buckeridge, M.S., Carpita, N.C., Eds.; Springer: New York, NY, USA, 2014; pp. 231–253. ISBN 978-1-4614-9328-0. [Google Scholar]
- Sarkar, D.; Bu, L.; Jakes, J.E.; Zieba, J.K.; Kaufman, I.D.; Crowley, M.F.; Ciesielski, P.N.; Vermaas, J.V. Diffusion in intact secondary cell wall models of plants at different equilibrium moisture content. Cell Surf. 2023, 9, 100105. [Google Scholar] [CrossRef]
- Jakes, J.E.; Zelinka, S.L.; Hunt, C.G.; Ciesielski, P.; Frihart, C.R.; Yelle, D.; Passarini, L.; Gleber, S.-C.; Vine, D.; Vogt, S. Measurement of moisture-dependent ion diffusion constants in wood cell wall layers using time-lapse micro X-ray fluorescence microscopy. Sci. Rep. 2020, 10, 9919. [Google Scholar] [CrossRef]
- Schneider, A. Beiträge zur Dimensionsstabilisierung des Holzes mit Polyäthylenglykol—Erste Mitteilung: Grundlegende Untersuchungen zur Dimensionsstabilisierung des Holzes mit Polyäthylenglykol. Holz Roh-Werkst. 1969, 27, 209–224. [Google Scholar] [CrossRef]
- Zhao, X.; Wu, R.; Liu, D. Evaluation of the mass transfer effects on delignification kinetics of atmospheric acetic acid fractionation of sugarcane bagasse with a shrinking-layer model. Bioresour. Technol. 2018, 261, 52–61. [Google Scholar] [CrossRef]
- Parthasarathi, R.; Romero, R.A.; Redondo, A.; Gnanakaran, S. Theoretical Study of the Remarkably Diverse Linkages in Lignin. J. Phys. Chem. Lett. 2011, 2, 2660–2666. [Google Scholar] [CrossRef]
- Kim, S.; Chmely, S.C.; Nimlos, M.R.; Bomble, Y.J.; Foust, T.D.; Paton, R.S.; Beckham, G.T. Computational Study of Bond Dissociation Enthalpies for a Large Range of Native and Modified Lignins. J. Phys. Chem. Lett. 2011, 2, 2846–2852. [Google Scholar] [CrossRef]
- Houston, R.W.; Abdoulmoumine, N.H. Investigation of the thermal deconstruction of β-β′ and 4-O-5 linkages in lignin model oligomers by density functional theory (DFT). RSC Adv. 2023, 13, 6181–6190. [Google Scholar] [CrossRef]
- Houston, R.W.; Elder, T.J.; Abdoulmoumine, N.H. Investigation into the Pyrolysis Bond Dissociation Enthalpies (BDEs) of a Model Lignin Oligomer Using Density Functional Theory (DFT). Energy Fuels 2022, 36, 1565–1573. [Google Scholar] [CrossRef]
- Younker, J.M.; Beste, A.; Buchanan, A.C. Computational study of bond dissociation enthalpies for lignin model compounds: β-5 Arylcoumaran. Chem. Phys. Lett. 2012, 545, 100–106. [Google Scholar] [CrossRef]
- Huang, J.; Wu, S.; Cheng, H.; Lei, M.; Liang, J.; Tong, H. Theoretical study of bond dissociation energies for lignin model compounds. J. Fuel Chem. Technol. 2015, 43, 429–436. [Google Scholar] [CrossRef]
- Elder, T. Bond Dissociation Enthalpies of a Dibenzodioxocin Lignin Model Compound. Energy Fuels 2013, 27, 4785–4790. [Google Scholar] [CrossRef]
- Elder, T.; Berstis, L.; Beckham, G.T.; Crowley, M.F. Density Functional Theory Study of Spirodienone Stereoisomers in Lignin. ACS Sustain. Chem. Eng. 2017, 5, 7188–7194. [Google Scholar] [CrossRef]
- Younker, J.M.; Beste, A.; Buchanan, A.C. Computational study of bond dissociation enthalpies for substituted β-O-4 lignin model compounds. Chemphyschem 2011, 12, 3556–3565. [Google Scholar] [CrossRef]
- Huang, J.; Liu, C.; Jin, Q.; Tong, H.; Li, W.; Wu, D. Density functional theory study on bond dissociation enthalpies for lignin dimer model compounds. J. Renew. Sustain. Energy 2014, 6, 033116. [Google Scholar] [CrossRef]
- Chen, J.; Lu, F.; Si, X.; Nie, X.; Chen, J.; Lu, R.; Xu, J. High Yield Production of Natural Phenolic Alcohols from Woody Biomass Using a Nickel-Based Catalyst. ChemSusChem 2016, 9, 3353–3360. [Google Scholar] [CrossRef]
- Valencia, D.; Vega, D.; Aburto, J. Electronic and topological analyses of C–O bonds in carbohydrates and lignin-carbohydrate complexes: An atomistic understanding of refractory molecules in agro-industrial waste. Renew. Energy 2024, 222, 119979. [Google Scholar] [CrossRef]
- Zhang, X.; Yang, W.; Blasiak, W. Modeling Study of Woody Biomass: Interactions of Cellulose, Hemicellulose, and Lignin. Energy Fuels 2011, 25, 4786–4795. [Google Scholar] [CrossRef]
- Anderson, E.M.; Stone, M.L.; Hülsey, M.J.; Beckham, G.T.; Román-Leshkov, Y. Kinetic Studies of Lignin Solvolysis and Reduction by Reductive Catalytic Fractionation Decoupled in Flow-Through Reactors. ACS Sustain. Chem. Eng. 2018, 6, 7951–7959. [Google Scholar] [CrossRef]
- Minami, E.; Kawamoto, H.; Saka, S. Reaction behavior of lignin in supercritical methanol as studied with lignin model compounds. J. Wood Sci. 2003, 49, 158–165. [Google Scholar] [CrossRef]
- Questell-Santiago, Y.M.; Galkin, M.V.; Barta, K.; Luterbacher, J.S. Stabilization strategies in biomass depolymerization using chemical functionalization. Nat. Rev. Chem. 2020, 4, 311–330. [Google Scholar] [CrossRef] [PubMed]
- Hillis, W.E.; Rozsa, A.N. The Softening Temperatures of Wood. Holzforschung 1978, 32, 68–73. [Google Scholar] [CrossRef]
- Hamdan, S.; Dwianto, W.; Morooka, T.; Norimoto, M. Softening Characteristics of Wet Wood under Quasi Static Loading. Holzforschung 2000, 54, 557–560. [Google Scholar] [CrossRef]
- Kelley, S.S.; Rials, T.G.; Glasser, W.G. Relaxation behaviour of the amorphous components of wood. J. Mater. Sci. 1987, 22, 617–624. [Google Scholar] [CrossRef]
- Kaliyan, N.; Morey, R.V. Natural binders and solid bridge type binding mechanisms in briquettes and pellets made from corn stover and switchgrass. Bioresour. Technol. 2010, 101, 1082–1090. [Google Scholar] [CrossRef]
- Börcsök, Z.; Pásztory, Z. The role of lignin in wood working processes using elevated temperatures: An abbreviated literature survey. Holz Roh-Werkst. 2021, 79, 511–526. [Google Scholar] [CrossRef]
- Dufour, A.; Castro-Diaz, M.; Brosse, N.; Bouroukba, M.; Snape, C. The origin of molecular mobility during biomass pyrolysis as revealed by in situ (1)H NMR spectroscopy. ChemSusChem 2012, 5, 1258–1265. [Google Scholar] [CrossRef]
- Dufour, A.; Castro-Díaz, M.; Marchal, P.; Brosse, N.; Olcese, R.; Bouroukba, M.; Snape, C. In Situ Analysis of Biomass Pyrolysis by High Temperature Rheology in Relations with 1 H NMR. Energy Fuels 2012, 26, 6432–6441. [Google Scholar] [CrossRef]
- Jakab, E.; Faix, O.; Till, F.; Székely, T. Thermogravimetry/mass spectrometry study of six lignins within the scope of an international round robin test. J. Anal. Appl. Pyrolysis 1995, 35, 167–179. [Google Scholar] [CrossRef]
- Sharma, R.K.; Wooten, J.B.; Baliga, V.L.; Lin, X.; Geoffrey Chan, W.; Hajaligol, M.R. Characterization of chars from pyrolysis of lignin. Fuel 2004, 83, 1469–1482. [Google Scholar] [CrossRef]
- Haw, J.F.; Schultz, T.P. Carbon-13 CP/MAS NMR and FT-IR Study of Low-Temperature Lignin Pyrolysis. Holzforschung 1985, 39, 289–296. [Google Scholar] [CrossRef]
- Kawamoto, H.; Horigoshi, S.; Saka, S. Pyrolysis reactions of various lignin model dimers. J. Wood Sci. 2007, 53, 168–174. [Google Scholar] [CrossRef]
- Nakamura, T.; Kawamoto, H.; Saka, S. Pyrolysis behavior of Japanese cedar wood lignin studied with various model dimers. J. Anal. Appl. Pyrolysis 2008, 81, 173–182. [Google Scholar] [CrossRef]
- Kawamoto, H. Lignin pyrolysis reactions. J. Wood Sci. 2017, 63, 117–132. [Google Scholar] [CrossRef]
- Li, L.; van de Vijver, R.; Eschenbacher, A.; Dogu, O.; van Geem, K.M. Primary Thermal Decomposition Pathways of Hydroxycinnamaldehydes. Energy Fuels 2021, 35, 12216–12226. [Google Scholar] [CrossRef]
- Ponomarev, D.A. Formation of quinone methides: An alternative pathway of thermal degradation of some β-O-4-ethers as compounds modeling lignin. Russ. J. Appl. Chem. 1997, 70, 846–866. [Google Scholar]
- Nakamura, T.; Kawamoto, H.; Saka, S. Condensation Reactions of Some Lignin Related Compounds at Relatively Low Pyrolysis Temperature. J. Wood Chem. Technol. 2007, 27, 121–133. [Google Scholar] [CrossRef]
- Funaoka, M.; Kako, T.; Abe, I. Condensation of lignin during heating of wood. Wood Sci. Technol. 1990, 24, 277–288. [Google Scholar] [CrossRef]
- Brosse, N.; El Hage, R.; Chaouch, M.; Pétrissans, M.; Dumarçay, S.; Gérardin, P. Investigation of the chemical modifications of beech wood lignin during heat treatment. Polym. Degrad. Stab. 2010, 95, 1721–1726. [Google Scholar] [CrossRef]
- Kotake, T.; Kawamoto, H.; Saka, S. Pyrolysis reactions of coniferyl alcohol as a model of the primary structure formed during lignin pyrolysis. J. Anal. Appl. Pyrolysis 2013, 104, 573–584. [Google Scholar] [CrossRef]
- Li, H.; Song, G. Ru-Catalyzed Hydrogenolysis of Lignin: Base-Dependent Tunability of Monomeric Phenols and Mechanistic Study. ACS Catal. 2019, 9, 4054–4064. [Google Scholar] [CrossRef]
- Song, Q.; Wang, F.; Cai, J.; Wang, Y.; Zhang, J.; Yu, W.; Xu, J. Lignin depolymerization (LDP) in alcohol over nickel-based catalysts via a fragmentation–hydrogenolysis process. Energy Environ. Sci. 2013, 6, 994. [Google Scholar] [CrossRef]
- van den Bosch, S.; Renders, T.; Kennis, S.; Koelewijn, S.-F.; van den Bossche, G.; Vangeel, T.; Deneyer, A.; Depuydt, D.; Courtin, C.M.; Thevelein, J.M.; et al. Integrating lignin valorization and bio-ethanol production: On the role of Ni-Al 2 O 3 catalyst pellets during lignin-first fractionation. Green Chem. 2017, 19, 3313–3326. [Google Scholar] [CrossRef]
- Shuai, L.; Luterbacher, J. Organic Solvent Effects in Biomass Conversion Reactions. ChemSusChem 2016, 9, 133–155. [Google Scholar] [CrossRef] [PubMed]
- Balogh, D.T.; Curvelo, A.; de Groote, R. Solvent Effects on Organosolv Lignin from Pinus caribaea hondurensis. Holzforschung 1992, 46, 343–348. [Google Scholar] [CrossRef]
- Novo, L.P.; Curvelo, A.A.S. Hansen Solubility Parameters: A Tool for Solvent Selection for Organosolv Delignification. Ind. Eng. Chem. Res. 2019, 58, 14520–14527. [Google Scholar] [CrossRef]
- van Leuken, S.H.M.; van Osch, D.J.G.P.; Kouris, P.D.; Yao, Y.; Jedrzejczyk, M.A.; Cremers, G.J.W.; Bernaerts, K.V.; van Benthem, R.A.T.M.; Tuinier, R.; Boot, M.D.; et al. Quantitative prediction of the solvent fractionation of lignin. Green Chem. 2023, 25, 7534–7540. [Google Scholar] [CrossRef]
- Du, X.; Tricker, A.W.; Yang, W.; Katahira, R.; Liu, W.; Kwok, T.T.; Gogoi, P.; Deng, Y. Oxidative Catalytic Fractionation and Depolymerization of Lignin in a One-Pot Single-Catalyst System. ACS Sustain. Chem. Eng. 2021, 9, 7719–7727. [Google Scholar] [CrossRef]
- Ko, J.K.; Kim, Y.; Ximenes, E.; Ladisch, M.R. Effect of liquid hot water pretreatment severity on properties of hardwood lignin and enzymatic hydrolysis of cellulose. Biotechnol. Bioeng. 2015, 112, 252–262. [Google Scholar] [CrossRef]
- Schutyser, W.; van den Bosch, S.; Renders, T.; de Boe, T.; Koelewijn, S.-F.; Dewaele, A.; Ennaert, T.; Verkinderen, O.; Goderis, B.; Courtin, C.M.; et al. Influence of bio-based solvents on the catalytic reductive fractionation of birch wood. Green Chem. 2015, 17, 5035–5045. [Google Scholar] [CrossRef]
- Ferrini, P.; Rinaldi, R. Catalytic biorefining of plant biomass to non-pyrolytic lignin bio-oil and carbohydrates through hydrogen transfer reactions. Angew. Chem. Int. Ed. Engl. 2014, 53, 8634–8639. [Google Scholar] [CrossRef] [PubMed]
- Lancefield, C.S.; Panovic, I.; Deuss, P.J.; Barta, K.; Westwood, N.J. Pre-treatment of lignocellulosic feedstocks using biorenewable alcohols: Towards complete biomass valorisation. Green Chem. 2017, 19, 202–214. [Google Scholar] [CrossRef]
- Bauer, S.; Sorek, H.; Mitchell, V.D.; Ibáñez, A.B.; Wemmer, D.E. Characterization of Miscanthus giganteus lignin isolated by ethanol organosolv process under reflux condition. J. Agric. Food Chem. 2012, 60, 8203–8212. [Google Scholar] [CrossRef]
- Yong, K.J.; Wu, T.Y. Recent advances in the application of alcohols in extracting lignin with preserved β-O-4 content from lignocellulosic biomass. Bioresour. Technol. 2023, 384, 129238. [Google Scholar] [CrossRef]
- Zijlstra, D.S.; Lahive, C.W.; Analbers, C.A.; Figueirêdo, M.B.; Wang, Z.; Lancefield, C.S.; Deuss, P.J. Mild Organosolv Lignin Extraction with Alcohols: The Importance of Benzylic Alkoxylation. ACS Sustain. Chem. Eng. 2020, 8, 5119–5131. [Google Scholar] [CrossRef]
- Luo, H.; Abu-Omar, M.M. Lignin extraction and catalytic upgrading from genetically modified poplar. Green Chem. 2018, 20, 745–753. [Google Scholar] [CrossRef]
- Li, H.; Song, G. Paving the Way for the Lignin Hydrogenolysis Mechanism by Deuterium-Incorporated β-O-4 Mimics. ACS Catal. 2020, 10, 12229–12238. [Google Scholar] [CrossRef]
- Kumaniaev, I.; Subbotina, E.; Sävmarker, J.; Larhed, M.; Galkin, M.V.; Samec, J.S.M. Lignin depolymerization to monophenolic compounds in a flow-through system. Green Chem. 2017, 19, 5767–5771. [Google Scholar] [CrossRef]
- Kishimoto, T.; Sano, Y. Delignification Mechanism during High-Boiling Solvent Pulping. Part 1. Reaction of Guaiacylglycerol-β-Guaiacyl Ether. Holzforschung 2001, 55, 611–616. [Google Scholar] [CrossRef]
- Kishimoto, T.; Sano, Y. Delignification Mechanism during High-Boiling Solvent Pulping. Part 2. Homolysis of Guaiacylglycerol-β-Guaiacyl Ether. Holzforschung 2002, 56, 623–631. [Google Scholar] [CrossRef]
- Nagel, E.; Zhang, C. Hydrothermal Decomposition of a Lignin Dimer under Neutral and Basic Conditions: A Mechanism Study. Ind. Eng. Chem. Res. 2019, 58, 18866–18880. [Google Scholar] [CrossRef]
- Giummarella, N.; Lawoko, M. Structural Basis for the Formation and Regulation of Lignin–Xylan Bonds in Birch. ACS Sustain. Chem. Eng. 2016, 4, 5319–5326. [Google Scholar] [CrossRef]
- Giummarella, N.; Zhang, L.; Henriksson, G.; Lawoko, M. Structural features of mildly fractionated lignin carbohydrate complexes (LCC) from spruce. RSC Adv. 2016, 6, 42120–42131. [Google Scholar] [CrossRef]
- Ferreira, J.A.; Taherzadeh, M.J. Improving the economy of lignocellulose-based biorefineries with organosolv pretreatment. Bioresour. Technol. 2020, 299, 122695. [Google Scholar] [CrossRef]
- Anderson, E.M.; Katahira, R.; Reed, M.; Resch, M.G.; Karp, E.M.; Beckham, G.T.; Román-Leshkov, Y. Reductive Catalytic Fractionation of Corn Stover Lignin. ACS Sustain. Chem. Eng. 2016, 4, 6940–6950. [Google Scholar] [CrossRef]
- Steinbrecher, T.; Sherbi, M.; Bonk, F.; Lüdtke, O.; Albert, J.; Kaltschmitt, M. Reductive Catalytic Fractionation of straw digestates for the production of biogenic aromatic monomers. Biomass Bioenergy 2024, 183, 107136. [Google Scholar] [CrossRef]
- Lan, W.; Luterbacher, J.S. Preventing Lignin Condensation to Facilitate Aromatic Monomer Production. Chimia 2019, 73, 591–598. [Google Scholar] [CrossRef]
- Li, J.; Henriksson, G.; Gellerstedt, G. Lignin depolymerization/repolymerization and its critical role for delignification of aspen wood by steam explosion. Bioresour. Technol. 2007, 98, 3061–3068. [Google Scholar] [CrossRef]
- Karlsson, M.; Romson, J.; Elder, T.; Emmer, Å.; Lawoko, M. Lignin Structure and Reactivity in the Organosolv Process Studied by NMR Spectroscopy, Mass Spectrometry, and Density Functional Theory. Biomacromolecules 2023, 24, 2314–2326. [Google Scholar] [CrossRef]
- Steinbrecher, T.; Bonk, F.; Scherzinger, M.; Lüdtke, O.; Kaltschmitt, M. Fractionation of Lignocellulosic Fibrous Straw Digestate by Combined Hydrothermal and Enzymatic Treatment. Energies 2022, 15, 6111. [Google Scholar] [CrossRef]
- Huang, X.; Korányi, T.I.; Boot, M.D.; Hensen, E.J.M. Ethanol as capping agent and formaldehyde scavenger for efficient depolymerization of lignin to aromatics. Green Chem. 2015, 17, 4941–4950. [Google Scholar] [CrossRef]
- Zhou, Z.; Liu, D.; Zhao, X. Conversion of lignocellulose to biofuels and chemicals via sugar platform: An updated review on chemistry and mechanisms of acid hydrolysis of lignocellulose. Renew. Sustain. Energy Rev. 2021, 146, 111169. [Google Scholar] [CrossRef]
- Zhang, Y.-H.P.; Cui, J.; Lynd, L.R.; Kuang, L.R. A transition from cellulose swelling to cellulose dissolution by o-phosphoric acid: Evidence from enzymatic hydrolysis and supramolecular structure. Biomacromolecules 2006, 7, 644–648. [Google Scholar] [CrossRef]
- Conrad, M.; Häring, H.; Smirnova, I. Design of an industrial autohydrolysis pretreatment plant for annual lignocellulose. Biomass Conv. Bioref. 2021, 11, 2293–2310. [Google Scholar] [CrossRef]
- Oriez, V.; Peydecastaing, J.; Pontalier, P.-Y. Lignocellulosic Biomass Fractionation by Mineral Acids and Resulting Extract Purification Processes: Conditions, Yields, and Purities. Molecules 2019, 24, 4273. [Google Scholar] [CrossRef]
- Donohoe, B.S.; Decker, S.R.; Tucker, M.P.; Himmel, M.E.; Vinzant, T.B. Visualizing lignin coalescence and migration through maize cell walls following thermochemical pretreatment. Biotechnol. Bioeng. 2008, 101, 913–925. [Google Scholar] [CrossRef]
- Yao, K.; Wu, Q.; An, R.; Meng, W.; Ding, M.; Li, B.; Yuan, Y. Hydrothermal pretreatment for deconstruction of plant cell wall: Part I. Effect on lignin-carbohydrate complex. AIChE J. 2018, 64, 1938–1953. [Google Scholar] [CrossRef]
- Sturgeon, M.R.; Kim, S.; Lawrence, K.; Paton, R.S.; Chmely, S.C.; Nimlos, M.; Foust, T.D.; Beckham, G.T. A Mechanistic Investigation of Acid-Catalyzed Cleavage of Aryl-Ether Linkages: Implications for Lignin Depolymerization in Acidic Environments. ACS Sustain. Chem. Eng. 2014, 2, 472–485. [Google Scholar] [CrossRef]
- Yokoyama, T. Revisiting the Mechanism of β- O -4 Bond Cleavage During Acidolysis of Lignin. Part 6: A Review. J. Wood Chem. Technol. 2015, 35, 27–42. [Google Scholar] [CrossRef]
- Košíková, B.; Joniak, D.; Kosáková, L. On the Properties of Benzyl Ether Bonds in the Lignin-Saccharidic Complex Isolated from Spruce. Holzforschung 1979, 33, 11–14. [Google Scholar] [CrossRef]
- Schlee, P.; Tarasov, D.; Rigo, D.; Balakshin, M. Advanced NMR Characterization of Aquasolv Omni (AqSO) Biorefinery Lignins/Lignin-Carbohydrate Complexes. ChemSusChem 2023, 16, e202300549. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Tu, M.; Paice, M.G. Routes to Potential Bioproducts from Lignocellulosic Biomass Lignin and Hemicelluloses. Bioenerg. Res. 2011, 4, 246–257. [Google Scholar] [CrossRef]
- Cheremisinoff, N.P.; Rosenfeld, P.E. Sources of air emissions from pulp and paper mills. In Handbook of Pollution Prevention and Cleaner Production; Cheremisinoff, N.P., Rosenfeld, P.E., Eds.; Elsevier: Amsterdam, The Netherlands, 2010; pp. 179–259. ISBN 9780080964461. [Google Scholar]
- Ek, M.; Gellerstedt, G.; Henriksson, G. (Eds.) Pulping Chemistry and Technology; Walter de Gruyter: Berlin, Germany, 2009; ISBN 978-3-11-021341-6. [Google Scholar]
- Roberts, V.M.; Stein, V.; Reiner, T.; Lemonidou, A.; Li, X.; Lercher, J.A. Towards quantitative catalytic lignin depolymerization. Chemistry 2011, 17, 5939–5948. [Google Scholar] [CrossRef]
- Gierer, J. Chemical aspects of kraft pulping. Wood Sci. Technol. 1980, 14, 241–266. [Google Scholar] [CrossRef]
- Komatsu, T.; Yamauchi, K.; Yokoyama, T. Promotive or suppressive effect of co-existing nucleophiles on lignin condensation in alkaline pulping processes. J. Wood Chem. Technol. 2023, 43, 67–77. [Google Scholar] [CrossRef]
- Deshpande, R.; Sundvall, L.; Grundberg, H.; Lawoko, M.; Henriksson, G. Lignin carbohydrate complex studies during kraft pulping for producing paper grade pulp from birch. TAPPI Press. 2020, 19, 447–460. [Google Scholar] [CrossRef]
- Tanbda, H.; Nakano, J.; Hosoya, S.; Chang, H.-M. Stability of α-Ether type Model Compounds During Chemical Pulping Processes. J. Wood Chem. Technol. 1987, 7, 485–497. [Google Scholar] [CrossRef]
- Lawoko, M.; Henriksson, G.; Gellerstedt, G. New Method for Quantitative Preparation of Lignin- Carbohydrate Complex from Unbleached Softwood Kraft Pulp: Lignin-Polysaccharide Networks I. Holzforschung 2003, 57, 69–74. [Google Scholar] [CrossRef]
- Balakshin, M.Y.; Capanema, E.A.; Chang, H. MWL fraction with a high concentration of lignin-carbohydrate linkages: Isolation and 2D NMR spectroscopic analysis. Holzforschung 2007, 61, 1–7. [Google Scholar] [CrossRef]
- Lawther, J.; Sun, R.-C.; Banks, W.B. Rapid isolation and structural characterization of alkali-soluble lignins during alkaline treatment and atmospheric refining of wheat straw. Ind. Crops Prod. 1996, 5, 97–105. [Google Scholar] [CrossRef]
- He, Y.; Pang, Y.; Liu, Y.; Li, X.; Wang, K. Physicochemical Characterization of Rice Straw Pretreated with Sodium Hydroxide in the Solid State for Enhancing Biogas Production. Energy Fuels 2008, 22, 2775–2781. [Google Scholar] [CrossRef]
- Grabber, J.H.; Hatfield, R.D.; Lu, F.; Ralph, J. Coniferyl ferulate incorporation into lignin enhances the alkaline delignification and enzymatic degradation of cell walls. Biomacromolecules 2008, 9, 2510–2516. [Google Scholar] [CrossRef]
- Omori, S.; Aoyama, M.; Sakakibara, A. Hydrolysis of Lignin with Dioxane-Water XIX. Reaction of ß-O-4 Lignin Model Compounds in the Presence of Carbohydrates. Holzforschung 1998, 52, 391–397. [Google Scholar] [CrossRef]
- Hossain, M.A.; Phung, T.K.; Rahaman, M.S.; Tulaphol, S.; Jasinski, J.B.; Sathitsuksanoh, N. Catalytic cleavage of the β-O-4 aryl ether bonds of lignin model compounds by Ru/C catalyst. Appl. Catal. A Gen. 2019, 582, 117100. [Google Scholar] [CrossRef]
- Galkin, M.V.; Dahlstrand, C.; Samec, J.S.M. Mild and Robust Redox-Neutral Pd/C-Catalyzed Lignol β-O-4′ Bond Cleavage Through a Low-Energy-Barrier Pathway. ChemSusChem 2015, 8, 2187–2192. [Google Scholar] [CrossRef]
- Nichols, J.M.; Bishop, L.M.; Bergman, R.G.; Ellman, J.A. Catalytic C-O bond cleavage of 2-aryloxy-1-arylethanols and its application to the depolymerization of lignin-related polymers. J. Am. Chem. Soc. 2010, 132, 12554–12555. [Google Scholar] [CrossRef]
- Lu, J.; Wang, M.; Zhang, X.; Heyden, A.; Wang, F. β-O-4 Bond Cleavage Mechanism for Lignin Model Compounds over Pd Catalysts Identified by Combination of First-Principles Calculations and Experiments. ACS Catal. 2016, 6, 5589–5598. [Google Scholar] [CrossRef]
- Liu, Z.; Li, H.; Gao, X.; Guo, X.; Wang, S.; Fang, Y.; Song, G. Rational highly dispersed ruthenium for reductive catalytic fractionation of lignocellulose. Nat. Commun. 2022, 13, 4716. [Google Scholar] [CrossRef]
- Galkin, M.V.; Smit, A.T.; Subbotina, E.; Artemenko, K.A.; Bergquist, J.; Huijgen, W.J.J.; Samec, J.S.M. Hydrogen-free catalytic fractionation of woody biomass. ChemSusChem 2016, 9, 3280–3287. [Google Scholar] [CrossRef]
- Sun, Z.; Bottari, G.; Afanasenko, A.; Stuart, M.C.A.; Deuss, P.J.; Fridrich, B.; Barta, K. Complete lignocellulose conversion with integrated catalyst recycling yielding valuable aromatics and fuels. Nat. Catal. 2018, 1, 82–92. [Google Scholar] [CrossRef]
- Li, Y.; Yu, Y.; Lou, Y.; Zeng, S.; Sun, Y.; Liu, Y.; Yu, H. Hydrogen-Transfer Reductive Catalytic Fractionation of Lignocellulose: High Monomeric Yield with Switchable Selectivity. Angew. Chem. Int. Ed. Engl. 2023, 62, e202307116. [Google Scholar] [CrossRef]
- Ullah, N.; Odda, A.H.; Liang, K.; Kombo, M.A.; Sahar, S.; Ma, L.-B.; Fang, X.-X.; Xu, A.-W. Metal–acid nanoplate-supported ultrafine Ru nanoclusters for efficient catalytic fractionation of lignin into aromatic alcohols. Green Chem. 2019, 21, 2739–2751. [Google Scholar] [CrossRef]
- Chen, L.; van Muyden, A.P.; Cui, X.; Fei, Z.; Yan, N.; Laurenczy, G.; Dyson, P.J. Lignin First: Confirming the Role of the Metal Catalyst in Reductive Fractionation. JACS Au J. 2021, 1, 729–733. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Wu, S.; Zhang, H.; Xiao, R. Catalytic oxidation of lignin to valuable biomass-based platform chemicals: A review. Fuel Process. Technol. 2019, 191, 181–201. [Google Scholar] [CrossRef]
- Parkås, J.; Brunow, G.; Lundquist, K. Quantitative lignin analysis based on permanganate oxidation. BioResources 2007, 2, 169–178. [Google Scholar] [CrossRef]
- Vangeel, T.; Schutyser, W.; Renders, T.; Sels, B.F. Perspective on Lignin Oxidation: Advances, Challenges, and Future Directions. Top. Curr. Chem. 2018, 376, 30. [Google Scholar] [CrossRef]
- Lancefield, C.S.; Ojo, O.S.; Tran, F.; Westwood, N.J. Isolation of functionalized phenolic monomers through selective oxidation and C-O bond cleavage of the β-O-4 linkages in lignin. Angew. Chem. Int. Ed. Engl. 2015, 54, 258–262. [Google Scholar] [CrossRef]
- Yu, X.; Wei, Z.; Lu, Z.; Pei, H.; Wang, H. Activation of lignin by selective oxidation: An emerging strategy for boosting lignin depolymerization to aromatics. Bioresour. Technol. 2019, 291, 121885. [Google Scholar] [CrossRef]
- Rahimi, A.; Ulbrich, A.; Coon, J.J.; Stahl, S.S. Formic-acid-induced depolymerization of oxidized lignin to aromatics. Nature 2014, 515, 249–252. [Google Scholar] [CrossRef]
- Zhang, C.; Li, H.; Lu, J.; Zhang, X.; MacArthur, K.E.; Heggen, M.; Wang, F. Promoting Lignin Depolymerization and Restraining the Condensation via an Oxidation−Hydrogenation Strategy. ACS Catal. 2017, 7, 3419–3429. [Google Scholar] [CrossRef]
- Zhu, G.; Qiu, X.; Zhao, Y.; Qian, Y.; Pang, Y.; Ouyang, X. Depolymerization of lignin by microwave-assisted methylation of benzylic alcohols. Bioresour. Technol. 2016, 218, 718–722. [Google Scholar] [CrossRef]
- Guo, H.; Miles-Barrett, D.M.; Zhang, B.; Wang, A.; Zhang, T.; Westwood, N.J.; Li, C. Is oxidation–reduction a real robust strategy for lignin conversion? A comparative study on lignin and model compounds. Green Chem. 2019, 21, 803–811. [Google Scholar] [CrossRef]
- Wang, M.; Lu, J.; Zhang, X.; Li, L.; Li, H.; Luo, N.; Wang, F. Two-Step, Catalytic C–C Bond Oxidative Cleavage Process Converts Lignin Models and Extracts to Aromatic Acids. ACS Catal. 2016, 6, 6086–6090. [Google Scholar] [CrossRef]
- Rinesch, T.; Mottweiler, J.; Puche, M.; Concepción, P.; Corma, A.; Bolm, C. Mechanistic Investigation of the Catalyzed Cleavage for the Lignin β-O-4 Linkage: Implications for Vanillin and Vanillic Acid Formation. ACS Sustain. Chem. Eng. 2017, 5, 9818–9825. [Google Scholar] [CrossRef]
- Dabral, S.; Hernández, J.G.; Kamer, P.C.J.; Bolm, C. Organocatalytic Chemoselective Primary Alcohol Oxidation and Subsequent Cleavage of Lignin Model Compounds and Lignin. ChemSusChem 2017, 10, 2707–2713. [Google Scholar] [CrossRef]
- Hu, Y.; Li, S.; Zhao, X.; Wang, C.; Zhang, X.; Liu, J.; Ma, L.; Chen, L.; Zhang, Q. Catalytic oxidation of native lignin to phenolic monomers: Insight into aldehydes formation and stabilization. Catal. Commun. 2022, 172, 106532. [Google Scholar] [CrossRef]
- Ishikawa, A.; Hosoya, T.; Miyafuji, H. Pathways for vanillin production through alkaline aerobic oxidation of a phenolic lignin model compound, guaiacylglycerol-β-guaiacyl ether, in concentrated aqueous alkali. RSC Sustain. 2024, 2, 1936–1947. [Google Scholar] [CrossRef]
- Schutyser, W.; Kruger, J.S.; Robinson, A.M.; Katahira, R.; Brandner, D.G.; Cleveland, N.S.; Mittal, A.; Peterson, D.J.; Meilan, R.; Román-Leshkov, Y.; et al. Revisiting alkaline aerobic lignin oxidation. Green Chem. 2018, 20, 3828–3844. [Google Scholar] [CrossRef]
- Mathieu, Y.; Vidal, J.D.; Arribas Martínez, L.; Abad Fernández, N.; Iborra, S.; Corma, A. Molecular Oxygen Lignin Depolymerization: An Insight into the Stability of Phenolic Monomers. ChemSusChem 2020, 13, 4743–4758. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Du, X.; Liu, W.; Tricker, A.W.; Dai, H.; Deng, Y. Highly Efficient Lignin Depolymerization via Effective Inhibition of Condensation during Polyoxometalate-Mediated Oxidation. Energy Fuels 2019, 33, 6483–6490. [Google Scholar] [CrossRef]
- Robinson, J.M. Lignin, land plants, and fungi: Biological evolution affecting Phanerozoic oxygen balance. Geology 1990, 18, 607–610. [Google Scholar] [CrossRef]
- Kirk, T.K.; Farrell, R.L. Enzymatic “combustion”: The microbial degradation of lignin. Annu. Rev. Microbiol. 1987, 41, 465–505. [Google Scholar] [CrossRef]
- Eastwood, D.C.; Floudas, D.; Binder, M.; Majcherczyk, A.; Schneider, P.; Aerts, A.; Asiegbu, F.O.; Baker, S.E.; Barry, K.; Bendiksby, M.; et al. The plant cell wall-decomposing machinery underlies the functional diversity of forest fungi. Science 2011, 333, 762–765. [Google Scholar] [CrossRef]
- Labatut, R.A.; Angenent, L.T.; Scott, N.R. Characterizing the influence of wastewater composition and lignin content on anaerobic biodegradability. Environ. Sci. Water Res. Technol. 2022, 8, 1507–1520. [Google Scholar] [CrossRef]
- Vikman, M.; Karjomaa, S.; Kapanen, A.; Wallenius, K.; Itävaara, M. The influence of lignin content and temperature on the biodegradation of lignocellulose in composting conditions. Appl. Microbiol. Biotechnol. 2002, 59, 591–598. [Google Scholar] [CrossRef]
- Kögel-Knabner, I. The macromolecular organic composition of plant and microbial residues as inputs to soil organic matter. Soil. Biol. Biochem. 2002, 34, 139–162. [Google Scholar] [CrossRef]
- Thevenot, M.; Dignac, M.-F.; Rumpel, C. Fate of lignins in soils: A review. Soil. Biol. Biochem. 2010, 42, 1200–1211. [Google Scholar] [CrossRef]
- Hall, S.J.; Huang, W.; Timokhin, V.I.; Hammel, K.E. Lignin lags, leads, or limits the decomposition of litter and soil organic carbon. Ecology 2020, 101, e03113. [Google Scholar] [CrossRef]
- Shevchenko, S.M.; Bailey, G.W. Life after death: Lignin-humic relationships reexamined. Crit. Rev. Environ. Sci. Technol. 1996, 26, 95–153. [Google Scholar] [CrossRef]
- Gerke, J. Concepts and Misconceptions of Humic Substances as the Stable Part of Soil Organic Matter: A Review. Agronomy 2018, 8, 76. [Google Scholar] [CrossRef]
- Atiwesh, G.; Parrish, C.C.; Banoub, J.; Le, T.-A.T. Lignin degradation by microorganisms: A review. Biotechnol. Prog. 2022, 38, e3226. [Google Scholar] [CrossRef] [PubMed]
- Cragg, S.M.; Beckham, G.T.; Bruce, N.C.; Bugg, T.D.H.; Distel, D.L.; Dupree, P.; Etxabe, A.G.; Goodell, B.S.; Jellison, J.; McGeehan, J.E.; et al. Lignocellulose degradation mechanisms across the Tree of Life. Curr. Opin. Chem. Biol. 2015, 29, 108–119. [Google Scholar] [CrossRef]
- Pollegioni, L.; Tonin, F.; Rosini, E. Lignin-degrading enzymes. FEBS J. 2015, 282, 1190–1213. [Google Scholar] [CrossRef]
- Martínez, A.T.; Speranza, M.; Ruiz-Dueñas, F.J.; Ferreira, P.; Camarero, S.; Guillén, F.; Martínez, M.J.; Gutiérrez, A.; Del Río, J.C. Biodegradation of lignocellulosics: Microbial, chemical, and enzymatic aspects of the fungal attack of lignin. Int. Microbiol. 2005, 8, 195–204. [Google Scholar]
- Bugg, T.D.H. The chemical logic of enzymatic lignin degradation. Chem. Commun. 2024, 60, 804–814. [Google Scholar] [CrossRef]
- Arantes, V.; Goodell, B. Current Understanding of Brown-Rot Fungal Biodegradation Mechanisms: A Review. In Deterioration and Protection of Sustainable Biomaterials; Schultz, T.P., Goodell, B., Nicholas, D.D., Eds.; American Chemical Society: Washington, DC, USA, 2014; pp. 3–21. ISBN 9780841230040. [Google Scholar]
- Arantes, V.; Jellison, J.; Goodell, B. Peculiarities of brown-rot fungi and biochemical Fenton reaction with regard to their potential as a model for bioprocessing biomass. Appl. Microbiol. Biotechnol. 2012, 94, 323–338. [Google Scholar] [CrossRef]
- Goodell, B.; Jellison, J.; Liu, J.; Daniel, G.; Paszczynski, A.; Fekete, F.; Krishnamurthy, S.; Jun, L.; Xu, G. Low molecular weight chelators and phenolic compounds isolated from wood decay fungi and their role in the fungal biodegradation of wood. J. Biotechnol. 1997, 53, 133–162. [Google Scholar] [CrossRef]
- Yelle, D.J.; Wei, D.; Ralph, J.; Hammel, K.E. Multidimensional NMR analysis reveals truncated lignin structures in wood decayed by the brown rot basidiomycete Postia placenta. Environ. Microbiol. 2011, 13, 1091–1100. [Google Scholar] [CrossRef]
- Hofrichter, M.; Scheibner, K.; Bublitz, F.; Schneegaß, I.; Ziegenhagen, D.; Martens, R.; Fritsche, W. Depolymerization of Straw Lignin by Manganese Peroxidase from Nematoloma frowardii is Accompanied by Release of Carbon Dioxide. Holzforschung 1999, 53, 161–166. [Google Scholar] [CrossRef]
- Hofrichter, M.; Vares, K.; Scheibner, K.; Galkin, S.; Sipilä, J.; Hatakka, A. Mineralization and solubilization of synthetic lignin by manganese peroxidases from Nematoloma frowardii and Phlebia radiata. J. Biotechnol. 1999, 67, 217–228. [Google Scholar] [CrossRef]
- Gall, D.L.; Kontur, W.S.; Lan, W.; Kim, H.; Li, Y.; Ralph, J.; Donohue, T.J.; Noguera, D.R. In Vitro Enzymatic Depolymerization of Lignin with Release of Syringyl, Guaiacyl, and Tricin Units. Appl. Environ. Microbiol. 2018, 84, e02076-17. [Google Scholar] [CrossRef] [PubMed]
- Marinović, M.; Nousiainen, P.; Dilokpimol, A.; Kontro, J.; Moore, R.; Sipilä, J.; de Vries, R.P.; Mäkelä, M.R.; Hildén, K. Selective Cleavage of Lignin β-O-4 Aryl Ether Bond by β-Etherase of the White-Rot Fungus Dichomitus squalens. ACS Sustain. Chem. Eng. 2018, 6, 2878–2882. [Google Scholar] [CrossRef]
- Linger, J.G.; Vardon, D.R.; Guarnieri, M.T.; Karp, E.M.; Hunsinger, G.B.; Franden, M.A.; Johnson, C.W.; Chupka, G.; Strathmann, T.J.; Pienkos, P.T.; et al. Lignin valorization through integrated biological funneling and chemical catalysis. Proc. Natl. Acad. Sci. USA 2014, 111, 12013–12018. [Google Scholar] [CrossRef]
- Fuchs, G.; Boll, M.; Heider, J. Microbial degradation of aromatic compounds—From one strategy to four. Nat. Rev. Microbiol. 2011, 9, 803–816. [Google Scholar] [CrossRef]
- Del Cerro, C.; Erickson, E.; Dong, T.; Wong, A.R.; Eder, E.K.; Purvine, S.O.; Mitchell, H.D.; Weitz, K.K.; Markillie, L.M.; Burnet, M.C.; et al. Intracellular pathways for lignin catabolism in white-rot fungi. Proc. Natl. Acad. Sci. USA 2021, 118, e2017381118. [Google Scholar] [CrossRef]
- Hori, C.; Gaskell, J.; Igarashi, K.; Kersten, P.; Mozuch, M.; Samejima, M.; Cullen, D. Temporal alterations in the secretome of the selective ligninolytic fungus Ceriporiopsis subvermispora during growth on aspen wood reveal this organism’s strategy for degrading lignocellulose. Appl. Environ. Microbiol. 2014, 80, 2062–2070. [Google Scholar] [CrossRef]
- Jeffries, T.W. Biodegradation of lignin-carbohydrate complexes. Biodegradation 1990, 1, 163–176. [Google Scholar] [CrossRef]
- van den Brink, J.; de Vries, R.P. Fungal enzyme sets for plant polysaccharide degradation. Appl. Microbiol. Biotechnol. 2011, 91, 1477–1492. [Google Scholar] [CrossRef]
- Arnling Bååth, J.; Giummarella, N.; Klaubauf, S.; Lawoko, M.; Olsson, L. A glucuronoyl esterase from Acremonium alcalophilum cleaves native lignin-carbohydrate ester bonds. FEBS Lett. 2016, 590, 2611–2618. [Google Scholar] [CrossRef]
- Agger, J.W.; Isaksen, T.; Várnai, A.; Vidal-Melgosa, S.; Willats, W.G.T.; Ludwig, R.; Horn, S.J.; Eijsink, V.G.H.; Westereng, B. Discovery of LPMO activity on hemicelluloses shows the importance of oxidative processes in plant cell wall degradation. Proc. Natl. Acad. Sci. USA 2014, 111, 6287–6292. [Google Scholar] [CrossRef] [PubMed]
- Vaaje-Kolstad, G.; Westereng, B.; Horn, S.J.; Liu, Z.; Zhai, H.; Sørlie, M.; Eijsink, V.G.H. An Oxidative Enzyme Boosting the Enzymatic Conversion of Recalcitrant Polysaccharides. Science 2010, 330, 219–222. [Google Scholar] [CrossRef] [PubMed]
- Bar-Lev, S.S.; Kirk, T.K. Effects of molecular oxygen on lignin degradation by Phanerochaete chrysosporium. Biochem. Biophys. Res. Commun. 1981, 99, 373–378. [Google Scholar] [CrossRef]
- Kirk, T.K.; Schultz, E.; Connors, W.J.; Lorenz, L.F.; Zeikus, J.G. Influence of culture parameters on lignin metabolism byPhanerochaete chrysosporium. Arch. Microbiol. 1978, 117, 277–285. [Google Scholar] [CrossRef]
- Reid, I.D.; Seifert, K.A. Effect of an atmosphere of oxygen on growth, respiration, and lignin degradation by white-rot fungi. Can. J. Bot. 1982, 60, 252–260. [Google Scholar] [CrossRef]
- Colberg, P.J.; Young, L.Y. Aromatic and Volatile Acid Intermediates Observed during Anaerobic Metabolism of Lignin-Derived Oligomers. Appl. Environ. Microbiol. 1985, 49, 350–358. [Google Scholar] [CrossRef]
- Zeikus, J.G.; Wellstein, A.L.; Kirk, T.K. Molecular basis for the biodegradative recalcitrance of lignin in anaerobic environments. FEMS Microbiol. Lett. 1982, 15, 193–197. [Google Scholar] [CrossRef]
- Lankiewicz, T.S.; Choudhary, H.; Gao, Y.; Amer, B.; Lillington, S.P.; Leggieri, P.A.; Brown, J.L.; Swift, C.L.; Lipzen, A.; Na, H.; et al. Lignin deconstruction by anaerobic fungi. Nat. Microbiol. 2023, 8, 596–610. [Google Scholar] [CrossRef]
- Dumond, L.; Lam, L.P.Y.; van Erven, G.; Kabel, M.; Mounet, F.; Grima-Pettenati, J.; Tobimatsu, Y.; Hernandez-Raquet, G. Termite Gut Microbiota Contribution to Wheat Straw Delignification in Anaerobic Bioreactors. ACS Sustain. Chem. Eng. 2021, 9, 2191–2202. [Google Scholar] [CrossRef]
- Yu, T.; Wu, W.; Liang, W.; Lever, M.A.; Hinrichs, K.-U.; Wang, F. Growth of sedimentary Bathyarchaeota on lignin as an energy source. Proc. Natl. Acad. Sci. USA 2018, 115, 6022–6027. [Google Scholar] [CrossRef]
- DeWeerd, K.A.; Saxena, A.; Nagle, D.P.; Suflita, J.M. Metabolism of the 18O-methoxy substituent of 3-methoxybenzoic acid and other unlabeled methoxybenzoic acids by anaerobic bacteria. Appl. Environ. Microbiol. 1988, 54, 1237–1242. [Google Scholar] [CrossRef]
- Barbosa, F.C.; Silvello, M.A.; Goldbeck, R. Cellulase and oxidative enzymes: New approaches, challenges and perspectives on cellulose degradation for bioethanol production. Biotechnol. Lett. 2020, 42, 875–884. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Davaritouchaee, M. Nature-inspired pretreatment of lignocellulose—Perspective and development. Bioresour. Technol. 2023, 369, 128456. [Google Scholar] [CrossRef] [PubMed]
- Hegnar, O.A.; Goodell, B.; Felby, C.; Johansson, L.; Labbé, N.; Kim, K.; Eijsink, V.G.H.; Alfredsen, G.; Várnai, A. Challenges and opportunities in mimicking non-enzymatic brown-rot decay mechanisms for pretreatment of Norway spruce. Wood Sci. Technol. 2019, 53, 291–311. [Google Scholar] [CrossRef]
- Klinger, G.E.; Zhou, Y.; Foote, J.A.; Wester, A.M.; Cui, Y.; Alherech, M.; Stahl, S.S.; Jackson, J.E.; Hegg, E.L. Nucleophilic Thiols Reductively Cleave Ether Linkages in Lignin Model Polymers and Lignin. ChemSusChem 2020, 13, 4394–4399. [Google Scholar] [CrossRef] [PubMed]
- Davaritouchaee, M.; Hiscox, W.C.; Martinez-Fernandez, J.; Fu, X.; Mancini, R.J.; Chen, S. Effect of reactive oxygen species on biomass structure in different oxidative processes. Ind. Crops Prod. 2019, 137, 484–494. [Google Scholar] [CrossRef]
- Sharma, H.K.; Xu, C.; Qin, W. Biological Pretreatment of Lignocellulosic Biomass for Biofuels and Bioproducts: An Overview. Waste Biomass Valor. 2019, 10, 235–251. [Google Scholar] [CrossRef]
- Todorovic, T.; Norström, E.; Khabbaz, F.; Brücher, J.; Malmström, E.; Fogelström, L. A fully bio-based wood adhesive valorising hemicellulose-rich sidestreams from the pulp industry. Green Chem. 2021, 23, 3322–3333. [Google Scholar] [CrossRef]
- Shuai, L.; Amiri, M.T.; Questell-Santiago, Y.M.; Héroguel, F.; Li, Y.; Kim, H.; Meilan, R.; Chapple, C.; Ralph, J.; Luterbacher, J.S. Formaldehyde stabilization facilitates lignin monomer production during biomass depolymerization. Science 2016, 354, 329–333. [Google Scholar] [CrossRef]
- Lan, W.; Amiri, M.T.; Hunston, C.M.; Luterbacher, J.S. Protection Group Effects During α,γ-Diol Lignin Stabilization Promote High-Selectivity Monomer Production. Angew. Chem. Int. Ed. Engl. 2018, 57, 1356–1360. [Google Scholar] [CrossRef]
- Usmani, Z.; Sharma, M.; Awasthi, A.K.; Lukk, T.; Tuohy, M.G.; Gong, L.; Nguyen-Tri, P.; Goddard, A.D.; Bill, R.M.; Nayak, S.; et al. Lignocellulosic biorefineries: The current state of challenges and strategies for efficient commercialization. Renew. Sustain. Energy Rev. 2021, 148, 111258. [Google Scholar] [CrossRef]
- Saini, R.; Osorio-Gonzalez, C.S.; Hegde, K.; Brar, S.K.; Magdouli, S.; Vezina, P.; Avalos-Ramirez, A. Lignocellulosic Biomass-Based Biorefinery: An Insight into Commercialization and Economic Standout. Curr. Sustain. Renew. Energy Rep. 2020, 7, 122–136. [Google Scholar] [CrossRef]
- Bartling, A.W.; Stone, M.L.; Hanes, R.J.; Bhatt, A.; Zhang, Y.; Biddy, M.J.; Davis, R.; Kruger, J.S.; Thornburg, N.E.; Luterbacher, J.S.; et al. Techno-economic analysis and life cycle assessment of a biorefinery utilizing reductive catalytic fractionation. Energy Environ. Sci. 2021, 14, 4147–4168. [Google Scholar] [CrossRef] [PubMed]
- Bhutto, A.W.; Qureshi, K.; Harijan, K.; Abro, R.; Abbas, T.; Bazmi, A.A.; Karim, S.; Yu, G. Insight into progress in pre-treatment of lignocellulosic biomass. Energy 2017, 122, 724–745. [Google Scholar] [CrossRef]
- Facas, G.G.; Brandner, D.G.; Bussard, J.R.; Román-Leshkov, Y.; Beckham, G.T. Interdependence of Solvent and Catalyst Selection on Low Pressure Hydrogen-Free Reductive Catalytic Fractionation. ACS Sustain. Chem. Eng. 2023, 11, 4517–4522. [Google Scholar] [CrossRef]
- Liu, M.-Y.; Zhang, Z.-H.; Wang, X.-Q.; Sun, Q.; Zhang, C.; Li, Y.; Sun, Z.; Barta, K.; Peng, F.; Yuan, T.-Q. Atmospheric reductive catalytic fractionation of lignocellulose integrated with one-pot catalytic conversion of carbohydrate yielding valuable lignin monomers and platform chemicals from corn straw. Green. Energy Environ. 2024, 10, 161–172. [Google Scholar] [CrossRef]
- Arts, W.; van Aelst, K.; Cooreman, E.; van Aelst, J.; van den Bosch, S.; Sels, B.F. Stepping away from purified solvents in reductive catalytic fractionation: A step forward towards a disruptive wood biorefinery process. Energy Environ. Sci. 2023, 16, 2518–2539. [Google Scholar] [CrossRef]
- Croes, T.; Dutta, A.; de Bie, R.; van Aelst, K.; Sels, B.; van der Bruggen, B. Extraction of monophenols and fractionation of depolymerized lignin oil with nanofiltration membranes. Chem. Eng. J. 2023, 452, 139418. [Google Scholar] [CrossRef]
- Sultan, Z.; Graça, I.; Li, Y.; Lima, S.; Peeva, L.G.; Kim, D.; Ebrahim, M.A.; Rinaldi, R.; Livingston, A.G. Membrane Fractionation of Liquors from Lignin-First Biorefining. ChemSusChem 2019, 12, 1203–1212. [Google Scholar] [CrossRef]
- Bourbiaux, D.; Pu, J.; Rataboul, F.; Djakovitch, L.; Geantet, C.; Laurenti, D. Reductive or oxidative catalytic lignin depolymerization: An overview of recent advances. Catal. Today 2021, 373, 24–37. [Google Scholar] [CrossRef]
- Liao, Y.; Koelewijn, S.-F.; van den Bossche, G.; van Aelst, J.; van den Bosch, S.; Renders, T.; Navare, K.; Nicolaï, T.; van Aelst, K.; Maesen, M.; et al. A sustainable wood biorefinery for low-carbon footprint chemicals production. Science 2020, 367, 1385–1390. [Google Scholar] [CrossRef]
- Koelewijn, S.-F.; Cooreman, C.; Renders, T.; Andecochea Saiz, C.; van den Bosch, S.; Schutyser, W.; de Leger, W.; Smet, M.; van Puyvelde, P.; Witters, H.; et al. Promising bulk production of a potentially benign bisphenol A replacement from a hardwood lignin platform. Green Chem. 2018, 20, 1050–1058. [Google Scholar] [CrossRef]
- Crestini, C.; Lange, H.; Sette, M.; Argyropoulos, D.S. On the structure of softwood kraft lignin. Green Chem. 2017, 19, 4104–4121. [Google Scholar] [CrossRef]







| Established Processes | Under Research at TRL ≥ 7 | Under Research at TRL ≤ 7 | ||||
|---|---|---|---|---|---|---|
| Kraft pulping (pp. 91–119), [227,328] | Sulfite pulping [227] (pp. 91–119) | Organosolv pulping [188,195,212] | Hydrothermal treatment [213,217,220] | Reductive catalytic fractionation (RCF) [128,186,209] | Aldehyde-assisted fractionation (AAF) [314] | |
| Primary focus | Removal of lignin | Removal of lignin | Removal of lignin | Removal of hemicellulose | Removal of lignin | Removal of lignin and hemicellulose |
| β-O-4 ether bond cleavage | Largely by alkaline conditions and application of strong nucleophiles | Largely by acidic or alkaline conditions and application of strong nucleophiles | To a certain extent, by solvolysis and often acidolysis | Slightly by (auto-)hydrolysis | Largely by solvolysis and hydrogenolysis | Not in first step due to stabilization strategy. Possible, e.g., by hydrogenolysis in second step. |
| Lignin solubility | Yes, in aqueous alkali | Yes, in water due to sulfonation | Yes, in organic solvent | No | Yes, typically in organic solvent | Yes, in organic solvent |
| Active lignin stabilization | None | None, but sulfonation leads to certain stabilization | None, but attachment of alcohols leads to certain stabilization | None | Yes, by hydrogenation | Yes, reactions with aldehydes lead to blocking of reactive positions and hinder ether cleavage |
| Modifications to lignin | Fragmentation, condensation, addition of thiol groups | Fragmentation, condensation, sulfonation | Fragmentation, condensation | Fragmentation, redeposition, condensation | Depolymerization to oligomers and monomers. Hydrogenation. Partial loss of aliphatic OH groups | Acetal formation and reaction of aldehydes with aromatic rings in first step. |
| Utilization of all fractions | Lignin typically burned. Hemicellulose partly retained in pulp, partly burned. Cellulose as pulp. | Lignin burned or low-value material use (e.g., as dispersant). Hemicellulose partly retained in pulp, partly burned. Cellulose as pulp. | Solid lignin recovered, potential use, e.g., for phenolic resins. Hemicellulose partly solubilized, partly retained together with cellulose as pulp. | Hemicellulose oligomers solubilized in water. Cellulose enzymatically hydrolysable. Solid lignin potentially usable, e.g., as filling material. | Lignin as oil with diverse potential uses as aromatic chemicals. Hemicellulose partly solubilized, partly retained together with cellulose as pulp. | Potential use of functionalized hemicellulose sugars and depolymerized lignin as chemicals. Cellulose as pulp. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Steinbrecher, T.; Albert, J.; Kaltschmitt, M. The Nature of Lignin and Implications for Its Technical Use as a Source for Biogenic Aromatics—A Review. Sustain. Chem. 2025, 6, 38. https://doi.org/10.3390/suschem6040038
Steinbrecher T, Albert J, Kaltschmitt M. The Nature of Lignin and Implications for Its Technical Use as a Source for Biogenic Aromatics—A Review. Sustainable Chemistry. 2025; 6(4):38. https://doi.org/10.3390/suschem6040038
Chicago/Turabian StyleSteinbrecher, Timo, Jakob Albert, and Martin Kaltschmitt. 2025. "The Nature of Lignin and Implications for Its Technical Use as a Source for Biogenic Aromatics—A Review" Sustainable Chemistry 6, no. 4: 38. https://doi.org/10.3390/suschem6040038
APA StyleSteinbrecher, T., Albert, J., & Kaltschmitt, M. (2025). The Nature of Lignin and Implications for Its Technical Use as a Source for Biogenic Aromatics—A Review. Sustainable Chemistry, 6(4), 38. https://doi.org/10.3390/suschem6040038