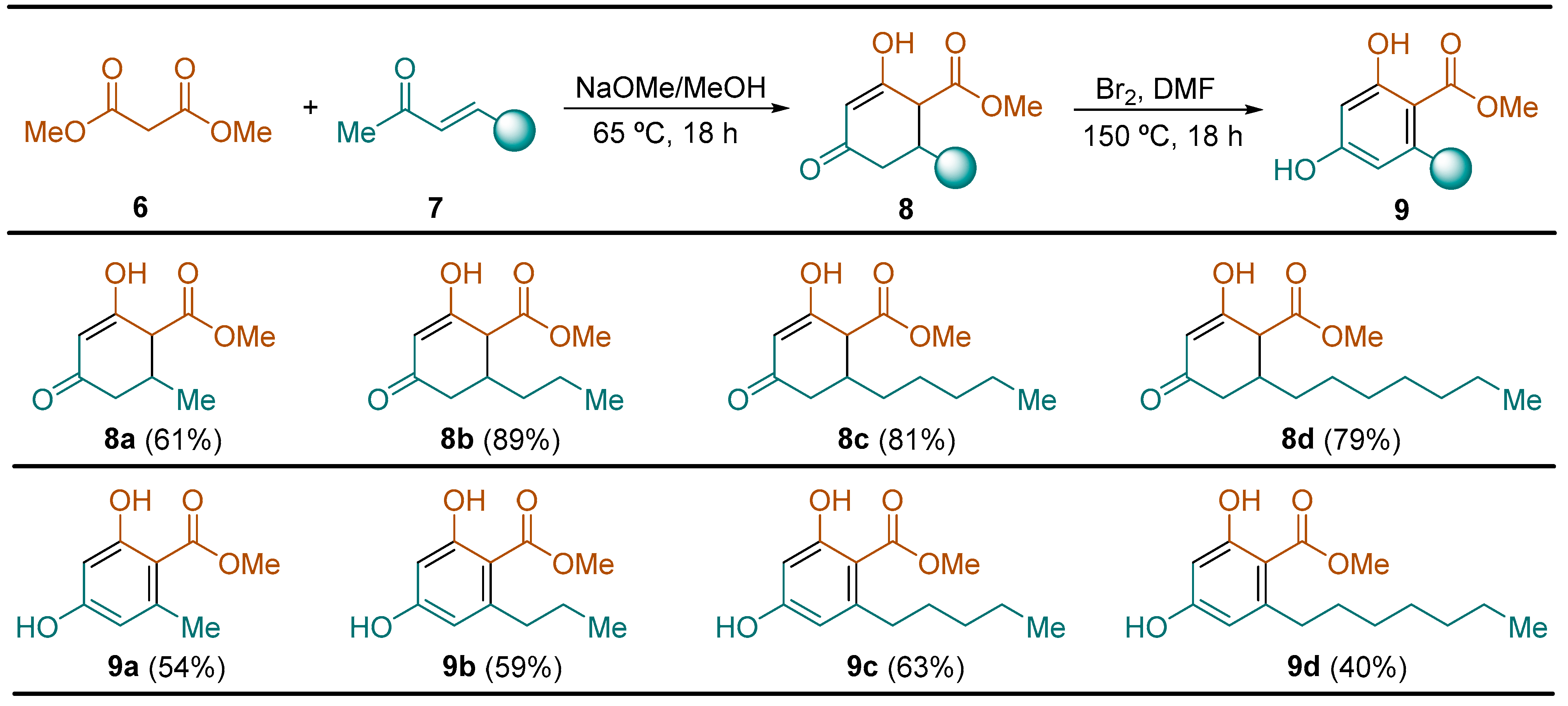
2.2.1. Synthesis of Hydroxycyclohexenone Derivatives 8
General Procedure. To a solution of dimethyl malonate 6 (13.2 g, 11.4 mL, 100.0 mmol) in methanol (80 mL) was successively added a 5.8 M solution of NaOMe in methanol (16.0 mL, 92.8 mmol) and the corresponding enone 7 (72.0 mmol) at 0 °C. After that, the resulting reaction mixture was heated at 65 °C for 18 h. Then, the solvent was evaporated under vacuum (15 Torr). The residue was dissolved first in 50 mL of dichloromethane, followed by the addition of 50 mL of water. After stirring for 15 min, the organic phase was discarded and the aqueous phase was acidified with a 3.0 M aqueous HCl solution to pH 2. The resulting turbid aqueous phase was extracted with dichloromethane (3 × 100 mL), dried over magnesium sulfate, and the solvent was evaporated (15 Torr), giving rise to the expected compounds 8 in high purity, which were used in the next reaction step without the need for further purification.
Methyl 2-Hydroxy-6-methyl-4-oxocyclohex-2-ene-1-carboxylate (
8a) [
18]: following the general procedure, compound 8a was obtained from pent-3-en-2-one (7a, 7.11 g, 8.25 mL, 72.0 mmol) as a yellow solid (8.00 g, 43.47 mmol, 61%): C
9H
12O
4; mp 127–128 °C (hexane/CH
2Cl
2, lit. 122–123 °C [
18]); [HPLC (Agilent IndinityLab Poroshell 120 EC-C18 1260 column, acetonitrile/formic acid = 99.9/0.1, 0.4 mL/min, 228 nm) t = 3.47 min (98.75%)];
Rf 0.22 (hexane/EtOAc 1:3);
1H NMR (300 MHz, DMSO-
d6) δ 11.44 (br s, 1H), 5.23 (s, 1H), 3.64 (s, 3H), 3.11 (d,
J = 10.9 Hz, 1H), 2.40–2.23 (m, 3H), 0.96 (d,
J = 6.1 Hz, 3H); LRMS (EI)
m/
z 184 (M
+, 15%), 169 (28), 153 (18), 125 (23), 114 (39), 101 (25), 84 (23), 69 (100), 55 (19), 43 (26); HRMS (EI-TOF) calcd. for C
9H
12O
4 184.0736; found 184.0736.
Methyl 2-Hydroxy-4-oxo-6-propylcyclohex-2-ene-1-carboxylate (8b): following the general procedure, compound 8b was obtained from hept-3-en-2-one (7b, 8.48 g, 10.05 mL, 72.0 mmol) as a yellow solid (13.51 g, 63.72 mmol, 88.5%): C11H16O4; mp 95–96 °C (hexane/CH2Cl2); [HPLC (Agilent IndinityLab Poroshell 120 EC-C18 1260 column, acetonitrile/formic acid = 99.9/0.1, 0.4 mL/min, 228 nm) t = 10.87 min (99.13%)]; Rf 0.27 (hexane/EtOAc 1:3); 1H NMR (300 MHz, DMSO-d6) δ 11.56 (br s, 1H), 5.23 (s, 1H), 3.64 (s, 3H), 3.16 (d, J = 10.8 Hz, 1H), 2.40 (dd, J = 16.0, 3.8 Hz, 1H), 2.33–2.18 (m, 2H), 1.33–1.19 (m, 4H), 0.84 (t, J = 7.0 Hz, 3H); LRMS (EI) m/z 212 (M+, 10%), 181 (20), 169 (100), 153 (11), 137 (73), 127 (33), 113 (24), 97 (82), 84 (29), 69 (31), 55 (41), 43 (44); HRMS (EI-TOF) calcd. for C11H16O4 212.1049; found 212.1047.
Methyl 2-Hydroxy-4-oxo-6-pentylcyclohex-2-ene-1-carboxylate (
8c) [
19]: following the general procedure, compound 8c was obtained from non-3-en-2-one (7c, 10.50 g, 12.38 mL, 72.0 mmol) as a pale yellow solid (14.00 g, 58.32 mmol, 81.0%): C
13H
20O
4; mp 82–83 °C (hexane/CH
2Cl
2, lit. 98–100 °C [
19]); [HPLC (Agilent IndinityLab Poroshell 120 EC-C18 1260 column, acetonitrile/formic acid = 99.9/0.1, 0.4 mL/min, 228 nm) t = 13.25 min (97.41%)];
Rf 0.35 (hexane/EtOAc 1:3);
1H NMR (300 MHz, DMSO-
d6) δ 6.09 (br s, 1H), 5.18 (s, 1H), 3.62 (s, 3H), 3.13 (d,
J = 10.7 Hz, 1H), 2.39 (dd,
J = 16.1, 3.9 Hz, 1H), 2.29–2.13 (m, 2H), 1.30–1.18 (m, 8H), 0.85 (t,
J = 6.9 Hz, 3H); LRMS (EI)
m/
z 240 (M
+, 3%), 169 (100), 157 (11), 137 (49), 125 (33), 95 (26), 84 (13), 69 (13), 55 (22), 43 (15); HRMS (EI-TOF) calcd. for C
13H
20O
4: 240.1362; found 240.1352.
Methyl 6-Heptyl-2-hydroxy-4-oxocyclohex-2-ene-1-carboxylate (
8d) [
20]: following the general procedure, compound 8d was obtained from undec-3-en-2-one (7d, 12.10 g, 13.44 mL, 72.0 mmol) as a pale yellow solid (15.24 g, 56.88 mmol, 79.0%): C
15H
24O
4; mp 71–72 °C (hexane/CH
2Cl
2, lit. 85–87 °C [
20]); [HPLC (Agilent IndinityLab Poroshell 120 EC-C18 1260 column, acetonitrile/formic acid = 99.9/0.1, 0.4 mL/min, 228 nm) t = 14.84 min (97.73%)];
Rf 0.29 (hexane/EtOAc 1:3);
1H NMR (300 MHz, DMSO-
d6) δ 11.38 (br s, 1H), 5.23 (s, 1H), 3.63 (s, 3H), 3.20–3.12 (m, 1H), 2.45–2.36 (m, 1H), 2.33–2.17 (m, 2H), 1.29–1.20 (m, 12H), 0.85 (t,
J = 6.7 Hz, 3H); LRMS (EI)
m/
z 268 (M
+, 2%),169 (100), 153 (12), 137 (43), 95 (20), 69 (12), 55 (12), 43 (19); HRMS (EI-TOF) calcd. for C
15H
24O
4: 268.1675; found 268.1669.
2.2.2. Synthesis of Resorcinol Methyl Ester Derivatives 9
General Procedure. To a solution of the corresponding enone 8 (40.0 mmol) in dimethyl formamide (40 mL) was added a solution of bromine (6.40 g, 2.05 mL, 40.0 mmol) in dimethyl formamide (20 mL) at 0 °C. The resulting reaction mixture was heated at 150 °C for 18 h. Then, it was cooled down to reach room temperature and after that, a 0.4 M solution of Na2S2O3 (12.64 g, 80.0 mmol) in water (200 mL) was added and stirred for 24 h at the same temperature. The reaction mixture was extracted with tert-butyl methyl ether (3 × 50 mL), and the combined organic phases were washed with water (50 mL) and brine (50 mL), dried over magnesium sulfate, and the solvent was evaporated under vacuum (15 Torr). The resulting residue was dissolved in a 1:1 hexane ethyl acetate solution (100 mL), filtered through a silica gel pad, and washed the 1:1 hexane ethyl acetate solution (3 × 100 mL). The solvent was evaporated under vacuum (15 Torr), and the residue was dissolved in dichloromethane (10 mL), warming up the solution to 30 °C, followed by the slow addition of hexane (80 mL). The resulting cloudy solution was cooled down to −10 °C, and after 1 h, the solid formed was filtered off and dried to yield pure resorcinol derivatives 9.
Methyl 2,4-Dihydroxy-6-methylbenzoate (
9a) [
21]: following the general procedure, compound 9a was obtained from enone
8a (7.36 g, 40.0 mmol) as a white solid (3.93 g, 21.6 mmol, 54%): C
9H
10O
4; mp 170–172 °C (hexane/CH
2Cl
2, lit. 173–174 °C [
21]);
Rf 0.21 (hexane/EtOAc 7:1);
1H NMR (300 MHz, CDCl
3) δ 11.82 (s, 1H), 6.30 (dd,
J = 2.6, 0.5 Hz, 1H), 6.25 (dd,
J = 2.6, 0.8 Hz, 1H), 4.93 (br s, 1H), 3.93 (s, 3H), 2.49 (s, 3H);
13C NMR (75 MHz, CDCl
3) δ 172.1 (C), 165.1 (C), 160.6 (C), 143.9 (C), 111.5 (CH), 105.5 (C), 101.3 (CH), 51.9 (CH
3), 24.2 (CH
3); LRMS (EI)
m/
z 182 (M
+, 45%), 150 (100), 122 (55), 94 (15), 69 (13), 43 (11); HRMS (EI-TOF) calcd. for C
9H
10O
4 182.0579; found 182.0575.
Methyl 2,4-Dihydroxy-6-propylbenzoate (
9b) [
21]. Following the general procedure, compound 9b was obtained from enone
8b (8.48 g, 40.0 mmol) as a white solid (4.95 g, 23.6 mmol, 59%): C
11H
14O
4; mp 152–153 °C (hexane/CH
2Cl
2, lit. 152–156 °C [
21]);
Rf 0.26 (hexane/EtOAc 7:1);
1H NMR (300 MHz, CDCl
3) δ 11.90 (s, 1H), 6.33 (d,
J = 2.6 Hz, 1H), 6.27 (d,
J = 2.6 Hz, 1H), 6.11 (br s, 1H), 3.94 (s, 3H), 2.87–2.76 (m, 2H), 1.59–1.51 (m, 2H), 0.95 (t,
J = 7.3 Hz, 3H);
13C NMR (75 MHz, CDCl
3) δ 172.1 (C), 164.8 (C), 160.6 (C), 148.8 (C), 111.2 (CH), 104.9 (C), 101.4 (CH), 52.1 (CH
3), 38.8 (CH
2), 24.8 (CH
2), 14.3 (CH
3); LRMS (EI)
m/
z 210 (M
+, 32%), 178 (100), 150 (30), 121 (31), 69 (11), 43 (13); HRMS (EI-TOF) calcd. for C
11H
14O
4 210.0892; found 210.0891.
Methyl 2,4-Dihydroxy-6-pentylbenzoate (
9c) [
21]: following the general procedure, compound 9c was obtained from enone
8c (9.60 g, 40.0 mmol) as a yellow solid (5.99 g, 25.2 mmol, 63%): C
13H
18O
4; mp 105–106 °C (hexane/CH
2Cl
2, lit. 105–106 °C [
21]);
Rf 0.28 (hexane/EtOAc 7:1);
1H NMR (300 MHz, CDCl
3) δ 11.91 (s, 1H), 6.59 (br s, 1H), 6.32 (d,
J = 2.6 Hz, 1H), 6.27 (d,
J = 2.6 Hz, 1H), 3.93 (s, 3H), 2.87–2.78 (m, 2H), 1.57–1.46 (m, 2H), 1.36–1.29 (m, 4H), 0.91 (t,
J = 7.2 Hz, 3H);
13C NMR (75 MHz, CDCl
3) δ 172.1 (C), 164.9 (C), 160.6 (C), 149.1 (C), 111.1 (CH), 104.9 (C), 101.4 (CH), 52.0 (CH
3), 36.8 (CH
2), 32.1 (CH
2), 31.5 (CH
2), 22.5 (CH
2), 14.1 (CH
3); LRMS (EI)
m/
z 238 (M
+, 48%), 206 (65), 182 (75), 150 (100), 121 (25), 94 (12), 69 (20), 43 (25); HRMS (EI-TOF) calcd. for C
13H
18O
4 238.1205; found 238.1199.
Methyl 2-Heptyl-2,4-dihydroxybenzoate (
9d) [
20]: following the general procedure, compound 9d was obtained from enone
8d (10.72 g, 40.0 mmol) as a yellow solid (4.25 g, 16.0 mmol, 40%): C
15H
22O
4; mp 95–96 °C (hexane/CH
2Cl
2, lit. 71.5–72 °C [
20]);
Rf 0.33 (hexane/EtOAc 7:1);
1H NMR (300 MHz, CDCl
3) δ 11.84 (s, 1H), 6.31 (d,
J = 2.5 Hz, 1H), 6.28 (br s, 1H), 6.26 (d,
J = 2.5 Hz, 1H), 3.93 (s, 3H), 2.88–2.78 (m, 2H), 1.57–1.45 (m, 2H), 1.35–1.27 (m, 8H), 0.90 (t,
J = 7.3 Hz, 3H);
13C NMR (75 MHz, CDCl
3) δ 172.0 (C), 165.0 (C), 160.6 (C), 149.0 (C), 110.9 (CH), 104.9 (C), 101.4 (CH), 51.9 (CH
3), 36.9 (CH
2), 31.85 (CH
2), 31.8 (CH
2), 29.8 (CH
2), 29.2 (CH
2), 22.7 (CH
2), 14.1 (CH
3); LRMS (EI)
m/
z 266 (M
+, 35%), 234 (29), 192 (12), 182 (100), 163 (37), 150 (59), 121 (18), 69 (17), 43 (39); HRMS (EI-TOF) calcd. for C
15H
22O
4 266.1522; found 266.1517.
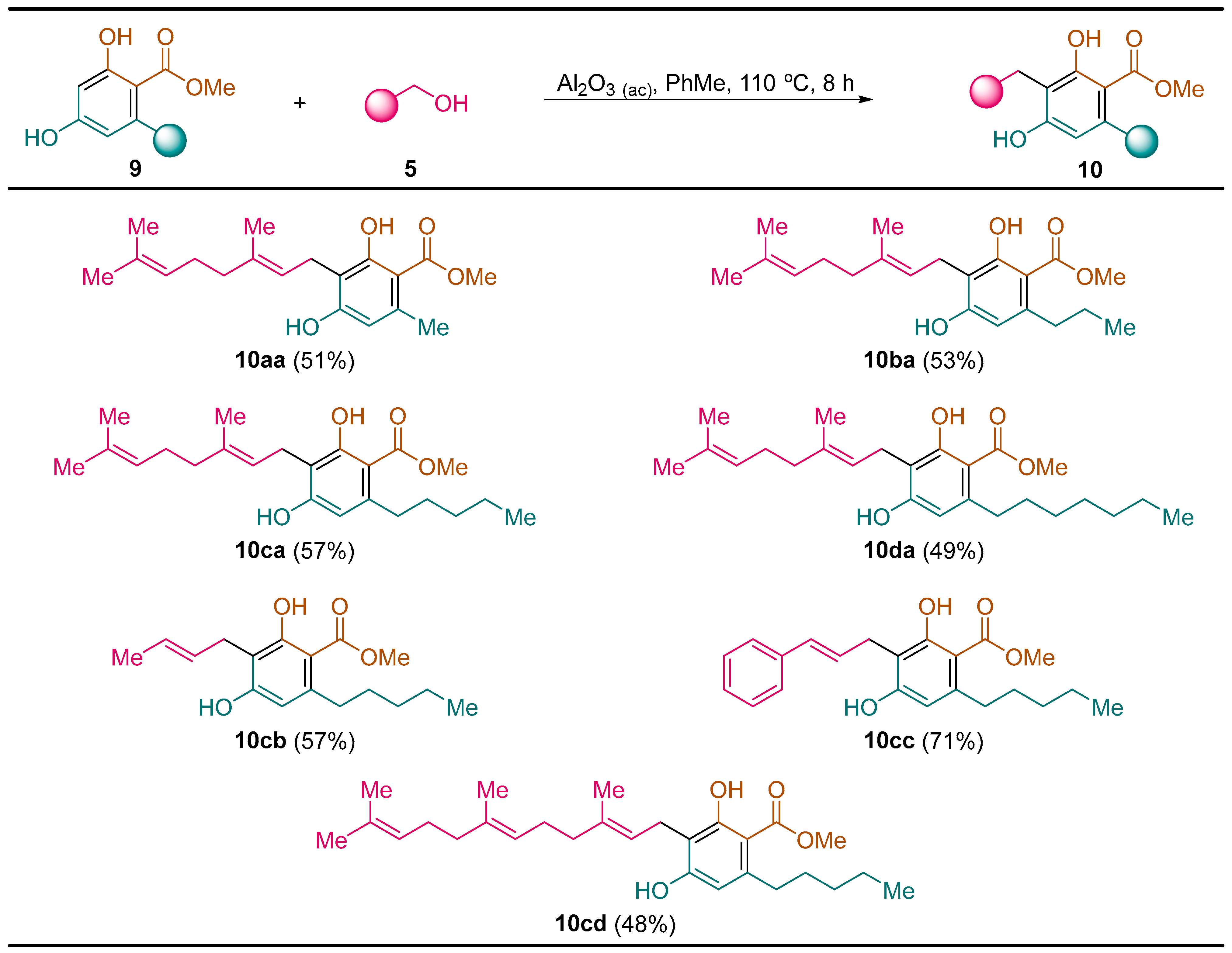
2.2.3. Synthesis of Cannabigerol Methyl Ester Derivatives 10
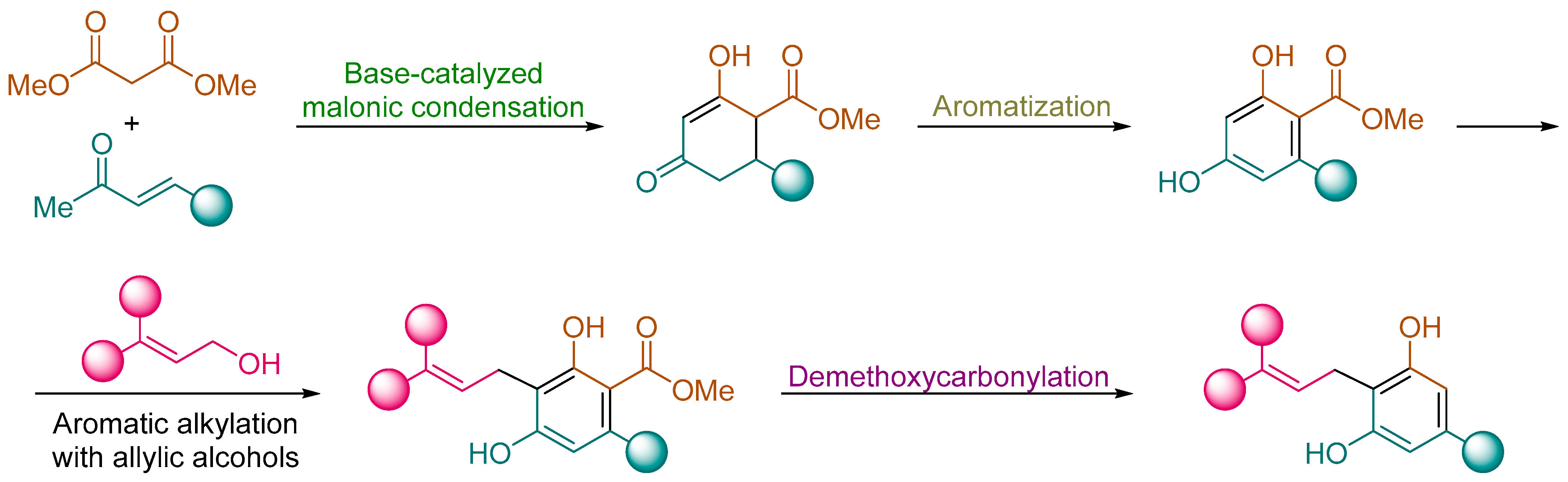
General Procedure. To a solution of the corresponding resorcinol derivative 9 (5.0 mmol) and alcohol 5 (5.00 mmol) in toluene (5.0 mL) was added acid alumina (10.0 g). The resulting reaction mixture was heated at 110 °C for 8 h. Then, acid alumina was filtered off and washed with ethyl acetate (3 × 25 mL). The solvent was evaporated under vacuum (15 Torr) and the residue was purified by column chromatography (silica gel, hexane/EtOAc) to yield pure compounds 10.
Methyl (
E)-2,4-Dihydroxy-3-(3,7-dimethylocta-2,6-dien-1-yl)-6-methylbenzoate (
10aa) [
22]. Following the general procedure, compound
10aa was obtained from resorcinol derivative
9a (0.91 g, 5.0 mmol) and geraniol (
5a, 0.772 g, 0.87 mL, 5.0 mmol) as a yellow solid (0.811 g, 2.55 mmol, 51%): C
19H
26O
4; mp 90–91 °C (hexane/CH
2Cl
2, lit. 46–47 °C [
22]);
Rf 0.75 (hexane/EtOAc 7:1);
1H NMR (300 MHz, CDCl
3) δ 12.13 (s, 1H), 6.24 (d,
J = 0.8 Hz, 1H), 5.82 (br s, 1H), 5.34–5.24 (m, 1H), 5.12–5.01 (m, 1H), 3.93 (s, 3H), 3.44 (d,
J = 7.2 Hz, 2H), 2.47 (s, 3H), 2.12–2.07 (m, 4H), 1.82 (s, 3H), 1.69 (s, 3H), 1.60 (s, 3H);
13C NMR (75 MHz, CDCl
3) δ 172.7 (C), 162.6 (C), 159.5 (C), 140.9 (C), 139.2 (C), 132.1 (C), 123.8 (CH), 121.4 (CH), 111.4 (CH), 111.3 (C), 105.1 (C), 51.9, 39.7 (CH
2), 26.4 (CH
2), 25.7 (CH
3), 24.2 (CH
3), 22.1 (CH
2), 17.7 (CH
3), 16.2 (CH
3); LRMS (EI)
m/
z 318 (M
+, <1%), 299 (55), 261 (20), 229 (100), 215 (11), 187 (13), 175 (42), 123 (13), 91 (11), 69 (48), 41 (35); HRMS (EI-TOF) calcd. for C
19H
26O
4 318.1831; found 318.1825.
Methyl (E)-2,4-Dihydroxy-3-(3,7-dimethylocta-2,6-dien-1-yl)-6-propylbenzoate (10ba): following the general procedure, compound 10ba was obtained from resorcinol derivative 9b (1.05 g, 5.0 mmol) and geraniol (5a, 0.772 g, 0.87 mL, 5.0 mmol) as a yellow solid (0.916 g, 2.65 mmol, 53%): C21H30O4; mp 82–83 °C (hexane/CH2Cl2); Rf 0.77 (hexane/EtOAc 7:1); 1H NMR (300 MHz, CDCl3) δ 12.05 (s, 1H), 6.25 (s, 1H), 5.86 (s, 1H), 5.35–5.25 (m, 1H), 5.12–5.03 (m, 1H), 3.94 (s, 3H), 3.45 (d, J = 7.3 Hz, 2H), 2.85–2.76 (m, 2H), 2.16–2.02 (m, 4H), 1.82 (q, J = 1.0 Hz, 3H), 1.69 (d, J = 1.3 Hz, 3H), 1.60 (dd, J = 1.5, 0.8 Hz, 3H), 1.58–1.52 (m, 2H), 0.96 (t, J = 7.3 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ 172.5 (C), 162.5 (C), 159.5 (C), 145.5 (C), 139.1 (C), 132.0 (C), 123.8 (CH), 121.5 (CH), 111.5 (C), 110.9 (CH), 104.6 (C) 51.8 (CH3), 39.7 (CH2), 38.8 (CH2), 26.4 (CH2), 25.7 (CH3), 24.9 (CH2), 22.1 (CH2), 17.7 (CH3), 16.2 (CH3), 14.3 (CH3); LRMS (EI) m/z 346 (M+, <1%), 314 (20), 271 (15), 245 (100), 223 (13), 191 (69), 123 (16), 69 (14), 41 (21); HRMS (EI-TOF) calcd. for C21H30O4 346.2144; found 346.2147.
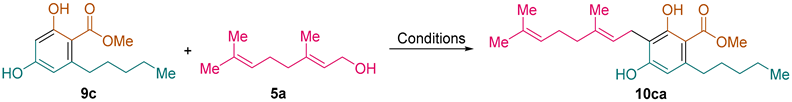
Methyl (
E)-2,4-Dihydroxy-3-(3,7-dimethylocta-2,6-dien-1-yl)-6-pentylbenzoate (
10ca) [
23]: following the general procedure, compound
10ca was obtained from resorcinol derivative
9c (1.19 g, 5.0 mmol) and geraniol (
5a, 0.772 g, 0.87 mL, 5.0 mmol) as a yellow solid (1.065 g, 2.85 mmol, 57%): C
23H
34O
4; mp 60–61 °C (hexane/CH
2Cl
2);
Rf 0.79 (hexane/EtOAc 7:1);
1H NMR (300 MHz, CDCl
3) δ 12.03 (s, 1H), 6.23 (s, 1H), 5.83 (s, 1H), 5.29–5.24 (m, 1H), 5.05 (m, 1H), 3.92 (s, 3H), 3.43 (d,
J = 7.2 Hz, 2H), 2.80 (t, J = 7.5 Hz, 2H), 2.13–2.02 (m, 4H), 1.81 (d,
J = 1.2 Hz, 3H), 1.67 (d,
J = 1.3 Hz, 3H), 1.59 (d,
J = 1.3 Hz, 3H), 1.59–1.56 (m, 2H), 1.37–1.29 (m, 4H), 0.90 (t,
J = 6.9 Hz, 3H);
13C NMR (75 MHz, CDCl
3) δ 172.5 (C), 162.5 (C), 159.5 (C), 145.8 (C), 138.9 (C), 131.9 (C), 123.8 (CH), 121.5 (CH), 111.5 (C), 110.8 (CH), 104.5 (C) 51.8 (CH
3), 39.7 (CH
2), 36.8 (CH
2), 32.1 (CH
2), 31.6 (CH
2), 26.4 (CH
2), 25.6 (CH
3), 22.5 (CH
2), 22.1 (CH
2), 17.7 (CH
3), 16.2 (CH
3), 14.1 (CH
3); LRMS (EI)
m/
z 373 (M
+-1, 6%), 342 (23), 299 (19), 273 (100), 251 (17), 219 (92), 123 (21), 91 (9), 69 (24), 41 (29); HRMS (EI-TOF) calcd. for C
23H
34O
4 374.2457; found 374.2453.
Methyl (E)-6-Heptyl-2,4-Dihydroxy-(3,7-dimethylocta-2,6-dien-1-yl)benzoate (10da): following the general procedure, compound 10da was obtained from resorcinol derivative 9d (1.33 g, 5.0 mmol) and geraniol (5a, 0.772 g, 0.87 mL, 5.0 mmol) as a yellow solid (0.984 g, 2.45 mmol, 49%): C25H38O4; mp 74–75 °C (hexane/CH2Cl2); Rf 0.80 (hexane/EtOAc 7:1); 1H NMR (300 MHz, CDCl3) δ 12.07 (s, 1H), 6.24 (s, 1H), 5.88 (s, 1H), 5.28 (dt, J = 6.4, 4.2 Hz, 1H), 5.14–5.01 (m, 1H), 3.93 (s, 3H), 3.44 (d, J = 7.1 Hz, 2H), 2.88–2.74 (m, 2H), 2.09 (q, J = 6.3 Hz, 4H), 1.82 (s, 3H), 1.68 (s, 3H), 1.60 (s, 3H), 1.53 (d, J = 6.9 Hz, 2H), 1.35–1.24 (m, 8H), 0.92 (t, J = 6.9 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ 172.5 (C), 162.5 (C), 159.5 (C), 145.8 (C), 139.1 (C), 132.0 (C), 123.8 (CH), 121.5 (CH), 111.4 (C), 110.8 (CH), 104.6 (C), 51.8 (CH3), 39.7 (CH2), 36.8 (CH2), 31.9 (CH2), 31.85 (CH2), 29.9 (CH2), 29.2 (CH2), 26.4 (CH2), 25.7 (CH3), 22.7 (CH2), 22.1 (CH2), 17.7 (CH3), 16.2 (CH3), 14.1 (CH3); LRMS (EI) m/z 402 (M+, 3%), 370 (8), 358 (11), 292 (18), 247 (100), 175 (15), 164 (15), 143 (47), 125 (22), 99 (13), 83 (15), 69 (19), 59 (25), 43 (73); HRMS (EI-TOF) calcd. For C25H38O4: 402.2770; found 402.2743.
Methyl (E)-3-(But-2-en-1-yl)-2,4-dihydroxy-6-pentylbenzoate (10cb): following the general procedure, compound 10cb was obtained from resorcinol derivative 9c (1.19 g, 5.0 mmol) and crotyl alcohol (5b, 0.360 g, 0.31 mL, 5.0 mmol) as a red solid (0.833 g, 2.85 mmol, 57%): C17H24O4; mp 38–40 °C (hexane/CH2Cl2); Rf 0.72 (hexane/EtOAc 7:1); 1H NMR (300 MHz, CDCl3) δ 12.04 (s, 1H), 6.26 (s, 1H), 5.97 (br s, 1H), 5.66–5.58 (m, 2H), 3.94 (s, 3H), 3.41 (q, J = 2.0 Hz, 2H), 2.86–2.77 (m, 2H), 1.69 (dt, J = 4.8, 1.6 Hz, 3H), 1.58–1.48 (m, 2H), 1.39–1.28 (m, 4H), 0.93 (t, J = 6.9 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ 172.5 (C), 162.6 (C), 159.2 (C), 146.0 (C), 128.4 (CH), 126.7 (CH), 110.9 (C), 110.7 (CH), 104.6 (CH3), 51.9 (CH2), 36.8 (CH2), 32.1 (CH2), 31.6 (CH2), 26.0 (CH2), 22.5 (CH2), 17.8 (CH3), 14.1 (CH3); LRMS (EI) m/z 292 (M+, 56%), 260 (100), 231 (95), 219 (48), 204 (17), 189 (21), 176 (77), 147 (46), 105 (10), 91 (21), 77 (18), 55 (23), 43 (30); HRMS (EI-TOF) calcd. for C17H24O4 292.1675; found 292.1663.
Methyl 3-Cinnamyl-2,4-dihydroxy-6-pentylbenzoate (10cc): following the general procedure, compound 10cc was obtained from resorcinol derivative 9c (1.19 g, 5.0 mmol) and cinnamyl alcohol (5c, 0.670 g, 0.64 mL, 5.0 mmol) as a yellow solid (1.25 g, 3.55 mmol, 71%): C22H26O4; mp 57–59 °C (hexane/CH2Cl2); Rf 0.78 (hexane/EtOAc 7:1); 1H NMR (300 MHz, CDCl3) δ 12.09 (s, 1H), 7.39–7.19 (m, 6H), 6.53–6.33 (m, 2H), 6.27 (s, 1H), 3.95 (s, 3H), 3.63 (dd, J = 6.3, 1.4 Hz, 2H), 2.91–2.76 (m, 2H), 1.61–1.49 (m, 2H), 1.42–1.30 (m, 4H), 0.98–0.91 (t, J = 6.8 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ 172.4 (C), 162.8 (C), 158.8 (C), 146.2 (C), 137.3 (C), 130.8 (CH), 128.6 (CH), 128.5 (CH), 127.6 (CH), 127.1 (CH), 126,5 (CH), 126.2 (CH), 110.7 (C), 110.6 (CH), 104.9 (C), 51.9 (CH3), 36.8 (CH2), 32.1 (CH2), 31.6 (CH2), 26.4 (CH2), 22.6 (CH2), 14.1 (CH3); LRMS (EI) m/z 354 (M+, 16%), 322 (30), 231 (100), 175 (11), 147 (13), 115 (11), 91 (26), 43 (12); HRMS (EI-TOF) calcd. for C22H26O4: 354.1831; found 354.1844.
Methyl 2,4-Dihydroxy-6-pentyl-3-((2E,6E)-3,7,11-trimethyldodeca-2,6,10-trien-1-yl)benzoate (10cd): following the general procedure, compound 10cd was obtained from resorcinol derivative 9c (1.19 g, 5.0 mmol) and farnesol (5d, 1.112 g, 1.25 mL, 5.0 mmol) as a yellow solid (1.060 g, 2.40 mmol, 48%): C28H42O4; mp 95–97 °C (hexane/CH2Cl2); Rf 0.74 (hexane/EtOAc 7:1); 1H NMR (300 MHz, CDCl3) δ 12.10 (s, 1H), 6.32–6.20 (m, 2H), 5.38–5.27 (m, 1H), 5.12 (dddq, J = 7.2, 4.5, 2.8, 1.5 Hz, 2H), 3.94 (s, 3H), 3.46 (d, J = 7.2 Hz, 2H), 2.86–2.77 (m, 2H), 2.17–1.98 (m, 8H), 1.87–1.82 (m, 3H), 1.71 (t, J = 1.4 Hz, 3H), 1.66–1.59 (m, 6H), 1.53 (tt, J = 7.0, 3.6 Hz, 2H), 1.42–1.31 (m, 4H), 0.93 (t, J = 6.9 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ 172.6 (C), 162.7 (C), 159.4 (C), 145.7 (C), 138.2 (C), 135.4 (C), 131.2 (C), 124.5 (CH), 123.8 (CH), 121.6 (CH), 111.8 (C), 110.8 (CH), 104.4 (C), 51.8 (CH3), 39.8 (CH2), 39.7 (CH2), 36.8 (CH2), 32.2 (CH2), 31.6 (CH2), 26.7 (CH2), 26.4 (CH2), 25.7 (CH3), 22.6 (CH2), 22.1 (CH2), 17.7 (CH3), 16.2 (CH3), 16.0 (CH3), 14.1 (CH3); LRMS (EI) m/z 442 (M+, 7%), 411 (13), 341 (36), 299 (13), 273 (99), 251 (43), 219 (100), 191 (30), 175 (13), 147 (15), 135 (14), 121 (25), 109 (14), 95 (11), 81 (20), 69 (53), 41 (22); HRMS (EI-TOF) calcd. For C28H42O4: 442.3083; found 442.3072.
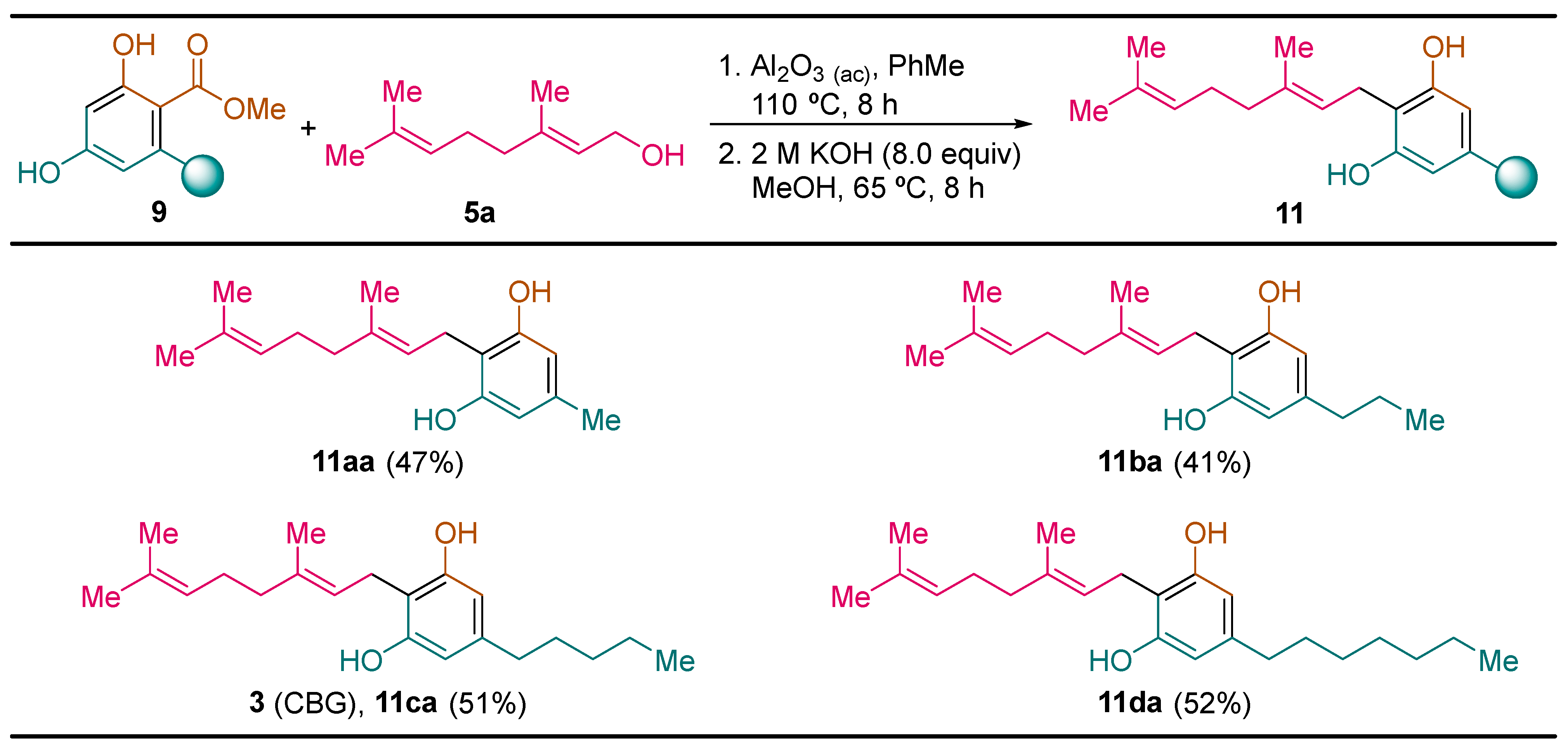
2.2.4. Synthesis of Cannabigerol Derivatives 11
General Procedure. To a solution of the corresponding resorcinol derivative 9 (1.0 mmol) and geraniol 5a (0.154 g, 0.173 mL, 1.00 mmol) in toluene (1.0 mL) was added acid alumina (2.0 g). The resulting reaction mixture was heated at 110 °C for 8 h. Then, acid alumina was filtered off and washed with ethyl acetate (3 × 10 mL). The solvent was evaporated under vacuum (15 Torr) and the resulting cannabigerol methyl ester derivative 10 was used in the next reaction step without further purification. Then, a solution of KOH (0.450 g, 8.0 mmol) in water (4.0 mL), and the corresponding cannabigerol methyl ester derivative 10 (1.0 mmol) in methanol (3.0 mL) was heated at 65 °C for 8 h, and after that, it was cooled down to room temperature and water (5 mL) and ethyl acetate (5 mL) was added. The resulting mixture was acidified with concentrated hydrochloric acid aqueous solution to pH 3. The reaction mixture was extracted with ethyl acetate (3 × 10 mL), and the combined organic phases were washed with water (20 mL) and brine (20 mL), dried over magnesium sulfate and the solvent was evaporated under vacuum (15 Torr). The resulting residue was dissolve in heptane (1.0 mL) at 80 °C, and after that, the solution was allowed to reach room temperature, and then it was cooled down at -10 °C. After 2 h, the crystalline solid formed was filtered off and dried to yield pure resorcinol derivatives 11.
(
E)-2-(3,7-Dimethylocta-2,6-dien-1-yl)-5-methylbenzene-1,3-diol (
11aa) [
15]: following the general procedure, compound
11aa was obtained from resorcinol methyl ester derivative
9a (0.128 g, 1.0 mmol) as a yellow solid (0.122 g, 0.47 mmol, 47%): C
17H
24O
2; mp 85–86 °C (heptane);
Rf 0.60 (hexane/EtOAc 7:1);
1H NMR (300 MHz, CDCl
3) δ 6.26 (s, 2H), 5.34–5.23 (m, 1H), 5.12–5.05 (m, 3H), 3.41 (d,
J = 7.1 Hz, 2H), 2.23 (s, 3H), 2.17–2.04 (m, 4H), 1.83 (s, 3H), 1.70 (s, 3H), 1.61 (s, 3H);
13C NMR (75 MHz, CDCl
3) δ 154.8 (C), 138.9 (C), 137.5 (C), 132.0 (C), 123.8 (CH), 121.7 (CH), 110.5 (C), 109.1 (CH), 39.7 (CH
2), 26.4 (CH
2), 25.7 (CH
3), 22.2 (CH
2), 21.0 (CH
3), 17.7 (CH
3), 16.2 (CH
2); LRMS (EI)
m/
z 260 (M
+, 14%), 217 (5), 163 (21), 149 (20), 137 (100), 123 (21), 69 (17), 61 (50); HRMS (EI-TOF) calcd. For C
17H
24O
2: 260.1776; found 260.1771.
(
E)-2-(3,7-Dimethylocta-2,6-dien-1-yl)-5-propylbenzene-1,3-diol (cannabigerovarin,
11ba) [
24]: following the general procedure, compound
11ba was obtained from resorcinol methyl ester derivative
9b (0.210 g, 1.0 mmol) as an orange solid (0.118 g, 0.41 mmol, 41%): C
19H
28O
2; mp 73–74 °C (heptane, lit. 52–53 °C [
24]);
Rf 0.63 (hexane/EtOAc 7:1);
1H NMR (300 MHz, CDCl
3) δ 6.27 (s, 2H), 5.32–5.26 (m, 1H), 5.10–4.97 (m, 3H), 3.41 (d,
J = 7.1 Hz, 2H), 2.45 (dd,
J = 8.6, 6.7 Hz, 2H), 2.10 (d,
J = 6.1 Hz, 4H), 1.83 (q,
J = 1.0 Hz, 3H), 1.69 (d,
J = 1.4 Hz, 3H), 1.63–1.52 (m, 5H), 0.94 (t,
J = 7.3 Hz, 3H);
13C NMR (75 MHz, CDCl
3) δ 154.8 (C), 142.5 (C), 139.0 (C), 132.1 (C), 123.8 (CH), 121.7 (CH), 110.6 (C), 108.4 (CH), 39.7 (CH
2), 37.6 (CH
2), 26.4 (CH
2), 25.7 (CH
3), 24.2 (CH
2), 22.3 (CH
2), 17.7 (CH
3), 16.2 (CH
3), 13.9 (CH
3); LRMS (EI)
m/
z 288 (M
+, 12%), 203 (27), 191 (14), 177 (10), 165 (100), 123 (20), 69 (14), 41 (19); HRMS (EI-TOF) calcd. For C
19H
28O
2: 288.2089; found 288.2095.
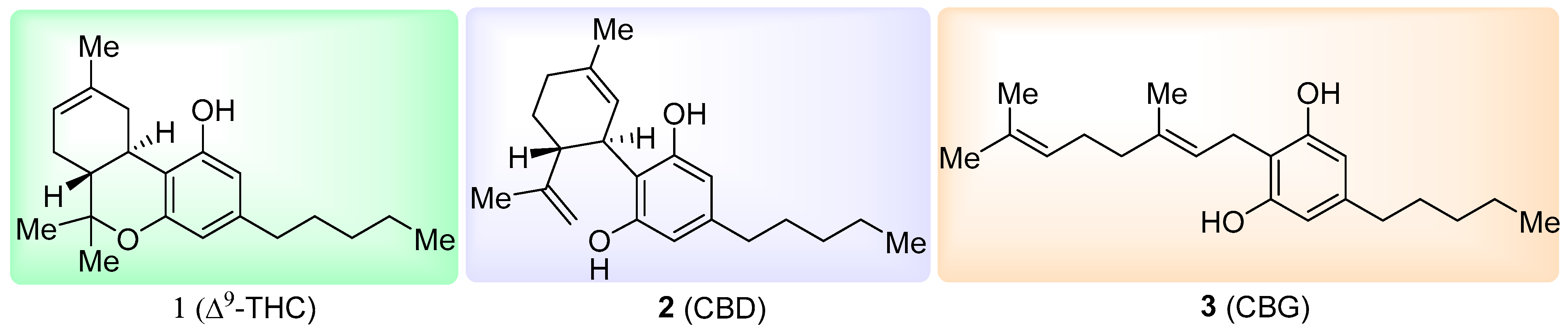
(
E)-2-(3,7-Dimethylocta-2,6-dien-1-yl)-5-pentylbenzene-1,3-diol (cannabigerol,
3) [
15]: following the general procedure, cannabigerol (
3) was obtained from resorcinol methyl ester derivative
9c (0.238 g, 1.0 mmol) as a yellow solid (0.161 g, 0.51 mmol, 51%): C
21H
32O
2; mp 49–50 °C (heptane);
Rf 0.61 (hexane/EtOAc 7:1);
1H NMR (300 MHz, CDCl
3) δ 6.24 (s, 2H), 5.31–5.23 (m, 1H), 5.16 (s, 2H), 5.09–5.01 (m, 1H), 3.39 (d,
J = 7.1 Hz, 2H), 2.48–2.40 (m, 2H), 2.17–2.00 (m, 4H), 1.80 (d,
J = 1.3 Hz, 3H), 1.67 (d,
J = 1.4 Hz, 3H), 1.61–1.48 (m, 5H), 1.34–1.25 (m, 4H), 0.88 (t,
J = 6.8 Hz, 3H);
13C NMR (75 MHz, CDCl
3) δ 154.8 (C), 142.8 (C), 139.0 (C), 132.1 (C), 123.8 (CH), 121.7 (CH), 110.6 (C), 108.4 (CH), 39.7 (CH
2), 35.5 (CH
2), 31.5 (CH
2), 30.8 (CH
2), 26.4 (CH
2), 25.7 (CH
3), 22.6 (CH
2), 22.3 (CH
2), 17.7 (CH
3), 16.2 (CH
3), 14.1 (CH
3); LRMS (EI)
m/
z 316 (M
+, 15%), 381 (30), 273 (5), 247 (16), 231 (31), 219 (11), 193 (100), 136 (10), 123 (15), 69 (9), 41 (9); HRMS (EI-TOF) calcd. For C
21H
32O
2: 316.2402; found 316.2384.
(E)-2-(3,7-dimethylocta-2,6-dien-1-yl)-5-heptylbenzene-1,3-diol (11da): following the general procedure, compound 11da was obtained from resorcinol methyl ester derivative 9d (0.266 g, 1.0 mmol) as a yellow solid (0.179 g, 0.52 mmol, 52%): C23H36O2; mp 62–63 °C (heptane); Rf 0.62 (hexane/EtOAc 7:1); 1H NMR (300 MHz, CDCl3) δ 6.27 (s, 2H), 5.33–5.22 (m, 3H), 5.12–5.04 (m, 1H), 3.41 (d, J = 7.1 Hz, 2H), 2.47 (dd, J = 8.8, 6.7 Hz, 2H), 2.20–2.04 (m, 4H), 1.83 (s, 3H), 1.70 (s, 3H), 1.61 (s, 3H), 1.56 (m, J = 8.1 Hz, 2H), 1.34–1.26 (m, 8H), 0.92 (t, J = 7.2 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ 154.8 (C), 142.8 (C), 139.0 (C), 132.1 (C), 123.78 (CH), 121.7 (CH), 110.6 (C), 108.3 (CH), 39.7 (CH2), 35.6 (CH2), 31.8 (CH2), 31.2 (CH2), 29.3 (CH2), 29.2 (CH2), 26.4 (CH2), 25.7 (CH3), 22.7 (CH2), 22.3 (CH2), 17.7 (CH3), 16.2 (CH3), 14.1 (CH3); LRMS (EI) m/z 344 (M+, 1%), 275 (14), 259 (42), 221 (100), 136 (13), 123 (22), 69 (14), 57 (11), 43 (21); HRMS (EI-TOF) calcd. For C23H36O2: 344.2715; found 344.2708.
Copies of
1H-NMR,
13C-NMR spectra of compounds
3,
8,
9,
10, and
11., in addition to HPLC chromatograms of compounds
8 are available in
Supplementary Materials.


 ,
, 



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}