Abstract
In this report, the catalytic potential of chalcogen-stabilized iron carbonyl clusters [Fe3E2(CO)9 (E = S, Se, Te)] for the photolytic hydration of alkynes has been explored. The iron chalcogenide clusters bring excellent transformations of terminal and internal alkynes to their respective keto products in just 25 min photolysis at −5 °C in inert free and aqueous conditions. After the completion of the reaction, the product can be extracted from organic solvent, and due to the lower solubility of the catalyst in water, it can also be isolated and further reused several times prior to any activation. The catalyst was also found to be active in thermal conditions and bring about the desired transformations with average to good catalytic efficiency. Moreover, during the thermal reaction, the catalyst decomposed and formed the nanoparticles of iron selenides, which worked as a single-source precursor for FeSe nanomaterials. The presented photolysis methodology was found to be most feasible, economical, instantly produce the desired product, and work for a wide range of internal and terminal alkynes; hence, all these features made this method superior to the other reported ones. This report also serves as the first catalytic report of chalcogen-stabilized iron carbonyl clusters for alkyne hydrations.
1. Introduction
The association of water and alkynes is difficult due to their different affinities. The chemical union of these molecules produces carbonyls in the form of ketone or aldehyde, depending upon the nature of the addition (markonikov or anti-markonikov) [1,2]. The hydration reaction is the most facile methodology to create a C–O bond, and the synthesis of various carbonyls is the crucial motif of the modern pharmaceutical and chemical industries [3]. Synthesizing ketones has wide-ranging fundamental importance for various pharmaceutical and API industries. The synthesis of fexofenadine, benperidol, droperidol, azaperone, and other related drugs required alkyne hydration as one of the crucial steps of synthesis [3]. Hydration reactions are slow even in the presence of a catalyst; it could be due to the irreconcilable polarities of the reagents used in the reaction. This demands a specific property of the catalyst to induce polarity in the alkyne used in the reaction [1,2]. Hydration of alkynes using mineral acids or organic acids has been known since the discovery of alkynes. Metal-catalyzed hydrations of alkynes are well established, and mercury-based complexes [4] are widely explored, but due to the toxicity of Hg metal and limited substrate scope, it is restricted to continue with this method. Hence, alternatively, the less toxic transition metal complexes were explored; in the 1960s, Kemp et al. reported the use of Ru (III) for the hydration of acetylenes [5,6]. During the 1990s, Grotjahn et al. and Wakatsuki et al. published several reports showing the catalytic applications of Ru(II) complexes for alkyne hydrations [7,8,9,10]. Pt(IV), Au(III), Pd(II), Cu(II), and Ag(I) were also used [2,11,12,13,14,15,16,17], but these catalytic methodologies were not found to be adequately efficient for alkyne hydration.
Tanaka et al. first reported a highly efficient Au(I) catalyst for alkyne hydration in 2002; this method also needs mineral acids to perform the reaction. However, the Au(I) complexes had emerged as a promising substitute for the previous toxic and inefficient catalysts. In the later years, Au(I) complexes with NHC and phosphine ligands were also reported for their exceptional catalytic ability towards alkyne hydrations (Scheme 1) [18,19,20,21,22]. However, these methods are associated with some drawbacks due to the prerequisite of a co-catalyst as a halide scavenger and the non-reusability of the catalyst. Heavy metals and expensive catalysts are unsustainable from the perspective of green chemistry. Therefore, many efforts toward the development of an economic catalyst have been made in recent years. Ag(I), Fe(III), Fe(II), and Co(III) are presented as valid alternatives for the previous expensive catalysts [23,24,25,26,27,28]. Naka et al. reported a Co(III) porphyrin complex for alkyne hydration, and the catalyst performed a reaction under a milder acidic condition that enables the catalyst to work with acid-labile functionalities [29]. Most of the catalysts cannot be reused further, while a few of them are highly sensitive metal-ligand complexes.
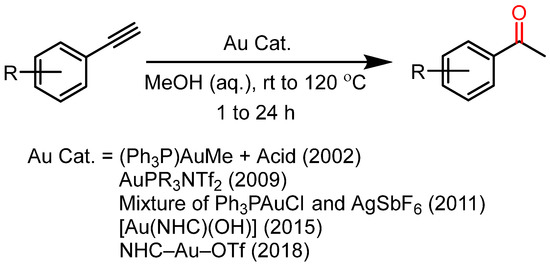
Scheme 1.
Showing the use of Au-based catalysts from the first report in 2002 to the most recent report in 2018.
To address the issue of the reusability of catalysts, several reports in the previous decade suggested the use of heterogeneous catalysts (Scheme 2) [30,31,32,33,34,35,36,37,38]. All the heterogeneous catalysts reported until 2016 are solid acid catalysts or require acidic conditions for catalysis. In 2016, Lin et al. used the CoIII (Porphyrin) Interpenetrating MOFs as a heterogeneous catalyst for acid-free catalysis [37]. Recently, our lab reported the FeOCN composite as a reusable heterogeneous catalyst for alkyne hydration under acid-free conditions [38]. However, in the absence of acids, these heterogeneous catalysts require a prolonged reaction time in the range of 12 h to 48 h. As an exception to all reaction methodologies in 2004, Vasudevan et al. reported a catalyst-free reaction methodology for alkyne hydration in superheated water under microwave irradiation [39]. In the year 2018, our lab reported a modified catalyst-free reaction methodology that was optimized up to the gram scale in an autoclave [40]. However, these methodologies need harsh reaction conditions (reaction temperature 150–200 °C; inert gas pressure 10–12 bar) for catalytic conversion.
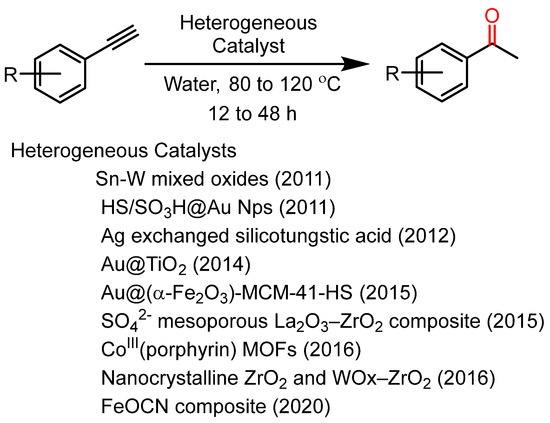
Scheme 2.
Heterogeneous reusable catalysts were reported for alkyne hydration in the previous decade.
To the best of our knowledge, there are only two reports that show transition metal-based photocatalysts for alkyne hydration (Scheme 3). Fu et al. and Xu et al. reported the use of Rh(III) porphyrin complex anf Cu(II) salt in mild acidic conditions for alkyne hydration under UV-visible irradiation [41,42]. Recently, work from our lab has been reported on the Rh(I) Schiff base complex for acid-free alkyne hydrations under UV irradiation [4]. These reactions use methanol as a solvent, and most of the synthesis was reported in aqueous methanol. Since the hydration reactions need water as one of the reactants, water is an appreciated green solvent. The non-toxic nature, abundance, and easy separation of organic compounds make water the most preferred solvent. Performing alkyne hydration reactions in neat water stems is a sustainable approach to methodology development. Metal carbonyls are well-implemented catalysts in the laboratory as well as in the commercial synthesis of various organic compounds [43,44,45,46,47,48,49,50,51,52]. Recently, Chikkali et al. reported an iron carbonyl-catalyzed hydroformylation reaction [53]. Alkoxy carbonylations of alkyl bromides and synthesis of γ-lactones using iron carbonyls as catalysts [54,55]. Mono-metal carbonyl complexes are sometimes considered toxic, but bi- or triatomic clusters are highly stable at ambient temperatures. Additionally, these metal carbonyls can be made stable at ambient conditions by introducing chalcogen-bridging ligands. Moreover, the use of chalcogen (especially S and Se) is well established for cluster growth reactions. As the literature suggests, metal carbonyls under UV irradiation form a catalytically active species [56]. Masuda et al. (1980) and Geoffroy et al. (1985) reported photo-induced alkyne polymerization using M(CO)6 of Cr, Mo, and W [57,58]. However, there are certain drawbacks to using M(CO)6 under photochemical conditions. These carbonyls may undergo some irreversible chemical changes during the reaction, and they are not soluble in aqueous solvents. The sensitivity of metal complexes and clusters to air and moisture remains a severe drawback when it comes to catalytic applications. However, it has been noted that the incorporation of chalcogens in metal complexes and clusters can abate this problem. Reports show that the inclusion of chalcogen in the complex increases its ability to withstand harsh conditions without losing catalytic activity. Metal chalcogenide complexes have efficiently catalyzed a variety of C–N coupling reactions, C–C coupling reactions, and some other organic transformation reactions [59,60,61,62].
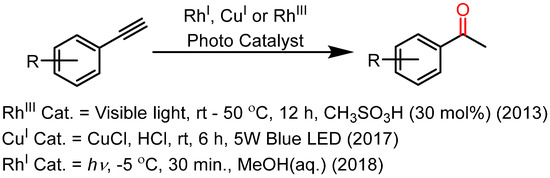
Scheme 3.
Photocatalysts were reported for alkyne hydrations.
In search of finding a stable catalyst that can perform reactions under aqueous and photochemical conditions. The chalcogen-stabilized metal carbonyl clusters could be a possible alternative. To the best of our knowledge, the catalytic properties of metal chalcogenide clusters are rare and have not been explored. In this script, we wish to report an iron chalcogenide carbonyl cluster for the hydration of alkyne, which works as a heterogeneous catalyst in aqueous media and produces excellent results under UV irradiation in very little time.
2. Results and Discussion
The initial test reaction of phenylacetylene and Fe(CO)5 in aqueous methanol under photolysis conditions does not produce any results. Other iron carbonyl clusters, Fe2(CO)9 and Fe3(CO)12, were also used for the trail reaction under the same reaction conditions but failed to produce any chemical change in the reaction. The shifting of iron carbonyl complexes to iron chalcogenides carbonyl clusters (Fe3E2(CO)9, E = S, Se, Te) in a water–methanol solution of phenylacetylene leads to instant success, and acetophenone forms as the sole reaction product; however, the yield of the product is considerably low. To improve productivity, different ratios of methanol and water were investigated, but they failed to improve the yield of the product. It was noted that the increased ratio of methanol in a methanol–water solvent mixture considerably reduced the formation of acetophenone, while, in contrast, the increasing ratio of water in a methanol–water solvent mixture significantly enhanced the yield of acetophenone. The Fe3Se2(CO)9 cluster was fully miscible in methanol but failed to initiate the reaction. While the catalyst is immiscible in water and floats over the aqueous layer and brings the desired transformations, it may perhaps work as a heterogeneous catalyst. Iron clusters of other chalcogens (S and Te) were also used for the catalysis, but the Se-containing cluster was found to be highly active while the S and Te-based clusters showed moderate to average reactivity. The reason could be the difference in the electron donor properties of S, Se, and Te. The literature suggests that going down the group, the HOMO–LUMO band gap decreases, which supports the higher activity of selenium. The reduced catalytic activity of the Te complex can be explained by the increased atomic size, which tends to reduce the electron density on the central metal atom [59,60,61,62,63].
An optimization investigation of the catalyst amount indicates that 5 mol% of Fe3Se2(CO)9 cluster brings about the significant transformation of the desired product (Table 1, entry 13). The 2 mol% of the Fe3Se2(CO)9 cluster yields the desired product only in traces (Table 1, entries 9–10), while gradually increasing the catalyst amount significantly improves the yield (Table 1, entries 10–14) and produces the maximum yield (86%) at 5 mol% of the catalyst. However, a further increase in catalyst amount does not affect the yield of the desired product (Table 1, entry 14). The scope of the reaction was explored concerning the temperature (Table 1, entries 15–17), and it was found that the reaction is sensitive to the temperature; at 0 °C, the reaction produces an average yield of the acetophenone (45%), while lowering the reaction temperature to −5 °C outstandingly shoots up the yield to 86%. No additional increase in product yield was achieved even after reducing the reaction temperature to −10 °C. Hence, all the experiments in the present hydration reaction were conducted at −5 °C. The UV irradiation optimization was also found to be one of the important aspects of these hydration reactions; a maximum 86% yield was recorded in 25 min of UV exposure (Table 1, entries 16–19). The initial 10-minute yield was 26%, while it went up to 59% and 76% in the next two consecutive 5-minute time spans, finally reaching the maximum 86% in the total 25-minute UV exposure. No, further significant changes in the yield were obtained beyond 25 min of photolysis (Table 1, entries 20–22). Using alternate reaction methodologies has extensive benefits over classical thermal reaction methods. Apart from the thermal heating methods, the alternate methods, including microwave and photolysis, are more efficient and timesaving for classical synthesis. In the present reaction, a 125 W mid-pressure Hg vapor lamp, which irradiates 289 nm wavelengths, has been used. This enables the pursuit of reactions in a very short duration; moreover, the same reaction mixture does not yield the desired product under sunlight for a long time.

Table 1.
Reaction optimization.
Substrate Scope. Next, the reaction was generalized towards the various alkynes at the optimized reaction conditions (Table 2). Alkynes with electron-donating and electron-withdrawing functionalities, irrespective of their position on the phenyl ring, react smoothly and form the desired keto product. The 4-tert-butyl-phenylacetylene (Table 2, entry 1b) produces the best yield (89%) of the reaction. The methyl-substituted phenylacetylenes, 4-methyl, 3-methyl, and 2-methyl phenylacetylene, also respond well to the reaction and yield 85%, 82%, and 84% of the respective keto products (Table 2, entries 1c–1e). Changing the alkyl groups, a slightly better transformation was recorded with 4-ethyl phenylacetylene (Table 2, entry 1f). The inert and difficult-to-activate molecules such as 4-methoxy and 2-methoxy substituted phenylacetylene showed efficient transformations of 87% and 84% for the desired product (Table 2, entries 1g and 1h) [64,65,66]. The halides, 4-fluoro, 4-chloro, and 4-bromophenylacetylene also reacted and showed 86, 80, and 74% transformations of the respective keto products (Table 2, entries 1i–1k). Here, the reaction was found to be more efficient for a strong withdrawing group; moreover, the position of the group also influences the rate of transformation, as a slightly reduced yield was observed with 2-chlorophenylacetylene (Table 2, entry 1l). The meta-substituted 3-aminophenylacetylene (1m) produces an 86% yield of the desired ketone. Ethynyl ferrocene also shows excellent transformation and an 85% yield was recorded (Table 2, entry 1n). The present method was also extended for the hydrations of internal alkynes: a symmetrical diphenylacetylene (Table 2, entry 1o), as well as two unsymmetrical internal acetylenes, (4-chloro)-diphenylacetylene and 4-methyl diphenylacetylene (Table 2, entries 1p and 1q) were investigated. The reactions work equally well at similar reaction parameters and produce excellent yields of their corresponding ketones. The symmetric internal alkyne produces 80% yield of its corresponding ketone, while substituted asymmetric internal alkynes, the 4-chloro and 4-methyl, produce 78% and 82% yield of their corresponding ketones, respectively.
Table 2.
Substrate scope.
Catalyst reusability. Since the catalyst is immiscible in water, it can be easily separated and reused for the next catalytic cycle. Moreover, the catalyst did not require any prior activations and was directly used for the next cycle. In a typical experiment, the water-insoluble iron-chalcogenide carbonyl cluster was recovered at the end of the reaction by filtration and directly used for a new catalytic run. The catalyst showed good recyclability and did not deactivate much up to six catalytic cycles; moreover, up to 50% product transformations were noted in the 8th catalytic cycle. We have used the same catalyst for up to 9 catalytic cycles, and 31% product formation was obtained in the 9th catalytic run. This reusability test indicates the highly robust and active catalytic nature of the clusters (Figure 1).
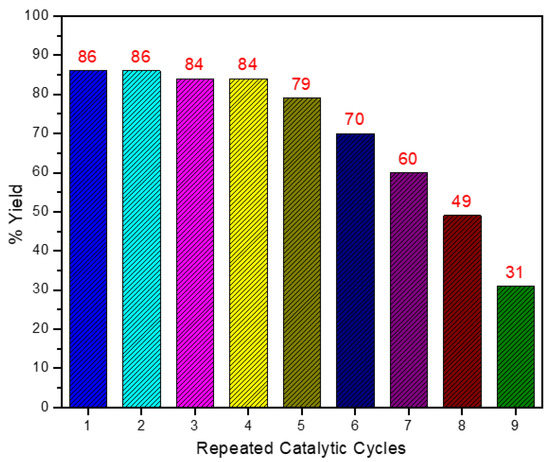
Figure 1.
Showing the percentage yield of the product obtained during catalyst reutilizations. Reaction Condition: Isolated yields: Phenylacetylene (1 equiv.) 204 mg, catalyst (5 mol%) 48 mg, water (80 mL), 25 min., −5 °C.
Catalytic investigations under thermal conditions. To check the robustness and activity of the catalyst, it was also examined under thermal conditions. An initial reaction of phenylacetylene in water and the catalytic presence of Fe3Se2(CO)9 at reflux temperature show inappreciable yields. Some recent reports of alkyne hydrations from our research group [4,38,40] indicate that a mixture of water and methanol solvent improves catalytic activity. So, using the directions based on the earlier optimizations, we moved onto the use of a water–methanol mixture solvent. The second trial of phenylacetylene in aqueous methanol (2:1) at 100 °C in the presence of a Fe3Se2(CO)9 catalyst shows considerable transformation (Scheme 4).
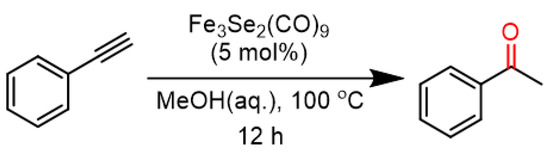
Scheme 4.
Hydration of phenylacetylene using iron selenide carbonyl clusters under thermal conditions.
Other iron carbonyl compounds, Fe(CO)5, Fe2(CO)9, and Fe3(CO)12, were not found suitable for this reaction, while Fe3S2(CO)9 and Fe3Te2(CO)9 show very low catalytic activity in comparison to the Se analogue. It was observed that during the thermal reaction, the catalyst decomposed and produced some solid insoluble material, which was identified as FeSe nanoparticles. The increased solubility of the cluster in aqueous methanol may be the reason for the catalyst’s decomposition during thermal heating. This may also reduce the catalytic efficiency of the catalyst, and a slightly reduced transformation was noted compared to the photochemical method. The substrate scope of the thermal method was also investigated and depicted in Table 3. To probe the role of nanoparticles in catalysis, a mercury drop test was conducted, which confirmed nanoparticles are not responsible for the catalysis. The formed nanoparticles were collected and characterized through various techniques to further study their bonding and surface properties.
Table 3.
Substrate scope.
Characterization of FeSe Nanoparticles. The powder XRD pattern of the collected nanoparticles shows the formation of tetragonal FeSe structures [54]. The powder XRD pattern is similar to JCPDS card no. 850735 [67,68,69,70]. The surface morphologies of the FeSe nanoparticles were investigated through FESEM images. The recorded images of FESEM show large clusters of nanoparticles (Figure 2a–d). Particle sizes were observed in the range of 9 nm to 85 nm. Additional insights into the morphology were collected through TEM images. Figure 2e,f illustrates that these particles are not spherical. HRTEM images reveal the interplaner distance is approximately 1.96 Å. Elemental mapping from TEM-EDX shows evenly distributed Se in Fe. SAED pattern details confirm the formation of FeSe nanoparticles (Supplementary Materials, Figure S4). The bonding properties of FeSe nanoparticles were characterized through XPS. The deconvolution of the binding energies of Fe (2p) and Se (3d) spectra shows the characteristic peaks, which confirm the presence of Fe and Se (Figure 3). The individual spectrum of binding energies in the region of Fe shows two FeSe bonding related characteristic peaks at 710.78 eV in Fe (2p1/2) and 725.00 eV in Fe (2p3/2) regions [71]. The corresponding peaks in the Se 3d regions similarly show two characteristic peaks of binding energies at 54.84 in Se 3d5/2 and 56.03 in Se 3d3/2, relevant to the FeSe bonds [72].
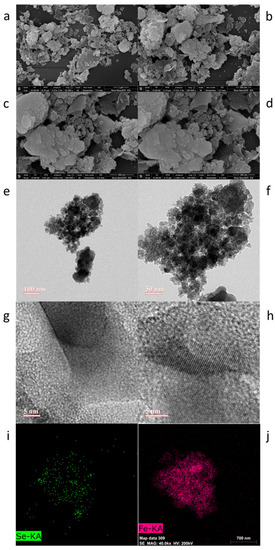
Figure 2.
FESEM images (a–d), TEM images (e,f), HRTEM images (g,h), and TEM–EDX (i,j). (See Supplementary Materials).
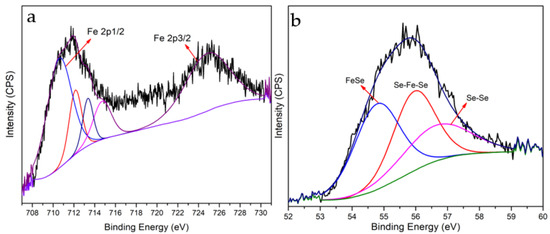
Figure 3.
(a) XPS spectrum of Fe (2p) and (b) XPS spectrum of Se (3d).
3. Materials and Methods
3.1. Photochemical Condition
In a 100-mL borosilicate immersion well of the UV reactor, 80 mL of water is used as solvent, and alkyne (1 mmol) and an Fe catalyst (5 mol%) were added. The reaction mixture is first cooled down to −5 °C, then exposed to the Hg vapor UV lamp, 125 W, 289 nm, for 25 min with continuous stirring. After completion of the reaction, the catalyst was separated through filtration, and the product was extracted through solvent extraction with ethyl acetate. The extracted solvent is dried over anhydrous Na2SO4, concentrated at reduced pressure, and purified by flash column chromatography with hexanes/ethyl acetate as eluent to obtain the corresponding product.
3.2. Thermal Condition
In a 100-mL round-bottom flask, a water and methanol mixture is used as a solvent, and alkyne (1 mmol) and an Fe catalyst (5 mol%) were added and heated at 100 °C with continuous stirring for 12 h. After completion of the reaction, the methanol was removed by a rotatory evaporator, and the product was extracted through solvent extraction with ethyl acetate. The extracted solvent is dried over anhydrous Na2SO4, concentrated at reduced pressure, and purified by flash column chromatography with hexanes/ethyl acetate as eluent to obtain the corresponding product.
4. Conclusions
We have shown a time-efficient photochemically assisted Fe3Se2(CO)9-catalyzed one-pot hydration method for both terminal and internal acetylenes. one pot hydration of terminal and internal acetylenes. The easy-to-prepare, economically viable, reusable, and highly robust nature of the chalcogen-stabilized Fe3Se2(CO)9 catalyst is superior to the existing precious metal catalysts. The present reaction fairly works with all functionally substituted aryl alkynes, irrespective of the nature of the functional group or substituted group and the position of the group on the benzene ring. Moreover, we are the first to report the iron chalcogenide carbonyl cluster as a heterogeneous catalyst for alkyne hydrogenations in fully aqueous media. This catalytic methodology is equally active for the internal alkynes, and the excellent yields show the compatibility with a wide range of functionalities. The presented catalyst brings about the transformation in just 25 min, and it can be reused several times without any prior activations; moreover, it can be easily separated through filtration. Moreover, the catalyst was further tested for catalytic activity under thermal conditions. The catalyst shows average to good catalytic activity, moreover, it also works as a single-source precursor for the in situ formation of FeSe nanoparticles.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/org4020020/s1. Figure S1: Photochemical reactor (Capacity 100 mL); Figure S2: Reaction setup for photochemical reactor; Figure S3: FTIR of Fe3Se2(CO)9 catalyst; Figure S4a–d: FESEM images of FeSe nanoparticles; Figure S5a–f: TEM images of FeSe nanoparticles; Figure S6: Elemental mapping of FeSe nanoparticles; Figure S7: SAED pattern FeSe nanoparticles; Table S1: Comparison Table. 1H NMR spectrum, 13C NMR spectrum, For known compounds (1a–1q). References [73,74,75,76,77,78,79,80,81,82,83,84] are cited in the Supplementary Materials.
Author Contributions
Conceptualization, methodology, investigation, and data curation, M.A.; writing—original draft preparation, A.K.S.; writing—review and editing, M.A. and N.S.U.; resources, N.S.; supervision, project administration, and funding acquisition, R.K.J. All authors have read and agreed to the published version of the manuscript.
Funding
CSIR (01(2996)/19/EMR-II), New Delhi, INDIA.
Data Availability Statement
Copies of spectral data for known compounds (1a–1q) and FTIR of Fe3Se2(CO)9 catalyst are available in the Supplementary Materials.
Acknowledgments
Raj K. Joshi thanks the CSIR (01(2996)/19/EMR-II) for their financial assistance. The authors also acknowledge the MRC and MNIT for providing characterization facilities.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Hintermann, L.; Labonne, A. Catalytic Hydration of Alkynes and Its Application in Synthesis. Synthesis 2007, 8, 1121. [Google Scholar] [CrossRef]
- Salvio, R.; Basseti, M. Sustainable hydration of alkynes promoted by first row transition metal complexes. Background, highlights and perspectives. Inorganica Chim. Acta 2021, 522, 120288. [Google Scholar] [CrossRef]
- Mizushima, E.; Sato, K.; Hayashi, T.; Tanaka, M. Highly Efficient AuI-Catalyzed Hydration of Alkynes. Angew. Chem. 2002, 114, 4745. [Google Scholar] [CrossRef]
- Ali, M.; Srivastava, A.K.; Siangwata, S.; Smith, G.S.; Joshi, R.K. Photo induced alkyne hydration reactions mediated by a water soluble, reusable Rhodium (I) catalyst. Catal. Commun. 2018, 115, 78. [Google Scholar] [CrossRef]
- Halpern, J.; James, B.R.; Kemp, A.L.W. Catalysis of The Hydration Of Acetylenic Compounds By Ruthenium(III) Chloride. J. Am. Chem. Soc. 1961, 83, 4097. [Google Scholar] [CrossRef]
- Halpern, J.; James, B.R.; Kemp, A.L.W. Formation and Properties of Some Chlorocarbonyl Complexes of Ruthenium(II) and Ruthenium(III). J. Am. Chem. Soc. 1966, 88, 5142. [Google Scholar] [CrossRef]
- Grotjahn, D.B.; Incarvito, C.D.; Rheingold, A.L. Combined Effects of Metal and Ligand Capable of Accepting a Proton or Hydrogen Bond Catalyze Anti-Markovnikov Hydration of Terminal Alkynes. Angew. Chem. Int. Ed. 2001, 40, 3884. [Google Scholar] [CrossRef]
- Tokunaga, M.; Wakatsuki, Y. The First Anti-Markovnikov Hydration of Terminal Alkynes: Formation of Aldehydes Catalyzed by a Ruthenium(II)/Phosphane Mixture. Angew. Chem. Int. Ed. 1998, 37, 2867. [Google Scholar] [CrossRef]
- Suzuki, T.; Tokunaga, M.; Wakatsuki, Y. Ruthenium Complex-Catalyzed anti-Markovnikov Hydration of Terminal Alkynes. Org. Lett. 2001, 3, 735. [Google Scholar] [CrossRef]
- Tokunaga, M.; Suzuki, T.; Koga, N.; Fukushima, T.; Horiuchi, A.; Wakatsuki, Y. Ruthenium-Catalyzed Hydration of 1-Alkynes to Give Aldehydes: Insight into anti-Markovnikov Regiochemistry. J. Am. Chem. Soc. 2001, 123, 11917. [Google Scholar] [CrossRef]
- Hiscox, W.; Jennings, P.W. Synthesis and reactions of nickel and palladium carbon-bound enolate complexes. Organometallics 1990, 9, 1997. [Google Scholar] [CrossRef]
- Baidossi, W.; Lahav, M.; Blum, J. Hydration of Alkynes by a PtCl4−CO Catalyst. J. Org. Chem. 1997, 62, 669. [Google Scholar] [CrossRef]
- Francisco, L.W.; Moreno, D.A.; Atwood, J.D. Synthesis, Characterization, and Reaction Chemistry of PtCl2[P(m-C6H4SO3Na)3]2, an Alkyne Hydration Catalyst. Organometallics 2001, 20, 4237. [Google Scholar] [CrossRef]
- Fukuda, Y.; Utimoto, K. Effective transformation of unactivated alkynes into ketones or acetals with a gold(III) catalyst. J. Org. Chem. 1991, 56, 3729. [Google Scholar] [CrossRef]
- Fukuda, Y.; Utimoto, K. Efficient transformation of methyl propargyl ethers into α, β-unsaturated ketones. Bull. Chem. Soc. Jpn. 1991, 64, 2013. [Google Scholar] [CrossRef]
- Imi, K.; Imai, K.; Utimoto, K. Regioselective hydration of alkynones by palladium catalysis. Tetrahedron Lett. 1987, 28, 3127. [Google Scholar] [CrossRef]
- Meier, K.; Marsella, J.A. Hydration of acetylenic compounds without using mercury. J. Mol. Catal. 1993, 78, 31. [Google Scholar] [CrossRef]
- Ghosh, N.; Nayak, S.; Sahoo, A.K. Gold-Catalyzed Regioselective Hydration of Propargyl Acetates Assisted by a Neighboring Carbonyl Group: Access to α-Acyloxy Methyl Ketones and Synthesis of (±)-Actinopolymorphol B. J. Org. Chem. 2011, 76, 500. [Google Scholar] [CrossRef]
- Leyva, A.; Corma, A. Isolable Gold(I) Complexes Having One Low-Coordinating Ligand as Catalysts for the Selective Hydration of Substituted Alkynes at Room Temperature without Acidic Promoters. J. Org. Chem. 2009, 74, 2067. [Google Scholar] [CrossRef]
- Nun, P.; Ramón, R.S.; Gaillard, S.; Nolan, S.P. Efficient silver-free gold (I)-catalyzed hydration of alkynes at low catalyst loading. J. Organomet. Chem. 2011, 696, 7. [Google Scholar] [CrossRef]
- Li, F.; Wang, N.; Lu, L.; Zhu, G. Regioselective Hydration of Terminal Alkynes Catalyzed by a Neutral Gold(I) Complex [(IPr)AuCl] and One-Pot Synthesis of Optically Active Secondary Alcohols from Terminal Alkynes by the Combination of [(IPr)AuCl] and Cp*RhCl[(R,R)-TsDPEN]. J. Org. Chem. 2015, 80, 3538. [Google Scholar] [CrossRef]
- Gatto, M.; Baratta, W.; Belanzoni, P.; Belpassi, L.; Zotto, A.D.; Tarantelli, F.; Zuccaccia, D. Hydration and alkoxylation of alkynes catalyzed by NHC–Au–OTf. Green Chem. 2018, 20, 2125. [Google Scholar] [CrossRef]
- Thuong, M.B.T.; Mann, A.; Wagner, A. Mild chemo-selective hydration of terminal alkynes catalysed by AgSbF6. Chem. Commun. 2012, 48, 434. [Google Scholar] [CrossRef] [PubMed]
- Damiano, J.P.; Pastel, M. FeCl3 H2O: A specific system for arylacetylene hydration. J. Organomet. Chem. 1996, 522, 303. [Google Scholar] [CrossRef]
- Wu, X.; Bezier, D.; Darcel, C. Development of the First Iron Chloride Catalyzed Hydration of Terminal Alkynes. Adv. Synth. Catal. 2009, 351, 367. [Google Scholar] [CrossRef]
- Antonino, J.R.C.; Perez, A.L.; Corma, A. Regioselective hydration of alkynes by iron (III) Lewis/Brønsted catalysis. Chem. Eur. J. 2012, 18, 11107. [Google Scholar] [CrossRef]
- Bassetti, M.; Ciceri, S.; Lancia, F.; Pasquini, C. Hydration of aromatic terminal alkynes catalyzed by iron (III) sulfate hydrate under chlorine-free conditions. Tetrahedron Lett. 2014, 55, 1608. [Google Scholar] [CrossRef]
- Hou, S.; Yang, H.; Cheng, B.; Zhai, H.; Li, Y. Cobaloxime-catalyzed hydration of terminal alkynes without acidic promoters. Chem. Commun. 2017, 53, 6926. [Google Scholar] [CrossRef]
- Tachinami, T.; Nishimura, T.; Ushimaru, R.; Noyori, R.; Naka, H. Hydration of terminal alkynes catalyzed by water-soluble cobalt porphyrin complexes. J. Am. Chem. Soc. 2013, 135, 50. [Google Scholar] [CrossRef]
- Jin, X.; Oishi, T.; Yamaguchi, K.; Mizuno, N. Heterogeneously catalyzed efficient hydration of alkynes to ketones by tin–tungsten mixed oxides. Chem. Eur. J. 2011, 17, 1261. [Google Scholar] [CrossRef]
- Zhu, F.; Wang, W.; Li, H. Water-Medium and Solvent-Free Organic Reactions over a Bifunctional Catalyst with Au Nanoparticles Covalently Bonded to HS/SO3H Functionalized Periodic Mesoporous Organosilica. J. Am. Chem. Soc. 2011, 133, 11632. [Google Scholar] [CrossRef]
- Venkateswara Rao, K.T.; Sai Prasad, P.S.; Lingaiah, N. Solvent-free hydration of alkynes over a heterogeneous silver exchanged silicotungstic acid catalyst. Green Chem. 2012, 14, 1507. [Google Scholar] [CrossRef]
- Liang, S.; Jasinski, J.; Hammond, G.B.; Xu, B. Supported gold nanoparticle-catalyzed hydration of alkynes under basic conditions. Org. Lett. 2015, 17, 162. [Google Scholar] [CrossRef]
- Rostamizadeh, H.; Estiri, S.; Azad, M. Au anchored to (α-Fe2O3)-MCM-41-HS as a novel magnetic nanocatalyst for water-medium and solvent-free alkyne hydration. Catal. Commun. 2014, 57, 29. [Google Scholar] [CrossRef]
- Zhao, Z.; Ran, J. Sulphated mesoporous La2O3–ZrO2 composite oxide as an efficient and reusable solid acid catalyst for alkenylation of aromatics with phenylacetylene. Appl. Catal. A Gen. 2015, 503, 77. [Google Scholar] [CrossRef]
- Lin, Z.; Zhang, Z.; Chen, Y.; Lin, W. Highly Efficient Cooperative Catalysis by CoIII(Porphyrin) Pairs in Interpenetrating Metal–Organic Frameworks. Angew. Chem. Int. Ed. 2016, 55, 13739. [Google Scholar] [CrossRef]
- Gonell, F.; Portehault, D.; Julián-López, K.; Vallé, B.; Sanchez, C.; Corma, A. One step microwave-assisted synthesis of nanocrystalline WO x–ZrO 2 acid catalysts. Catal. Sci. Technol. 2016, 6, 8257. [Google Scholar] [CrossRef]
- Srivastava, A.K.; Ali, M.; Siangwata, S.; Satrawala, N.; Smith, G.S.; Joshi, R.K. Multitasking FeOCN Composite as an Economic, Heterogeneous Catalyst for 1-Octene Hydroformylation and Hydration Reactions. Asian J. Org. Chem. 2020, 9, 377. [Google Scholar] [CrossRef]
- Vasudevan, A.; Verzal, M.K. Neutral, metal-free hydration of alkynes using microwave irradiation in superheated water. SYNLETT 2004, 4, 631. [Google Scholar] [CrossRef]
- Ali, M.; Srivastava, A.K.; Joshi, R.K. Metal/catalyst/reagent free hydration of alkynes up to gram scale under temperature and pressure controlled condition. Tetrahedron Lett. 2018, 59, 2075. [Google Scholar] [CrossRef]
- Liu, X.; Liu, L.; Wang, Z.; Fu, X. Visible light promoted hydration of alkynes catalyzed by rhodium (III) porphyrins. Chem. Commun. 2015, 51, 11896. [Google Scholar] [CrossRef] [PubMed]
- Niu, T.; Jiang, D.; Li, S.; Shu, X.; Li, H.; Zhang, A.; Xu, J.; Ni, B. Visible light promoted copper-catalyzed Markovnikov hydration of alkynes at room temperature. Tetrahedron Lett. 2017, 58, 1156. [Google Scholar] [CrossRef]
- Mathur, P.; Joshi, R.K.; Jha, B.; Singh, A.K.; Mobin, S.M. Towards the catalytic formation of α, β-vinylesters and alkoxy substituted γ-lactones. J. Organomet. Chem. 2010, 695, 2687. [Google Scholar] [CrossRef]
- Mathur, P.; Joshi, R.K.; Rai, B.; Jha, D.K.; Mobin, S.M. One pot synthesis of maleimide and hydantoin by Fe(CO)5 catalyzed [2 + 2 + 1] co-cyclization of acetylene, isocyanate and CO. Dalton Trans. 2012, 41, 5045. [Google Scholar] [CrossRef] [PubMed]
- Mathur, P.; Jha, B.; Raghuvanshi, A.; Joshi, R.K.; Mobin, S.M. Photolytic reaction of substituted (ethynyl) benzaldehyde and Fe(CO)5: Formation of indenone and chelated iron complexes. J. Organomet. Chem. 2012, 712, 7. [Google Scholar] [CrossRef]
- Mathur, P.; Rai, D.K.; Tauqeer, M.; Joshi, R.K.; Lahiri, G.K.; Mobin, S.M. Synthesis, structure and redox property of first 1, 2, 3-triselenole. J. Organomet. Chem. 2012, 721, 144. [Google Scholar] [CrossRef]
- Jha, B.; Raghuvanshi, A.; Joshi, R.K.; Mobin, S.M.; Mathur, P. A photochemical route to ferrocenyl-substituted ferrapyrrolinone complexes. Appl. Organomet.Chem. 2017, 31, e3805. [Google Scholar] [CrossRef]
- Joshi, R.K.; Satrawala, N. One pot synthesis of important retinoid synthon by the catalytic regioselective bi-functionalization of acetylenes, alcohol and carbon monoxide. Tetrahedron Lett. 2017, 58, 2931. [Google Scholar] [CrossRef]
- Lapidus, A.L.; Savelev, M.M. Metal carbonyl catalysts of the synthesis of organic compounds from carbon monoxide and molecular hydrogen. Russ. Chem. Rev. 1988, 57, 17. [Google Scholar] [CrossRef]
- Zhu, L.; Yempally, V.; Isrow, D.; Pellechia, P.J.; Captain, B. Selective benzylic C–H activation of solvent toluene and m-xylene by an iron–tin cluster complex: Fe2(μ-Sn )2(CO)8. J. Organomet. Chem. 2010, 695, 1. [Google Scholar] [CrossRef]
- Kaisare, A.A.; Jr, O.S.; Valente, E.J.; Gray, G.M. Metallacrown ethers with a symmetric bis(phosphite) ligand derived from 1,2-bis-(2-hydroxyethoxy)benzene: Synthesis, characterization and hydroformylation of styrene. J. Organomet. Chem. 2010, 695, 2658. [Google Scholar] [CrossRef]
- Tan, G.; Szilvási, T.; Inoue, S.; Blom, B.; Driess, M. An Elusive Hydridoaluminum(I) Complex for Facile C–H and C–O Bond Activation of Ethers and Access to Its Isolable Hydridogallium(I) Analogue: Syntheses, Structures, and Theoretical Studies. J. Am. Chem. Soc. 2014, 136, 9732. [Google Scholar] [CrossRef]
- Pandey, S.; Raj, K.V.; Shinde, D.R.; Vanka, K.; Kashyap, V.; Kurungot, S.; Vinod, C.P.; Chikkali, S.H. Iron catalyzed hydroformylation of alkenes under mild conditions: Evidence of an Fe (II) catalyzed process. J. Am. Chem. Soc. 2018, 140, 4430. [Google Scholar] [CrossRef]
- Li, Y.; Wu, X.F. Copper/iron co-catalyzed alkoxycarbonylation of unactivated alkyl bromides. Commun. Chem. 2018, 1, 39. [Google Scholar] [CrossRef]
- Iwasaki, M.; Miki, N.; Ikemoto, Y.; Ura, Y.; Nishihara, Y. Regioselective Synthesis of γ-Lactones by Iron-Catalyzed Radical Annulation of Alkenes with α-Halocarboxylic Acids and Their Derivatives. Org. Lett. 2018, 20, 3848. [Google Scholar] [CrossRef]
- Xu, K.; Peng, H.; Lam, J.W.Y.; Poon, T.W.H.; Dong, Y.; Xu, H.; Sun, Q.; Cheuk, K.K.L.; Salhi, F.; Lee, P.P.S.; et al. Transition metal carbonyl catalysts for polymerizations of substituted acetylenes. Macromolecules 2000, 33, 6918. [Google Scholar] [CrossRef]
- Masuda, T.; Kuwane, Y.; Yamamoto, K.; Higashimura, T. Polymerization of acetylene derivatives induced by UV irradiation via metal carbonyls. Polym. Bull. 1980, 2, 823. [Google Scholar] [CrossRef]
- Landon, S.J.; Shulman, P.M.; Geoffrey, G.L. Photoassisted polymerization of terminal alkynes by W (CO) 6 involving catalyst generation by an alkyne to vinylidene ligand rearrangement. J. Am. Chem. Soc. 1985, 107, 6739. [Google Scholar] [CrossRef]
- Sharma, K.N.; Satrawala, N.; Srivastava, A.K.; Ali, M.; Joshi, R.K. Palladium (ii) ligated with a selenated (Se, C NHC, N−)-type pincer ligand: An efficient catalyst for Mizoroki–Heck and Suzuki–Miyaura coupling in water. Org. Biomol. Chem. 2019, 17, 8969. [Google Scholar] [CrossRef]
- Sharma, K.N.; Satrawala, N.; Joshi, R.K. Thioether–NHC-Ligated PdII Complex for Crafting a Filtration-Free Magnetically Retrievable Catalyst for Suzuki–Miyaura Coupling in Water. Eur. J. Inorg. Chem. 2018, 1743. [Google Scholar] [CrossRef]
- Sharma, C.; Srivastava, A.K.; Sharma, K.N.; Joshi, R.K. Half-sandwich (η5-Cp*)Rh(iii) complexes of pyrazolated organo-sulfur/selenium/tellurium ligands: Efficient catalysts for base/solvent free C–N coupling of chloroarenes under aerobic conditions. Org. Biomol. Chem. 2020, 18, 3599. [Google Scholar] [CrossRef] [PubMed]
- Sharma, K.N.; Ali, M.; Srivastava, A.K.; Joshi, R.K. (η6-Benzene) Ru (II) half-sandwich complexes of pyrazolated chalcogenoethers for catalytic activation of aldehydes to amides transformation. J. Organomet. Chem. 2019, 879, 69. [Google Scholar] [CrossRef]
- Yao, Q.; Kinney, E.P.; Zheng, C. Selenium-ligated palladium (ii) complexes as highly active catalysts for carbon−carbon coupling reactions: The heck reaction. Org. Lett. 2004, 6, 2997. [Google Scholar] [CrossRef] [PubMed]
- Paris, S.I.M.; Lemke, F.R. Substituent effects in the ruthenium catalyzed hydrosilylation of para-substituted phenylacetylenes. Inorg. Chem. Commun. 2005, 8, 425. [Google Scholar] [CrossRef]
- Eckart, K.; Schwarz, H. Vinyl cation-induced cleavage of the oxygen-carbon bond in ortho-methoxy-substituted phenylacetylenes. J. Mass Spectrom. Ion Process. 1985, 66, 245. [Google Scholar] [CrossRef]
- Ramana, D.V.; Krishna, N.V.S.R. Ortho effects in organic molecules on electron impact: 18—Novel hydrogen transfer from the methoxy group to acetylenic carbon in 2-methoxyphenylacetylene and 2-methoxydiphenylacetylenes. J. Mass Spectrom. 1989, 24, 317. [Google Scholar]
- Dutta, A.K.; Maji, S.K.; Srivastava, D.N.; Mondal, A.; Biswas, P.; Paul, P.; Adhikary, B. Synthesis of FeS and FeSe Nanoparticles from a Single Source Precursor: A Study of Their Photocatalytic Activity, Peroxidase-Like Behavior, and Electrochemical Sensing of H2O2. ACS Appl. Mater. Interfaces 2012, 4, 1919. [Google Scholar] [CrossRef]
- Cong, B.; Sun, S.; Wang, B.; Lv, B.; Zhao, J.; Jin, F.; Jia, J.; Chen, G. Iron selenide nanoparticles-encapsulated within bamboo-like N-doped carbon nanotubes as composite anodes for superior lithium and sodium-ion storage. J. Chem. Eng. 2022, 435, 135185. [Google Scholar] [CrossRef]
- Hou, B.; Benito-Alifonso, D.; Webster, R.F.; Cherns, D.; Galan, M.C.; Fermín, D.J. Synthetic Mechanism Studies of Iron Selenides: An Emerging Class of Materials for Electrocatalysis. Catalysts 2021, 11, 681. [Google Scholar] [CrossRef]
- Oyetunde, T.; Omorogie, M.O.; O’Brien, P. Ferromagnetic FeSe2 from a mixed sulphur-selenium complex of iron [Fe{(SePPh2NPPh2S)2N}3] through pyrolysis. Heliyon 2020, 6, e03763. [Google Scholar] [CrossRef]
- Cho, J.S.; Park, J.; Kang, Y.C. Double-walled iron oxide nanotubes via selective chemical etching and Kirkendall process. Sci. Rep. 2020, 6, 38933. [Google Scholar] [CrossRef]
- Zheng, Q.; Cheng, X.; Li, H. Microwave Synthesis of High Activity FeSe2/C Catalyst toward Oxygen Reduction Reaction. Catalysts 2015, 5, 1079. [Google Scholar] [CrossRef]
- Hieber, W.; Gruber, J. Zur Kenntnis der Eisencarbonylchalkogenide. J. Inorg. Gen. Chem. 1958, 296, 91–103. [Google Scholar] [CrossRef]
- Polin, J.; Schottenberger, H. Conversion of methyl ketones into terminal acetylenes: Ethynylferrocene. Organic Syntheses 1998, 9, 411–414. [Google Scholar]
- Jacubert, M.; Provot, O.; Peyrat, J.F.; Hamze, A.; Brion, J.D.; Alami, M. p-Toluenesulfonic acid-promoted selective functionalization of unsymmetrical arylalkynes: A regioselective access to various arylketones and heterocycles. Tetrahedron 2010, 66, 3775–3787. [Google Scholar] [CrossRef]
- Nishizawa, M.; Skwarczynski, M.; Imagawa, H.; Sugihara, T. Mercuric triflate-TMU catalyzed hydration of terminal alkyne to give methyl ketone under mild conditions. Chem. Lett. 2002, 31, 12–13. [Google Scholar] [CrossRef]
- Jennings, P.W.; Hartman, J.W.; Hiscox, W.C. Alkyne hydration using Pt (II) catalysts. Inorg. Chim. Acta 1994, 222, 317–322. [Google Scholar] [CrossRef]
- Lumbroso, A.; Vautravers, N.R.; Breit, B. Rhodium-Catalyzed Selective anti-Markovnikov Addition of Carboxylic Acids to Alkynes. Org. Lett. 2010, 12, 5498–5501. [Google Scholar] [CrossRef]
- Park, Y.J.; Kwon, B.; Ahn, J.; Lee, H.; Jun, C. Chelation-assisted hydrative dimerization of 1-alkyne forming α, β-enones by an Rh (I) catalyst. J. Am. Chem. Soc. 2004, 126, 13892–13893. [Google Scholar] [CrossRef]
- Manikar, P.S.; Chippala, V.; Chegondi, R.; Chandrasekher, S. Ruthenium(II)-Catalyzed Hydration of Terminal Alkynes in PEG-400. Synlett 2016, 27, 1969–1972. [Google Scholar] [CrossRef]
- Kusakabe, T.; Ito, Y.; Kamimura, M.; Shirai, T.; Takahashi, K.; Mochida, T.; Kato, K. Palladium(II) Bis(oxazoline) Complexes that Catalyze the Hydration of Alkynes. Asian J. Org. Chem. 2017, 6, 1086–1090. [Google Scholar] [CrossRef]
- Marion, N.; Ramon, R.S.; Nolan, S.P. [(NHC)AuI]-Catalyzed Acid-Free Alkyne Hydration at Part-per-Million Catalyst Loadings. J. Am. Chem. Soc. 2009, 131, 448–449. [Google Scholar] [CrossRef] [PubMed]
- Casado, R.; Contel, M.; Laguna, M.; Romero, P.; Sanz, S. Organometallic gold (III) compounds as catalysts for the addition of water and methanol to terminal alkynes. J. Am. Chem. Soc. 2003, 125, 11925–11935. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Yeon, J.; Lee, P.H.; Lee, K. Iron-catalyzed indirect hydration of alkynes in presence of methanesulfonic acid. Tetrahedron Lett. 2013, 54, 4414–4417. [Google Scholar] [CrossRef]




















































Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).