
Stabilized Arylzinc Iodides in Negishi Acylative Cross-Coupling: A Modular Synthesis of Chalcones



Abstract
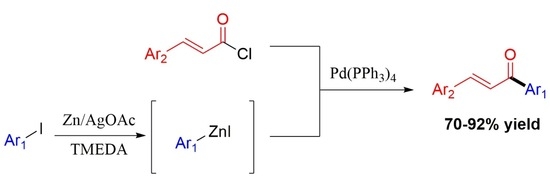
:
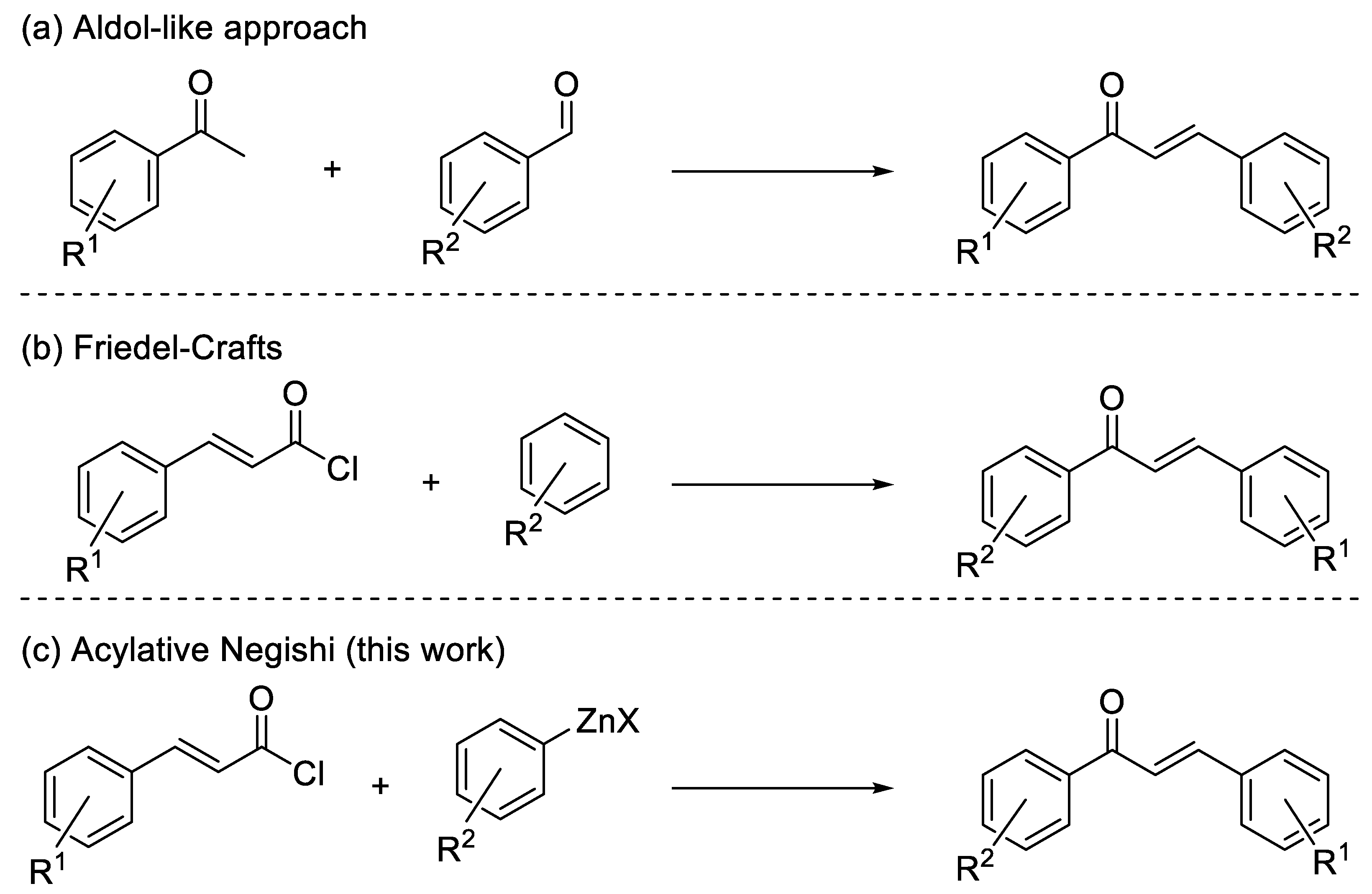
1. Introduction
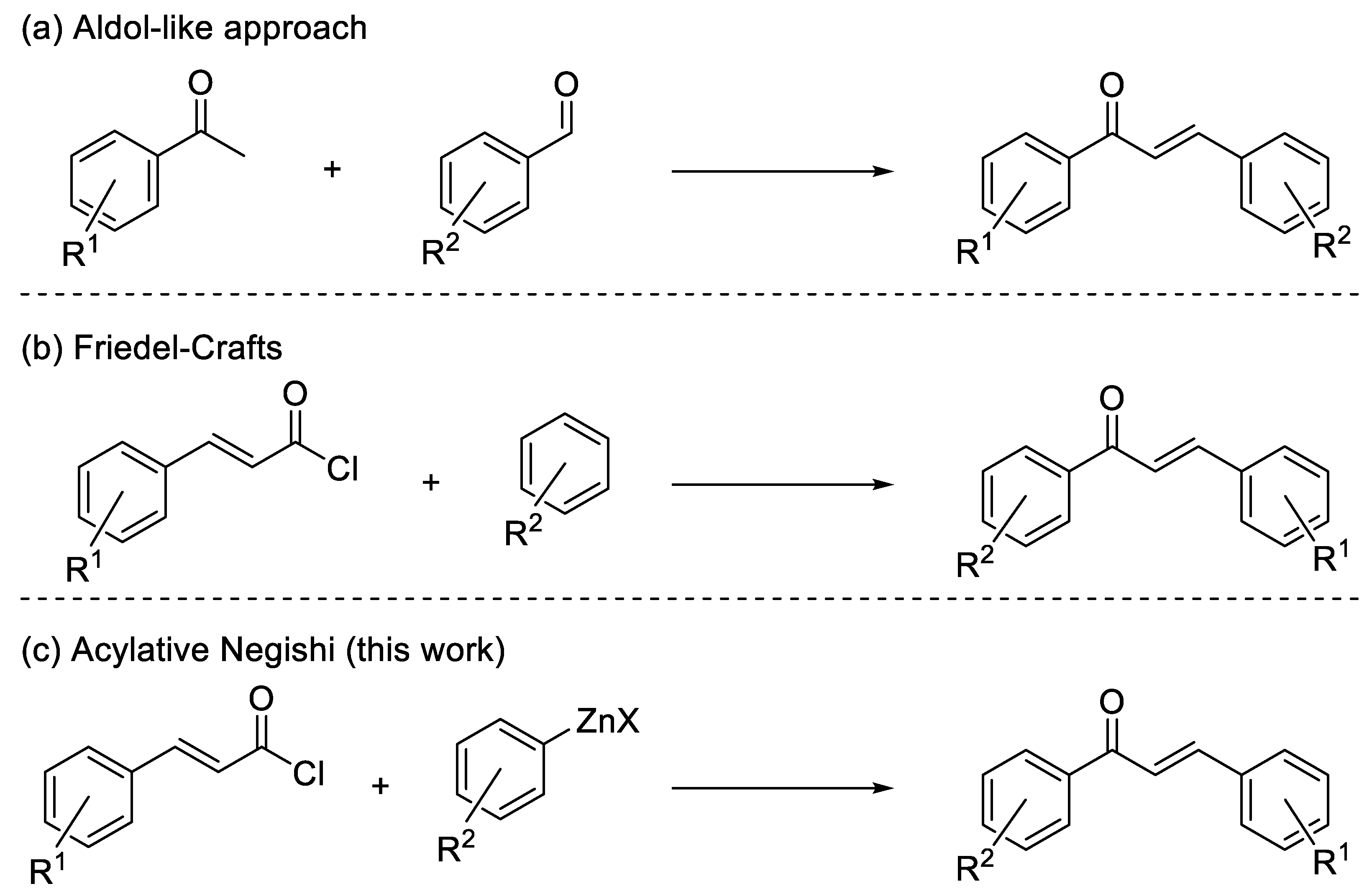
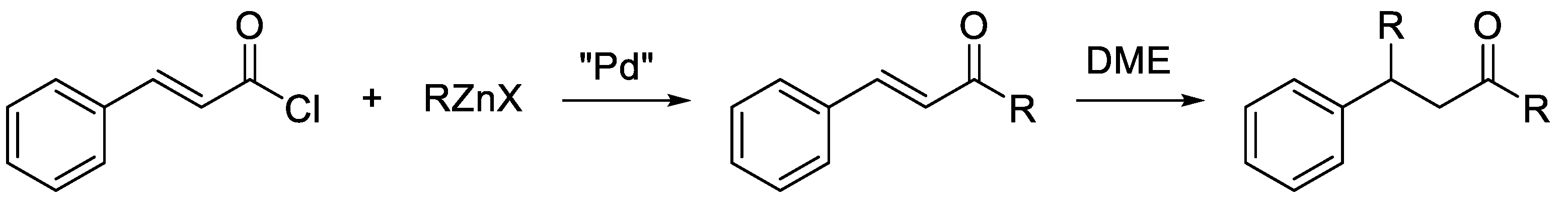
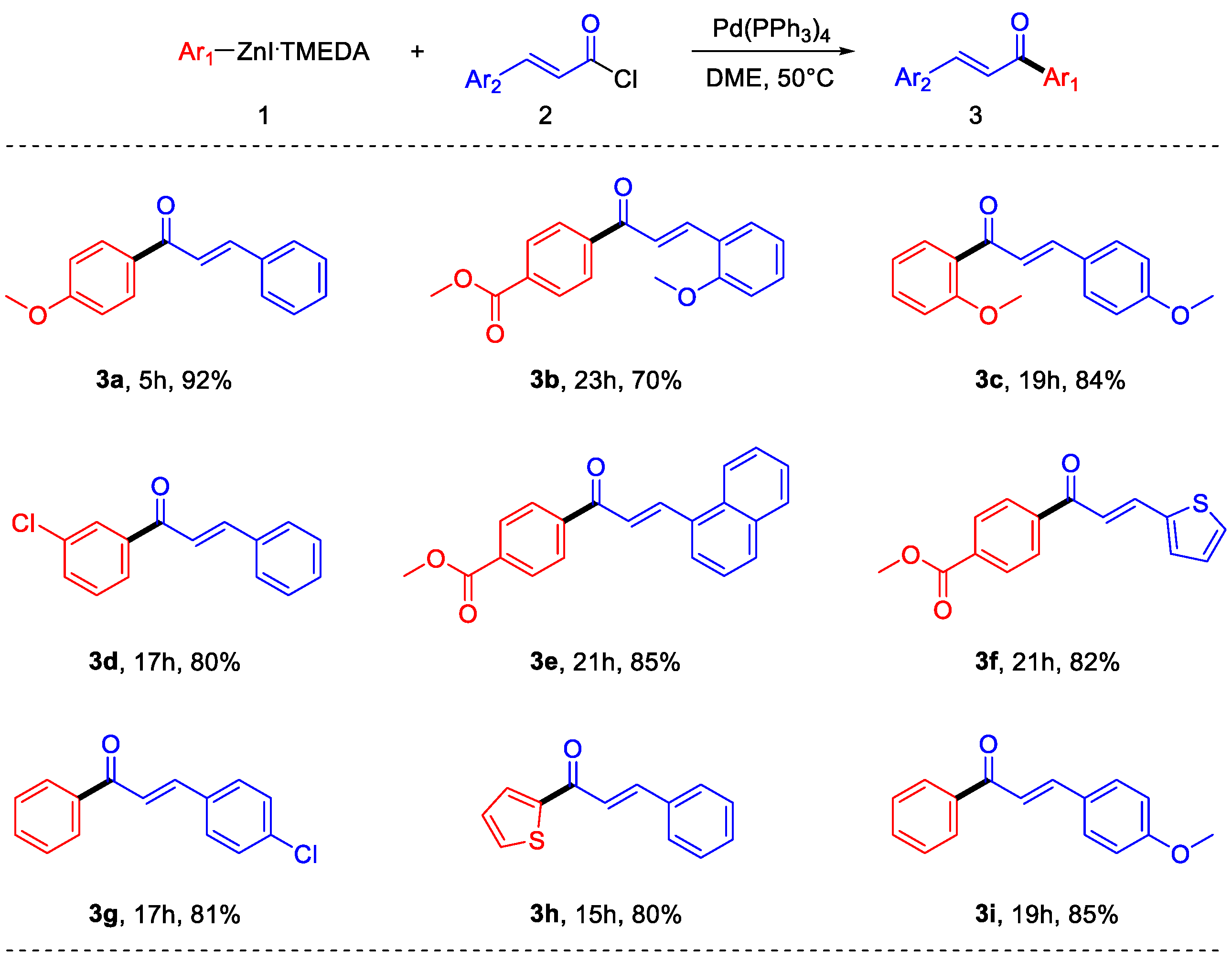
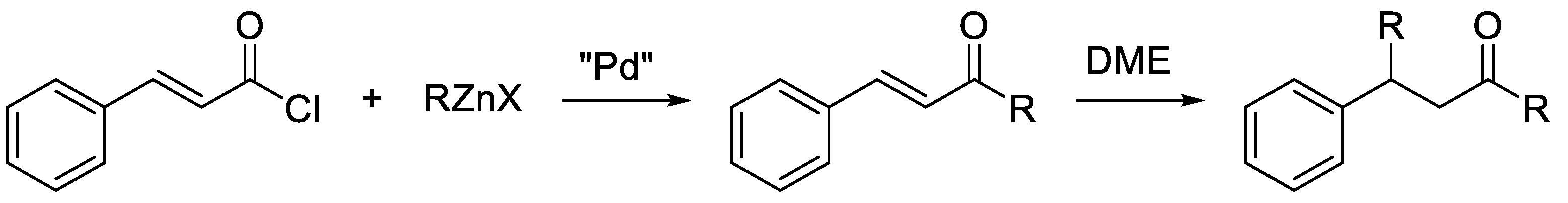
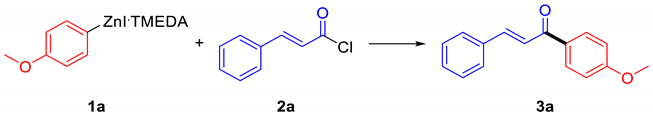
2. Results and Discussion
3. Conclusions
4. Experimental Section
4.1. General Information
4.2. General Procedure for the Synthesis of the Arylzinc Halides
4.3. General Procedure for the Synthesis of Chalcones
4.3.1. (E)-1-(4-Methoxyphenyl)-3-phenylprop-2-en-1-one (3a)
4.3.2. Methyl (E)-4-(3-(2-Methoxyphenyl)acryloyl)benzoate (3b)
4.3.3. (E)-1-(2-Methoxyphenyl)-3-(4-methoxyphenyl)prop-2-en-1-one (3c)
4.3.4. (E)-1-(3-Chlorophenyl)-3-phenylprop-2-en-1-one (3d)
4.3.5. Methyl (E)-4-(3-(Naphthalen-1-yl)acryloyl)benzoate (3e)
4.3.6. Methyl (E)-4-(3-(Thiophen-2-yl)acryloyl)benzoate (3f)
4.3.7. (E)-3-(4-Chlorophenyl)-1-phenylprop-2-en-1-one (3g)
4.3.8. (E)-3-Phenyl-1-(thiophen-2-yl)prop-2-en-1-one (3h)
4.3.9. (E)-3-(4-Methoxyphenyl)-1-phenylprop-2-en-1-one (3i)
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bukhari, S.N.A.; Jasamai, M.; Jantan, I.; Ahmad, W. Review of Methods and Various Catalyst Used for Chalcones Synthesis. Mini-Rev. Org. Chem. 2013, 10, 73–83. [Google Scholar] [CrossRef]
- Suwito, H.; Jumina, J.; Mustofa, M.; Kristanti, A.N.; Tri Puspaningsih, N.N. Chalcones: Synthesis, Structure Diversity and Pharmacological Aspects. J. Chem. Pharm. Res. 2014, 6, 1076. [Google Scholar] [CrossRef]
- Chavan, B.B.; Gadekar, A.S.; Mehta, P.P.; Vawhal, P.K.; Kolsure, A.K.; Chabukswar, A.R. Synthesis and Medicinal Significance of Chalcones—A Review. Asian J. Pharm. Biol. Res. 2016, 6, 1–7. [Google Scholar]
- Yazdan, S.K.; Sagar, D.V.; Shaik, A.B. Chemical and Biological Potentials of Chalcones: A Review. Org. Med. Chem. 2015, 1, 20–28. [Google Scholar] [CrossRef]
- Singh, P.; Anand, A.; Kumar, V. Recent Developments in Biological Activities of Chalcones: A Mini Review. Eur. J. Med. Chem. 2014, 85, 758–777. [Google Scholar] [CrossRef]
- Rozmer, Z.; Perjési, P. Naturally Occurring Chalcones and Their Biological Activities. Phytochem. Rev. 2016, 15, 87–120. [Google Scholar] [CrossRef]
- More, P.E.; Bandgar, B.P.; Kamble, V.T. Zinc Oxide as a Regioselective and Heterogeneous Catalyst for the Synthesis of Chalcones at Room Temperature. Catal. Commun. 2012, 27, 30–32. [Google Scholar] [CrossRef]
- Negishi, E.; Bagheri, V.; Chatterjee, S.; Luo, F.-T.; Miller, J.A.; Stoll, A.T. Palladium-Catalyzed Acylation of Organozincs and Other Organometallics as a Convenient Route to Ketones. Tetrahedron Lett. 1983, 24, 5181–5184. [Google Scholar] [CrossRef]
- Wang, D.; Zhang, Z. Palladium-Catalyzed Cross-Coupling Reactions of Carboxylic Anhydrides with Organozinc Reagents. Org. Lett. 2003, 5, 4645–4648. [Google Scholar] [CrossRef]
- Paul Knochel, P.J. Organozinc Reagents—A Practical Approach; Oxford University Press: Oxford, UK, 1999; ISBN 9780198501213. [Google Scholar]
- Knochel, P.; Millot, N.; Rodriguez, A.L.; Tucker, C.E. Preparation and Applications of Functionalized Organozinc Compounds. In Organic Reactions; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2001; pp. 417–759. ISBN 9783527615179. [Google Scholar]
- Krasovskiy, A.; Malakhov, V.; Gavryushin, A.; Knochel, P. Efficient Synthesis of Functionalized Organozinc Compounds by the Direct Insertion of Zinc into Organic Iodides and Bromides. Angew. Chem. Int. Ed. 2006, 45, 6040–6044. [Google Scholar] [CrossRef]
- Casotti, G.; Iuliano, A.; Carpita, A. Arylzinc Halides by Silver Catalyzed Zinc Insertion into Aryl Iodides. Eur. J. Org. Chem. 2019, 2019, 1021–1026. [Google Scholar] [CrossRef]
- Bhar, S.; Ranu, B.C. Zinc-Promoted Selective Cleavage of Ethers in Presence of Acyl Chloride. J. Org. Chem. 1995, 60, 745–747. [Google Scholar] [CrossRef]
- Casotti, G.; Ciancaleoni, G.; Lipparini, F.; Nieri, C.; Iuliano, A. Uncatalyzed Conjugate Addition of Organozinc Halides to Enones in DME: A Combined Experimental/Computational Study on the Role of the Solvent and the Reaction Mechanism. Chem. Sci. 2020, 11, 257–263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laudadio, G.; Fusini, G.; Casotti, G.; Evangelisti, C.; Angelici, G.; Carpita, A. Synthesis of Pterostilbene through Supported-Catalyst Promoted Mizoroki-Heck Reaction, and Its Transposition in Continuous Flow Reactor. J. Flow Chem. 2019, 9, 133–143. [Google Scholar] [CrossRef]
- Casotti, G.; Fusini, G.; Ferreri, M.; Pardini, L.F.; Evangelisti, C.; Angelici, G.; Carpita, A. Total Synthesis of Asparenydiol by Two Sonogashira Cross-Coupling Reactions Promoted by Supported Pd and Cu Catalysts. Synthesis 2020, 52, 1795–1803. [Google Scholar] [CrossRef] [Green Version]
- Fusini, G.; Barsanti, D.; Angelici, G.; Casotti, G.; Canale, A.; Benelli, G.; Lucchi, A.; Carpita, A. Identification and Synthesis of New Sex-Specific Components of Olive Fruit Fly (Bactrocera Oleae) Female Rectal Gland, through Original Negishi Reactions on Supported Catalysts. Tetrahedron 2018, 74, 4381–4389. [Google Scholar] [CrossRef]
- Casotti, G.; Rositano, V.; Iuliano, A. Enantioselective Conjugate Addition of Stabilized Arylzinc Iodide to Enones: An Improved Protocol of the Hayashi Reaction. Adv. Synth. Catal. 2021, 363, 1126–1131. [Google Scholar] [CrossRef]
- Culkin, D.A.; Hartwig, J.F. Carbon−Carbon Bond-Forming Reductive Elimination from Arylpalladium Complexes Containing Functionalized Alkyl Groups. Influence of Ligand Steric and Electronic Properties on Structure, Stability, and Reactivity. Organometallics 2004, 23, 3398–3416. [Google Scholar] [CrossRef]
- Han, C.; Buchwald, S.L. Negishi Coupling of Secondary Alkylzinc Halides with Aryl Bromides and Chlorides. J. Am. Chem Soc. 2009, 131, 7532–7533. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Oldenhuis, N.J.; Buchwald, S.L. Mild and General Conditions for Negishi Cross-Coupling Enabled by the Use of Palladacycle Precatalysts. Angew. Chem. Int. Ed. 2013, 52, 615–619. [Google Scholar] [CrossRef] [Green Version]
- D’Alterio, M.C.; Casals-Cruañas, È.; Tzouras, N.V.; Talarico, G.; Nolan, S.P.; Poater, A. Mechanistic Aspects of the Palladium-Catalyzed Suzuki-Miyaura Cross-Coupling Reaction. Chem. A Eur. J. 2021, 27, 13481–13493. [Google Scholar] [CrossRef] [PubMed]
- Tucker, C.E.; Majid, T.N.; Knochel, P. Preparation of Highly Functionalized Magnesium, Zinc, and Copper Aryl and Alkenyl Organometallics via the Corresponding Organolithiums. J. Am. Chem. Soc. 1992, 114, 3983–3985. [Google Scholar] [CrossRef]
- Qian, Y.; Zhang, H.-J.; Zhang, H.; Xu, C.; Zhao, J.; Zhu, H.-L. Synthesis, Molecular Modeling, and Biological Evaluation of Cinnamic Acid Metronidazole Ester Derivatives as Novel Anticancer Agents. Bioorganic Med. Chem. 2010, 18, 4991–4996. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Yan, W.; Zhang, C.; Zhang, Y.; Liang, M.; Chu, F.; Gong, Y.; Xu, B.; Wang, P.; Lei, H. New Synthesis Method for Sultone Derivatives: Synthesis, Crystal Structure and Biological Evaluation of S-CA. Molecules 2015, 20, 4307–4318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, X.-F.; Neumann, H.; Spannenberg, A.; Schulz, T.; Jiao, H.; Beller, M. Development of a General Palladium-Catalyzed Carbonylative Heck Reaction of Aryl Halides. J. Am. Chem. Soc. 2010, 132, 14596–14602. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, S.; Kapoor, R.; Yadav, L.D.S. Visible Light Activated Radical Denitrative Benzoylation of β -Nitrostyrenes: A Photocatalytic Approach to Chalcones. Adv. Synth. Catal. 2018, 360, 1407–1413. [Google Scholar] [CrossRef]
- Stevenson, S.M.; Higgins, R.F.; Shores, M.P.; Ferreira, E.M. Chromium Photocatalysis: Accessing Structural Complements to Diels–Alder Adducts with Electron-Deficient Dienophiles. Chem. Sci. 2017, 8, 654–660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shrestha, A.; Shrestha, A.; Park, P.; Lee, E. Hydroxyl- and Halogen-containing Chalcones for the Inhibition of LPS-stimulated ROS Production in RAW 264.7 Macrophages: Design, Synthesis and Structure–Activity Relationship Study. Bull. Korean Chem. Soc. 2019, 40, 729–734. [Google Scholar] [CrossRef]
- Mekky, A.E.M.; Sanad, S.M.H.; Said, A.Y.; Elneairy, M.A.A. Synthesis, Cytotoxicity, in-Vitro Antibacterial Screening and In-Silico Study of Novel Thieno[2,3-b]Pyridines as Potential Pim-1 Inhibitors. Synth. Commun. 2020, 50, 2376–2389. [Google Scholar] [CrossRef]
- Schmink, J.R.; Holcomb, J.L.; Leadbeater, N.E. Testing the Validity of Microwave-Interfaced, in Situ Raman Spectroscopy as a Tool for Kinetic Studies. Org. Lett. 2009, 11, 365–368. [Google Scholar] [CrossRef]
- Romanelli, G.; Pasquale, G.; Sathicq, Á.; Thomas, H.; Autino, J.; Vázquez, P. Synthesis of Chalcones Catalyzed by Aminopropylated Silica Sol–Gel under Solvent-Free Conditions. J. Mol. Catal. A Chem. 2011, 340, 24–32. [Google Scholar] [CrossRef]
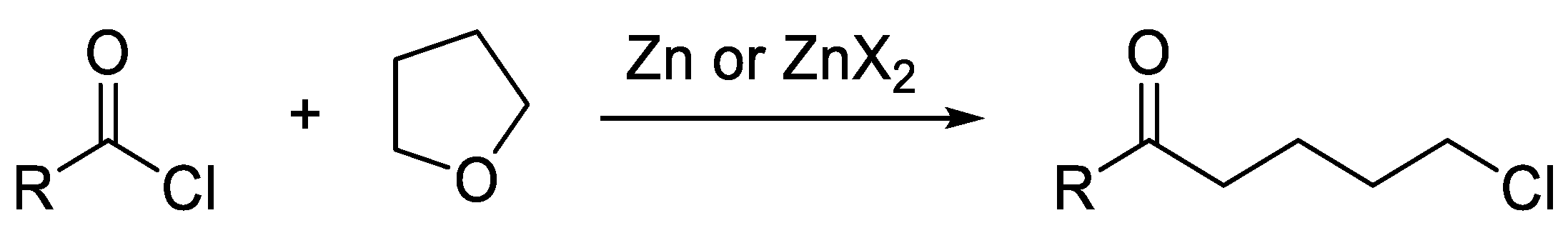

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Pre-Catalyst | Temperature | Solvent | Yield 1 |
|---|---|---|---|---|
| 1 | PdCl2(PPh3)2 | 50 °C | THF | 19% |
| 2 | PdCl2(PPh3)2 | 50 °C | DME | 46% |
| 3 | PdCl2(PPh3)2 | 70 °C | DME | 36% |
| 4 | Pd(dppf)Cl2 | 50 °C | DME | 51% |
| 5 | Pd(dppf)Cl2 | 70 °C | DME | 49% |
| 6 | PdCl2(PCy3)2 | 50 °C | DME | 22% |
| 7 | Pd(OAc)2 + SPhos | 50 °C | DME | 45% |
| 8 | Pd(OAc)2 + XPhos | 50 °C | DME | 25% |
| 9 | Pd2(dba)3 + SPhos | 50 °C | DME | 90% |
| 10 | Pd(PPh3)4 | 50 °C | DME | 92% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pierigé, M.; Iuliano, A.; Angelici, G.; Casotti, G. Stabilized Arylzinc Iodides in Negishi Acylative Cross-Coupling: A Modular Synthesis of Chalcones. Organics 2022, 3, 87-94. https://doi.org/10.3390/org3020006
Pierigé M, Iuliano A, Angelici G, Casotti G. Stabilized Arylzinc Iodides in Negishi Acylative Cross-Coupling: A Modular Synthesis of Chalcones. Organics. 2022; 3(2):87-94. https://doi.org/10.3390/org3020006
Chicago/Turabian StylePierigé, Michele, Anna Iuliano, Gaetano Angelici, and Gianluca Casotti. 2022. "Stabilized Arylzinc Iodides in Negishi Acylative Cross-Coupling: A Modular Synthesis of Chalcones" Organics 3, no. 2: 87-94. https://doi.org/10.3390/org3020006
APA StylePierigé, M., Iuliano, A., Angelici, G., & Casotti, G. (2022). Stabilized Arylzinc Iodides in Negishi Acylative Cross-Coupling: A Modular Synthesis of Chalcones. Organics, 3(2), 87-94. https://doi.org/10.3390/org3020006