
Recent Applications of Pd-Catalyzed Suzuki–Miyaura and Buchwald–Hartwig Couplings in Pharmaceutical Process Chemistry


1
Department of Chemistry and Biochemistry, University of California, Santa Barbara, CA 93106, USA
2
Department of Chemistry, Pomona College, Claremont, CA 91711, USA
*
Author to whom correspondence should be addressed.
Organics 2022, 3(1), 1-21; https://doi.org/10.3390/org3010001
Submission received: 1 November 2021
/
Revised: 20 December 2021
/
Accepted: 23 December 2021
/
Published: 18 January 2022
(This article belongs to the Special Issue New Reactions and Strategies for Natural Product Synthesis)
Abstract
:Cross-coupling reactions have changed the way complex molecules are synthesized. In particular, Suzuki–Miyaura and Buchwald–Hartwig amination reactions have given opportunities to elegantly make pharmaceutical ingredients. Indeed, these reactions are at the forefront of both the stages of drug development, medicinal chemistry, and process chemistry. On the one hand, these reactions have given medicinal chemists a resource to derivatize the core compound to arrive at scaffold rapidly. On the other hand, these cross couplings have offered the process chemists a smart tool to synthesize the development candidates safely, quickly, and efficiently. Generally, the application of cross-coupling reactions is broad. This review will specifically focus on their real (pharma) world applications in large-scale synthesis appearing in the last three years.
1. Introduction
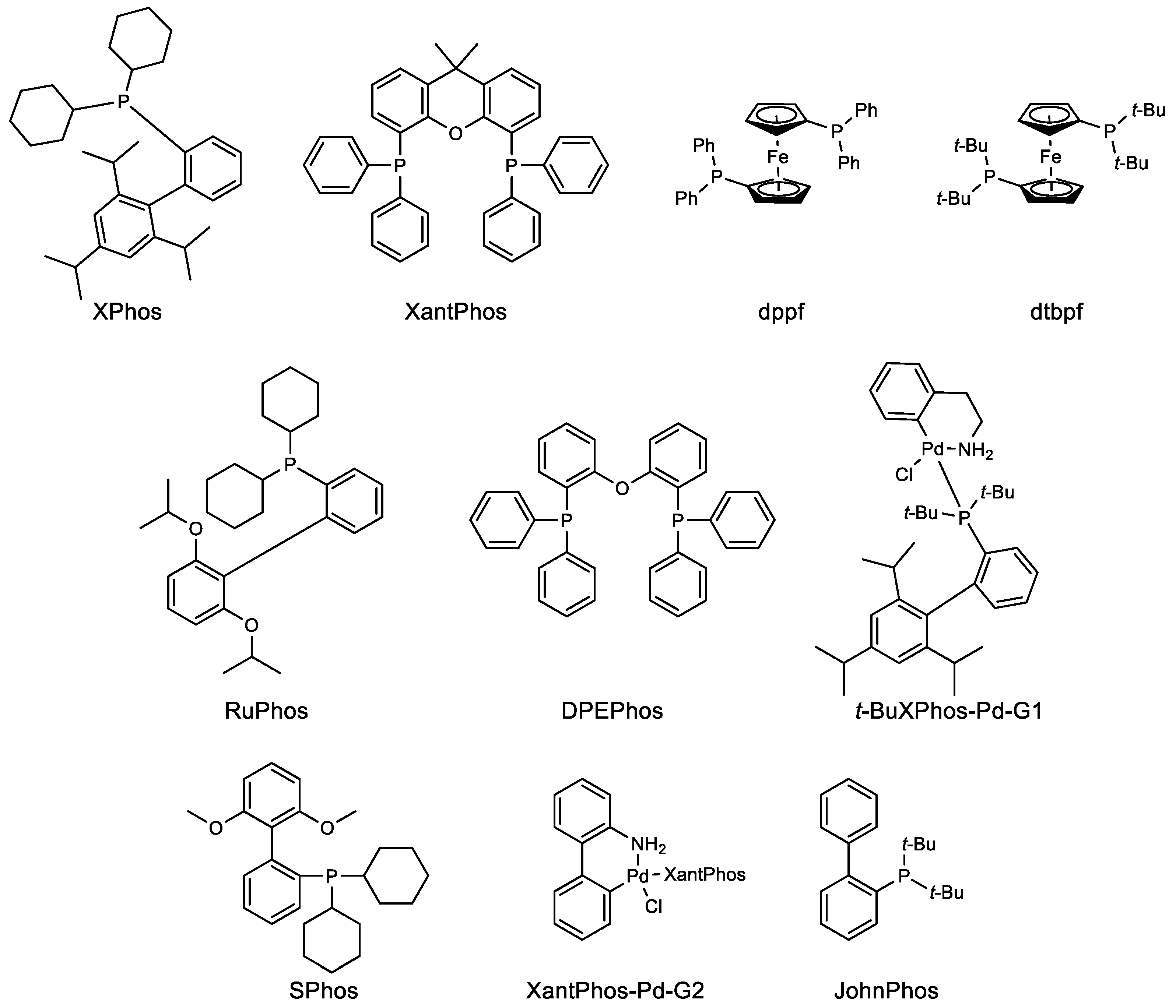
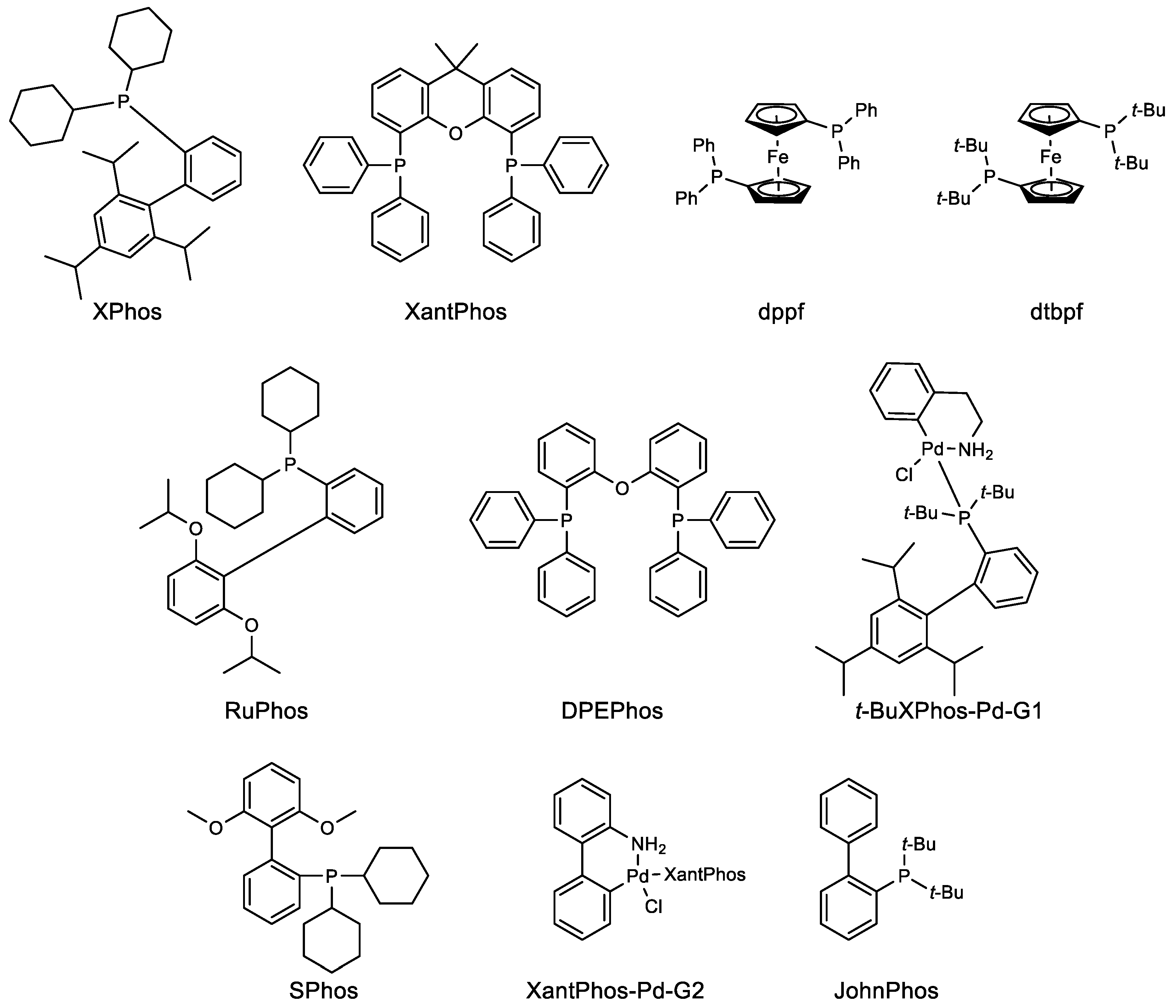
Metal catalyzed cross couplings have played a crucial role in different stages of drug development. For example, the drug discovery stage requires multiple compounds to be tested for the best biological activity, wherein rapid access to the chemical matter is very important. The ability of cross-coupling reactions to quickly derivatize the core compound results in the multiple compounds available for biological studies. The aim at this stage remains to get the compound in milligrams scale. This scenario changes rapidly as the compound moves to the next stage of development, where only a few out of many compounds are selected for toxicology and animal studies. The required amount of compound then increases to 10–100 g, and most of the time, the chemistry used to make the compound in milligram scale will be replicated in this case. Once the compound enters the development phase, several factors need to be considered. At this stage, “how” instead of “what” is important—how to make the compounds efficiently, quickly, and safely with the best quality is given prime importance. Nevertheless, process chemists were quite hesitant to use Pd-catalyzed reactions given the high toxicity of residual Pd content in the final drug substance. According to Food Drug Administration (FDA), ≤10 ppm Pd is allowed per dose [1,2]. This suggests that the old practice of cross-coupling reactions done using 5–10 mol% (50,000–100,000 ppm) Pd catalyst would most probably lead to very high residual Pd. Hence, a tedious and costly downstream Pd cleaning process would be required. The recent advances both in the actual development of efficient Pd catalysts [3,4] and Pd scavenging processes [5] have mostly solved this issue. How these two pathbreaking coupling reactions, Suzuki–Miyaura and Buchwald–Hartwig, are becoming the essence of drug development is discussed in this review. The detailed structure of ligands and catalysts used in the work described in this review is also given in Figure 1.
2. Suzuki–Miyaura Coupling
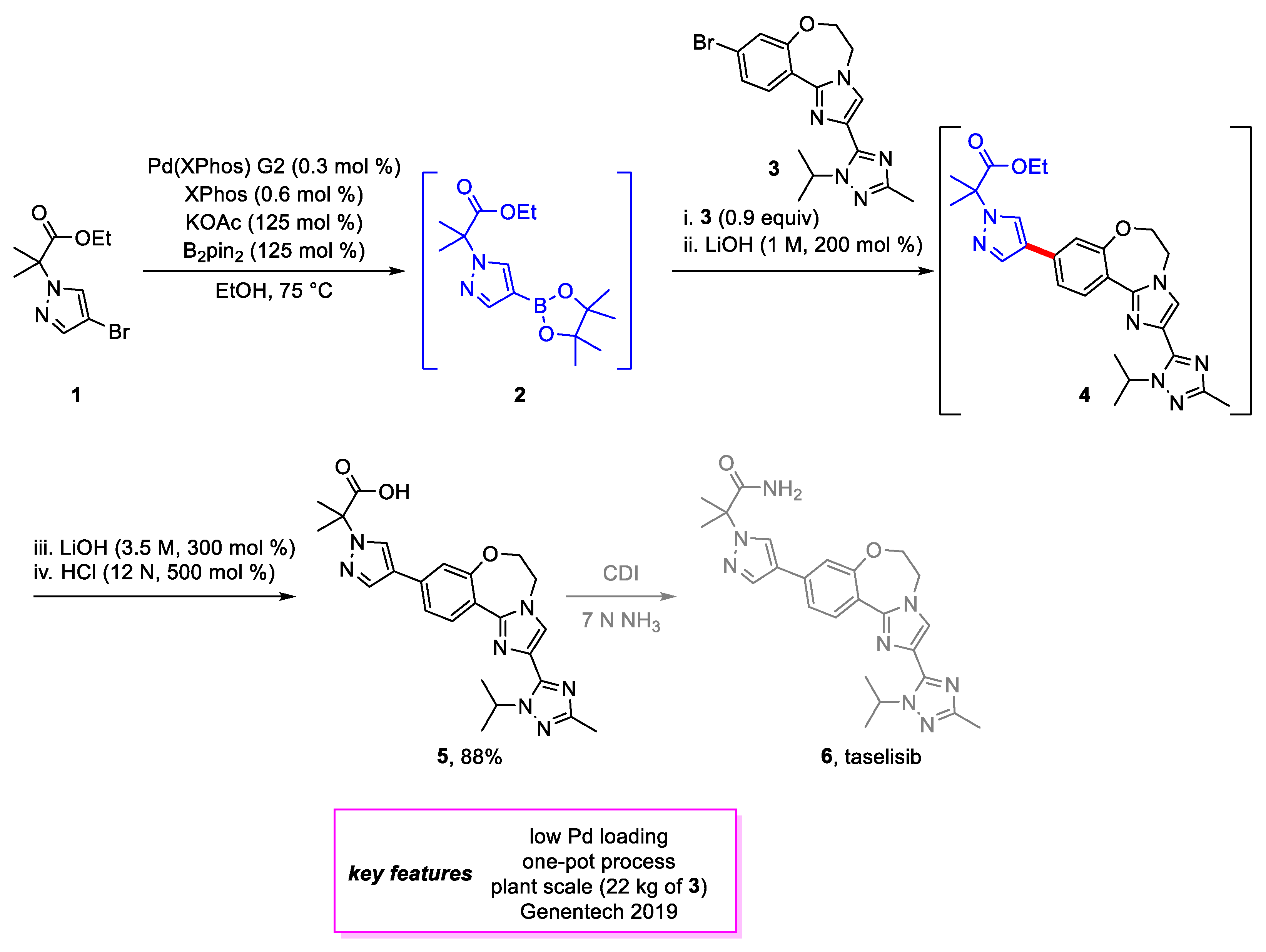
The Suzuki–Miyaura coupling remains a matured technology widely used in industrial synthesis. Although the initial discovery happened in 1979, the coupling’s real applicability was realized most recently, owing to the discovery of new metal catalysts and bulky ligands that could couple complex starting materials. This success led to the Nobel prize in 2010 [6]. This reaction is so fundamentally and mechanistically well understood that it is possible to forge any complex C-C bond from the respective aryl halide and aryl boron reagents. The Suzuki–Miyaura coupling is one of the widely used reactions in pharmaceutical synthesis. For example, St-Jean and coworkers [7] from Genentech developed an efficient late-stage synthetic method for PI3K β-Sparing Inhibitor taselisib 6 [8], which has been demonstrated to have increased activity against PIK3CA mutant breast cancer. A one-pot Miyaura borylation, Suzuki–Miyaura coupling, and saponification to produce 5 from 1 were described. Extensive palladium catalyst screening indicated only the Buchwald XPhos G1 and G2 pre-catalysts [9] were able to achieve full conversion to boronate ester 2, with G2 selected after further optimization; 90 mol% of 3, which was synthesized on a kilogram scale through four steps, and 200 mol% of 1 M LiOH were added directly to the reaction vessel following borylation to carry out the Suzuki–Miyaura coupling. Remarkably, only 0.3 mol% of Pd catalyst was used to perform these two steps. After full conversion to 4, more LiOH was added to carry out saponification. Acidification with the addition of aqueous HCl provided 5 as a solid. An 88% yield and >98 A% purity was achieved for 5 on a kilogram scale (Scheme 1). Overall, the development of this one-pot synthesis of 5 lowered the process mass intensity (PMI) of the entire route significantly from previous reports. Due to the corrosivity of HCl and foaming issues during acidification, the plant scale acidification was run in n-propanol and H2SO4. The use of n-propanol also helped to purge Pd to <40 ppm vs. ~400 ppm.
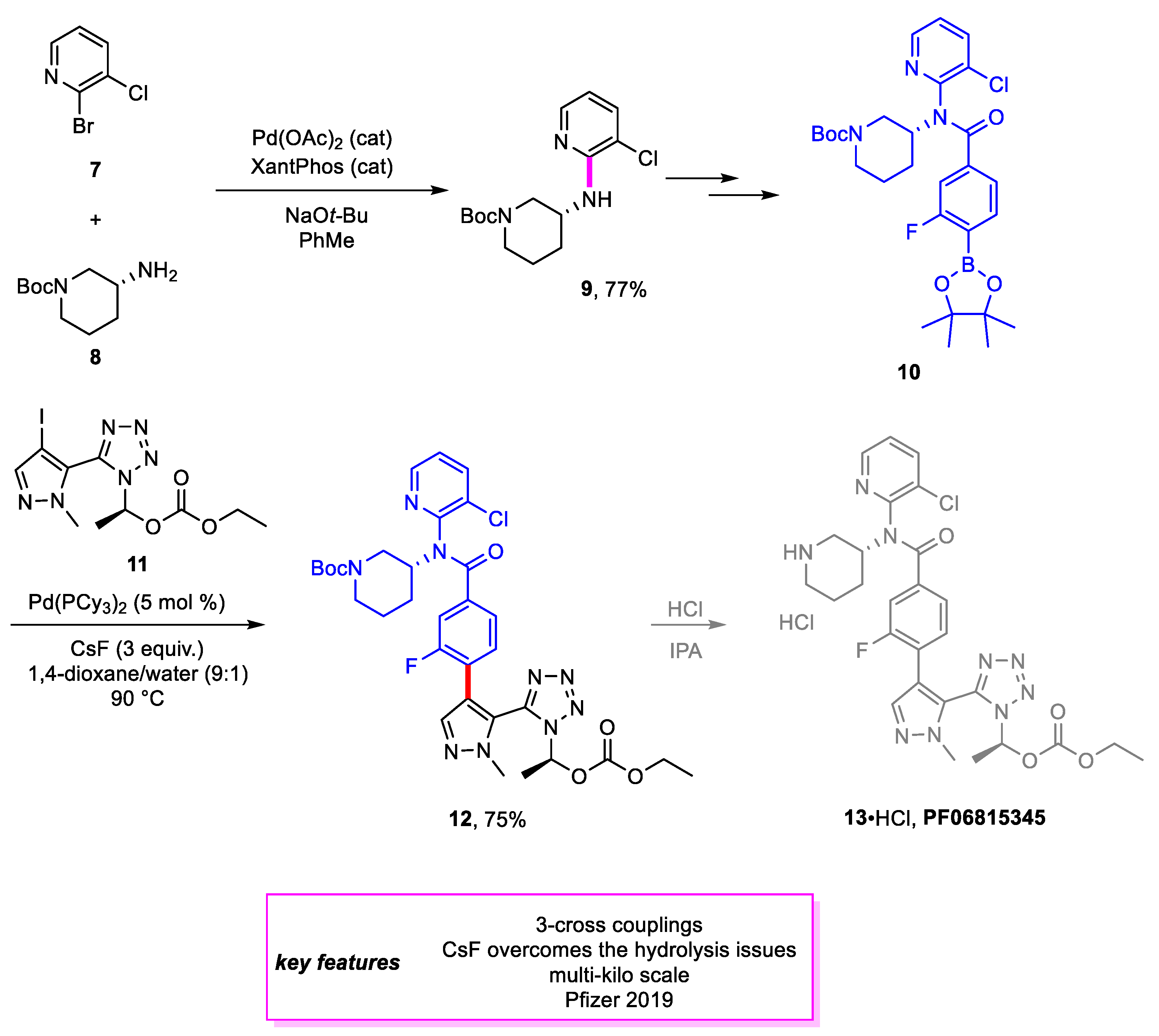
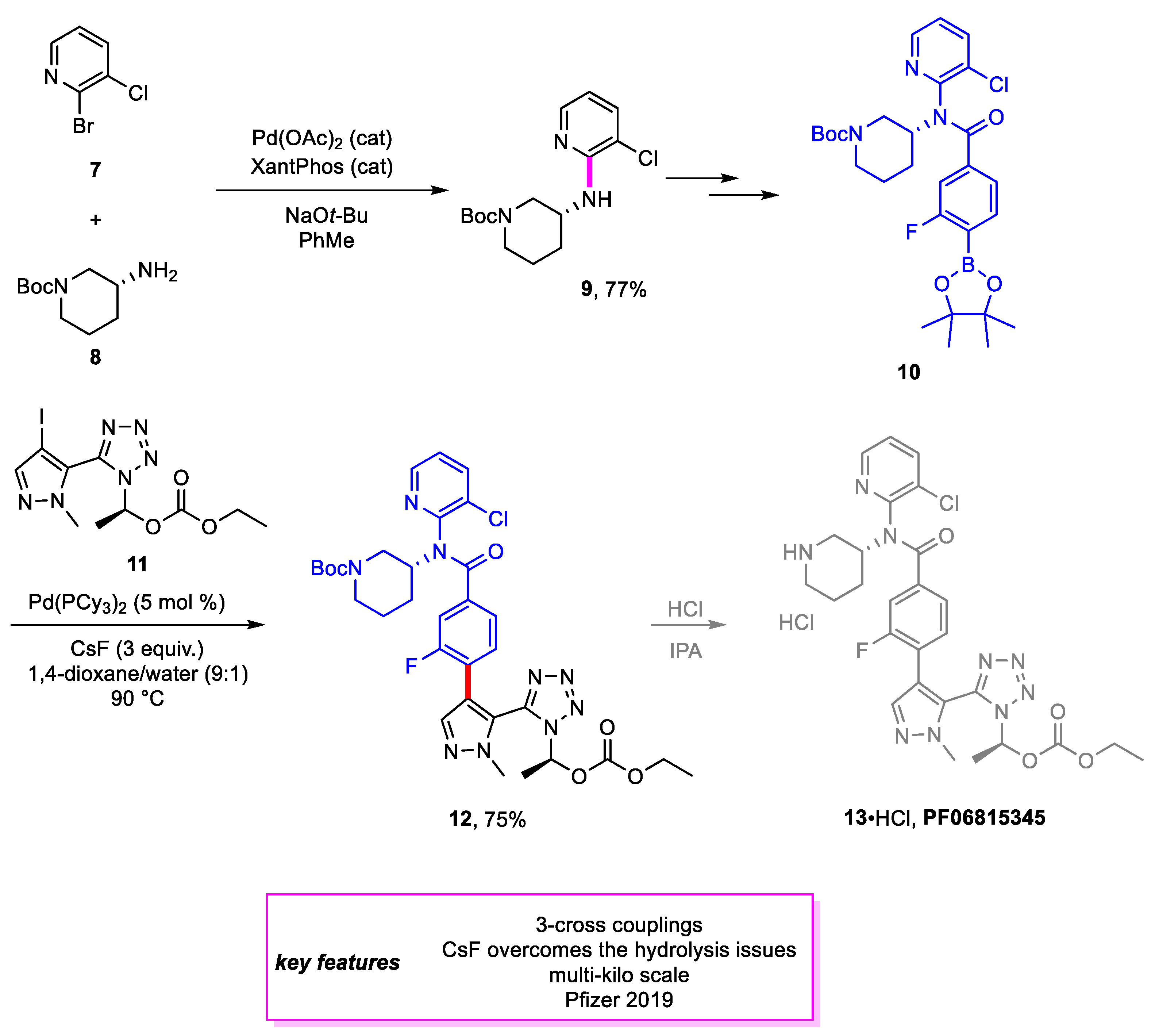
Akin and coworkers [10] from Pfizer designed a new synthetic method for proprotein convertase subtilisin/kexin type 9 (PCSK9) inhibitor PF-06815345 (13) [11], a potential treatment for decreasing serum LDL-cholesterol levels and reducing the risk of coronary heart disease. The precursor to 13•HCl is formed through a Suzuki–Miyaura coupling of intermediates 10 and 11. A Buchwald–Hartwig amination is involved in the first step towards synthesizing 9, and a yield of 77% was achieved for the amination product. Optimization of the Suzuki–Miyaura coupling focused on minimizing various side products formed. The most attention was paid to a homodimer side product that was caused by higher oxygen levels of the reaction solution. Additional precautions and adaptations were made to the reaction protocol, including nitrogen purging, installing oxygen sensors, and ordering palladium catalysts from vendors in presubdivided ampules, to minimize the formation of the side product. Extensive screening also indicated that Pd(0) catalysts, in general, performed better than their Pd(II) counterparts, with Pd(PCy3)2 achieving the best results. This reaction scaled up well, achieving >99% conversion on a kilogram scale (Scheme 2).
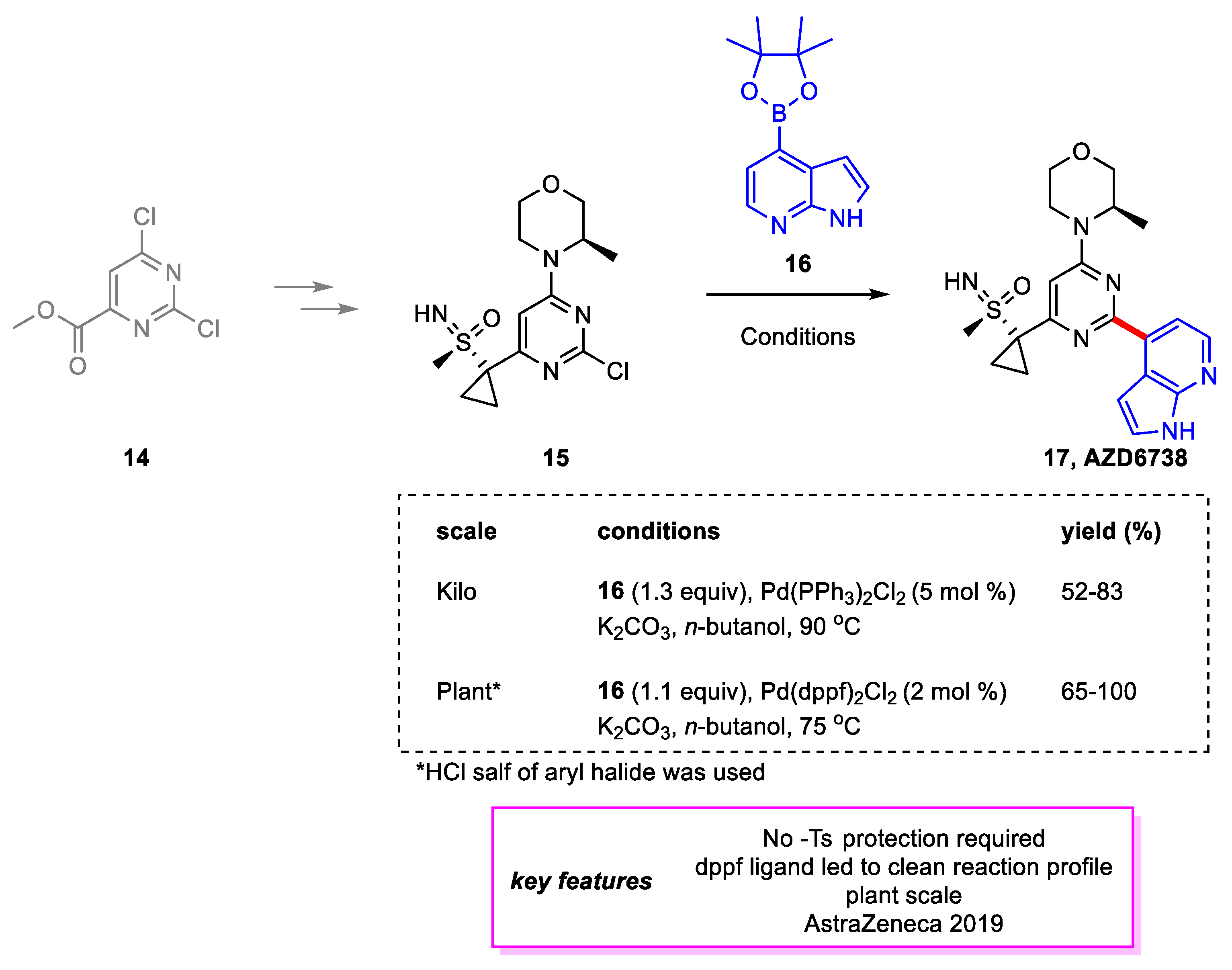
Goundry and coworkers [12] from AstraZeneca developed a large-scale synthetic route to ATR Inhibitor AZD6738 17 [13], a potential drug candidate in Phase I/II trials for treating both solid and hematological cancers. Due to time pressure, major changes were not made to the discovery route. Rather, the conditions for each chemical transformation were optimized and scaled up. The penultimate step in the synthesis of 17, which became the ultimate step in the new route due to the elimination of a deprotection step, is a Suzuki–Miyaura coupling to install the azaindole structure from boronate ester 16. On a kilogram scale, the catalyst system of (DTBPPS) palladium was tested as DTBPPS is a water-soluble ligand that would allow the palladium to be largely removed via aqueous washes. However, at 0.5 mol% of the catalyst, the reaction stalled at 5% conversion and could not be restarted by adding more catalyst or base. Therefore, the team reverted to Pd(PPh)3Cl2 conditions. On a plant scale, a screening of catalyst and ligands showed Pd(dppf)Cl2 to be a superior catalyst. The HCl salt of 15 was used, which necessitated the addition of an extra equivalent of the base. The reaction used ethanol as a solvent and achieved complete conversion in 2 h, with an average yield of 88% between three multi-kilogram batches. However, it was later found that n-butanol proved best for this cross coupling (Scheme 3).
Gontcharov and coworkers [14] from Pfizer developed a robust multi-kilogram scale synthetic route for PF-06263276 23 [15]. 23 is an inhaled pan-JAK inhibitor in clinical development for the treatment of chronic obstructive pulmonary disease. A Suzuki–Miyaura coupling is required between an aryl fragment and cyano-substituted indazole to produce a key intermediate for the new synthesis route. The use of a boronic acid coupling partner 19 instead of its boronate ester was an improvement from the medicinal chemistry synthetic route as 19 was easily prepared via Grignard reaction. This eliminated the need for one Pd-catalyzed step. Side product borinic acid 20 was found to be as reactive as the boronic acid in the future Suzuki–Miyaura coupling, and due to the low isolated yield of 19 (<60%), the solution of 19 and 20 were telescoped into the next step. The Suzuki–Miyaura coupling proceeded smoothly, although its product, intermediate 22, was not able to be isolated as a crystalline solid. Thus, it was decided to telescope the solution containing 22 into the next sequence of steps. The yield for 22 was 86% in situ (Scheme 4).
Baenziger and coworkers [16] from Novartis reported the development of a synthesis of dactolisib 27 [17], a dual kinase inhibitor of phosphatidylinositol 3-kinase (PI3K) and mammalian target of rapamycin (mTOR) currently investigated as the treatment of solid tumor forms. The coupling reaction between 25 and 26 afforded the desired product 27 and was optimized for commercial-scale manufacturing. The goal was to lower the palladium loading (6 mol%) to reduce cost and residual Pd levels. It was found that adding additional PPh3 (2 equiv. to Pd catalyst) as a supplementary ligand helped stabilize the catalytically active Pd(0) complex as well as act as a reducing agent for converting Pd(II) to Pd(0). On a laboratory scale, the Pd loading could be reduced to <0.2 mol% with a 2 Pd equiv. addition of PPh3 but was not reported on a commercial scale. The yield of crude 27 from the Suzuki–Miyaura coupling was >96%, and the overall yield for pure 27 was 85–90% with >99.5% purity on a multi-kilogram scale (Scheme 5).
Maltais and coworkers [18] from the Université Laval designed three new synthetic routes for PBRM, the first selective covalent inhibitor of 17β-hydroxysteroid dehydrogenase type 1 (17β-HSD1) [19]. 17β-HSD1 is involved in the last step of estrogen biosynthesis, making PBRM a leading candidate to treat breast cancer. Previous routes were only able to produce milligrams to a few grams of PBRM with long steps and low yield, so new routes that can reduce the number of steps and improve the overall yield are needed on a gram scale. The installation of the bromoethyl side chain was improved. Instead of arriving at 30 after multiple steps from 28, potassium (2-benzyloxyethyl) trifluoroborate was prepared from a published procedure and used as the coupling partner for 28 to arrive at 30 in one step. The coupling conditions followed previous conditions by Molander et al., who reported different examples of cross-coupling reactions of potassium alkyltrifluoroborate with aryl triflates. The yield for 30 (67%) was similar to those published (66–75%) (Scheme 6). The scale-up of this reaction to a multi-gram scale performed just as well as the small-scale assay. Following the development of this step, the entire synthesis of PBRM was optimized, particularly regarding reducing the amount of purification needed and improving the efficiency of other reactions in the route.
Zhao and coworkers [20] of Lexicon Pharmaceuticals developed two large-scale synthetic routes for Tryptophan Hydroxylase Inhibitor LX1031, a drug candidate for treating irritable bowel syndrome (IBS) in clinical trials [21].
The first-generation process route aimed to construct the LX1031 API via coupling two penultimate intermediates pyrimidine chloride 35 and biaryl chiral alcohol 38. Intermediate 35 required a Suzuki–Miyaura coupling of boronate 33 and aminopyridine 34. Extensive screening and optimization of palladium catalyst and base were conducted to minimize the formation of various impurities. The solvent was also screened to find a relatively safe and environmentally benign solvent system that was able to perform well in the reaction. A 65% yield with 95 A% purity was achieved on a 50 kg scale, although many batches had higher residual Pd levels than lab batches and required further recrystallization. The Suzuki–Miyaura coupling for intermediate 38 achieved a 93% yield with >99% purity on a 120 kg batch without further optimization (Scheme 7, top equation).
The second-generation process route aimed to reduce the cost of production even further by avoiding the tedious process to produce 35 on a modest yield. Therefore, the task became optimizing the Suzuki–Miyaura coupling to form 41, the penultimate intermediate, which is converted to LX1031 via deprotection of the chiral amine. Extensive screening and optimization of the conditions improved the conversion of the sluggish reaction, and an 83% yield with ~98% purity was achieved on a multi-kilogram scale. However, residual Pd levels proved to be an issue again, but it was found that extending the aging time by 3–5 h lowered the residual Pd levels drastically (Scheme 7, bottom equation).
Hardouin and coworkers [22] from Oril Industries designed a multi-kilogram scale synthesis of 45, a key intermediate towards the synthesis of 46, a Potent Dual Bcl-2/Bcl-xL Antagonist in clinical trials to treat Bcl-2 dependent malignancies such as leukemias and lymphomas [23]. An improved Suzuki–Miyaura coupling using Pd/C was explored, and optimal conditions for the reaction were determined following the extensive screening. This process had several advantages over the previous method. First, the purity of 44 was improved from 60–70% to 97% after extraction with toluene. Second, issues associated with intellectual property related to the phosphine ligand were avoided. Third, being able to conduct the reaction in water improved the environmental impact of the production. Overall, the yield of a penultimate intermediate required to make 46 via sulfonamide coupling has improved from 64 to 83% with the new synthetic method (Scheme 8).
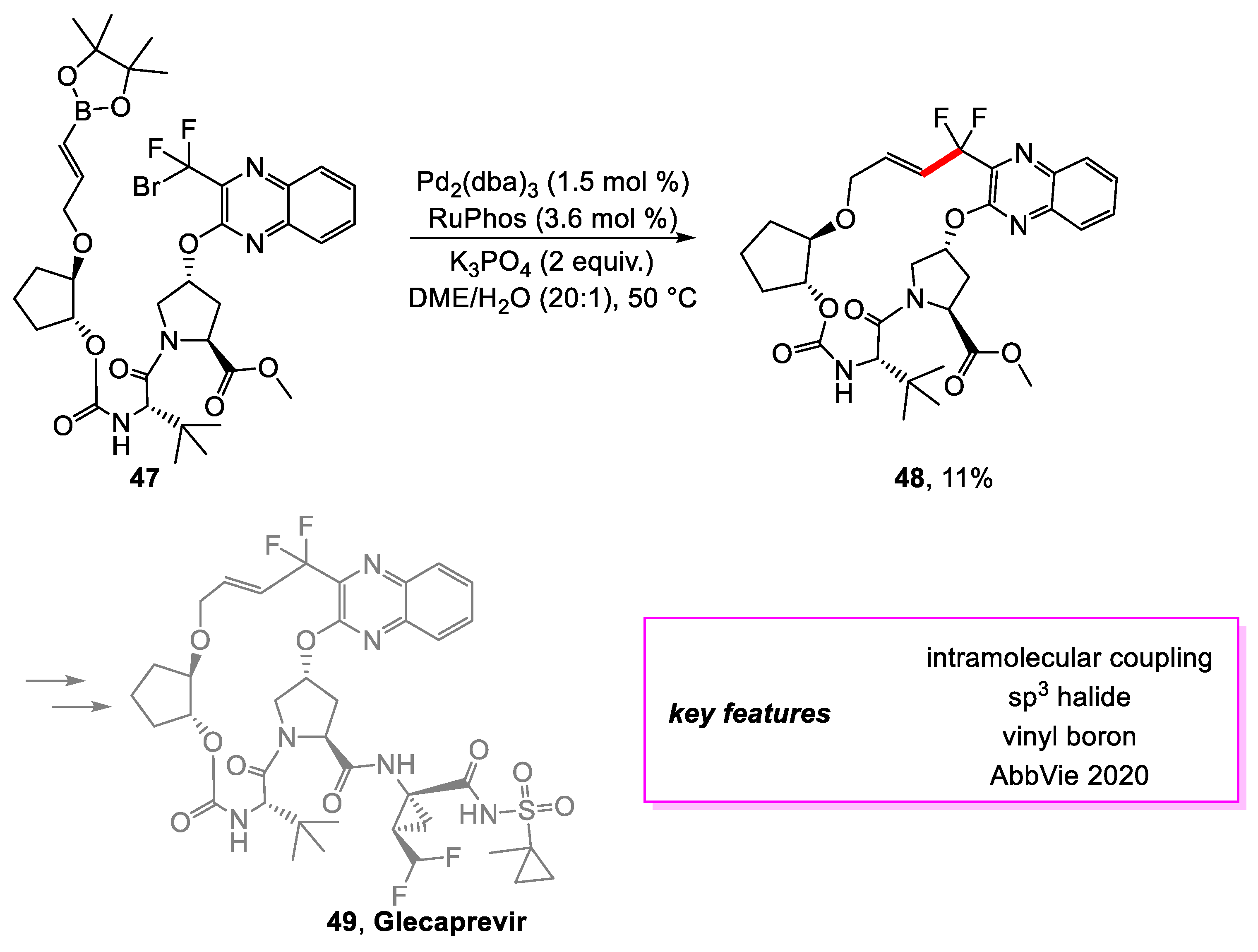
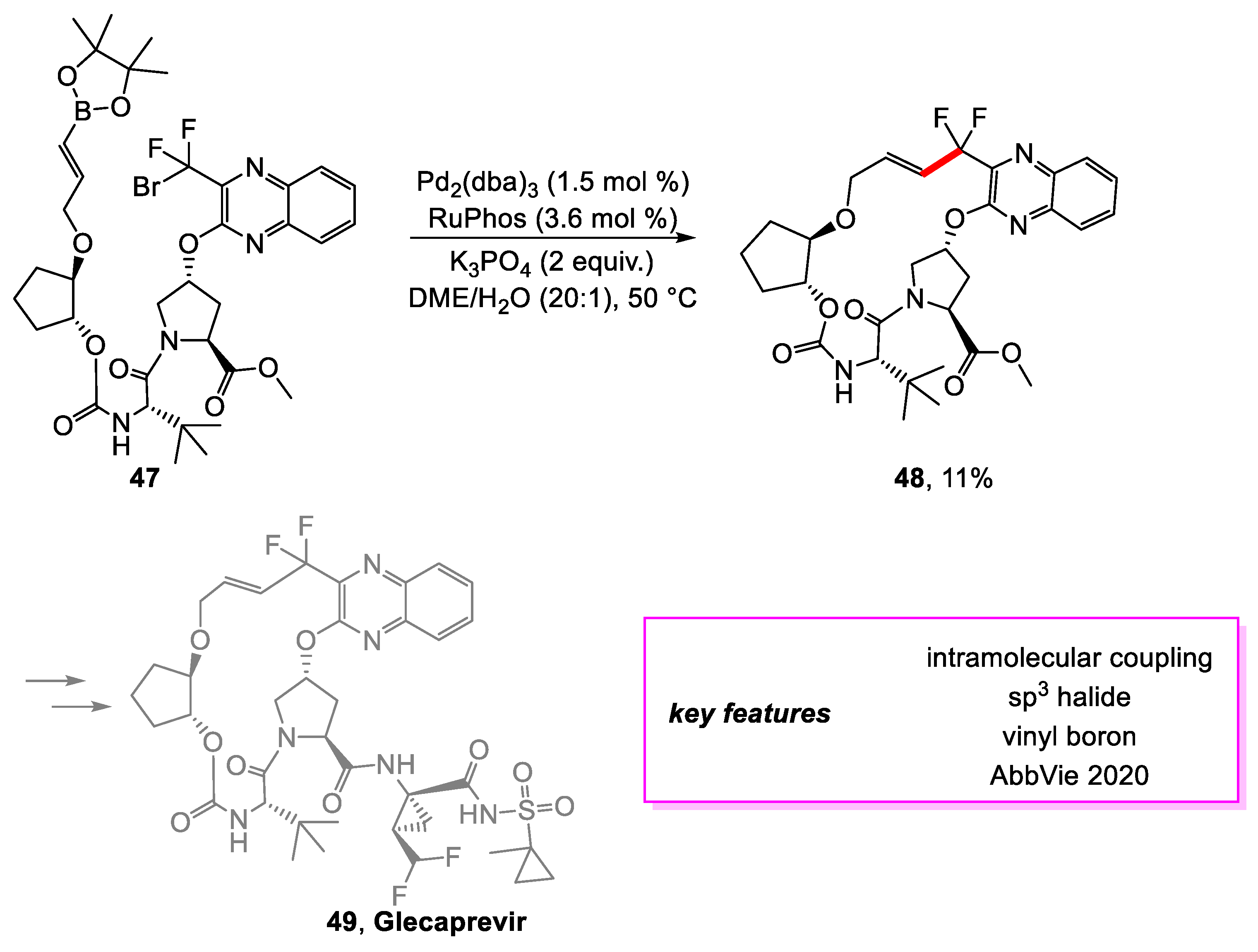
Kallemeyn and coworkers [24] from AbbVie set out to design a large-scale synthetic method for a macrocycle intermediate towards the synthesis of Glecaprevir [25], a potent hepatitis C virus (HCV) NS3/4A protease inhibitor. Two ring-closing options for the desired macrocycle were considered. The first uses a coupling reaction, and the second uses an etherification reaction. While exploring the first option, an extensive screening was conducted for a Suzuki–Miyaura coupling of 47, and the optimal conditions were determined. However, this method was only able to achieve an 11% yield of 48 due to the tendency of 48 to decompose rapidly due to the instability of the allylic difluoride functionality in the presence of palladium and ligand (Scheme 9). Therefore, it would be necessary to determine conditions for forming the C-C bond at a faster rate than the product’s decomposition. This option was abandoned as the instability of the coupling product proved to be a liability for large-scale production.
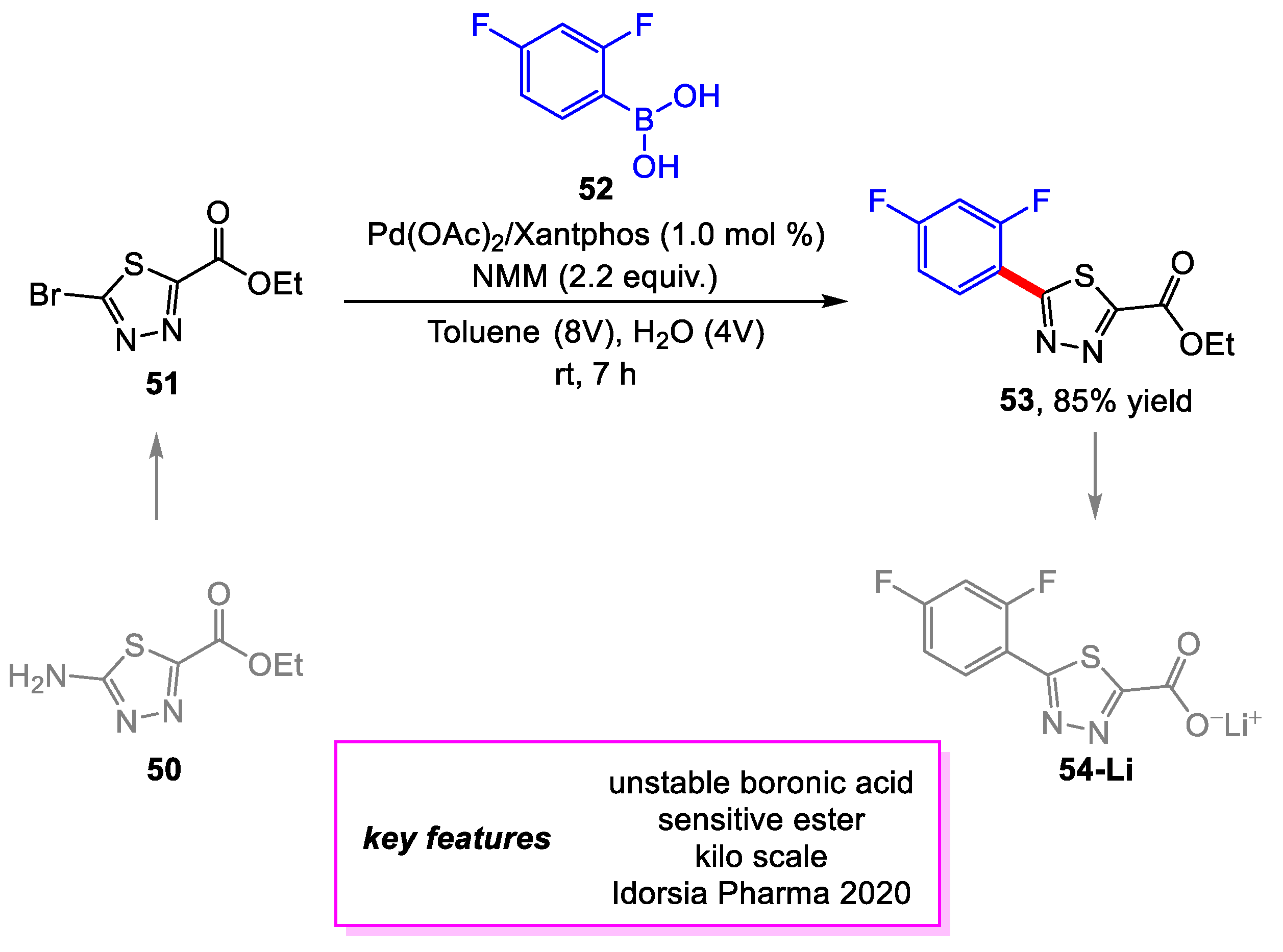
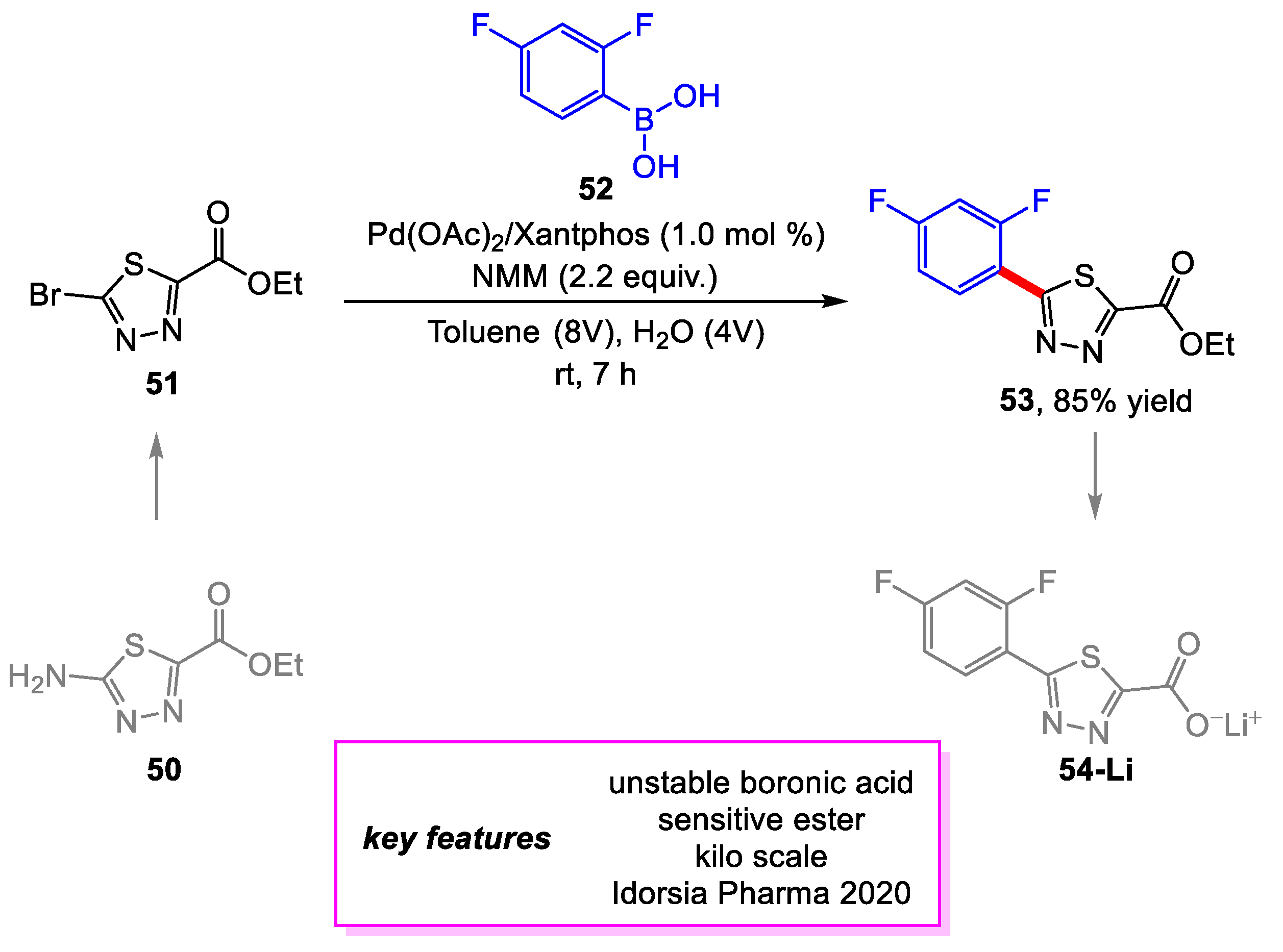
Schäfer and coworkers [26] from Idorsia Pharmaceuticals developed a scalable synthetic route for the thiadiazole building block 53 to be used for a project at Idorsia Pharmaceuticals. The first-generation synthesis of 53 used Lawesson’s reagent to produce the desired thiadiazole moiety, which hindered the scalability of the route. The new route starts with 50, a bench-stable compound with commercial availability on a kilogram scale. The final product was afforded following a Sandmeyer bromination and Suzuki–Miyaura coupling. Screening of the Suzuki–Miyaura coupling determined the optimal conditions for the reaction on a gram scale, which, notably, includes the ability to perform the reaction at room temperature. Upon scale-up to a 1.0 kg scale, it was discovered that a greater loading (3.0 mol%) of Pd(OAc)2/XantPhos and longer reaction time (24 h) was needed to achieve full conversion of the 51. This was due to time pressure for the project, which prevented performing full purification of the kg scale batch of 51 to remove residual Cu catalysts from the Sandmeyer bromination, in turn interfering with the Pd catalyst system. On the kg scale, a 61% yield was achieved for the Suzuki–Miyaura coupling, while with additional purification of 51, an 85% yield was achieved on a 200 g scale with the screening conditions (vide supra) (Scheme 10).
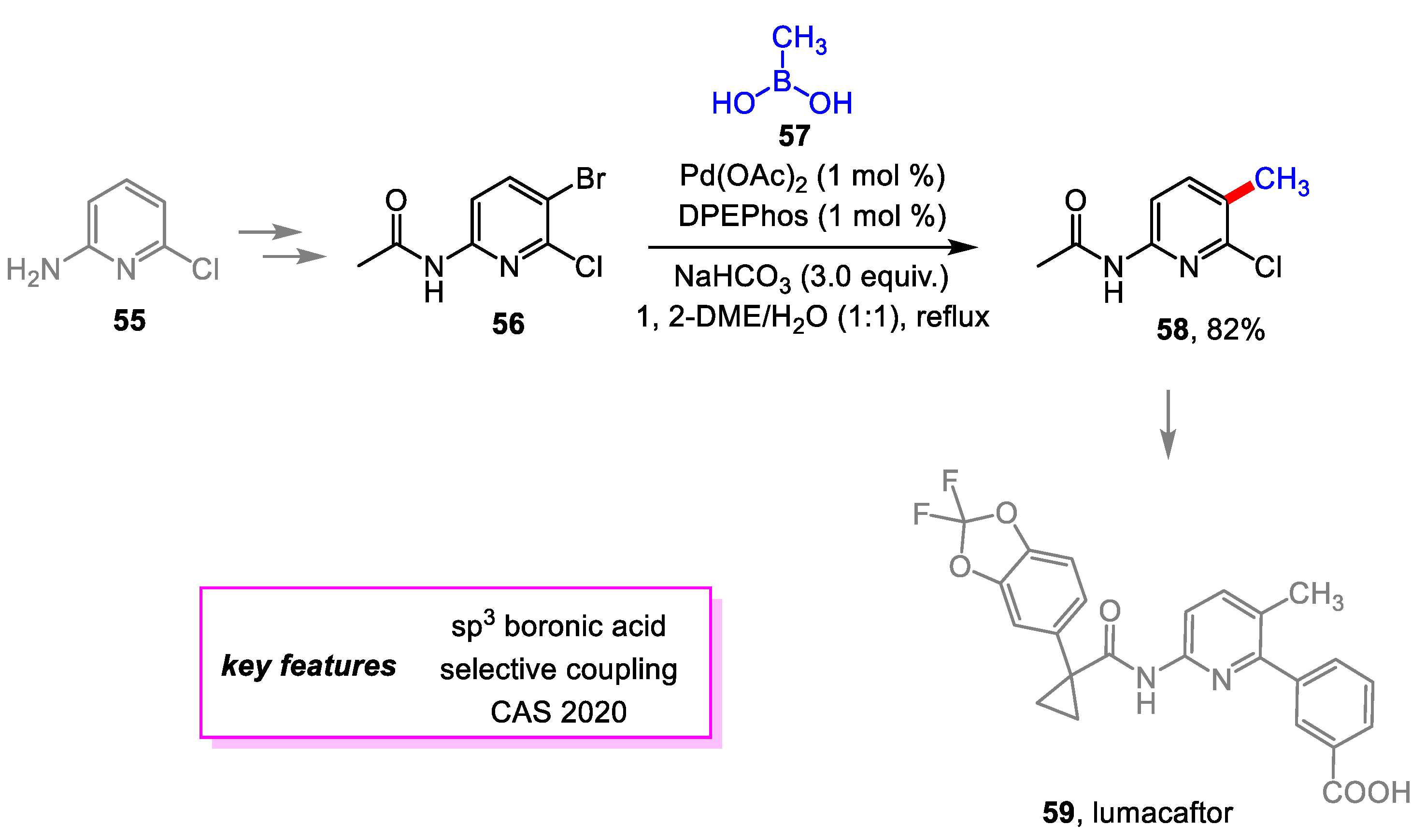
Zhang and coworkers [27] from the Chinese Academy of Science designed a novel method for synthesizing crucial intermediate to lumacaftor 59 [28], the active ingredient of treatment for cystic fibrosis, that increases the overall yield and decreases the environmental impact. Instead of introducing chlorines and amines like previous methods, the starting material 55 in the new route already contained the chlorine and amine in their proper positions. A Suzuki–Miyaura coupling is then used to introduce the 5-methyl group with regioselectivity. An extensive screening was conducted to determine the optimal conditions for minimizing impurities as a result of hydrolysis of acetyl groups on 56 and 58 or debromination of 58. The coupling step achieved an 82% yield on a scale of over 300 g (Scheme 11).
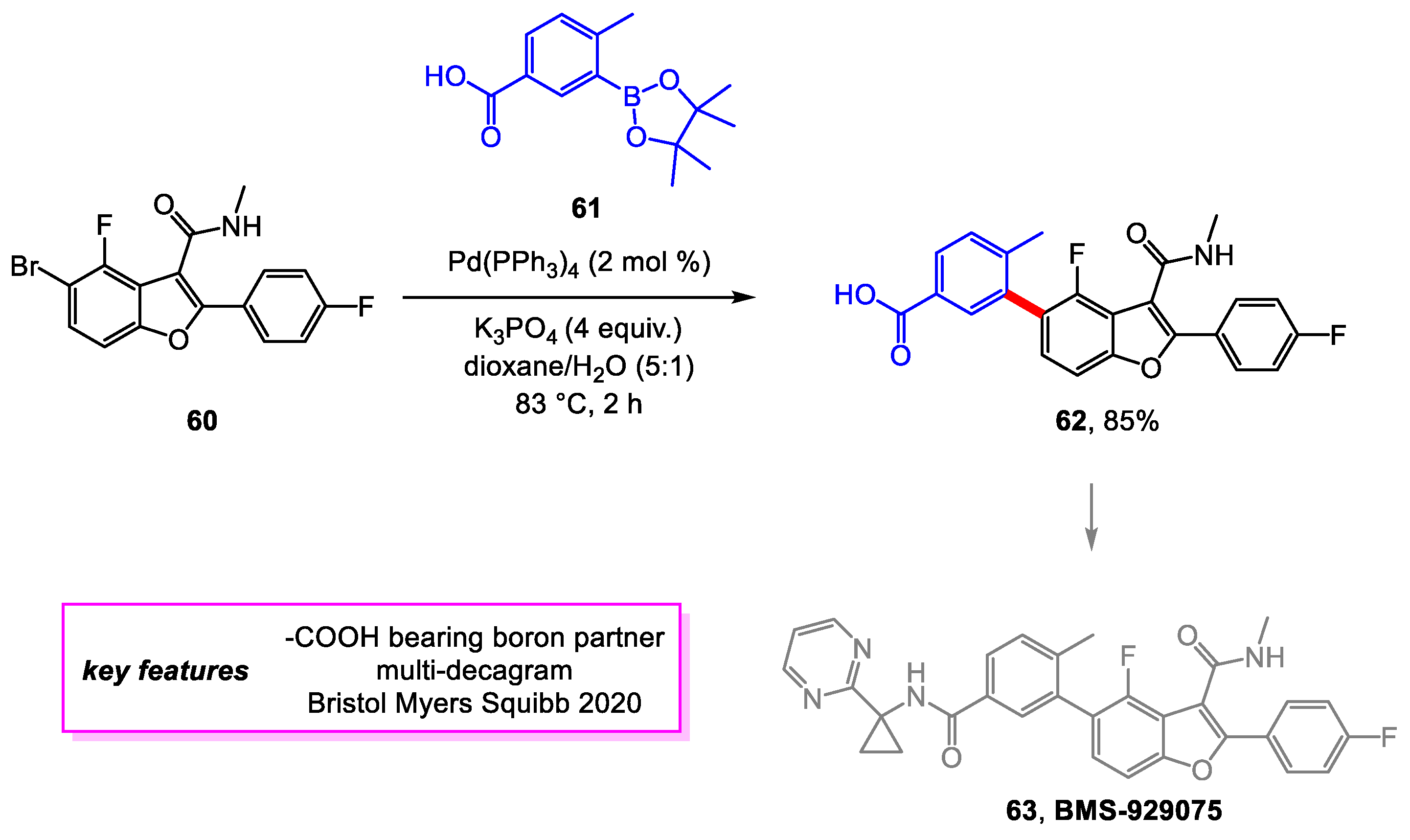
Smith and coworkers [29] from Bristol Myers Squibb developed a scalable synthetic method for BMS-929075, 63, a therapy drug in clinical trials for the hepatitis C virus infection [30]. Among the improvements was a search for an alternate coupling partner to 61 that would elevate the yield of late-stage intermediate 62. Bromo benzofuran 60 was found to be an ideal coupling partner. The above Suzuki–Miyaura coupling produced intermediate 62 in an 85% yield, an increase from 58% of the previous method, on a decagram scale. Following an amide coupling, 62 is converted to BMS-929075, 63. The overall yield for this route was 30%, producing 110 g of high-quality BMS-929075 (Scheme 12).
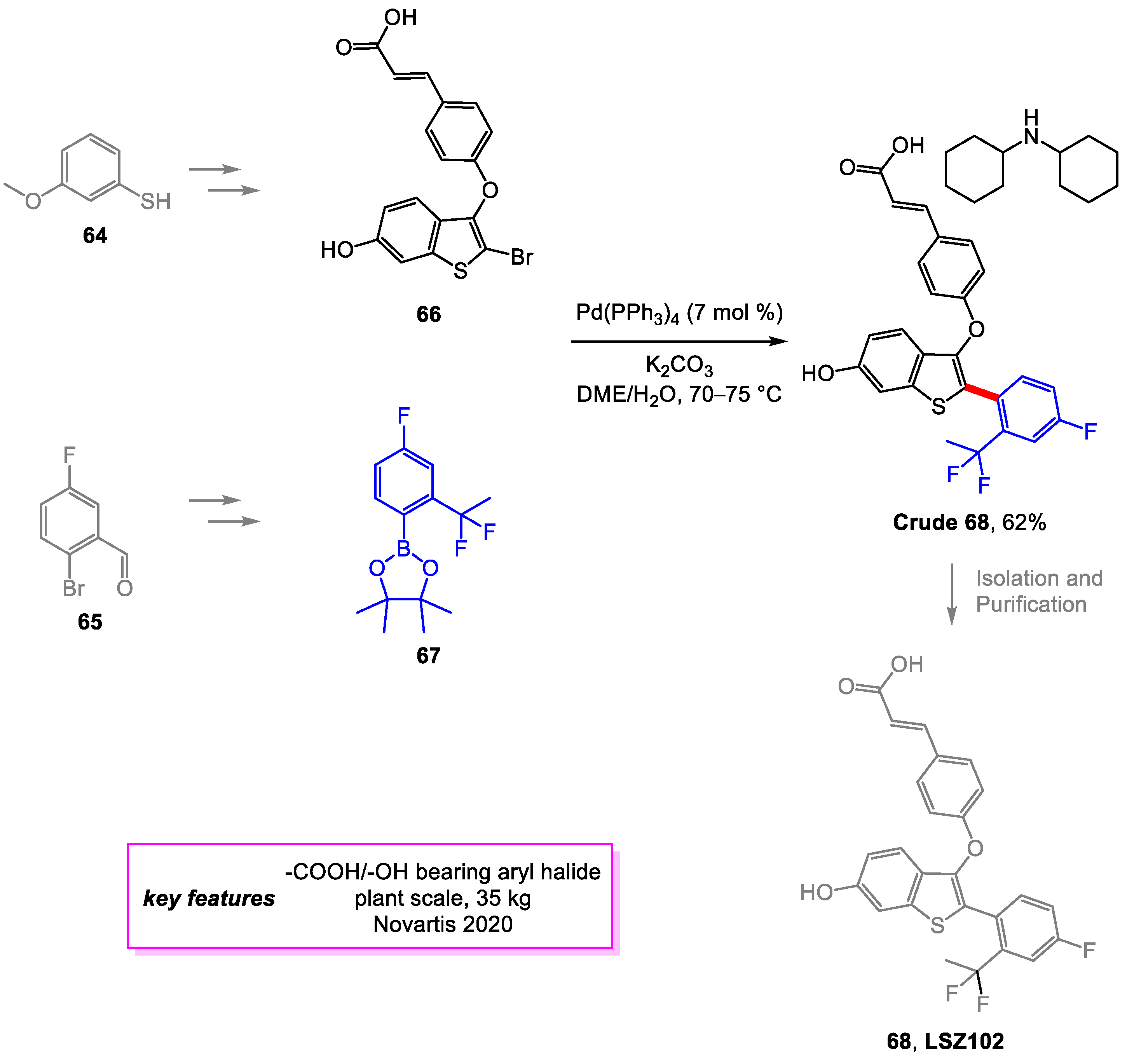
Baenziger and coworkers [31] from Novartis developed synthetic methods for streamlined large-scaled manufacturing of LSZ102 [32], a selective estrogen receptor degrader drug candidate for treating breast cancer. The new methods significantly improved the overall yield and reduced the cost. The important Suzuki–Miyaura coupling, also the final step in the original synthesis route, was optimized to minimize the formation of the main side product by reducing the temperature of the reaction and changing the solvent. The extensive screening was also conducted for a superior catalyst system, but none was found to perform better than the original. A 62% yield on this Suzuki–Miyaura coupling product 68 was achieved. Alternate routes to intermediates 66 and 67 were also described (Scheme 13).
Parmentier and coworkers [33] from Novartis developed a kilogram-scale protocol for the final step in the synthesis of LSZ102 [32], a drug candidate for treating breast cancer. The Suzuki–Miyaura coupling can now be carried out using the designer surfactant TPGS-750-M [34] to reduce the environmental impact and cost of this reaction. Less Palladium catalyst and non-toxic organic solvents are needed. Further, the use of acetone as a co-solvent and the addition of LiBr lowered the required excess loading of boronate ester 67 to achieve desirable conversion. The main impurity was also controlled effectively, reduced to <0.1% compared to the usual 7–8% found in the crude mixture. Notably, the palladium loading was also lowered to 1.5 mol% of PdCl2(dtbpf) from 7.5 mol% of Pd(PPh3)4 (Scheme 14).
Graham and coworkers [35] from AstraZeneca developed an improved manufacturing route to ceralasertib 71 [13], an ATR inhibitor currently tested as a drug for treating cancer. The new method features an improved Suzuki–Miyaura coupling between 70 and 16 that simplifies the workup and crystallization required by simply performing a solvent swap to 2-methyltetrahydrofuran (2-MeTHF). This Suzuki–Miyaura coupling and crystallization process achieved a 70% yield of ceralasertib with great control of residual Palladium (<3 ppm in all batches) and impurities on a scale of 32 kg. The overall yield of this route was 16% (Scheme 15).
Xie and coworkers [36] from Sunshine Lake Pharma developed a new synthetic process for emitasvir (DAG181) 76, a novel hepatitis C treatment drug that can be applied to the kilogram scale and increases the overall yield of the target active pharmaceutical ingredients from 17 to 40%. The above Suzuki–Miyaura coupling reaction produces the important intermediate 74 in the synthesis route and a side product 75, which is easily converted to the desired hydrolysis product 75 via the addition of K2CO3. Extensive screening of conditions showed that mild bases provided better conversion than strong bases due to the tendency of strong bases to hydrolyze 72 into a compound that could not participate in the Suzuki–Miyaura coupling. The effective yield (74 + 75) was 91%. Similar conditions are used for a later Suzuki–Miyaura coupling, with the only exception being using K2CO3 instead of NaHCO3 as the base (Scheme 16).
3. Buchwald–Hartwig Coupling
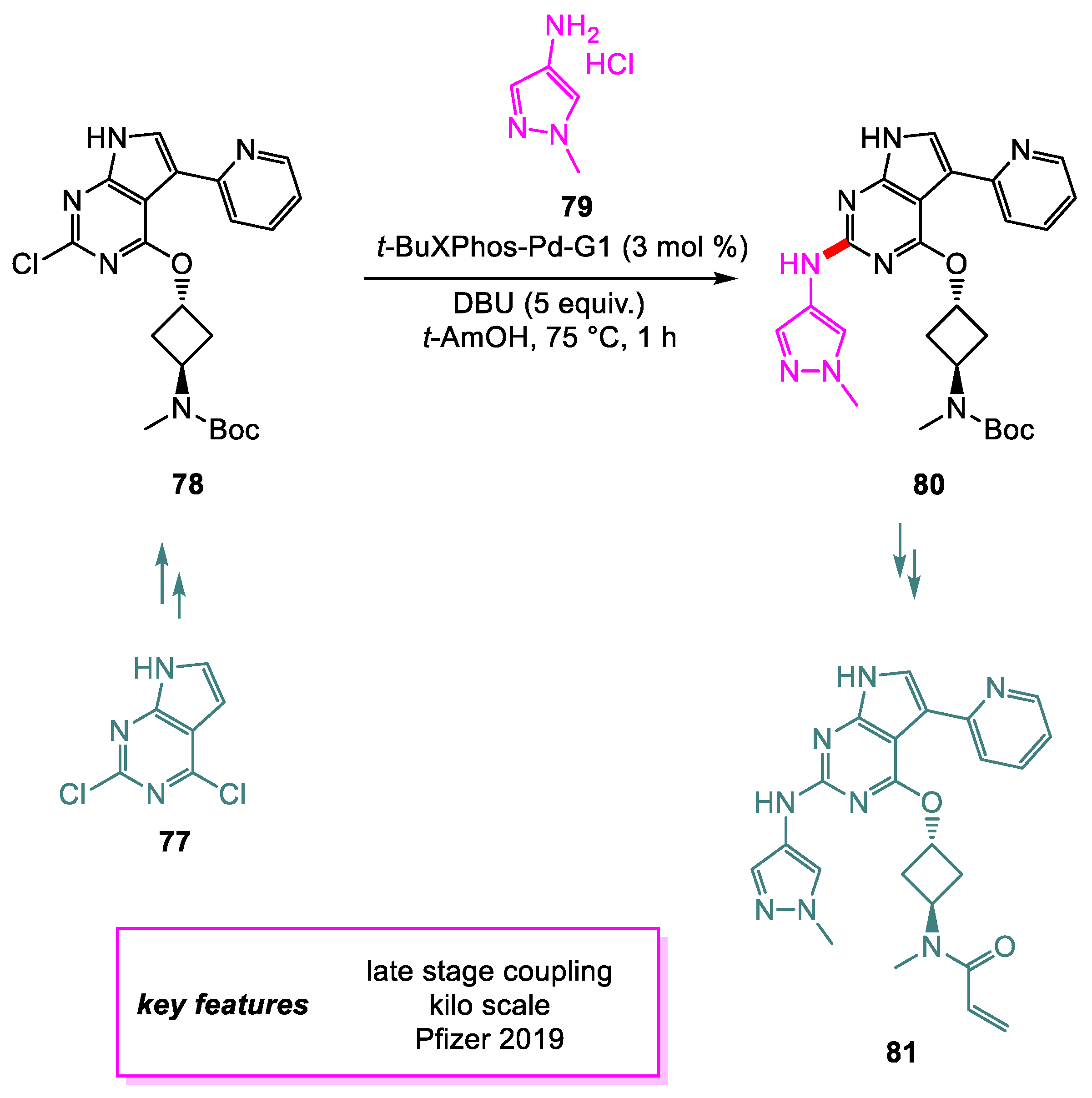
The amines are an integral part of pharmaceutical compounds [37]. Traditionally, such C(sp2)-N bonds are formed using copper-catalyzed Goldberg [38] and Ullmann processes [39] or SNAr (if the aryl halide is highly electrophilic). There are several pitfalls in these processes, especially poor substrate scope. These processes mostly fail to reproduce in making complex pharmaceuticals that bear several Lewis basic atoms (such as N, O, S, etc.). SNAr reactions require special activated reactants and, most of the time, polar aprotic solvents such as DMF. Although Migita discovered [40] the initial Pd catalyzed C-N bond formation, it was not greatly explored due to poor substrate scope and the use of toxic amino stannanes. In 1994, Buchwald [41] and Hartwig [42] independently reported Pd catalyzed C-N bond formation. This breakthrough and the subsequent utilization of this reaction led to a well-known name reaction “Buchwald–Hartwig amination”. The utility of this reaction is so broad that scientists can confidently forge the C-N bond at the late stage of the API synthesis. For instance, Tao and coworkers from Pfizer [43] have developed an improved synthetic route to Irreversible Epidermal Growth Factor Receptor (EGFR) T790 M [44] Inhibitor 81, a lead candidate for treating non-small cell lung cancer. A thorough small-scale screening of catalysts, bases, and solvents for the Buchwald–Hartwig Amination to form intermediate 80 was conducted. Out of four catalysts screened, t-BuXPhos Palladacycle was the only catalyst that achieved full conversion with few impurities. t-AmOH was shown to be the optimal solvent and DBU to be the optimal base for achieving full conversion in 2 h. Catalyst loading was lowered to 3 mol% upon final optimization, and full conversion of starting material was achieved in 1 h under a nitrogen atmosphere. A 75% isolated yield of 80 isopropanol solvate (1:1) was achieved with 94% UPLC purity on a hundred-gram scale (Scheme 17).
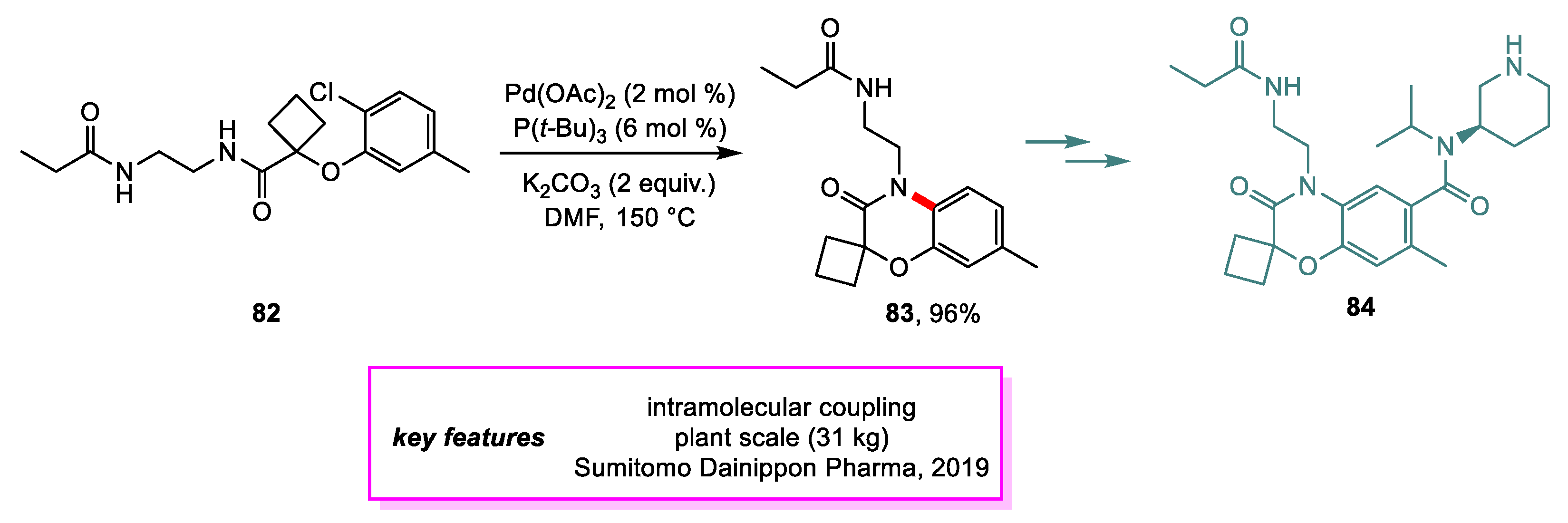
Nonoyama and coworkers from Sumitomo Dainippon Pharma [45] have developed several new routes to 84, a potential renin inhibitor and precursor to its more membrane-permeable prodrug, that improved upon the efficiency and cost of the discovery route. Plasma renin activity (PRA) plays an important role in the pathogenesis of hypertension, and inhibition of PRA can lower the risk of myocardial infarction in patients suffering from hypertension. In the second-generation route, the formation of the benzoxazine core was accomplished through an intermolecular Buchwald–Hartwig coupling. Extensive screening of reaction conditions was conducted to minimize the formation of side products while maximizing the conversion of 82. Pd(PtBu3)2 as a catalyst, K2CO3 as a base, and DMF as a solvent were selected. Upon scale-up, however, it was discovered that different lots of Pd(PtBu3)2 performed differently, so 5.0 mol% of catalyst was used instead of 2.5 mol% to prepare the needed material. This reaction achieved 6.4 kg of 83 in 75% isolated yield with >99% HPLC purity. The team then investigated the formation of Pd(Pt-Bu3)2 in situ to address the inconsistency of catalytic activity with different lots. Pd(OAc)2 and 3 Pd equiv. of Pt-Bu3 in xylene was found to achieve the optimal catalytic activity. A 96% yield of 83 on a multi-kilogram scale with >99% HPLC purity was achieved with this protocol (Scheme 18). However, future generations of synthetic routes were developed to improve the overall yield of 84 and address issues with other steps in this generation. The Buchwald–Hartwig coupling step was ultimately eliminated in favor of other ways to achieve the benzoxazine core.
Carroll and coworkers from Eli Lilly [46] have developed a synthetic route towards 89. It is an important intermediate for the synthesis of abemaciclib 90 (trade name Verzenio), a small-molecule CDK4/6 inhibitor approved for the treatment of hormone-receptor-positive, HER2-negative metastatic, or advanced-stage breast cancer as a monotherapy, in combination with fulvestrant, or a nonsteroidal aromatase inhibitor. The aim was primarily to avoid expensive starting material, harsh amination conditions, and many recrystallizations leading to low yield. Following optimizing the SN2 reaction to form 87, the Buchwald–Hartwig Amination to form 89 by using LiHMDS was explored to install a protected amine, preventing the formation of undesired impurities. It was found after the extensive screening that phosphine ligands with substituents in the ortho positions of the lower aryl ring performed better than those that lacked those substituents. RuPhos was the only such ligand screened that provided the desired conversion with lower loadings of palladium. A screening of the Pd(II) source with RuPhos showed that PdCl2(PhCN)2 was capable of providing complete conversion. RuPhos/PdCl2(PhCN)2 was selected over Pd-171 (Johnson Matthey precatalyst, RuPhos Pd(crotyl)Cl), which achieved full conversion at an even lower loading of palladium (0.25 mol%) due to the former’s availability. Following deprotection of 89, the respected 2-amino pyridine was produced on a kilogram scale with a 75% yield (Scheme 19).
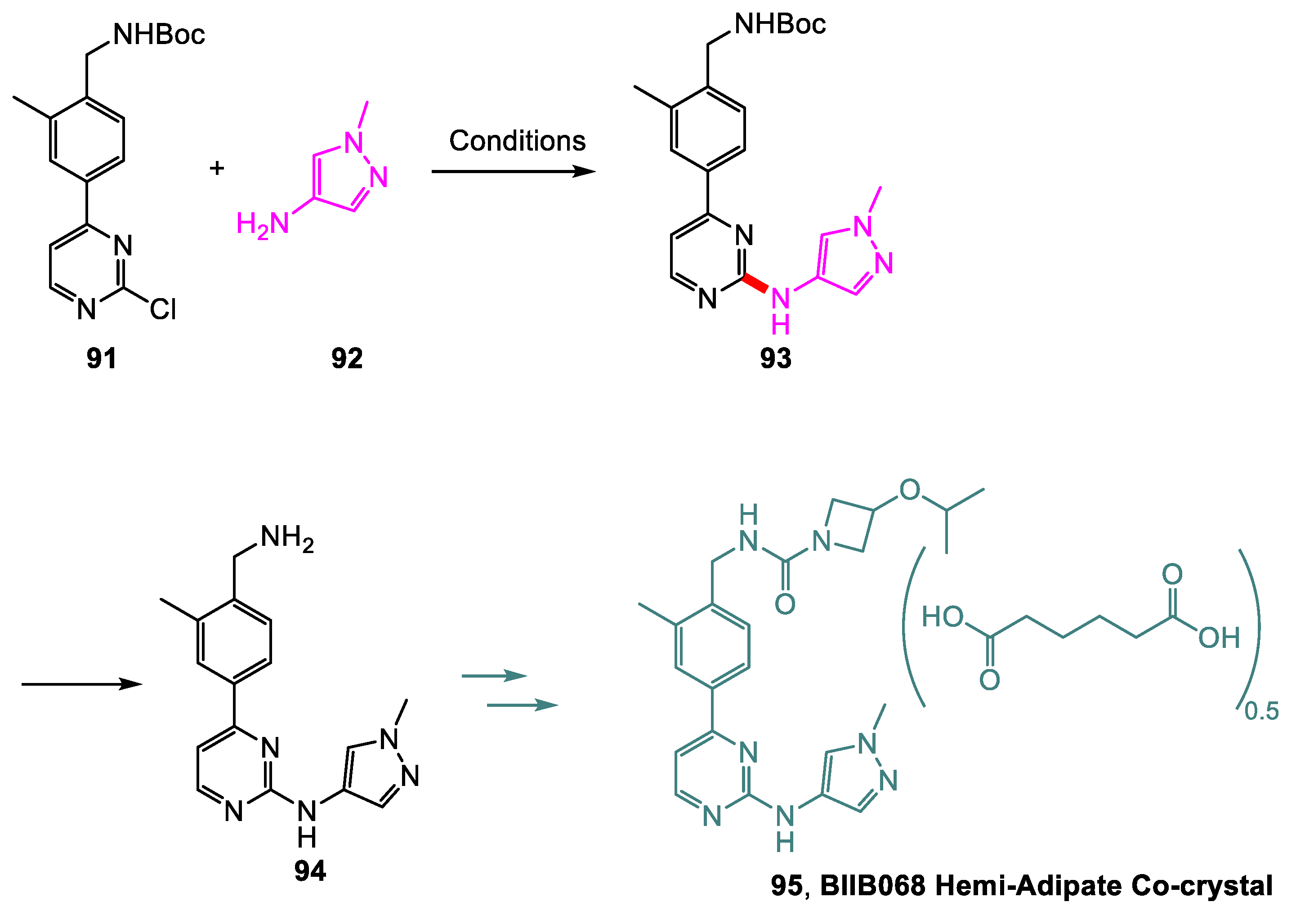
Li and coworkers [47] from Biogen have developed a large-scale synthetic method for BIIB068 95, a potent Bruton’s tyrosine kinase (BTK) that can provide therapy to patients with autoimmune diseases, as a Hemi-Adipate Co-crystal. The process development improved on various aspects of the discovery route, the first being the formation of the C-N bond between intermediates 91 and 92 (Scheme 20). The discovery route employed a Buchwald–Hartwig amination (same reagents as Table 1, entry 1) and carried out deprotection of 93 in a subsequent step following chromatography. After screening, it was discovered that neither the palladium catalyst nor 1,4-dioxane as a solvent was needed for the coupling of 91 with 92. Upon optimization, organic bases such as DIEA afforded 93 in ~60% yield (entry 2). However, it was also found that, under acidic conditions, the coupling reaction and the deprotection of 93 to form 94 can be carried out in one pot, and a 91% yield of 94 was achieved with 3 equiv. of H3PO4 (entry 3). Using the HCl salt of 92 instead of free base 92 was also examined, and it was discovered that the HCl generated from 92•HCl was sufficient to drive the deprotection reaction. Further optimization also revealed that using a mixture of 2-butanol and water as a solvent eliminated the butylated impurity. The optimal conditions achieved a 95% yield of 94 (entry 4).
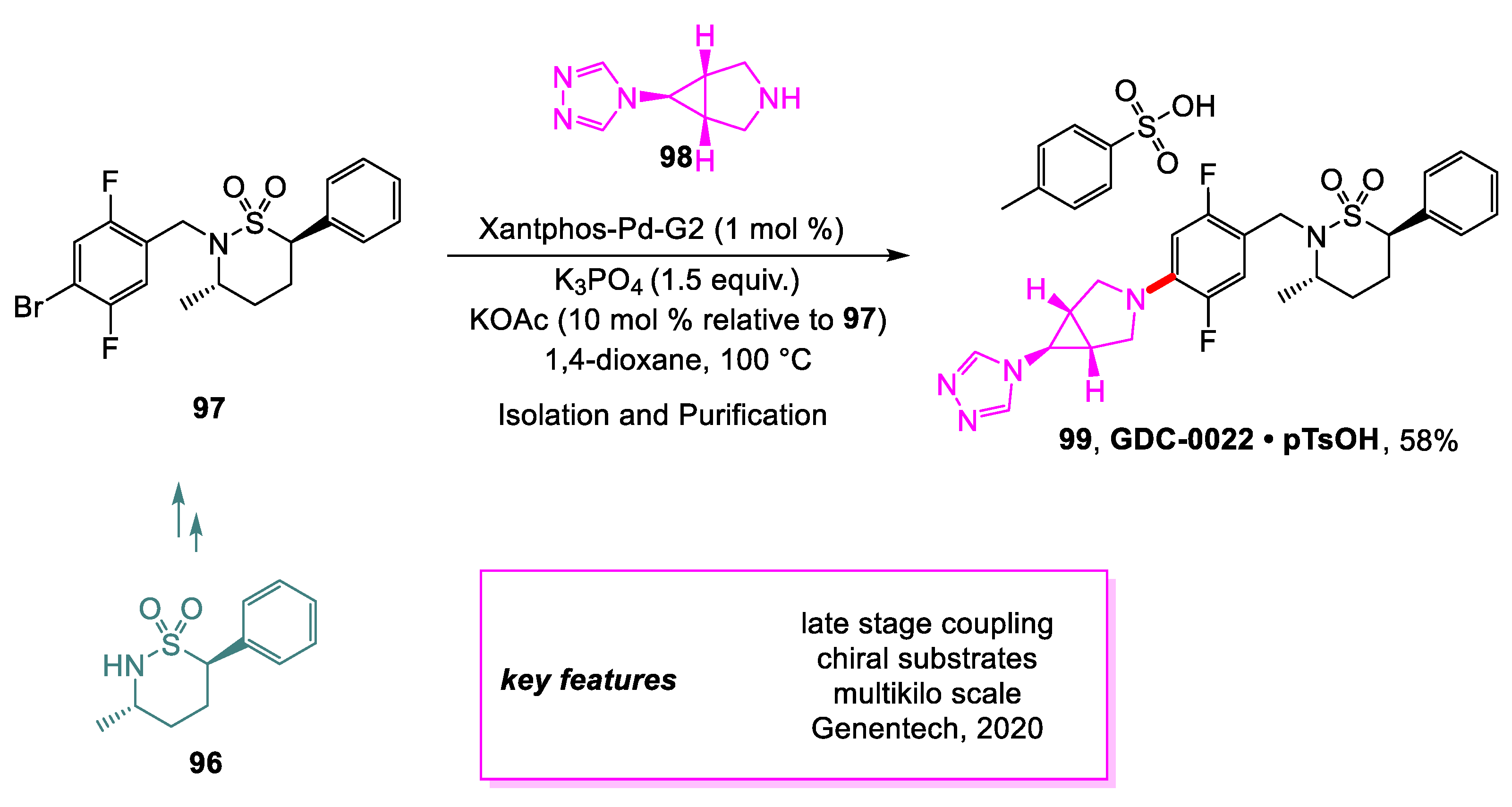
Sirois and coworkers [48] from Genentech have developed a method for producing drug candidate GDC-0022 99, an inhibitor of the Retinoid-related orphan receptor γ (RORc), on a multi-kilogram scale. One of the major problems that was addressed from the discovery synthesis route was improving the late-stage Buchwald–Hartwig amination between 97 and 98 to preserve the stereochemical purity of the sultam moiety while improving the overall conversion. Using free base 98 instead of its bis-HCl salt afforded less undesired diastereomer impurities. Extensive screening and optimization of the coupling conditions also improved the purity of the desired product and overall conversion. It was found that the addition of KOAc allowed the reaction to be performed with lower loadings of palladium. The behavior of the reaction was studied with reaction progress kinetic analysis with interesting findings regarding the role of KOAc and the effects of biphosphine monoxide ligands. The final yield of > 5 kg of GDC-0022•pTsOH 99 synthesized was 58% with >98 A% purity (Scheme 21).
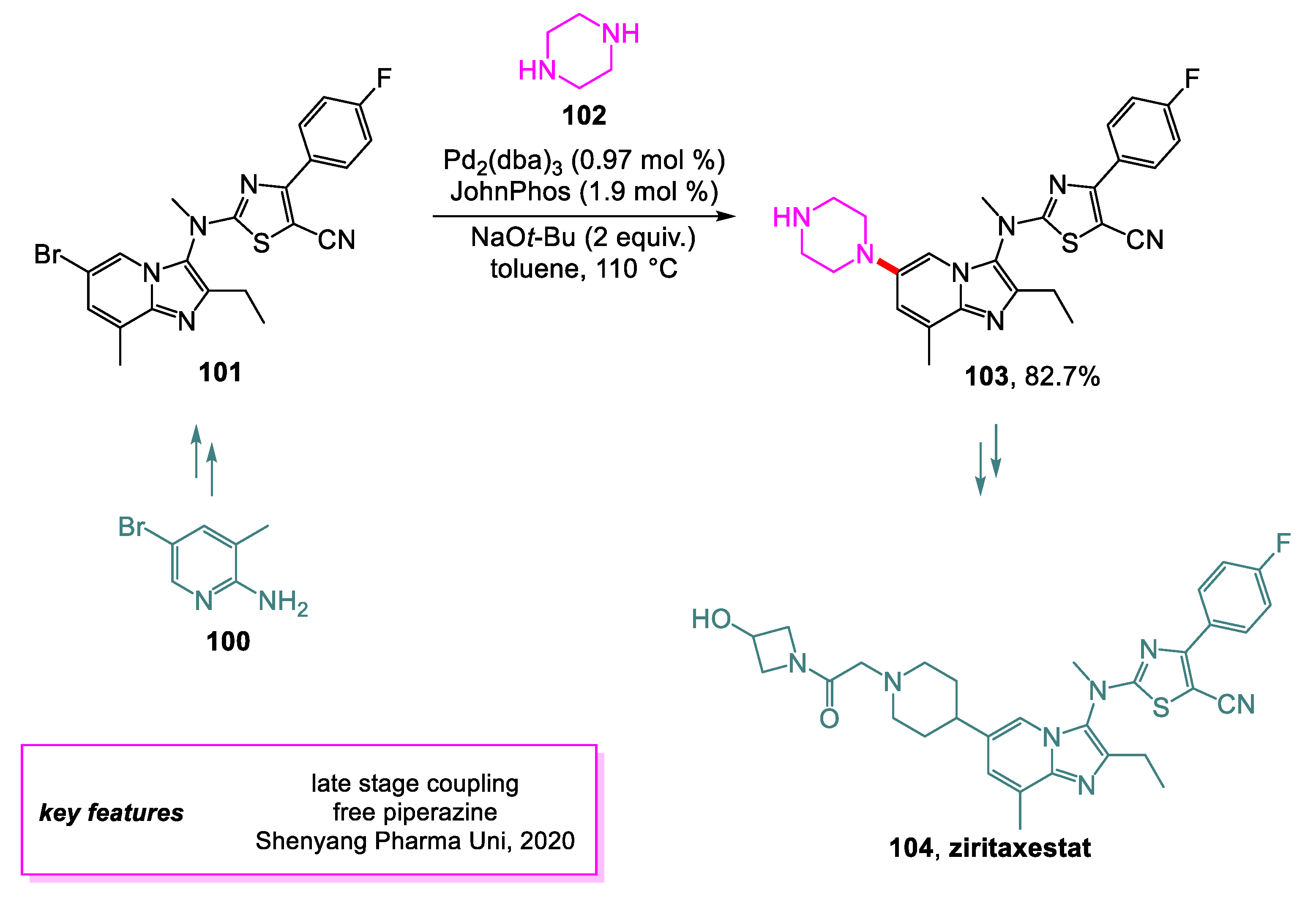
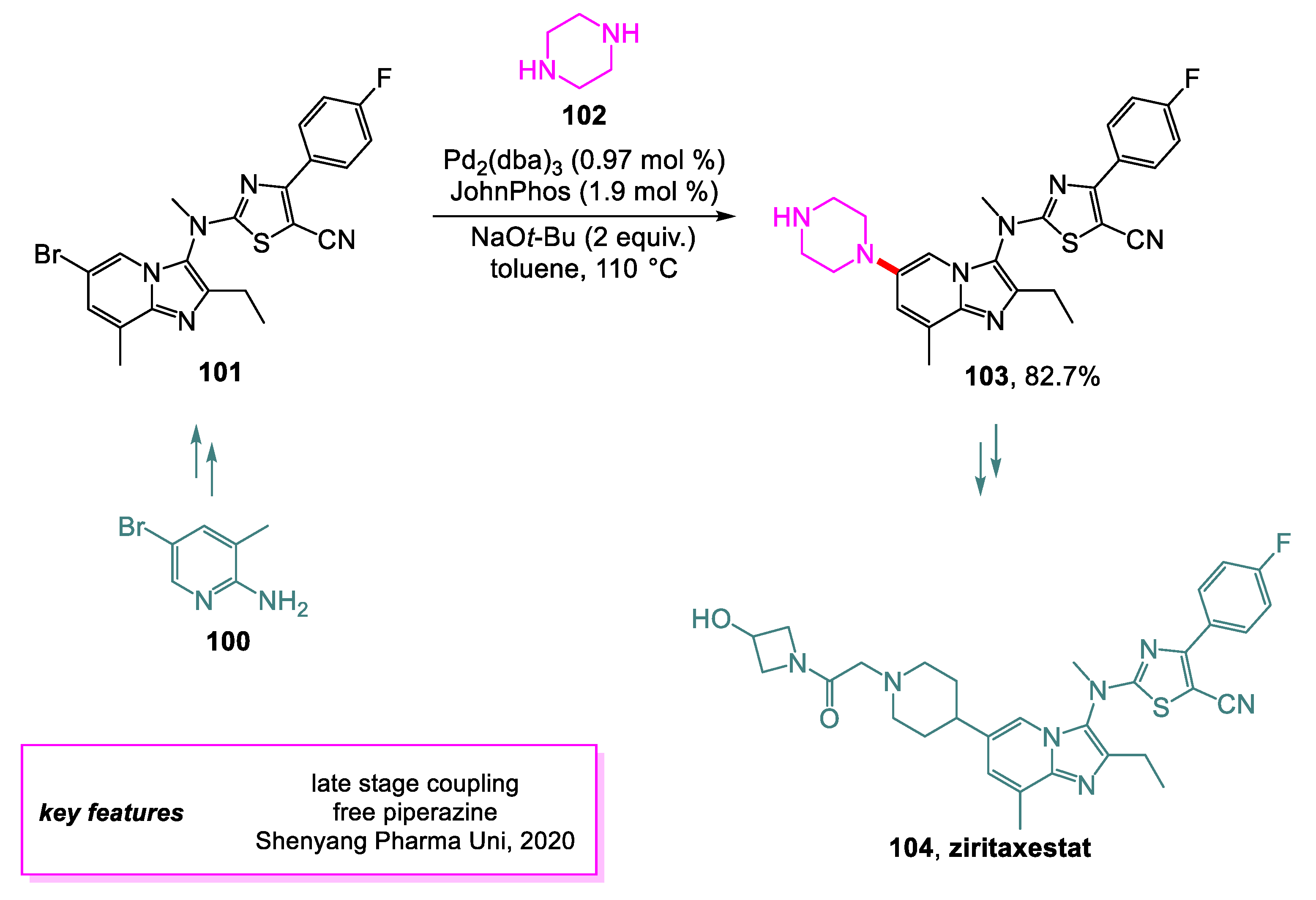
Lei and coworkers [49] from the Shenyang Pharmaceutical University have developed a novel method for producing autotaxin inhibitor ziritaxestat 104, a drug used to treat patients with idiopathic pulmonary fibrosis or chronic obstructive pulmonary diseases. Various aspects of the previous synthetic routes were improved upon, aiming to lower the cost of production. The Buchwald–Hartwig amination to achieve penultimate intermediate 101 was modified by using free piperazine 102 instead of N-Boc piperazine [50] to skip the deprotection step and improve the overall yield of 103. It was found that the original conditions for the coupling were optimal for the new coupling after extensive screening. This step of the new synthesis route could be achieved on a 300–400 g scale (Scheme 22).
4. Conclusions
With the advent of emerging cross-coupling technologies such as Suzuki–Miyaura and Buchwald–Hartwig reactions, pharmaceutical synthesis has become quickly possible. The vast studies performed to understand their mechanisms and reactivity support further development of ligands and catalyst for the couplings of highly complex molecules. Extending the applications of Pd-catalyzed cross coupling from medicinal chemistry to process chemistry has significantly reduced the associated time, cost, and waste in the synthesis of active pharmaceutical ingredients. As highlighted in this review, both cross-coupling reactions are the epitome of drug development, and their utility will only keep growing.
Author Contributions
Conceptualization, B.S.T. methodology, B.S.T., F.-Y.K. and R.R.T.; writing—original draft preparation, B.S.T., F.-Y.K. and R.R.T.; writing—review and editing, B.S.T., F.-Y.K. and R.R.T.; supervision, B.S.T. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
Fan-Yi Kong graciously acknowledges the Remote Alternative Independent Summer Experience program for financial support and Nicholas D. Ball for mentorship on this project.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Garrett, C.E.; Prasad, K. The Art of Meeting Palladium Specifications in Active Pharmaceutical Ingredients Produced by Pd-Catalyzed Reactions. Adv. Synth. Catal. 2004, 346, 889–900. [Google Scholar] [CrossRef]
- Phillips, S.; Holdsworth, D.; Kauppinen, P.; Mac Namara, C. Palladium Impurity Removal from Active Pharmaceutical Ingredient Process Streams. Johns. Matthey Technol. Rev. 2016, 60, 277–286. [Google Scholar] [CrossRef]
- Thakore, R.R.; Iyer, K.S.; Lipshutz, B.H. Sustainable Routes to Amines in Recyclable Water Using Ppm Pd Catalysis. Curr. Opin. Green Sustain. Chem. 2021, 31, 100493. [Google Scholar] [CrossRef]
- Takale, B.S.; Thakore, R.R.; Handa, S.; Gallou, F.; Reilly, J.; Lipshutz, B.H. A New, Substituted Palladacycle for Ppm Level Pd-Catalyzed Suzuki–Miyaura Cross Couplings in Water. Chem. Sci. 2019, 10, 8825–8831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phillips, S.; Kauppinen, P. Final Analysis: The Use of Metal Scavengers for Recovery of Palladium Catalyst from Solution. Platin. Met. Rev. 2010, 54, 69–70. [Google Scholar] [CrossRef]
- Johansson Seechurn, C.C.C.; Kitching, M.O.; Colacot, T.J.; Snieckus, V. Palladium-Catalyzed Cross-Coupling: A Historical Contextual Perspective to the 2010 Nobel Prize. Angew. Chem. Int. Ed. 2012, 51, 5062–5085. [Google Scholar] [CrossRef]
- St-Jean, F.; Remarchuk, T.; Angelaud, R.; Carrera, D.E.; Beaudry, D.; Malhotra, S.; McClory, A.; Kumar, A.; Ohlenbusch, G.; Schuster, A.M.; et al. Manufacture of the PI3K β-Sparing Inhibitor Taselisib. Part 2: Development of a Highly Efficient and Regioselective Late-Stage Process. Org. Process Res. Dev. 2019, 23, 783–793. [Google Scholar] [CrossRef]
- Ndubaku, C.O.; Heffron, T.P.; Staben, S.T.; Baumgardner, M.; Blaquiere, N.; Bradley, E.; Bull, R.; Do, S.; Dotson, J.; Dudley, D.; et al. Discovery of 2-{3-[2-(1-Isopropyl-3-Methyl-1H-1,2–4-Triazol-5-Yl)-5,6-Dihydrobenzo[f]Imidazo[1,2-d][1,4]Oxazepin-9-Yl]-1H-Pyrazol-1-Yl}-2-Methylpropanamide (GDC-0032): A β-Sparing Phosphoinositide 3-Kinase Inhibitor with High Unbound Exposure and Robust in Vivo Antitumor Activity. J. Med. Chem. 2013, 56, 4597–4610. [Google Scholar] [CrossRef] [PubMed]
- Biscoe, M.R.; Fors, B.P.; Buchwald, S.L. A New Class of Easily Activated Palladium Precatalysts for Facile C–N Cross-Coupling Reactions and the Low Temperature Oxidative Addition of Aryl Chlorides. J. Am. Chem. Soc. 2011, 133, 16707. [Google Scholar] [CrossRef]
- Akin, A.; Barilla, M.T.; Brandt, T.A.; Brennan, J.; Henegar, K.E.; Hoagland, S.; Kumar, R.; Magano, J.; McInturff, E.L.; Nematalla, A.; et al. Overcoming the Challenges of Making a Single Enantiomer N-1 Substituted Tetrazole Prodrug Using a Tin-Mediated Alkylation and Enzymatic Resolution. Org. Process Res. Dev. 2019, 23, 1167–1177. [Google Scholar] [CrossRef]
- McClure, K.F.; Piotrowski, D.W.; Petersen, D.; Wei, L.; Xiao, J.; Londregan, A.T.; Kamlet, A.S.; Dechert-Schmitt, A.-M.; Raymer, B.; Ruggeri, R.B.; et al. Liver-Targeted Small-Molecule Inhibitors of Proprotein Convertase Subtilisin/Kexin Type 9 Synthesis. Angew. Chem. Int. Ed. 2017, 56, 16218–16222. [Google Scholar] [CrossRef] [PubMed]
- Goundry, W.R.F.; Dai, K.; Gonzalez, M.; Legg, D.; O’Kearney-McMullan, A.; Morrison, J.; Stark, A.; Siedlecki, P.; Tomlin, P.; Yang, J. Development and Scale-up of a Route to ATR Inhibitor AZD6738. Org. Process Res. Dev. 2019, 23, 1333–1342. [Google Scholar] [CrossRef]
- Foote, K.M.; Nissink, J.W.M.; McGuire, T.; Turner, P.; Guichard, S.; Yates, J.W.T.; Lau, A.; Blades, K.; Heathcote, D.; Odedra, R.; et al. Discovery and Characterization of AZD6738, a Potent Inhibitor of Ataxia Telangiectasia Mutated and Rad3 Related (ATR) Kinase with Application as an Anticancer Agent. J. Med. Chem. 2018, 61, 9889–9907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gontcharov, A.; Magano, J.; Samp, L.; Houck, T.L.; Rose, P.R.; Rane, A.; Coe, J.W.; Kortum, S.W.; Chung, S.; Jones, P.; et al. Development of a Scalable Synthesis for an Inhaled Pan-JAK Inhibitor. Org. Process Res. Dev. 2019, 23, 1990–2000. [Google Scholar] [CrossRef]
- Jones, P.; Storer, R.I.; Sabnis, Y.A.; Wakenhut, F.M.; Whitlock, G.A.; England, K.S.; Mukaiyama, T.; Dehnhardt, C.M.; Coe, J.W.; Kortum, S.W.; et al. Design and Synthesis of a Pan-Janus Kinase Inhibitor Clinical Candidate (PF-06263276) Suitable for Inhaled and Topical Delivery for the Treatment of Inflammatory Diseases of the Lungs and Skin. J. Med. Chem. 2017, 60, 767–786. [Google Scholar] [CrossRef]
- Baenziger, M.; Pachinger, W.; Stauffer, F.; Zaugg, W. Development of a Robust Synthesis of Dactolisib on a Commercial Manufacturing Scale. Org. Process Res. Dev. 2019, 23, 1908–1917. [Google Scholar] [CrossRef]
- Maira, S.-M.; Stauffer, F.; Brueggen, J.; Furet, P.; Schnell, C.; Fritsch, C.; Brachmann, S.; Chène, P.; De Pover, A.; Schoemaker, K.; et al. Identification and Characterization of NVP-BEZ235, a New Orally Available Dual Phosphatidylinositol 3-Kinase/Mammalian Target of Rapamycin Inhibitor with Potent in Vivo Antitumor Activity. Mol. Cancer Ther. 2008, 7, 1851–1863. [Google Scholar] [CrossRef] [Green Version]
- Maltais, R.; Poirier, D. Development of a Gram-Scale Synthesis of PBRM, an Irreversible Inhibitor of 17β-Hydroxysteroid Dehydrogenase Type 1. Org. Process Res. Dev. 2019, 23, 2323–2335. [Google Scholar] [CrossRef]
- Maltais, R.; Ayan, D.; Trottier, A.; Barbeau, X.; Lagüe, P.; Bouchard, J.-E.; Poirier, D. Discovery of a Non-Estrogenic Irreversible Inhibitor of 17β-Hydroxysteroid Dehydrogenase Type 1 from 3-Substituted-16β-(m-Carbamoylbenzyl)-Estradiol Derivatives. J. Med. Chem. 2014, 57, 204–222. [Google Scholar] [CrossRef]
- Zhao, M.M.; Zhang, H.; Iimura, S.; Bednarz, M.S.; Kanamarlapudi, R.C.; Yan, J.; Lim, N.-K.; Wu, W. Process Development of Tryptophan Hydroxylase Inhibitor LX1031, a Drug Candidate for the Treatment of Irritable Bowel Syndrome. Org. Process Res. Dev. 2020, 24, 261–273. [Google Scholar] [CrossRef]
- Shi, Z.-C.; Devasagayaraj, A.; Gu, K.; Jin, H.; Marinelli, B.; Samala, L.; Scott, S.; Stouch, T.; Tunoori, A.; Wang, Y.; et al. Modulation of Peripheral Serotonin Levels by Novel Tryptophan Hydroxylase Inhibitors for the Potential Treatment of Functional Gastrointestinal Disorders. J. Med. Chem. 2008, 51, 3684–3687. [Google Scholar] [CrossRef]
- Hardouin, C.; Baillard, S.; Barière, F.; Copin, C.; Craquelin, A.; Janvier, S.; Lemaitre, S.; Le Roux, S.; Russo, O.; Samson, S. Multikilogram Synthesis of a Potent Dual Bcl-2/Bcl-x L Antagonist. 1. Manufacture of the Acid Moiety and Development of Some Key Reactions. Org. Process Res. Dev. 2020, 24, 652–669. [Google Scholar] [CrossRef]
- Leverson, J.D.; Phillips, D.C.; Mitten, M.J.; Boghaert, E.R.; Diaz, D.; Tahir, S.K.; Belmont, L.D.; Nimmer, P.; Xiao, Y.; Ma, X.M.; et al. Exploiting Selective BCL-2 Family Inhibitors to Dissect Cell Survival Dependencies and Define Improved Strategies for Cancer Therapy. Sci. Transl. Med. 2015, 7, ra40–ra279. [Google Scholar] [CrossRef] [PubMed]
- Kallemeyn, J.M.; Engstrom, K.M.; Pelc, M.J.; Lukin, K.A.; Morrill, W.H.; Wei, H.; Towne, T.B.; Henle, J.; Nere, N.K.; Welch, D.S.; et al. Development of a Large-Scale Route to Glecaprevir: Synthesis of the Macrocycle via Intramolecular Etherification. Org. Process Res. Dev. 2020, 24, 1373–1392. [Google Scholar] [CrossRef]
- Zeuzem, S.; Foster, G.R.; Wang, S.; Asatryan, A.; Gane, E.; Feld, J.J.; Asselah, T.; Bourlière, M.; Ruane, P.J.; Wedemeyer, H.; et al. Glecaprevir–Pibrentasvir for 8 or 12 Weeks in HCV Genotype 1 or 3 Infection. N. Engl. J. Med. 2018, 378, 354–369. [Google Scholar] [CrossRef] [PubMed]
- Schäfer, G.; Fleischer, T.; Ahmetovic, M.; Abele, S. Development of a Scalable Route for a Key Thiadiazole Building Block via Sequential Sandmeyer Bromination and Room-Temperature Suzuki–Miyaura Coupling. Org. Process Res. Dev. 2020, 24, 228–234. [Google Scholar] [CrossRef]
- Zhang, J.; Zhu, F.; Tian, G.; Jiang, X.; Shen, J. Improved Synthesis of 6-Chloro-5-Methylpyridin-2-Amine: A Key Intermediate for Making Lumacaftor. Org. Process Res. Dev. 2020, 24, 1175–1179. [Google Scholar] [CrossRef]
- Bulloch, M.N.; Hanna, C.; Giovane, R. Lumacaftor/Ivacaftor, a Novel Agent for the Treatment of Cystic Fibrosis Patients Who Are Homozygous for the F580del CFTR Mutation. Expert Rev. Clin. Pharmacol. 2017, 10, 1055–1072. [Google Scholar] [CrossRef]
- Smith, D.; Krishnananthan, S.; Meanwell, N.A.; Mathur, A.; Li, J. Multigram Synthesis of BMS-929075, an Allosteric, Palm Site Inhibitor of HCV NS5B Replicase, Involving the Synthesis of a Highly Functionalized Benzofuran through a Telescoped Process. Org. Process Res. Dev. 2020, 24, 1157–1163. [Google Scholar] [CrossRef]
- Yeung, K.-S.; Beno, B.R.; Parcella, K.; Bender, J.A.; Grant-Young, K.A.; Nickel, A.; Gunaga, P.; Anjanappa, P.; Bora, R.O.; Selvakumar, K.; et al. Discovery of a Hepatitis C Virus NS5B Replicase Palm Site Allosteric Inhibitor (BMS-929075) Advanced to Phase 1 Clinical Studies. J. Med. Chem. 2017, 60, 4369–4385. [Google Scholar] [CrossRef] [PubMed]
- Baenziger, M.; Baierl, M.; Devanathan, K.; Eswaran, S.; Fu, P.; Gschwend, B.; Haller, M.; Kasinathan, G.; Kovacic, N.; Langlois, A.; et al. Synthesis Development of the Selective Estrogen Receptor Degrader (SERD) LSZ102 from a Suzuki Coupling to a C–H Activation Strategy. Org. Process Res. Dev. 2020, 24, 1405–1419. [Google Scholar] [CrossRef]
- Tria, G.S.; Abrams, T.; Baird, J.; Burks, H.E.; Firestone, B.; Gaither, L.A.; Hamann, L.G.; He, G.; Kirby, C.A.; Kim, S.; et al. Discovery of LSZ102, a Potent, Orally Bioavailable Selective Estrogen Receptor Degrader (SERD) for the Treatment of Estrogen Receptor Positive Breast Cancer. J. Med. Chem. 2018, 61, 2837–2864. [Google Scholar] [CrossRef]
- Parmentier, M.; Wagner, M.; Wickendick, R.; Baenziger, M.; Langlois, A.; Gallou, F. A General Kilogram Scale Protocol for Suzuki–Miyaura Cross-Coupling in Water with TPGS-750-M Surfactant. Org. Process Res. Dev. 2020, 24, 1536–1542. [Google Scholar] [CrossRef]
- Lipshutz, B.H.; Ghorai, S.; Abela, A.R.; Moser, R.; Nishikata, T.; Duplais, C.; Krasovskiy, A.; Gaston, R.D.; Gadwood, R.C. TPGS-750-M: A Second-Generation Amphiphile for Metal-Catalyzed Cross-Couplings in Water at Room Temperature. J. Org. Chem. 2011, 76, 4379–4391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Graham, M.A.; Askey, H.; Campbell, A.D.; Chan, L.; Cooper, K.G.; Cui, Z.; Dalgleish, A.; Dave, D.; Ensor, G.; Galan Espinosa, M.R.; et al. Development and Scale-Up of an Improved Manufacturing Route to the ATR Inhibitor Ceralasertib. Org. Process Res. Dev. 2021, 25, 43–56. [Google Scholar] [CrossRef]
- Xie, H.; Liu, H.; Zhang, Y.; Huang, E.; Feng, Y.; Xiang, X.; Fang, Q.; Peng, Z.; Dong, W.; An, D. Development of a Synthesis Process for a Novel HCV NS5A Inhibitor, Emitasvir. Org. Process Res. Dev. 2021, 25, 838–848. [Google Scholar] [CrossRef]
- Schlummer, B.; Scholz, U. Palladium-Catalyzed C-N and C-O Coupling–A Practical Guide from an Industrial Vantage Point. Adv. Synth. Catal. 2004, 346, 1599–1626. [Google Scholar] [CrossRef]
- Ueber Phenylirungen bei Gegenwart von Kupfer als Katalysator—Goldberg—1906—Berichte der Deutschen Chemischen Gesellschaft—Wiley Online Library. Available online: https://chemistry-europe.onlinelibrary.wiley.com/doi/abs/10.1002/cber.19060390298 (accessed on 23 October 2021).
- Ullmann, F. Ueber Eine Neue Bildungsweise von Diphenylaminderivaten. Ber. Dtsch. Chem. Ges. 1903, 36, 2382–2384. [Google Scholar] [CrossRef] [Green Version]
- Kosugi, M.; Kameyama, M.; Migita, T. Palladium-Catalyzed Aromatic Amination of Aryl Bromides with n,n-Di-Ethylamino-Tributyltin. Chem. Lett. 1983, 12, 927–928. [Google Scholar] [CrossRef] [Green Version]
- Guram, A.S.; Buchwald, S.L. Palladium-Catalyzed Aromatic Aminations with in Situ Generated Aminostannanes. J. Am. Chem. Soc. 1994, 116, 7901–7902. [Google Scholar] [CrossRef]
- Paul, F.; Patt, J.; Hartwig, J.F. Palladium-Catalyzed Formation of Carbon-Nitrogen Bonds. Reaction Intermediates and Catalyst Improvements in the Hetero Cross-Coupling of Aryl Halides and Tin Amides. J. Am. Chem. Soc. 1994, 116, 5969–5970. [Google Scholar] [CrossRef]
- Tao, Y.; Keene, N.F.; Wiglesworth, K.E.; Sitter, B.; McWilliams, J.C. Early Process Development of an Irreversible Epidermal Growth Factor Receptor (EGFR) T790 M Inhibitor. Org. Process Res. Dev. 2019, 23, 382–388. [Google Scholar] [CrossRef]
- Cheng, H.; Nair, S.K.; Murray, B.W.; Almaden, C.; Bailey, S.; Baxi, S.; Behenna, D.; Cho-Schultz, S.; Dalvie, D.; Dinh, D.M.; et al. Discovery of 1-{(3R,4R)-3-[({5-Chloro-2-[(1-Methyl-1H-Pyrazol-4-Yl)Amino]-7H-Pyrrolo[2,3-d]Pyrimidin-4-Yl}oxy)Methyl]-4-Methoxypyrrolidin-1-Yl}prop-2-En-1-One (PF-06459988), a Potent, WT Sparing, Irreversible Inhibitor of T790M-Containing EGFR Mutants. J. Med. Chem. 2016, 59, 2005–2024. [Google Scholar] [CrossRef]
- Nonoyama, A.; Nakai, Y.; Lee, S.; Suzuki, S.; Ando, T.; Fukuda, N.; Tanaka, H.; Takahashi, K. Process Development of an Efficient and Cost-Effective Telescoping Route to a Key Synthetic Precursor for the Preparation of a Renin Inhibitor. Org. Process Res. Dev. 2019, 23, 499–511. [Google Scholar] [CrossRef]
- Carroll, M.P.; Moloney, H.; Gowran, O.; O’Connor, A.; Wilson, E.M.; Murray, M.M.; Pietz, M.A.; Kjell, D.P.; Held, C.B.; Frederick, M.O. Development of an Improved Route for the Synthesis of an Abemaciclib Intermediate. Org. Process Res. Dev. 2019, 23, 2549–2555. [Google Scholar] [CrossRef]
- Li, C.; Franklin, L.; Chen, R.; Mack, T.; Humora, M.; Ma, B.; Hopkins, B.T.; Guzowski, J.; Zheng, F.; MacPhee, M.; et al. Process Development and Large-Scale Synthesis of BTK Inhibitor BIIB068. Org. Process Res. Dev. 2020, 24, 1199–1206. [Google Scholar] [CrossRef]
- Sirois, L.E.; Lao, D.; Xu, J.; Angelaud, R.; Tso, J.; Scott, B.; Chakravarty, P.; Malhotra, S.; Gosselin, F. Process Development Overcomes a Challenging Pd-Catalyzed C–N Coupling for the Synthesis of RORc Inhibitor GDC-0022. Org. Process Res. Dev. 2020, 24, 567–578. [Google Scholar] [CrossRef]
- Lei, H.; Yang, Y.; Li, C.; Jia, F.; Jiang, N.; Gong, P.; Zhai, X. Catalyst-Free Cyclization- and Curtius Rearrangement-Induced Functional Group Transformation: An Improved Synthetic Strategy of First-in-Class ATX Inhibitor Ziritaxestat (GLPG-1690). Org. Process Res. Dev. 2020, 24, 997–1005. [Google Scholar] [CrossRef]
- Joncour, A.; Desroy, N.; Housseman, C.; Bock, X.; Bienvenu, N.; Cherel, L.; Labeguere, V.; Peixoto, C.; Annoot, D.; Lepissier, L.; et al. Discovery, Structure–Activity Relationship, and Binding Mode of an Imidazo[1,2-a]Pyridine Series of Autotaxin Inhibitors. J. Med. Chem. 2017, 60, 7371–7392. [Google Scholar] [CrossRef]
Figure 1.
Structures of ligand and catalysts used in this review.
Scheme 1.
Synthesis of an intermediate en route to taselisib.
Scheme 2.
Synthesis of an intermediate en route to PF06815345.
Scheme 3.
Synthesis of AZD6738.
Scheme 4.
Synthesis of an intermediate en route to PF06263276.
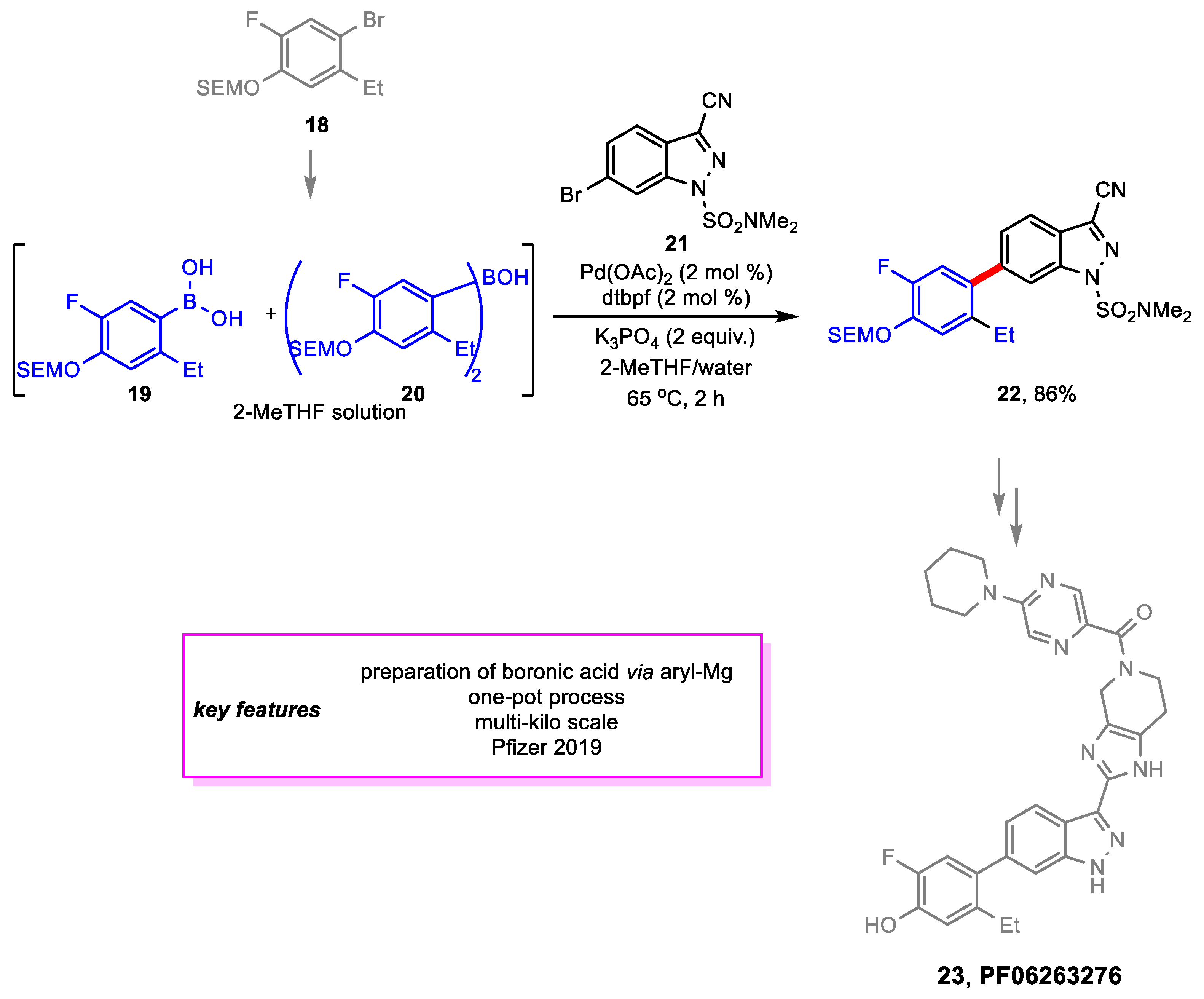
Scheme 5.
Synthesis of an intermediate en route to dactolisib.

Scheme 6.
Synthesis of an intermediate en route to PBRM.
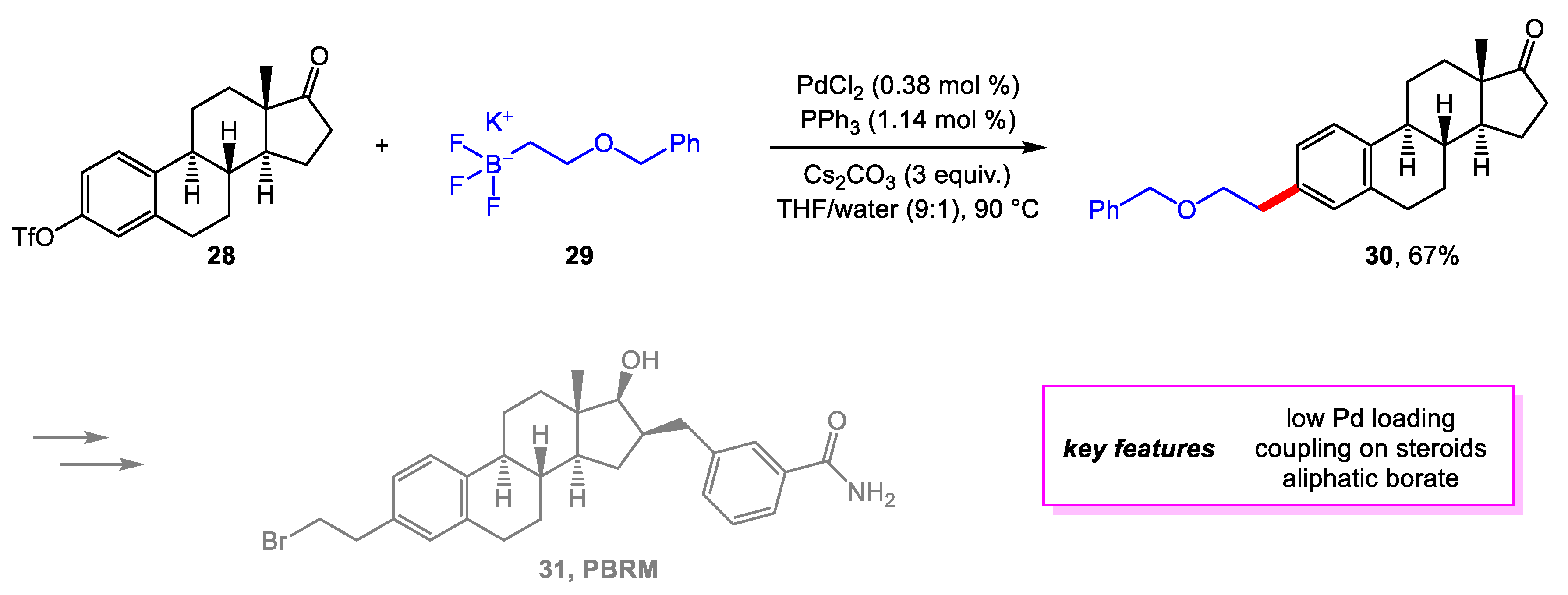
Scheme 7.
Synthesis of an intermediate en route to LX1031.
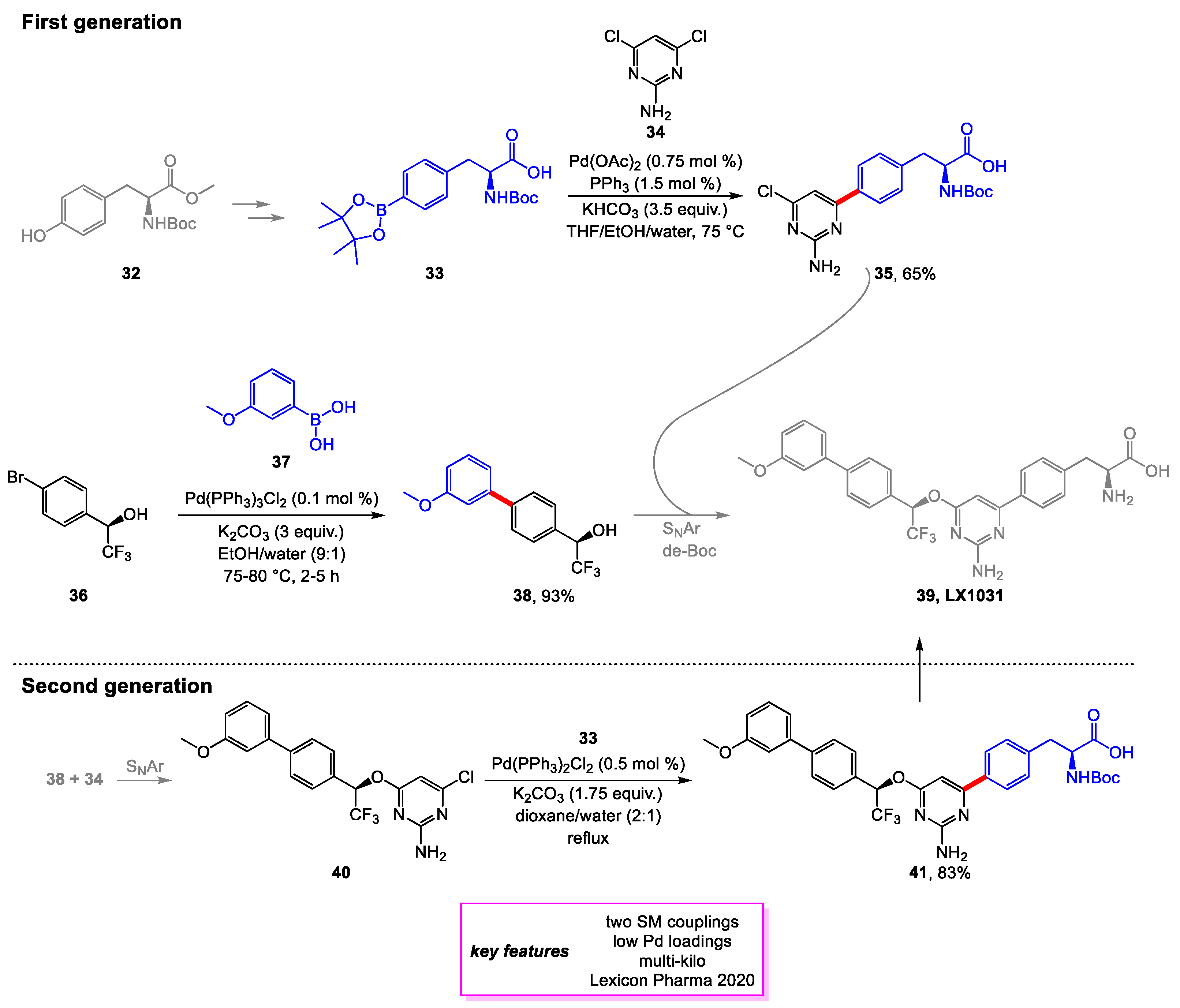
Scheme 8.
Synthesis of an intermediate en route to BCL inhibitor.
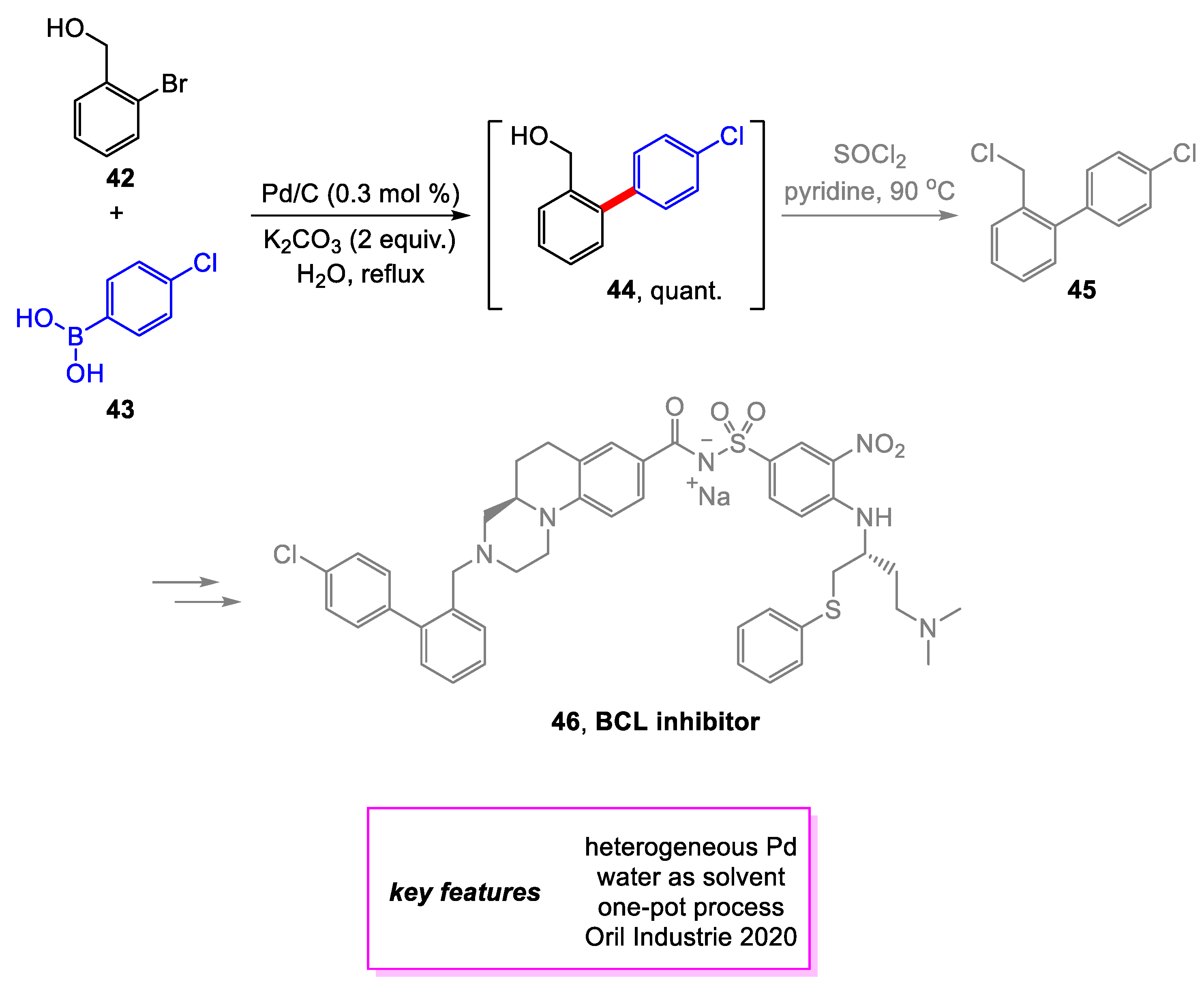
Scheme 9.
Synthesis of an intermediate en route to glecaprevir.
Scheme 10.
Synthesis of thiadiazole intermediate.
Scheme 11.
Synthesis of an intermediate en route to lumacaftor.
Scheme 12.
Synthesis of an intermediate en route to BMS-929075.
Scheme 13.
Synthesis of LSZ102.
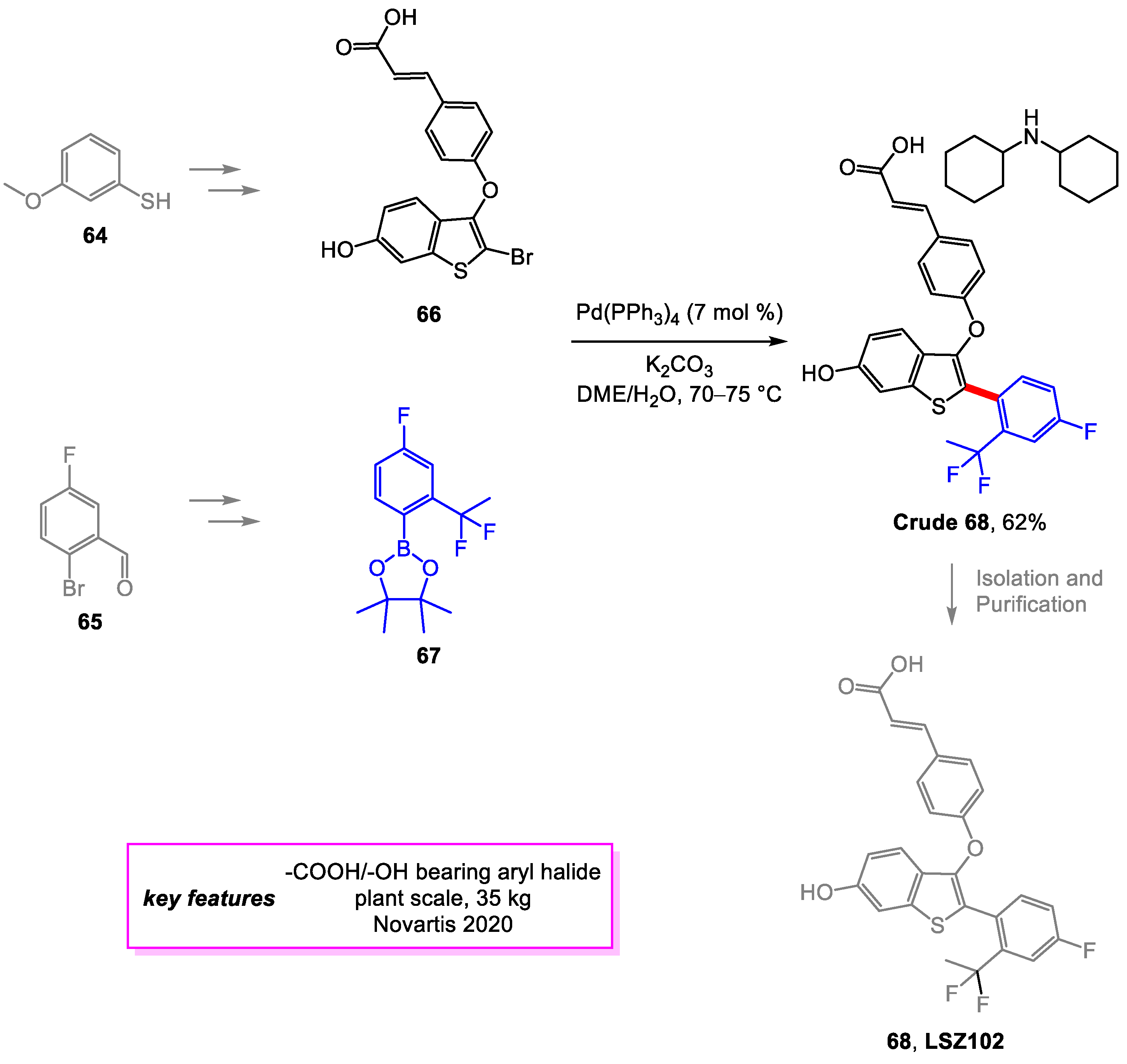
Scheme 14.
Synthesis of LSZ102 in aqueous surfactant.
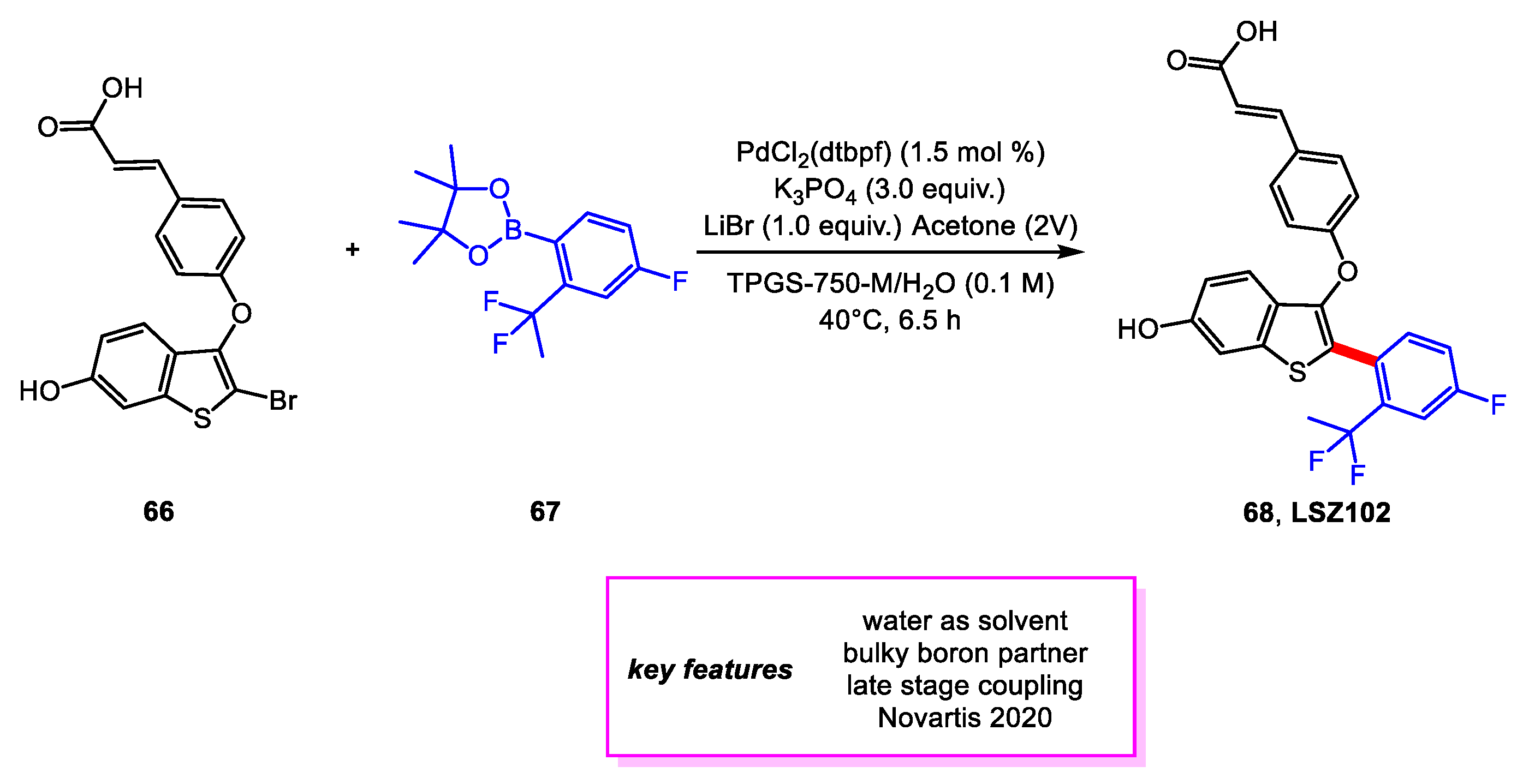
Scheme 15.
Synthesis of ceralasertib.
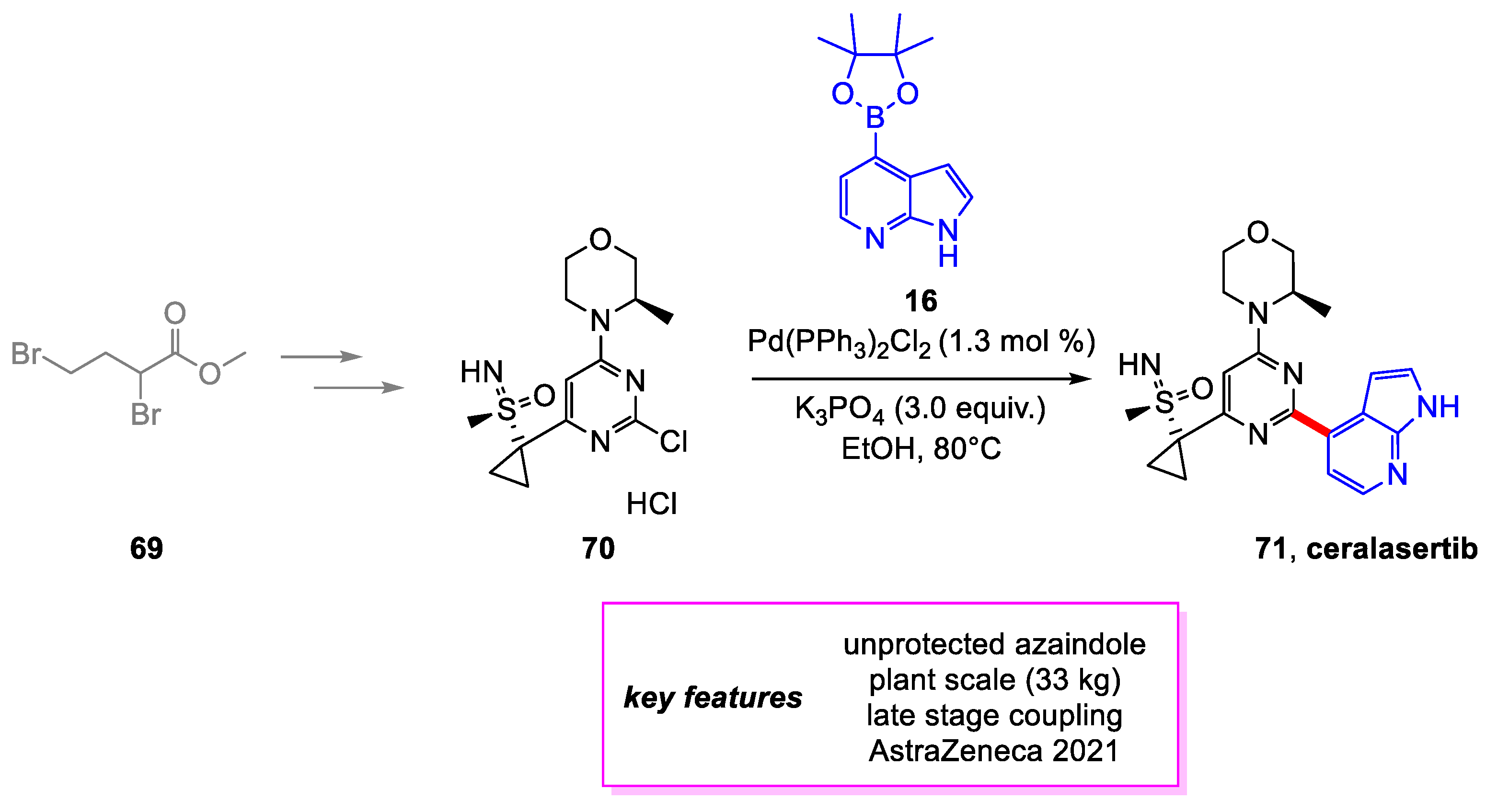
Scheme 16.
Synthesis of an intermediate en route to emitasvir.

Scheme 17.
Synthesis of an intermediate en route to T790 M.
Scheme 18.
Synthesis of an intermediate en route to 84.

Scheme 19.
Synthesis of an intermediate en route to abemaciclib.

Scheme 20.
Synthesis of an intermediate en route to BIIB068.
Scheme 21.
Synthesis of an intermediate en route to GDC-0022.

Scheme 22.
Synthesis of an intermediate en route to ziritaxestat.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Selected screening results for the C-N coupling of 91 with 92.
| Yields (HPLC Area %) | |||||
|---|---|---|---|---|---|
| Entry | Reagents | Solvent | T (°C) | 93 | 94 |
| 1 | Pd2(dba)3 (0.2 equiv.), S-Phos (0.1 equiv.), Cs2CO3 (2 equiv.) | 1,4-dioxane | 100 | 58 | 0 |
| 2 | DIEA (2 equiv.) | 1-butanol | 100 | 61 | <1 |
| 3 | H3PO4 (3 equiv.) | 1-butanol | 100 | 0 | 91 |
| 4 a | None | 2-butanol/water (2:3) | 85 | 0 | 95 |
a92•HCl was used instead of 92.
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Takale, B.S.; Kong, F.-Y.; Thakore, R.R. Recent Applications of Pd-Catalyzed Suzuki–Miyaura and Buchwald–Hartwig Couplings in Pharmaceutical Process Chemistry. Organics 2022, 3, 1-21. https://doi.org/10.3390/org3010001
AMA Style
Takale BS, Kong F-Y, Thakore RR. Recent Applications of Pd-Catalyzed Suzuki–Miyaura and Buchwald–Hartwig Couplings in Pharmaceutical Process Chemistry. Organics. 2022; 3(1):1-21. https://doi.org/10.3390/org3010001
Chicago/Turabian StyleTakale, Balaram S., Fan-Yi Kong, and Ruchita R. Thakore. 2022. "Recent Applications of Pd-Catalyzed Suzuki–Miyaura and Buchwald–Hartwig Couplings in Pharmaceutical Process Chemistry" Organics 3, no. 1: 1-21. https://doi.org/10.3390/org3010001