
Design, Synthesis, and Utility of Defined Molecular Scaffolds


Abstract
1. Introduction
2. Hub Molecules and Rational Chemistry
- Published within the past 10–20 years.
- Represent the characteristics of the hub in molecular design and synthesis.
- Have an elaborate molecular structure and corresponding graph.
- Are prepared by a distinctive (sometimes non-obvious) synthesis.
- Are prepared by selective (convergent), unique, and efficient synthetic methods.
- Have reliable experimental data such as synthetic yield and characterization.
3. Graph Theory in Design of Multifunctional Molecules
- Count all the edge-forming reaction steps of the graph. For a single hub, counting proceeds sequentially with each distinct (A, B, C, etc.) unit. An example is shown in Figure 5A. When there are ≥2 hubs, counting starts at the parent hub in linear sequential fashion. For a convergent synthesis step, wherein multiple identical groups become attached to one hub, the number of edge-forming reaction steps is tallied as one. If the edge has been formed in a commercially available building block, the counting for the edge is labeled as “0”. An example is shown in Figure 5B, which contains a scaffold composed of two hubs and A3BC substituents.
- Identify the parent hub. If the molecule contains ≥2 hubs, the number of edge-forming reaction steps starting at each hub is compared. The hub designated as the parent has the largest number of edge-forming reaction steps. If the steps are identical, four-arm hubs override three-arm hubs. If both the steps and the arm-numbers are identical, the parent hub is selected arbitrarily, considering the complexity of the molecular structures and the length of the synthetic process. An example is shown in Figure 5B, which contains two hubs in the scaffold, a three-arm and a four-arm hub. The three-arm hub is the parent because there are three distinct substituents (e.g., A, B and the four-arm hub) whereas the four-arm hub contains three identical substituents and the three-arm hub.
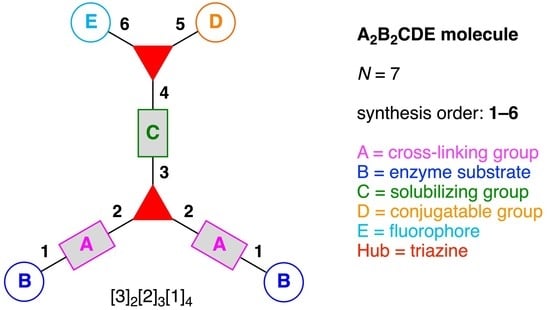
- Denote the synthesis order. The synthesis order is not unique to a particular tree but reflects the choices for a particular instantiation of a given tree to reach a particular target molecule. An example is provided in Figure 5C for the tree [3][2][1]3. Here, the three-arm hub is sequentially derivatized (Steps 1 and 2), the functional linker is derivatized with the functional unit Xd (Step 3), and the other terminus of the functional linker is joined to the third arm of the three-arm hub (Step 4). This terminology embodies the synthesis order, which for hubs, linkers, and functional units as molecular building blocks allows concise representation of a large quantity of information without expression of all the intricate details typical of a synthetic scheme.
- Assign the functional units. If the molecule contains ≥2 identical functional units, the units are named alphabetically in order of the number of the identical units. Thus, A2BC is preferred versus AB2C. If the number is identical, the units conjugated to the parent hub in the earliest step have the highest priority. For a subunit (a group of units), the unit with the least distance (edge number) from the parent hub has the highest priority. The example shown in Figure 6 is the tree with graph [3][2]3[1]3, functional units ABCDEF, and a synthesis that assembles A–E in a sequential manner to build the two arms of the three-arm hub and then attaches the functional unit F in the final step.
4. Solubilizing Groups
“The fact that other polyethers such as poly(oxymethylene) (–OCH2–)m, poly(oxytrimethylene) (–OCH2CH2CH2–)m and poly(oxypropylene) [–OCH2CH(CH3)–]m are insoluble in water implies that the distance between the neighboring ether oxygen atoms along the chain and the hydrophobicity and size of the alkylene spacer between the ether oxygens are important factors of the water solubility. As the spatial distance between the ether oxygens depends directly on the conformation of the polymer chain, the phase behavior of the poly(oxyethylene)–water system should be closely related to the poly(oxyethylene) chain conformation.”
5. Linear Functional Molecules, No Hubs (13)
6. Amino Acid and Peptide Hubs—General Features
7. Lysine Hubs (21)
8. Other Amino Acid Hubs (17)
9. Triazine Hubs (16)
- For a triazine that bears two identical substituents (X) and an alkylamino group (II), rotamers are not possible by symmetry (Figure 34B). Nonetheless, distinguishable signals in the NMR spectrum are possible because the two X substituents experience non-equivalent environments with respect to the orientation of the substituents on the attached nitrogen atom.
- For a triazine that bears two different substituents (X and Y) and one alkylamino group, a pair of rotamers (i.e., IIIa and IIIb) is possible (Figure 34C).
- For a triazine compound that bears two nonidentical alkylamino groups, four rotamers (IVa–IVd) are possible (Figure 34D).
- Possible tautomers IIa–c can in principle arise from Compound II (Figure 34E). Such tautomers are generally not detectable and ignored. Indeed, the tautomer equilibria between triazine and N-alkylimine species were excluded on the basis of different substituted triazines as reported in the literature. These types of equilibrium always shift towards triazine forms [201,204,205,206].
- Introduction of at least one amino group on the triazine hub is recommended. The amino group stabilizes the triazine hub and prevents undesired further nucleophilic substitution such as hydrolytic decomposition.
- The order of nucleophiles used toward cyanuric chloride should be as follows: alcohols first, phenols or thiols next, then anilines, and finally aliphatic amines.
- Alcohols and amines are preferred nucleophiles for the triazine hub. Building blocks containing hydroxy and amino groups are synthetically and commercially abundant. In particular, useful bifunctionalized linkers such as PEG linkers [41] are readily available with terminal alcohol and amine groups. Furthermore, unlike phenol- and aniline-based nucleophiles, aliphatic alcohols and amines do not contain a hydrophobic phenyl ring, which may impart water-solubility problems in biological applications.
- Cyanuric chloride (9a) undergoes successive substitution with an amine to give the monoamino (9a-7), diamino (9a-8), and triamino (9a-9) derivatives. A triaminotriazine (9a-9) is a representative triazine hub (Figure 35A, top). Each amino substitution reaction can be carried out in a straightforward manner by treating the chlorotriazine with the corresponding amine. However, base-labile functionalities could be damaged because the third amino substitution requires elevated temperature (typically >70 °C). If the first nucleophile is an alcohol, affording the monoalkoxy derivative (9a-10), the second substitution with an amine affords the monoalkoxy-monoamino derivative (9a-11), and a third substitution with an amine affords the monoalkoxy-diamino derivative (9a-12) (Figure 35A, middle). In this case, both the first alkoxy and second amino substitution occur at temperatures lower than room temperature. A general method for preparation of unsymmetric dialkoxychlorotriazines (9a-13) has not yet been established, precluding use of dialkoxy-aminotriazines (9a-14) as common hub designs (Figure 35A, bottom). Limited examples have adopted conditions at >100 °C for the second alkoxy substitution; the conditions entail 2,6-lutidine [207] or K2CO3/18-crown-6 [208] as a base, or solvolysis conditions in MeOH [209]. Note that primary alcohols are employed in Figure 35A. The alkoxy substitution with secondary alcohols proceeds slower than primary alcohols and thus was carried out under harsher heating conditions [210]. Tertiary alcohols generally require prior conversion to the corresponding metal alkoxide to react with cyanuric chloride [211].
- Substitution reactions of the chlorotriazine are typically carried out in the presence of an amine base. The most common bases are listed below along with the pKa value of the conjugate acid in water:The base strengths are solvent dependent; for example, the pKa value of 2,6-lutidine in H2O is 6.77 [213], whereas that in 50% EtOH-H2O is 5.77 [216]. Regardless, the trend shown here provides a general framework for selecting an appropriate base.The chloro-triazine substitutions are typically done in the presence of a hindered amine base such as Et3N and i-Pr2EtN. Since these tertiary amines are sufficiently basic, deactivation of the amine nucleophile by protonation with HCl is prevented, given that such protonation otherwise would occur during the chlorotriazine substitution. In contrast, Et3N is not employed for the cyanuric chloride substitution with an alcohol to prepare alkoxydichlorotriazines, due to a side reaction forming (N,N-diethyl)aminotriazine byproducts [217,218]. Instead, i-Pr2EtN is commonly used as the base to promote this alkoxy substitution at 0 °C, although the same type of side reactions could compete [210,219]. Alternatively, weaker bases such as 2,4,6-collidine [210] and 1,10-phenanthroline [207] can be used over the temperature range of 0 °C to room temperature to strictly suppress the side reaction forming the aminotriazine byproducts or the undesired second alkoxy substitution. The aromatic heterocyclic 1,10-phenanthroline is also an effective base for double-alkoxy substitution of cyanuric chloride to synthesize symmetric dialkoxychlorotriazines [209], although severe heating conditions (reflux in xylene) were employed. Recently, we introduced use of pempidine to this reaction [220]. Owing to the bulkiness and high basicity of this tertiary amine, the double-alkoxy substitution of cyanuric chloride proceeded at lower temperature (60 °C) without forming the (N,N-dialkyl)aminotriazine byproducts. Structures and the pKa values (in H2O) of amine bases employed in the chlorotriazine substitution are shown in Figure 35B.
- Rotamerism (vide supra) can present a nuisance for interpretation of NMR spectra, and the rotamers might potentially engender distinct features in biological systems. Rotamerism can be sidestepped through the use of cyclic amines. Thus, 4-substituted piperidinyl, 4-substituted piperazinyl, and 3-substituted azetidinyl groups are symmetric dialkylamino groups that do not give rise to triazine rotamers (Figure 35C). Such groups may serve as attractive linkers.
10. Benzene Hubs (11)
11. Nitrogen Hubs (12)
12. Carbon Hubs (5)
13. Functional Hubs (12)
14. Outlook
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AAZTA | 1,4-bis(carboxymethyl)-6-[bis(carboxymethyl)]amino-6-methylperhydro-1,4-diazepine |
| ADC | antibody–drug conjugate |
| 5-AVA | 5-aminovaleric acid |
| All | Allyl |
| Alloc | allyloxylcarbonyl |
| AMP | adenosine monophosphate |
| BBN | bombesin peptide |
| BHQ | black hole quencher |
| Bn | benzyl |
| BNCT | boron neutron capture therapy |
| Boc | tert-butyloxycarbonyl |
| BODIPY | boron-dipyrrin |
| BP | benzophenone |
| C2Am | C2A domain of synaptotagmin-I |
| CA | carbonic anhydrase |
| CBT | 2-cyanobenzothiazole |
| CDR | complementarity determining region |
| CES | carboxyesterase |
| Cha | cyclohexylalanine |
| Cit | citrulline |
| Cp* | pentamethylcyclopentadienyl |
| CSAN | chemically self-assembled nanoring |
| CT | computed tomography |
| CuAAC | copper-promoted [3 + 2]-azide–alkyne cycloaddition |
| Dabcyl | 4-dimethylaminophenylazobenzene-4-carboxylic acid |
| Dansyl | 5-(dimethylamino)naphthalene-1-sulfonyl |
| DCC | N,N′-dicyclohexylcarbodiimide |
| Dde | 1-(4,4-dimethyl-2,6-dioxocyclohexylidene)ethyl |
| DEAC | 7-diethylaminocoumarin |
| DHP | 3,4-dihydro-2H-pyran |
| DIBAL-H | diisobutylaluminum hydride |
| DIBO | 5,6-didehydro-11,12-dihydrodibenzo[a,e]cyclooctene |
| DIC | N,N′-diisopropylcarbodiimide |
| DIEA | diisopropylethylamine |
| DKP | 2,5-diketopiperazine |
| DMAP | 4-(N,N-dimethylamino)pyridine |
| DMED | N,N′-dimethylethylenediamine |
| DMF | N,N-dimethylformamide |
| DMPA | 2,2-dimethoxy-2-phenylacetophenone |
| DMSO | dimethylsulfoxide |
| DOTA | 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid |
| DPPA | diphenylphosphoryl azide |
| DTBPY | 4,4′-di-tert-butyl-2,2′-bipyridyl |
| DTT | 1,4-dithio-d-threitol |
| EDA | ethylenediamine |
| EDC | 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride |
| EDT | Ethanedithiol |
| EPR | enhanced permeation and retention |
| ESI-MS | electrospray ionization mass spectrometry |
| FITC | fluorescein isothiocyanate |
| Fmoc | 9-fluorenylmethoxycarbonyl |
| FR | folate receptor |
| FRET | Förster resonance energy transfer |
| 5-FU | 5-fluorouracil |
| GABA | γ-aminobutyric acid |
| GFP | green fluorescent protein |
| GLUT | glucose transporter |
| GRPR | gastrin-releasing peptide receptor |
| HATU | O-(1H-7-azabenzotriazol-1-yl)-1,1,3,3 -tetramethyluronium hexafluorophosphate |
| HBTU | O-(1H-benzotriazol-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate |
| hGC | heteroglycocluster |
| HCTU | O-(1H-6-chlorobenzotriazol-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate |
| HMTTA | 1,1,4,7,10,10-hexamethyltriethylenetetramine |
| HOAt | 1-hydroxy-7-azabenzotriazole |
| HOBt | 1-hydroxybenzotriazole |
| Hof | homophenylalanine |
| IBX | 2-iodoxybenzoic acid |
| ICP-MS | Inductively coupled plasma mass spectrometry |
| iEDDA | inverse electron-demand Diels–Alder |
| KAHA | α-ketoacid–hydroxylamine |
| LHRH | luteinizing hormone releasing hormone |
| MALDI-TOF-MS | matrix-assisted laser desorption ionization time-of-flight mass spectrometry |
| MBHA | 4-methylbenzhydrylamine |
| MCA | 7-methoxycoumarinyl-4-acetic acid |
| MMAE | monomethylauristatin E |
| MMP | matrix metalloproteinase |
| mPDA | m-phenylenediamine |
| MRI | magnetic resonance imaging |
| Mtt | 4-methyltrityl |
| MTX | methotrexate |
| nanoSIMS | nanoscale secondary ion mass spectrometry |
| NaPi | sodium phosphate |
| NBS | N-bromosuccinimide |
| NCL | native chemical ligation |
| NCS | N-chlorosuccinimide |
| NGF | nerve growth factor |
| NHS | N-hydroxysuccinimide |
| NHS ester | N-hydroxysuccinimidyl ester |
| NIQ | naphthylisoquinoline |
| NIR | near infrared |
| NMM | N-methylmorpholine |
| NMP | N-methyl-2-pyrrolidone |
| NMR | Nuclear magnetic resonance |
| NOTA | 1,4,7-triazacyclononane-triacetic acid |
| NP | nanoparticle |
| NPY(Y1)R | neuropeptide Y receptor subtype 1 |
| OA | oleanolic acid |
| Pag | propargylglycine |
| PAL | photoaffinity labeling |
| Pam | palmitoyl |
| PBS | Phosphate-buffered saline |
| PDT | photodynamic therapy |
| PET | positron emission tomography |
| PFTase | protein farnesyl transferase |
| PKA | AMP-dependent protein kinase |
| PKI | protein kinase inhibitor |
| PMDETA | N,N,N′,N″,N″-pentamethyldiethylenetriamine |
| PpIX | protoporphyrin IX |
| PPTS | pyridinium p-toluenesulfonate |
| PTSA | p-toluenesulfonic acid |
| PyBOP | benzotriazol-1-yloxytripyrrolidinophosphonium hexafluorophosphate |
| qABP | quenched activity-based probe |
| Q-LDT | quenched ligand-directed tosylate |
| RGD | Arg-Gly-Asp |
| ROS | reactive oxygen species |
| RT | room temperature |
| SASRIN | super acid sensitive resin |
| SDS-PAGE | sodium dodecylsulfate-polyacrylamide gel electrophoresis |
| siRNA | small interfering RNA |
| SPAAC | strain-promoted [3 + 2]-azide–alkyne cycloaddition |
| SPECT | single photon emission computed tomography |
| SPPS | solid-phase peptide synthesis |
| TACTV | tris-tertiary amine cyclotriveratrylene |
| TAMRA | tetramethylrhodamine |
| TBAF | tetrabutylammonium fluoride |
| TBS | tert-butyldimethylsilyl |
| TBTA | tris[(1-benzyl-1H-1,2,3-triazole-4-yl)methyl]amine |
| TCEP | tris(2-carboxyethyl)phosphine |
| TES | triethylsilyl |
| TFA | trifluoroacetic acid |
| TFAA | trifluoroacetic anhydride |
| THF | tetrahydrofuran |
| THP | 2-tetrahydropyranyl |
| THPTA | tris(3-hydroxypropyltriazolylmethyl)amine |
| TIPS | triisopropylsilyl |
| TIS | triisopropylsilane |
| TLR | Toll-like receptor |
| TMS | trimethylsilyl |
| Trt | trityl |
| Tz-Cy3 | 6-methyl-tetrazine-sulfo-Cy3 |
| UDQ | universal dark quencher |
| uPA | urokinase-like plasminogen activator |
| UV | ultraviolet |
References
- Landsteiner, K. The Specificity of Serological Reactions, Revised; Dover Publications: New York, NY, USA, 1962. [Google Scholar]
- King, T.P. Chemical and Biological Properties of Some Atopic Allergens. In Advances in Immunology Volume 23; Elsevier: Amsterdam, The Netherlands, 1976; pp. 77–105. [Google Scholar]
- Mayer, R.L. The Significance of Cross-Links in the Formation of Hapten-Carrier Complexes. Int. Arch. Allergy Immunol. 1956, 8, 115–129. [Google Scholar] [CrossRef] [PubMed]
- Plescia, O.J. The role of the carrier in antibody formation. Curr. Top. Microbiol. Immunol. 1969, 50, 78–106. [Google Scholar] [CrossRef] [PubMed]
- Kontiainen, S.; Makela, O.; Hurme, M. Immune responses to hapten conjugates in vitro. Q. Rev. Biophys. 1975, 8, 507–522. [Google Scholar] [CrossRef] [PubMed]
- King, T.P.; Kochoumian, L.; Ishizaka, K.; Lichtenstein, L.M.; Norman, P.S. Immunochemical studies of dextran coupled ragweed pollen allergen, antigen E. Arch. Biochem. Biophys. 1975, 169, 464–473. [Google Scholar] [CrossRef]
- Carter, J.M. Techniques for conjugation of synthetic peptides to carrier molecules. Methods Mol. Biol. 1994, 36, 155–191. [Google Scholar] [CrossRef] [PubMed]
- Harris, J.R.; Markl, J. Keyhole limpet hemocyanin (KLH): A biomedical review. Micron 1999, 30, 597–623. [Google Scholar] [CrossRef]
- Bundle, D.R.; Paszkiewicz, E.; Elsaidi, H.R.H.; Mandal, S.S.; Sarkar, S. A Three Component Synthetic Vaccine Containing a betab-Mannan T-Cell Peptide Epitope and a beta-Glucan Dendritic Cell Ligand. Molecules 2018, 23, 1961. [Google Scholar] [CrossRef]
- Molina, N.; Martin-Serrano, A.; Fernandez, T.D.; Tesfaye, A.; Najera, F.; Torres, M.J.; Mayorga, C.; Vida, Y.; Montanez, M.I.; Perez-Inestrosa, E. Dendrimeric Antigens for Drug Allergy Diagnosis: A New Approach for Basophil Activation Tests. Molecules 2018, 23, 997. [Google Scholar] [CrossRef]
- Louie, A. Multimodality imaging probes: Design and challenges. Chem. Rev. 2010, 110, 3146–3195. [Google Scholar] [CrossRef]
- Beal, D.M.; Jones, L.H. Molecular scaffolds using multiple orthogonal conjugations: Applications in chemical biology and drug discovery. Angew. Chem. Int. Ed. 2012, 51, 6320–6326. [Google Scholar] [CrossRef]
- Kanfar, N.; Bartolami, E.; Zelli, R.; Marra, A.; Winum, J.Y.; Ulrich, S.; Dumy, P. Emerging trends in enzyme inhibition by multivalent nanoconstructs. Org. Biomol. Chem. 2015, 13, 9894–9906. [Google Scholar] [CrossRef]
- Zhao, J.; Chen, J.; Ma, S.; Liu, Q.; Huang, L.; Chen, X.; Lou, K.; Wang, W. Recent developments in multimodality fluorescence imaging probes. Acta Pharm. Sin. B 2018, 8, 320–338. [Google Scholar] [CrossRef]
- Clave, G.; Boutal, H.; Hoang, A.; Perraut, F.; Volland, H.; Renard, P.Y.; Romieu, A. A novel heterotrifunctional peptide-based cross-linking reagent for facile access to bioconjugates. Applications to peptide fluorescent labelling and immobilisation. Org. Biomol. Chem. 2008, 6, 3065–3078. [Google Scholar] [CrossRef]
- Batschelet, E. Introduction to Mathematics for Life Scientists, 3rd ed.; Springer: Berlin/Heidelberg, Germany; New York, NY, USA, 1979; pp. 446–452. [Google Scholar]
- Jiang, J.; Chen, C.-Y.; Zhang, N.; Vairaprakash, P.; Lindsey, J.S. Polarity-Tunable and Wavelength-Tunable Bacteriochlorins Bearing a Single Carboxylic Acid or NHS Ester. Use in a Protein Bioconjugation Model System. New J. Chem. 2015, 39, 403–419. [Google Scholar] [CrossRef]
- Bondy, J.A.; Murty, U.S.R. Graph Theory with Applications; North Holland: New York, NY, USA, 1976. [Google Scholar]
- Garcia-Domenech, R.; Galvez, J.; de Julian-Ortiz, J.V.; Pogliani, L. Some new trends in chemical graph theory. Chem. Rev. 2008, 108, 1127–1169. [Google Scholar] [CrossRef] [PubMed]
- Amigo, J.M.; Galvez, J.; Villar, V.M. A review on molecular topology: Applying graph theory to drug discovery and design. Naturwissenschaften 2009, 96, 749–761. [Google Scholar] [CrossRef]
- Choi, K.Y.; Swierczewska, M.; Lee, S.; Chen, X. Protease-activated drug development. Theranostics 2012, 2, 156–178. [Google Scholar] [CrossRef] [PubMed]
- Ni, S.; Zhang, H.; Huang, W.; Zhou, J.; Qian, H.; Chen, W. The application of an aryl hydrazine linker prevents beta-elimination side products in the SPPS of C-terminal cysteine peptides. J. Pept. Sci. 2010, 16, 309–313. [Google Scholar] [CrossRef]
- Price, E.W.; Orvig, C. Matching chelators to radiometals for radiopharmaceuticals. Chem. Soc. Rev. 2014, 43, 260–290. [Google Scholar] [CrossRef]
- Fu, Y.; Finney, N.S. Small-molecule fluorescent probes and their design. RSC Adv. 2018, 8, 29051–29061. [Google Scholar] [CrossRef]
- MacPherson, D.S.; Fung, K.; Cook, B.E.; Francesconi, L.C.; Zeglis, B.M. A brief overview of metal complexes as nuclear imaging agents. Dalton Trans. 2019, 48, 14547–14565. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Wang, F.; Hyun, J.Y.; Wei, T.; Qiang, J.; Ren, X.; Shin, I.; Yoon, J. Recent progress in the development of fluorescent, luminescent and colorimetric probes for detection of reactive oxygen and nitrogen species. Chem. Soc. Rev. 2016, 45, 2976–3016. [Google Scholar] [CrossRef]
- Murale, D.P.; Hong, S.C.; Haque, M.M.; Lee, J.S. Photo-affinity labeling (PAL) in chemical proteomics: A handy tool to investigate protein-protein interactions (PPIs). Proteome Sci. 2017, 15, 14. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Cheng, Y.; Zhao, X.; Luo, Y.; Chen, J.; Yuan, W.E. Advances in redox-responsive drug delivery systems of tumor microenvironment. J. Nanobiotechnol. 2018, 16, 74–83. [Google Scholar] [CrossRef] [PubMed]
- Gisbert-Garzarán, M.; Manzano, M.; Vallet-Regí, M. Self-immolative chemistry in nanomedicine. Chem. Eng. J. 2018, 340, 24–31. [Google Scholar] [CrossRef]
- Alouane, A.; Labruere, R.; Le Saux, T.; Schmidt, F.; Jullien, L. Self-immolative spacers: Kinetic aspects, structure-property relationships, and applications. Angew. Chem. Int. Ed. 2015, 54, 7492–7509. [Google Scholar] [CrossRef] [PubMed]
- Roth, M.E.; Green, O.; Gnaim, S.; Shabat, D. Dendritic, Oligomeric, and Polymeric Self-Immolative Molecular Amplification. Chem. Rev. 2016, 116, 1309–1352. [Google Scholar] [CrossRef]
- Dumoulin, F.; Durmuş, M.; Ahsen, V.; Nyokong, T. Synthetic pathways to water-soluble phthalocyanines and close analogs. Coord. Chem. Rev. 2010, 254, 2792–2847. [Google Scholar] [CrossRef]
- Pisarek, S.; Maximova, K.; Gryko, D. Strategies toward the synthesis of amphiphilic porphyrins. Tetrahedron 2014, 70, 6685–6715. [Google Scholar] [CrossRef]
- Luciano, M.; Bruckner, C. Modifications of Porphyrins and Hydroporphyrins for Their Solubilization in Aqueous Media. Molecules 2017, 22, 980. [Google Scholar] [CrossRef]
- Ogata, F.; Nagaya, T.; Maruoka, Y.; Akhigbe, J.; Meares, A.; Lucero, M.Y.; Satraitis, A.; Fujimura, D.; Okada, R.; Inagaki, F.; et al. Activatable Near-Infrared Fluorescence Imaging Using PEGylated Bacteriochlorin-Based Chlorin and BODIPY-Dyads as Probes for Detecting Cancer. Bioconjug. Chem. 2019, 30, 169–183. [Google Scholar] [CrossRef]
- Matsumoto, N.; Taniguchi, M.; Lindsey, J.S. Bioconjugatable synthetic chlorins rendered water-soluble with three PEG-12 groups via click chemistry. J. Porphyr. Phthalocyanines 2020, 24, 362–378. [Google Scholar] [CrossRef]
- Liu, R.; Liu, S.J.; Hu, G.F.; Lindsey, J.S. Aqueous solubilization of hydrophobic tetrapyrrole macrocycles by attachment to an amphiphilic single-chain nanoparticle (SCNP). New J. Chem. 2020, 44, 21293–21308. [Google Scholar] [CrossRef]
- Liu, S.; Rong, J.; Liu, R.; Lindsey, J.S. Single-Fluorophore Single-Chain Nanoparticle Undergoes Fluorophore-Driven Assembly with Fluorescence Features Retained in Physiological Milieu. ACS Appl. Polym. Mater. 2021, 3, 1767–1776. [Google Scholar] [CrossRef]
- Harris, J.M. Poly(ethylene glycol) Chemistry: Biotechnical and Biomedical Applications; Plenum Press: New York, NY, USA, 1992; pp. 1–14. [Google Scholar]
- Zalipsky, S.; Harris, J.M. (Eds.) Introduction to Chemistry and Biological Applications of Poly(ethylene glycol). In Poly(ethylene glycol) Chemistry and Biological Applications; ACS Symposium Series; American Chemical Society: Washington, DC, USA, 1997; Volume 680, pp. 1–13. [Google Scholar]
- Thompson, M.S.; Vadala, T.P.; Vadala, M.L.; Lin, Y.; Riffle, J.S. Synthesis and applications of heterobifunctional poly(ethylene oxide) oligomers. Polymer 2008, 49, 345–373. [Google Scholar] [CrossRef]
- Larson, N.; Ghandehari, H. Polymeric conjugates for drug delivery. Chem. Mater. 2012, 24, 840–853. [Google Scholar] [CrossRef] [PubMed]
- Herzberger, J.; Niederer, K.; Pohlit, H.; Seiwert, J.; Worm, M.; Wurm, F.R.; Frey, H. Polymerization of Ethylene Oxide, Propylene Oxide, and Other Alkylene Oxides: Synthesis, Novel Polymer Architectures, and Bioconjugation. Chem. Rev. 2016, 116, 2170–2243. [Google Scholar] [CrossRef]
- Kolate, A.; Baradia, D.; Patil, S.; Vhora, I.; Kore, G.; Misra, A. PEG—A versatile conjugating ligand for drugs and drug delivery systems. J. Control. Release 2014, 192, 67–81. [Google Scholar] [CrossRef]
- Begum, R.; Matsuura, H. Conformational properties of short poly(oxyethylene) chains in water studied by IR spectroscopy. J. Chem. Soc. Faraday Trans. 1997, 93, 3839–3848. [Google Scholar] [CrossRef]
- Lang, W.; Yuan, C.; Zhu, B.; Pan, S.; Liu, J.; Luo, J.; Nie, S.; Zhu, Q.; Lee, J.S.; Ge, J. Expanding the “minimalist” small molecule tagging approach to different bioactive compounds. Org. Biomol. Chem. 2019, 17, 3010–3017. [Google Scholar] [CrossRef]
- Zhang, L.; Zhang, Y.; Dong, J.; Liu, J.; Zhang, L.; Sun, H. Design and synthesis of novel photoaffinity probes for study of the target proteins of oleanolic acid. Bioorg. Med. Chem. Lett. 2012, 22, 1036–1039. [Google Scholar] [CrossRef]
- Sharma, A.; Lee, M.G.; Won, M.; Koo, S.; Arambula, J.F.; Sessler, J.L.; Chi, S.G.; Kim, J.S. Targeting Heterogeneous Tumors Using a Multifunctional Molecular Prodrug. J. Am. Chem. Soc. 2019, 141, 15611–15618. [Google Scholar] [CrossRef] [PubMed]
- Devalapally, H.; Navath, R.S.; Yenamandra, V.; Akkinepally, R.R.; Devarakonda, R.K. Beta-galactoside prodrugs of doxorubicin for application in antibody directed enzyme prodrug therapy/prodrug monotherapy. Arch. Pharm. Res. 2007, 30, 723–732. [Google Scholar] [CrossRef] [PubMed]
- Jouanno, L.A.; Chevalier, A.; Sekkat, N.; Perzo, N.; Castel, H.; Romieu, A.; Lange, N.; Sabot, C.; Renard, P.Y. Kondrat’eva ligation: Diels-Alder-based irreversible reaction for bioconjugation. J. Org. Chem. 2014, 79, 10353–10366. [Google Scholar] [CrossRef]
- van der Meer, B.W. Kappa-squared: From nuisance to new sense. J. Biotechnol. 2002, 82, 181–196. [Google Scholar] [CrossRef]
- Taniguchi, M.; Du, H.; Lindsey, J.S. PhotochemCAD 3: Diverse Modules for Photophysical Calculations with Access to Multiple Spectral Databases. Photochem. Photobiol. 2017, 94, 277–289. [Google Scholar] [CrossRef] [PubMed]
- Qi, Q.; Taniguchi, M.; Lindsey, J.S. Heuristics from modeling of spectral overlap in forster resonance energy transfer (FRET). J. Chem. Inf. Model. 2019, 59, 652–667. [Google Scholar] [CrossRef]
- Lindsey, J.S.; Taniguchi, M.; Bocian, D.F.; Holten, D. The fluorescence quantum yield parameter in Förster resonance energy transfer (FRET)—Meaning, misperception, and molecular design. Chem. Phys. Rev. 2021, 2, 011302. [Google Scholar] [CrossRef]
- Jouanno, L.-A.; Renault, K.; Sabot, C.; Renard, P.-Y. 5-Alkoxyoxazole—A Versatile Building Block in (Bio)organic Synthesis. Eur. J. Org. Chem. 2016, 2016, 3264–3281. [Google Scholar] [CrossRef]
- Shi, J.Y.; Liu, T.W.B.; Chen, J.; Green, D.; Jaffray, D.; Wilson, B.C.; Wang, F.; Zheng, G. Transforming a Targeted Porphyrin Theranostic Agent into a PET Imaging Probe for Cancer. Theranostics 2011, 1, 363–370. [Google Scholar] [CrossRef]
- Benov, L. Photodynamic therapy: Current status and future directions. Med. Princ. Pract. 2015, 24, 14–28. [Google Scholar] [CrossRef]
- dos Santos, A.F.; de Almeida, D.R.Q.; Terra, L.F.; Baptista, M.S.; Labriola, L. Photodynamic therapy in cancer treatment—An update review. J. Cancer Metastasis Treat. 2019, 5, 25–44. [Google Scholar] [CrossRef]
- Ametamey, S.M.; Honer, M.; Schubiger, P.A. Molecular imaging with PET. Chem. Rev. 2008, 108, 1501–1516. [Google Scholar] [CrossRef] [PubMed]
- Mukai, H.; Watanabe, Y. Review: PET imaging with macro- and middle-sized molecular probes. Nucl. Med. Biol. 2021, 92, 156–170. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.-Y.; Sun, E.; Fan, D.; Taniguchi, M.; McDowell, B.E.; Yang, E.; Diers, J.R.; Bocian, D.F.; Holten, D.; Lindsey, J.S. Synthesis and Physicochemical Properties of Metallobacteriochlorins. Inorg. Chem. 2012, 51, 9443–9464. [Google Scholar] [CrossRef] [PubMed]
- Yang, E.; Diers, J.R.; Huang, Y.-Y.; Hamblin, M.R.; Lindsey, J.S.; Bocian, D.F.; Holten, D. Molecular Electronic Tuning of Photosensitizers to Enhance Photodynamic Therapy: Synthetic Dicyanobacteriochlorins as a Case Study. Photochem. Photobiol. 2013, 89, 605–618. [Google Scholar] [CrossRef]
- Fuwa, H.; Takahashi, Y.; Konno, Y.; Watanabe, N.; Miyashita, H.; Sasaki, M.; Natsugari, H.; Kan, T.; Fukuyama, T.; Tomita, T.; et al. Divergent synthesis of multifunctional molecular probes to elucidate the enzyme specificity of dipeptidic gamma-secretase inhibitors. ACS Chem. Biol. 2007, 2, 408–418. [Google Scholar] [CrossRef]
- Albright, C.F.; Graciani, N.; Han, W.; Yue, E.; Stein, R.; Lai, Z.; Diamond, M.; Dowling, R.; Grimminger, L.; Zhang, S.Y.; et al. Matrix metalloproteinase-activated doxorubicin prodrugs inhibit HT1080 xenograft growth better than doxorubicin with less toxicity. Mol. Cancer Ther. 2005, 4, 751–760. [Google Scholar] [CrossRef]
- Verwilst, P.; Han, J.; Lee, J.; Mun, S.; Kang, H.G.; Kim, J.S. Reconsidering azobenzene as a component of small-molecule hypoxia-mediated cancer drugs: A theranostic case study. Biomaterials 2017, 115, 104–114. [Google Scholar] [CrossRef] [PubMed]
- McKeown, S.R. Defining normoxia, physoxia and hypoxia in tumours-implications for treatment response. Br. J. Radiol. 2014, 87, 20130676. [Google Scholar] [CrossRef]
- Song, X.; Han, X.; Yu, F.; Zhang, X.; Chen, L.; Lv, C. Polyamine-Targeting Gefitinib Prodrug and its Near-Infrared Fluorescent Theranostic Derivative for Monitoring Drug Delivery and Lung Cancer Therapy. Theranostics 2018, 8, 2217–2228. [Google Scholar] [CrossRef]
- Yuan, Y.; Kwok, R.T.; Zhang, R.; Tang, B.Z.; Liu, B. Targeted theranostic prodrugs based on an aggregation-induced emission (AIE) luminogen for real-time dual-drug tracking. Chem. Commun. 2014, 50, 11465–11468. [Google Scholar] [CrossRef] [PubMed]
- Qian, J.; Tang, B.Z. AIE Luminogens for Bioimaging and Theranostics: From Organelles to Animals. Chem 2017, 3, 56–91. [Google Scholar] [CrossRef]
- Wang, D.; Lee, M.M.S.; Xu, W.; Kwok, R.T.K.; Lam, J.W.Y.; Tang, B.Z. Theranostics based on AIEgens. Theranostics 2018, 8, 4925–4956. [Google Scholar] [CrossRef] [PubMed]
- van Scherpenzeel, M.; Moret, E.E.; Ballell, L.; Liskamp, R.M.; Nilsson, U.J.; Leffler, H.; Pieters, R.J. Synthesis and evaluation of new thiodigalactoside-based chemical probes to label galectin-3. ChemBioChem 2009, 10, 1724–1733. [Google Scholar] [CrossRef]
- Weinstain, R.; Savariar, E.N.; Felsen, C.N.; Tsien, R.Y. In vivo targeting of hydrogen peroxide by activatable cell-penetrating peptides. J. Am. Chem. Soc. 2014, 136, 874–877. [Google Scholar] [CrossRef] [PubMed]
- Beal, D.M.; Albrow, V.E.; Burslem, G.; Hitchen, L.; Fernandes, C.; Lapthorn, C.; Roberts, L.R.; Selby, M.D.; Jones, L.H. Click-enabled heterotrifunctional template for sequential bioconjugations. Org. Biomol. Chem. 2012, 10, 548–554. [Google Scholar] [CrossRef]
- Akram, M.; Asif, H.M.; Uzair, M.; Akhtar, N.; Madni, A.; Shah, S.M.A.; ul Hasan, Z.; Ullah, A. Amino acids: A review article. J. Med. Plants Res. 2011, 5, 3997–4000. [Google Scholar] [CrossRef]
- Tang, W.; Becker, M.L. “Click” reactions: A versatile toolbox for the synthesis of peptide-conjugates. Chem. Soc. Rev. 2014, 43, 7013–7039. [Google Scholar] [CrossRef]
- Liu, S.P.; Zhou, L.; Lakshminarayanan, R.; Beuerman, R.W. Multivalent Antimicrobial Peptides as Therapeutics: Design Principles and Structural Diversities. Int. J. Pept. Res. Ther. 2010, 16, 199–213. [Google Scholar] [CrossRef]
- He, R.; Finan, B.; Mayer, J.P.; DiMarchi, R.D. Peptide Conjugates with Small Molecules Designed to Enhance Efficacy and Safety. Molecules 2019, 24, 1855. [Google Scholar] [CrossRef]
- Lin, Y.; Mazo, M.M.; Skaalure, S.C.; Thomas, M.R.; Schultz, S.R.; Stevens, M.M. Activatable cell-biomaterial interfacing with photo-caged peptides. Chem. Sci. 2019, 10, 1158–1167. [Google Scholar] [CrossRef] [PubMed]
- Poreba, M. Protease-activated prodrugs: Strategies, challenges, and future directions. FEBS J. 2020, 287, 1936–1969. [Google Scholar] [CrossRef] [PubMed]
- Kovalova, A.; Pohl, R.; Vrabel, M. Stepwise triple-click functionalization of synthetic peptides. Org. Biomol. Chem. 2018, 16, 5960–5964. [Google Scholar] [CrossRef] [PubMed]
- Jaradat, D.M.M. Thirteen decades of peptide synthesis: Key developments in solid phase peptide synthesis and amide bond formation utilized in peptide ligation. Amino Acids 2018, 50, 39–68. [Google Scholar] [CrossRef] [PubMed]
- Martin, V.; Egelund, P.H.G.; Johansson, H.; Thordal Le Quement, S.; Wojcik, F.; Sejer Pedersen, D. Greening the synthesis of peptide therapeutics: An industrial perspective. RSC Adv. 2020, 10, 42457–42492. [Google Scholar] [CrossRef]
- Al-Warhi, T.I.; Al-Hazimi, H.M.A.; El-Faham, A. Recent development in peptide coupling reagents. J. Saudi Chem. Soc. 2012, 16, 97–116. [Google Scholar] [CrossRef]
- El-Faham, A.; Albericio, F. Peptide coupling reagents, more than a letter soup. Chem. Rev. 2011, 111, 6557–6602. [Google Scholar] [CrossRef]
- Montalbetti, C.A.G.N.; Falque, V. Amide bond formation and peptide coupling. Tetrahedron 2005, 61, 10827–10852. [Google Scholar] [CrossRef]
- Isidro-Llobet, A.; Alvarez, M.; Albericio, F. Amino acid-protecting groups. Chem. Rev. 2009, 109, 2455–2504. [Google Scholar] [CrossRef]
- Behrendt, R.; White, P.; Offer, J. Advances in Fmoc solid-phase peptide synthesis. J. Pept. Sci. 2016, 22, 4–27. [Google Scholar] [CrossRef]
- Coin, I.; Dolling, R.; Krause, E.; Bienert, M.; Beyermann, M.; Sferdean, C.D.; Carpino, L.A. Depsipeptide methodology for solid-phase peptide synthesis: Circumventing side reactions and development of an automated technique via depsidipeptide units. J. Org. Chem. 2006, 71, 6171–6177. [Google Scholar] [CrossRef]
- Huang, Y.C.; Guan, C.J.; Tan, X.L.; Chen, C.C.; Guo, Q.X.; Li, Y.M. Accelerated Fmoc solid-phase synthesis of peptides with aggregation-disrupting backbones. Org. Biomol. Chem. 2015, 13, 1500–1506. [Google Scholar] [CrossRef]
- Paradís-Bas, M.; Tulla-Puche, J.; Albericio, F. The road to the synthesis of “difficult peptides”. Chem. Soc. Rev. 2016, 45, 631–654. [Google Scholar] [CrossRef]
- Pícha, J.; Buděšínský, M.; Macháčková, K.; Collinsová, M.; Jiráček, J. Optimized syntheses of Fmoc azido amino acids for the preparation of azidopeptides. J. Pept. Sci. 2017, 23, 202–214. [Google Scholar] [CrossRef]
- Weis, S.M.; Cheresh, D.A. Tumor angiogenesis: Molecular pathways and therapeutic targets. Nat. Med. 2011, 17, 1359–1370. [Google Scholar] [CrossRef] [PubMed]
- Dou, X.; Nomoto, T.; Takemoto, H.; Matsui, M.; Tomoda, K.; Nishiyama, N. Effect of multiple cyclic RGD peptides on tumor accumulation and intratumoral distribution of IRDye 700DX-conjugated polymers. Sci. Rep. 2018, 8, 8126–8137. [Google Scholar] [CrossRef]
- Renault, K.; Fredy, J.W.; Renard, P.Y.; Sabot, C. Covalent Modification of Biomolecules through Maleimide-Based Labeling Strategies. Bioconjug. Chem. 2018, 29, 2497–2513. [Google Scholar] [CrossRef] [PubMed]
- Sun, B.; Luo, C.; Yu, H.; Zhang, X.; Chen, Q.; Yang, W.; Wang, M.; Kan, Q.; Zhang, H.; Wang, Y.; et al. Disulfide Bond-Driven Oxidation- and Reduction-Responsive Prodrug Nanoassemblies for Cancer Therapy. Nano Lett. 2018, 18, 3643–3650. [Google Scholar] [CrossRef] [PubMed]
- Postma, T.M.; Albericio, F. Disulfide Formation Strategies in Peptide Synthesis. Eur. J. Org. Chem. 2014, 2014, 3519–3530. [Google Scholar] [CrossRef]
- Fujiwara, Y.; Akaji, K.; Kiso, Y. Racemization-free synthesis of C-terminal cysteine-peptide using 2-chlorotrityl resin. Chem. Pharm. Bull. 1994, 42, 724–726. [Google Scholar] [CrossRef]
- Lukszo, J.; Patterson, D.; Albericio, F.; Kates, S.A. 3-(1-Piperidinyl)alanine formation during the preparation of C-terminal cysteine peptides with the Fmoc/t-Bu strategy. Lett. Pept. Sci. 1996, 3, 157–166. [Google Scholar] [CrossRef]
- Gongora-Benitez, M.; Mendive-Tapia, L.; Ramos-Tomillero, I.; Breman, A.C.; Tulla-Puche, J.; Albericio, F. Acid-labile Cys-protecting groups for the Fmoc/tBu strategy: Filling the gap. Org. Lett. 2012, 14, 5472–5475. [Google Scholar] [CrossRef]
- Stathopoulos, P.; Papas, S.; Pappas, C.; Mousis, V.; Sayyad, N.; Theodorou, V.; Tzakos, A.G.; Tsikaris, V. Side reactions in the SPPS of Cys-containing peptides. Amino Acids 2013, 44, 1357–1363. [Google Scholar] [CrossRef]
- Lelievre, D.; Terrier, V.P.; Delmas, A.F.; Aucagne, V. Native Chemical Ligation Strategy to Overcome Side Reactions during Fmoc-Based Synthesis of C-Terminal Cysteine-Containing Peptides. Org. Lett. 2016, 18, 920–923. [Google Scholar] [CrossRef]
- Tsuda, S.; Masuda, S.; Yoshiya, T. Epimerization-Free Preparation of C-Terminal Cys Peptide Acid by Fmoc SPPS Using Pseudoproline-Type Protecting Group. J. Org. Chem. 2020, 85, 1674–1679. [Google Scholar] [CrossRef] [PubMed]
- Albericio, F.; El-Faham, A. Choosing the Right Coupling Reagent for Peptides: A Twenty-Five-Year Journey. Org. Process Res. Dev. 2018, 22, 760–772. [Google Scholar] [CrossRef]
- Valeur, E.; Bradley, M. Amide bond formation: Beyond the myth of coupling reagents. Chem. Soc. Rev. 2009, 38, 606–631. [Google Scholar] [CrossRef] [PubMed]
- Prasad, K.; Bharathi, K.; Haseena, B.B. Applications of peptide coupling reagents—An update. Intl. J. Pharm. Sci. Rev. Res. 2011, 8, 108–119. [Google Scholar]
- Tsakos, M.; Schaffert, E.S.; Clement, L.L.; Villadsen, N.L.; Poulsen, T.B. Ester coupling reactions--an enduring challenge in the chemical synthesis of bioactive natural products. Nat. Prod. Rep. 2015, 32, 605–632. [Google Scholar] [CrossRef] [PubMed]
- Dunetz, J.R.; Magano, J.; Weisenburger, G.A. Large-Scale Applications of Amide Coupling Reagents for the Synthesis of Pharmaceuticals. Org. Process Res. Dev. 2016, 20, 140–177. [Google Scholar] [CrossRef]
- Sakakura, A.; Kawajiri, K.; Ohkubo, T.; Kosugi, Y.; Ishihara, K. Widely useful DMAP-catalyzed esterification under auxiliary base- and solvent-free conditions. J. Am. Chem. Soc. 2007, 129, 14775–14779. [Google Scholar] [CrossRef]
- Renard, E.; Dancer, P.A.; Portal, C.; Denat, F.; Prignon, A.; Goncalves, V. Design of Bimodal Ligands of Neurotensin Receptor 1 for Positron Emission Tomography Imaging and Fluorescence-Guided Surgery of Pancreatic Cancer. J. Med. Chem. 2020, 63, 2426–2433. [Google Scholar] [CrossRef] [PubMed]
- Lelle, M.; Peneva, K. An amino acid-based heterofunctional cross-linking reagent. Amino Acids 2014, 46, 1243–1251. [Google Scholar] [CrossRef] [PubMed]
- Krall, N.; Pretto, F.; Neri, D. A bivalent small molecule-drug conjugate directed against carbonic anhydrase IX can elicit complete tumour regression in mice. Chem. Sci. 2014, 5, 3640–3644. [Google Scholar] [CrossRef]
- Lee, H.M.; Priestman, M.A.; Lawrence, D.S. Light-mediated spatial control via photolabile fluorescently quenched peptide cassettes. J. Am. Chem. Soc. 2010, 132, 1446–1447. [Google Scholar] [CrossRef] [PubMed]
- Lock, L.L.; Tang, Z.; Keith, D.; Reyes, C.; Cui, H. Enzyme-Specific Doxorubicin Drug Beacon as Drug-Resistant Theranostic Molecular Probes. ACS Macro Lett. 2015, 4, 552–555. [Google Scholar] [CrossRef]
- Yan, X.; Li, Z.; Liang, Y.; Yang, L.; Zhang, B.; Wang, Q. A chemical “hub” for absolute quantification of a targeted protein: Orthogonal integration of elemental and molecular mass spectrometry. Chem. Commun. 2014, 50, 6578–6581. [Google Scholar] [CrossRef] [PubMed]
- Tsukiji, S.; Wang, H.; Miyagawa, M.; Tamura, T.; Takaoka, Y.; Hamachi, I. Quenched ligand-directed tosylate reagents for one-step construction of turn-on fluorescent biosensors. J. Am. Chem. Soc. 2009, 131, 9046–9054. [Google Scholar] [CrossRef]
- Huang, R.; Wang, X.; Wang, D.; Liu, F.; Mei, B.; Tang, A.; Jiang, J.; Liang, G. Multifunctional fluorescent probe for sequential detections of glutathione and caspase-3 in vitro and in cells. Anal. Chem. 2013, 85, 6203–6207. [Google Scholar] [CrossRef]
- Anami, Y.; Xiong, W.; Gui, X.; Deng, M.; Zhang, C.C.; Zhang, N.; An, Z.; Tsuchikama, K. Enzymatic conjugation using branched linkers for constructing homogeneous antibody-drug conjugates with high potency. Org. Biomol. Chem. 2017, 15, 5635–5642. [Google Scholar] [CrossRef] [PubMed]
- Oriana, S.; Cai, Y.; Bode, J.W.; Yamakoshi, Y. Synthesis of tri-functionalized MMP2 FRET probes using a chemo-selective and late-stage modification of unprotected peptides. Org. Biomol. Chem. 2017, 15, 1792–1800. [Google Scholar] [CrossRef] [PubMed]
- Mishra, P.; Nayak, B.; Dey, R.K. PEGylation in anti-cancer therapy: An overview. Asian J. Pharm. Sci. 2016, 11, 337–348. [Google Scholar] [CrossRef]
- Page-McCaw, A.; Ewald, A.J.; Werb, Z. Matrix metalloproteinases and the regulation of tissue remodelling. Nat. Rev. Mol. Cell Biol. 2007, 8, 221–233. [Google Scholar] [CrossRef]
- Hu, J.; Van den Steen, P.E.; Sang, Q.X.; Opdenakker, G. Matrix metalloproteinase inhibitors as therapy for inflammatory and vascular diseases. Nat. Rev. Drug Discov. 2007, 6, 480–498. [Google Scholar] [CrossRef]
- Bode, J.W. Chemical Protein Synthesis with the alpha-Ketoacid-Hydroxylamine Ligation. Acc. Chem. Res. 2017, 50, 2104–2115. [Google Scholar] [CrossRef]
- Zuo, C.; Zhang, B.; Yan, B.; Zheng, J.S. One-pot multi-segment condensation strategies for chemical protein synthesis. Org. Biomol. Chem. 2019, 17, 727–744. [Google Scholar] [CrossRef]
- Arbour, C.A.; Kondasinghe, T.D.; Saraha, H.Y.; Vorlicek, T.L.; Stockdill, J.L. Epimerization-free access to C-terminal cysteine peptide acids, carboxamides, secondary amides, and esters via complimentary strategies. Chem. Sci. 2018, 9, 350–355. [Google Scholar] [CrossRef]
- Zuo, C.; Yan, B.J.; Zhu, H.Y.; Shi, W.W.; Xi, T.K.; Shi, J.; Fang, G.M. Robust synthesis of C-terminal cysteine-containing peptide acids through a peptide hydrazide-based strategy. Org. Biomol. Chem. 2019, 17, 5698–5702. [Google Scholar] [CrossRef]
- Mariani, G.; Bruselli, L.; Kuwert, T.; Kim, E.E.; Flotats, A.; Israel, O.; Dondi, M.; Watanabe, N. A review on the clinical uses of SPECT/CT. Eur. J. Nucl. Med. Mol. Imaging 2010, 37, 1959–1985. [Google Scholar] [CrossRef]
- Israel, O.; Pellet, O.; Biassoni, L.; De Palma, D.; Estrada-Lobato, E.; Gnanasegaran, G.; Kuwert, T.; la Fougere, C.; Mariani, G.; Massalha, S.; et al. Two decades of SPECT/CT—The coming of age of a technology: An updated review of literature evidence. Eur. J. Nucl. Med. Mol. Imaging 2019, 46, 1990–2012. [Google Scholar] [CrossRef]
- Gao, H.; Luo, C.; Yang, G.; Du, S.; Li, X.; Zhao, H.; Shi, J.; Wang, F. Improved in Vivo Targeting Capability and Pharmacokinetics of (99m)Tc-Labeled isoDGR by Dimerization and Albumin-Binding for Glioma Imaging. Bioconjug. Chem. 2019, 30, 2038–2048. [Google Scholar] [CrossRef]
- Chen, P.; Kuang, W.; Zheng, Z.; Yang, S.; Liu, Y.; Su, L.; Zhao, K.; Liang, G. Carboxylesterase-Cleavable Biotinylated Nanoparticle for Tumor-Dual Targeted Imaging. Theranostics 2019, 9, 7359–7369. [Google Scholar] [CrossRef] [PubMed]
- Li, S.Y.; Cheng, H.; Xie, B.R.; Qiu, W.X.; Song, L.L.; Zhuo, R.X.; Zhang, X.Z. A ratiometric theranostic probe for tumor targeting therapy and self-therapeutic monitoring. Biomaterials 2016, 104, 297–309. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.M.; Xu, W.; Lawrence, D.S. Construction of a photoactivatable profluorescent enzyme via propinquity labeling. J. Am. Chem. Soc. 2011, 133, 2331–2333. [Google Scholar] [CrossRef][Green Version]
- Hai, Z.; Wu, J.; Saimi, D.; Ni, Y.; Zhou, R.; Liang, G. Smart Dual Quenching Strategy Enhances the Detection Sensitivity of Intracellular Furin. Anal. Chem. 2018, 90, 1520–1524. [Google Scholar] [CrossRef] [PubMed]
- Lai, C.H.; Chang, T.C.; Chuang, Y.J.; Tzou, D.L.; Lin, C.C. Stepwise orthogonal click chemistry toward fabrication of paclitaxel/galactose functionalized fluorescent nanoparticles for HepG2 cell targeting and delivery. Bioconjug. Chem. 2013, 24, 1698–1709. [Google Scholar] [CrossRef]
- Lai, C.H.; Lin, Y.C.; Chou, F.I.; Liang, C.F.; Lin, E.W.; Chuang, Y.J.; Lin, C.C. Design of multivalent galactosyl carborane as a targeting specific agent for potential application to boron neutron capture therapy. Chem. Commun. 2012, 48, 612–614. [Google Scholar] [CrossRef]
- Lee, D.L.; Chin, H.-L.M.; Knudsen, C.G.; Mayers, G.L.; Rose, D.S.; Skogstrom, R.K.; Palzkill, T.; Fujita, H.; Zhang, Y.; Wu, Z.; et al. Peptide-based scaffolds for in vivo immobilization and enzyme attachment in therapeutic applications. Proc. SPIE 2020, 11477, 1147708. [Google Scholar] [CrossRef]
- Yao, Z.; Borbas, K.E.; Lindsey, J.S. Soluble Precipitable Porphyrins for Use in Targeted Molecular Brachytherapy. New J. Chem. 2008, 32, 436–451. [Google Scholar] [CrossRef]
- Wu, X.; Burden-Gulley, S.M.; Yu, G.P.; Tan, M.; Lindner, D.; Brady-Kalnay, S.M.; Lu, Z.R. Synthesis and evaluation of a peptide targeted small molecular Gd-DOTA monoamide conjugate for MR molecular imaging of prostate cancer. Bioconjug. Chem. 2012, 23, 1548–1556. [Google Scholar] [CrossRef]
- Lee, M.R.; Jung, D.W.; Williams, D.; Shin, I. Efficient solid-phase synthesis of trifunctional probes and their application to the detection of carbohydrate-binding proteins. Org. Lett. 2005, 7, 5477–5480. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Garcia, M.; Benito, J.M.; Rodriguez-Lucena, D.; Yu, J.X.; Chmurski, K.; Ortiz Mellet, C.; Gutierrez Gallego, R.; Maestre, A.; Defaye, J.; Garcia Fernandez, J.M. Probing secondary carbohydrate-protein interactions with highly dense cyclodextrin-centered heteroglycoclusters: The heterocluster effect. J. Am. Chem. Soc. 2005, 127, 7970–7971. [Google Scholar] [CrossRef]
- Jimenez Blanco, J.L.; Ortiz Mellet, C.; Garcia Fernandez, J.M. Multivalency in heterogeneous glycoenvironments: Hetero-glycoclusters, -glycopolymers and -glycoassemblies. Chem. Soc. Rev. 2013, 42, 4518–4531. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Cuesta, M.; Ortiz Mellet, C.; Garcia Fernandez, J.M. Carbohydrate supramolecular chemistry: Beyond the multivalent effect. Chem. Commun. 2020, 56, 5207–5222. [Google Scholar] [CrossRef] [PubMed]
- Muller, C.; Despras, G.; Lindhorst, T.K. Organizing multivalency in carbohydrate recognition. Chem. Soc. Rev. 2016, 45, 3275–3302. [Google Scholar] [CrossRef] [PubMed]
- Goyard, D.; Thomas, B.; Gillon, E.; Imberty, A.; Renaudet, O. Heteroglycoclusters With Dual Nanomolar Affinities for the Lectins LecA and LecB From Pseudomonas aeruginosa. Front. Chem. 2019, 7, 666–675. [Google Scholar] [CrossRef] [PubMed]
- Galan, M.C.; Dumy, P.; Renaudet, O. Multivalent glyco(cyclo)peptides. Chem. Soc. Rev. 2013, 42, 4599–4612. [Google Scholar] [CrossRef]
- Pifferi, C.; Berthet, N.; Renaudet, O. Cyclopeptide scaffolds in carbohydrate-based synthetic vaccines. Biomater. Sci. 2017, 5, 953–965. [Google Scholar] [CrossRef]
- Richard, M.; Ariztia, J.; Lamandé-Langle, S.; Pellegrini Moïse, N. Sugar γ-Amino Acids as Building Blocks for the Synthesis of Cyclic Neoglycopeptides. ChemistrySelect 2018, 3, 9121–9126. [Google Scholar] [CrossRef]
- Thomas, B.; Fiore, M.; Daskhan, G.C.; Spinelli, N.; Renaudet, O. A multi-ligation strategy for the synthesis of heterofunctionalized glycosylated scaffolds. Chem. Commun. 2015, 51, 5436–5439. [Google Scholar] [CrossRef]
- Chelius, D.; Shaler, T.A. Capture of peptides with N-terminal serine and threonine: A sequence-specific chemical method for peptide mixture simplification. Bioconjug. Chem. 2003, 14, 205–211. [Google Scholar] [CrossRef] [PubMed]
- Spears, R.J.; Fascione, M.A. Site-selective incorporation and ligation of protein aldehydes. Org. Biomol. Chem. 2016, 14, 7622–7638. [Google Scholar] [CrossRef] [PubMed]
- El-Mahdi, O.; Melnyk, O. alpha-Oxo aldehyde or glyoxylyl group chemistry in peptide bioconjugation. Bioconjug. Chem. 2013, 24, 735–765. [Google Scholar] [CrossRef]
- Mettu, R.; Chen, C.Y.; Wu, C.Y. Synthetic carbohydrate-based vaccines: Challenges and opportunities. J. Biomed. Sci. 2020, 27, 9. [Google Scholar] [CrossRef] [PubMed]
- Bringmann, G.; Gampe, C.M.; Reichert, Y.; Bruhn, T.; Faber, J.H.; Mikyna, M.; Reichert, M.; Leippe, M.; Brun, R.; Gelhaus, C. Synthesis and pharmacological evaluation of fluorescent and photoactivatable analogues of antiplasmodial naphthylisoquinolines. J. Med. Chem. 2007, 50, 6104–6115. [Google Scholar] [CrossRef]
- Liu, Q.H.; Yan, X.L.; Guo, J.C.; Wang, D.H.; Li, L.; Yan, F.Y.; Chen, L.G. Spectrofluorimetric determination of trace nitrite with a novel fluorescent probe. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2009, 73, 789–793. [Google Scholar] [CrossRef]
- Carrigan, C.N.; Imperiali, B. The engineering of membrane-permeable peptides. Anal. Biochem. 2005, 341, 290–298. [Google Scholar] [CrossRef]
- Fan, D.; Wang, K.; Gao, H.; Luo, Q.; Wang, X.; Li, X.; Tong, W.; Zhang, X.; Luo, C.; Yang, G.; et al. A (64)Cu-porphyrin-based dual-modal molecular probe with integrin αvβ3 targeting function for tumour imaging. J. Label. Compd. Radiopharm. 2020, 63, 212–221. [Google Scholar] [CrossRef]
- Ruhl, T.; Volke, D.; Stembera, K.; Hatanaka, Y.; Hennig, H.; Schumer, F.; Welzel, P. Isoserine-based biotinylated photoaffinity probes that interact with penicillin-binding protein 1b. Chem. Commun. 2002, 1630–1631. [Google Scholar] [CrossRef]
- Dijkgraaf, I.; Van de Vijver, P.; Dirksen, A.; Hackeng, T.M. Synthesis and application of cNGR-containing imaging agents for detection of angiogenesis. Bioorg. Med. Chem. 2013, 21, 3555–3564. [Google Scholar] [CrossRef]
- Seibold, U.; Wangler, B.; Schirrmacher, R.; Wangler, C. Bimodal imaging probes for combined PET and OI: Recent developments and future directions for hybrid agent development. BioMed Res. Int. 2014, 2014, 153741. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Ding, J.; Xing, W.; Gai, Y.; Sheng, J.; Zeng, D. Novel Strategy for Preparing Dual-Modality Optical/PET Imaging Probes via Photo-Click Chemistry. Bioconjug. Chem. 2016, 27, 1200–1204. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Guo, N.; Li, Q.; Ma, Y.; Jacboson, O.; Lee, S.; Choi, H.S.; Mansfield, J.R.; Niu, G.; Chen, X. Dynamic PET and Optical Imaging and Compartment Modeling using a Dual-labeled Cyclic RGD Peptide Probe. Theranostics 2012, 2, 746–756. [Google Scholar] [CrossRef]
- Hawala, I.; De Rosa, L.; Aime, S.; D’Andrea, L.D. An innovative approach for the synthesis of dual modality peptide imaging probes based on the native chemical ligation approach. Chem. Commun. 2020, 56, 3500–3503. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, S.S.; Sayers, J.; Premdjee, B.; Payne, R.J. Rapid and efficient protein synthesis through expansion of the native chemical ligation concept. Nat. Rev. Chem. 2018, 2, 0122–0147. [Google Scholar] [CrossRef]
- Agouridas, V.; El Mahdi, O.; Diemer, V.; Cargoet, M.; Monbaliu, J.M.; Melnyk, O. Native Chemical Ligation and Extended Methods: Mechanisms, Catalysis, Scope, and Limitations. Chem. Rev. 2019, 119, 7328–7443. [Google Scholar] [CrossRef]
- Thapa, P.; Zhang, R.Y.; Menon, V.; Bingham, J.P. Native chemical ligation: A boon to peptide chemistry. Molecules 2014, 19, 14461–14483. [Google Scholar] [CrossRef]
- Debie, P.; Hernot, S. Emerging Fluorescent Molecular Tracers to Guide Intra-Operative Surgical Decision-Making. Front. Pharmacol. 2019, 10, 510–529. [Google Scholar] [CrossRef]
- Trester-Zedlitz, M.; Kamada, K.; Burley, S.K.; Fenyo, D.; Chait, B.T.; Muir, T.W. A modular cross-linking approach for exploring protein interactions. J. Am. Chem. Soc. 2003, 125, 2416–2425. [Google Scholar] [CrossRef]
- Lelle, M.; Kaloyanova, S.; Freidel, C.; Theodoropoulou, M.; Musheev, M.; Niehrs, C.; Stalla, G.; Peneva, K. Octreotide-Mediated Tumor-Targeted Drug Delivery via a Cleavable Doxorubicin-Peptide Conjugate. Mol. Pharm. 2015, 12, 4290–4300. [Google Scholar] [CrossRef]
- Abu Ajaj, K.; Biniossek, M.L.; Kratz, F. Development of protein-binding bifunctional linkers for a new generation of dual-acting prodrugs. Bioconjug. Chem. 2009, 20, 390–396. [Google Scholar] [CrossRef]
- Sinisi, R.; Morales, A.R.M.; Dubikovskaya, E.A.; Singh, R. Azacyanine Dyes and Use Thereof. U.S. Patent US2018/0079906 A1, 22 March 2018. [Google Scholar]
- Nunez, J.; Renslow, R.; Cliff, J.B., 3rd; Anderton, C.R. NanoSIMS for biological applications: Current practices and analyses. Biointerphases 2018, 13, 03B301-301–303B301-326. [Google Scholar] [CrossRef]
- Kabatas, S.; Agui-Gonzalez, P.; Saal, K.A.; Jahne, S.; Opazo, F.; Rizzoli, S.O.; Phan, N.T.N. Boron-Containing Probes for Non-optical High-Resolution Imaging of Biological Samples. Angew. Chem. Int. Ed. 2019, 58, 3438–3443. [Google Scholar] [CrossRef]
- Chang, T.C.; Adak, A.K.; Lin, T.W.; Li, P.J.; Chen, Y.J.; Lai, C.H.; Liang, C.F.; Chen, Y.J.; Lin, C.C. A photo-cleavable biotin affinity tag for the facile release of a photo-crosslinked carbohydrate-binding protein. Bioorg. Med. Chem. 2016, 24, 1216–1224. [Google Scholar] [CrossRef]
- Kim, E.; Koo, H. Biomedical applications of copper-free click chemistry: In vitro, in vivo, and ex vivo. Chem. Sci. 2019, 10, 7835–7851. [Google Scholar] [CrossRef] [PubMed]
- Brodersen, N.; Arbuzova, A.; Herrmann, A.; Egger, H.; Liebscher, J. Synthesis of novel amphiphilic conjugates with a biological recognition function for developing targeted triggered liposomal delivery systems. Tetrahedron 2011, 67, 7763–7774. [Google Scholar] [CrossRef]
- Cheng, Z.; Wu, Y.; Xiong, Z.; Gambhir, S.S.; Chen, X. Near-infrared fluorescent RGD peptides for optical imaging of integrin αvβ3 expression in living mice. Bioconjug. Chem. 2005, 16, 1433–1441. [Google Scholar] [CrossRef] [PubMed]
- Ragupathi, G.; Koide, F.; Livingston, P.O.; Cho, Y.S.; Endo, A.; Wan, Q.; Spassova, M.K.; Keding, S.J.; Allen, J.; Ouerfelli, O.; et al. Preparation and evaluation of unimolecular pentavalent and hexavalent antigenic constructs targeting prostate and breast cancer: A synthetic route to anticancer vaccine candidates. J. Am. Chem. Soc. 2006, 128, 2715–2725. [Google Scholar] [CrossRef]
- Giacomelli, G.; Porcheddu, A.; De Luca, L. [1,3,5]-triazine: A versatile heterocycle in current applications of organic chemistry. Curr. Org. Chem. 2004, 8, 1497–1519. [Google Scholar] [CrossRef]
- Bretterbauer, K.; Schwarzinger, C. Melamine Derivatives—A Review on Synthesis and Application. Curr. Org. Synth. 2012, 9, 342–356. [Google Scholar] [CrossRef]
- Shinde, R.S. Review on Synthesis and Biological Study of Triazines Derivatives. United J. Chem. 2018, 1, 92–100. [Google Scholar]
- Blotny, G. Recent applications of 2,4,6-trichloro-1,3,5-triazine and its derivatives in organic synthesis. Tetrahedron 2006, 62, 9507–9522. [Google Scholar] [CrossRef]
- Thurston, J.T.; Schaefer, F.C.; Dudley, J.R.; Holmhansen, D. Cyanuric Chloride Derivatives. V. Reaction of Alkoxy-S-Triazines and Aryloxy-Ss-Triazines with Amines. J. Am. Chem. Soc. 1951, 73, 2992–2996. [Google Scholar] [CrossRef]
- Yamada, K.; Fujita, H.; Kunishima, M. A novel acid-catalyzed O-benzylating reagent with the smallest unit of imidate structure. Org. Lett. 2012, 14, 5026–5029. [Google Scholar] [CrossRef]
- Kunishima, M.; Kawachi, C.; Iwasaki, F.; Terao, K.; Tani, S. Synthesis and characterization of 4-(4,6-dimethoxy-1,3,5-triazin-2-yl)-4-methylmorpholinium chloride. Tetrahedron Lett. 1999, 40, 5327–5330. [Google Scholar] [CrossRef]
- Mibu, N.; Yokomizo, K.; Aki, H.; Ota, N.; Fujii, H.; Yuzuriha, A.; Saneyoshi, S.; Tanaka, A.; Koga, A.; Zhou, J.; et al. Synthesis and Antiviral Evaluation of Some C(3)-Symmetrical Trialkoxy-Substituted 1,3,5-Triazines and Their Molecular Geometry. Chem. Pharm. Bull. 2015, 63, 935–944. [Google Scholar] [CrossRef]
- Kunishima, M.; Ujigawa, T.; Nagaoka, Y.; Kawachi, C.; Hioki, K.; Shiro, M. Study on 1,3,5-triazine chemistry in dehydrocondensation: Gauche effect on the generation of active triazinylammonium species. Chem. Eur. J. 2012, 18, 15856–15867. [Google Scholar] [CrossRef]
- Cuthbertson, W.W.; Moffatt, J.S. 115. Contributions to the chemistry of synthetic antimalarials. Part VI. Some derivatives of 1:3:5-triazine. J. Chem. Soc. 1948, 561–564. [Google Scholar] [CrossRef]
- Goi, M. Reactivities of Cyanuric Chloride Derivatives. II Displacement Reactions of 2-Chloro-4-substituted-6-anilino-S-triazines with Benzylamine. J. Synth. Org. Chem. Jpn. 1960, 18, 332–336. [Google Scholar] [CrossRef]
- Fukushima, Y.; Hashida, Y.; Matsui, K. Substituent Effects in 1,3,5-Triazine Derivatives. Nippon. Kagaku Kaishi 1972, 629–634. [Google Scholar] [CrossRef]
- Hansch, C.; Leo, A.; Taft, R.W. A Survey of Hammett Substituent Constants and Resonance and Field Parameters. Chem. Rev. 1991, 91, 165–195. [Google Scholar] [CrossRef]
- Fries, H.H. XXXI.—Contributions to a knowledge of cyanuric derivatives. J. Chem. Soc. Trans. 1886, 49, 314–316. [Google Scholar] [CrossRef][Green Version]
- Diels, O. Zur Kenntniss der Cyanurverbindungen. Ber. Dtsch. Chem. Ges. 1899, 32, 691–702. [Google Scholar] [CrossRef]
- Steffensen, M.B.; Simanek, E.E. Chemoselective building blocks for dendrimers from relative reactivity data. Org. Lett. 2003, 5, 2359–2361. [Google Scholar] [CrossRef]
- Simanek, E.E.; Abdou, H.; Lalwani, S.; Lim, J.; Mintzer, M.; Venditto, V.J.; Vittur, B. The 8 year thicket of triazine dendrimers: Strategies, targets and applications. Proc. R. Soc. A Math. Phys. Eng. Sci. 2010, 466, 1445–1468. [Google Scholar] [CrossRef]
- Goi, M. Reactivities of Cyanuric Chloride Derivatives II Displacement Reactions of 2-Amino-4,6-dichloro-S-triazines with Aromatic Amines. J. Synth. Org. Chem. Jpn. 1960, 18, 337–342. [Google Scholar] [CrossRef]
- Moreno, K.X.; Simanek, E.E. Identification of Diamine Linkers with Differing Reactivity and their Application in the Synthesis of a Melamine Dendrimers. Tetrahedron Lett. 2008, 49, 1152–1154. [Google Scholar] [CrossRef]
- Sharma, A.; El-Faham, A.; de la Torre, B.G.; Albericio, F. Exploring the Orthogonal Chemoselectivity of 2,4,6-Trichloro-1,3,5-Triazine (TCT) as a Trifunctional Linker With Different Nucleophiles: Rules of the Game. Front. Chem. 2018, 6, 516–526. [Google Scholar] [CrossRef]
- Sharma, A.; Sheyi, R.; Kumar, A.; El-Faham, A.; de la Torre, B.G.; Albericio, F. Investigating Triorthogonal Chemoselectivity. Effect of Azide Substitution on the Triazine Core. Org. Lett. 2019, 21, 7888–7892. [Google Scholar] [CrossRef]
- Maes, B.; Sheyi, R.; Sharma, A.; Kumar, A.; El-Faham, A.; Torre, B.G.d.l.; Albericio, F. 1,3,5-Triazine as core for the preparation of dendrons. Arkivoc 2020, 2020, 64–73. [Google Scholar] [CrossRef]
- Sharma, A.; Kumar, A.; El-Faham, A.; de la Torre, B.G.; Albericio, F. Exploiting azido-dichloro-triazine as a linker for regioselective incorporation of peptides through their N, O, S functional groups. Bioorg. Chem. 2020, 104, 104334. [Google Scholar] [CrossRef]
- Fujita, H.; Zhang, Y.; Wu, Z.; Lindsey, J.S. Chromogenic agents built around a multifunctional double-triazine framework for enzymatically triggered cross-linking under physiological conditions. New J. Chem. 2020, 44, 3856–3867. [Google Scholar] [CrossRef]
- Bourguet, E.; Correia, I.; Dorgeret, B.; Chassaing, G.; Sicsic, S.; Ongeri, S. Synthesis and conformational studies of pseudopeptides containing an unsymmetrical triazine scaffold. J. Pept. Sci. 2008, 14, 596–609. [Google Scholar] [CrossRef]
- Amm, M.; Platzer, N.; Guilhem, J.; Bouchet, J.P.; Volland, J.P. Structural and conformational study of substituted triazines by N-15 NMR and x-ray analysis. Magn. Reson. Chem. 1998, 36, 587–596. [Google Scholar] [CrossRef]
- Katritzky, A.R.; Ghiviriga, I.; Steel, P.J.; Oniciu, D.C. Restricted rotations in 4,6-bis- and 2,4,6-tris-(N,N-dialkylamino)-s-triazines. J. Chem. Soc. Perk. Trans. 2 1996, 443–447. [Google Scholar] [CrossRef]
- Birkett, H.E.; Harris, R.K.; Hodgkinson, P.; Carr, K.; Charlton, M.H.; Cherryman, J.C.; Chippendale, A.M.; Glover, R.P. NMR studies of exchange between triazine rotamers. Magn. Reson. Chem. 2000, 38, 504–511. [Google Scholar] [CrossRef]
- Birkett, H.E.; Cherryman, J.C.; Chippendale, A.M.; Hazendonk, P.; Harris, R.K. Molecular modelling studies of side-chain rotation in substituted triazine rings. J. Mol. Struct. 2002, 602, 59–70. [Google Scholar] [CrossRef]
- Birkett, H.E.; Cherryman, J.C.; Chippendale, A.M.; Evans, J.S.O.; Harris, R.K.; James, M.; King, I.J.; McPherson, G.J. Structural investigations of three triazines: Solution-state NMR studies of internal rotation and structural information from solid-state NMR, plus a full structure determination from powder x-ray diffraction in one case. Magn. Reson. Chem. 2003, 41, 324–336. [Google Scholar] [CrossRef]
- Kunishima, M.; Asao, R.; Yamada, K.; Kitamura, M.; Fujita, H. Development of acid-catalyzed fluorous benzylating reagents based on a triazinedione core. J. Fluor. Chem. 2016, 190, 68–74. [Google Scholar] [CrossRef]
- Menicagli, R.; Malanga, C.; Peluso, P. Selective Mono- or Dialkoxylation of 2,4,6-Trichloro-1,3,5-triazine in Solid-Liquid Phase Transfer Conditions. Synth. Commun. 1994, 24, 2153–2158. [Google Scholar] [CrossRef]
- Fujita, H.; Hayakawa, N.; Kunishima, M. Study of the Reactivities of Acid-Catalyzed O-Benzylating Reagents Based on Structural Isomers of 1,3,5-Triazine. J. Org. Chem. 2015, 80, 11200–11205. [Google Scholar] [CrossRef] [PubMed]
- Mibu, N.; Yokomizo, K.; Koga, A.; Honda, M.; Mizokami, K.; Fujii, H.; Ota, N.; Yuzuriha, A.; Ishimaru, K.; Zhou, J.R.; et al. Synthesis and Antiviral Activities of Some 2,4,6-Trisubstituted 1,3,5-Triazines. Chem. Pharm. Bull. 2014, 62, 1032–1040. [Google Scholar] [CrossRef] [PubMed]
- Fujita, H.; Kakuyama, S.; Fukuyoshi, S.; Hayakawa, N.; Oda, A.; Kunishima, M. Triazine-Based Cationic Leaving Group: Synergistic Driving Forces for Rapid Formation of Carbocation Species. J. Org. Chem. 2018, 83, 4568–4580. [Google Scholar] [CrossRef]
- Robb, W.; Nicholson, C.G. Kinetics of Exchange-Reactions of Ethylenediamine with a Series of Cationic Rhodium(I)Complexes. S. Afr. J. Chem. 1978, 31, 1–5. [Google Scholar] [CrossRef]
- Clarke, K.; Rothwell, K. A Kinetic Study of the Effect of Substituents on the Rate of Formation of Alkylpyridinium Halides in Nitromethane Solution. J. Chem. Soc. 1960, 1885–1895. [Google Scholar] [CrossRef]
- Hall, H.K. Steric Effects on the Base Strengths of Cyclic Amines. J. Am. Chem. Soc. 1957, 79, 5444–5447. [Google Scholar] [CrossRef]
- Fujii, T.; Nishida, H.; Abiru, Y.; Yamamoto, M.; Kise, M. Studies on Synthesis of the Antibacterial Agent Nm441. II. Selection of a Suitable Base for Alkylation of 1-Substituted Piperazine with 4-(Bromomethyl)-5-methyl-1,3-dioxol-2-one. Chem. Pharm. Bull. 1995, 43, 1872–1877. [Google Scholar] [CrossRef]
- Brown, H.C.; Kanner, B. Preparation and Reactions of 2,6-Di-t-butylpyridine and Related Hindered Bases. A Case of Steric Hindrance toward the Proton. J. Am. Chem. Soc. 1966, 88, 986–992. [Google Scholar] [CrossRef]
- Kober, E.; Ratz, R. Reaction of Tertiary Amines with Halo-S-Triazines and Halopyrimidines. J. Org. Chem. 1962, 27, 2509–2514. [Google Scholar] [CrossRef]
- Kolesinska, B.; Kaminski, Z.J. The umpolung of substituent effect in nucleophilic aromatic substitution. A new approach to the synthesis of N,N-disubstituted melamines (triazine triskelions) under mild reaction conditions. Tetrahedron 2009, 65, 3573–3576. [Google Scholar] [CrossRef]
- Mannion, J.C.; Dax, S.L.; Colder, F.J.; Macintyre, D.E.; Mcleod, J.; Ozola, V.; Suna, E.; Shubin, K.; Mencel, J.J.; Peng, S.X. Novel Orally Bioavailable Breathing Control Modulating Compounds, and Methods of Using Same. International Patent Application No WO2014078575A2, 22 May 2014. [Google Scholar]
- Fujita, H.; Dou, J.; Matsumoto, N.; Wu, Z.; Lindsey, J.S. Enzymatically triggered chromogenic cross-linking agents under physiological conditions. New J. Chem. 2020, 44, 719–743. [Google Scholar] [CrossRef]
- Brahimi, F.; Liu, J.; Malakhov, A.; Chowdhury, S.; Purisima, E.O.; Ivanisevic, L.; Caron, A.; Burgess, K.; Saragovi, H.U. A monovalent agonist of TrkA tyrosine kinase receptors can be converted into a bivalent antagonist. Biochim. Biophys. Acta 2010, 1800, 1018–1026. [Google Scholar] [CrossRef]
- Calvete, M.J.F.; Pinto, S.M.A.; Burrows, H.D.; Castro, M.M.C.A.; Geraldes, C.F.G.C.; Pereira, M.M. Multifunctionalization of cyanuric chloride for the stepwise synthesis of potential multimodal imaging chemical entities. Arabian J. Chem. 2020, 13, 2517–2525. [Google Scholar] [CrossRef]
- Gonzalez, P.; Pota, K.; Turan, L.S.; da Costa, V.C.P.; Akkaraju, G.; Green, K.N. Synthesis, Characterization, and Activity of a Triazine Bridged Antioxidant Small Molecule. ACS Chem. Neurosci. 2017, 8, 2414–2423. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Chung, B.; Ko, D.; Lim, H.S. A solid-phase method for synthesis of dimeric and trimeric ligands: Identification of potent bivalent ligands of 14-3-3sigma. Bioorg. Chem. 2019, 91, 103141. [Google Scholar] [CrossRef]
- Lönnberg, T.; Hutchinson, M.; Rokita, S. Selective Alkylation of C-Rich Bulge Motifs in Nucleic Acids by Quinone Methide Derivatives. Chem. Eur. J. 2015, 21, 13127–13136. [Google Scholar] [CrossRef]
- Li, H.; Zhou, H.; Krieger, S.; Parry, J.J.; Whittenberg, J.J.; Desai, A.V.; Rogers, B.E.; Kenis, P.J.; Reichert, D.E. Triazine-based tool box for developing peptidic PET imaging probes: Syntheses, microfluidic radiolabeling, and structure-activity evaluation. Bioconjug. Chem. 2014, 25, 761–772. [Google Scholar] [CrossRef]
- Zong, H.; Goonewardena, S.N.; Chang, H.N.; Otis, J.B.; Baker, J.R., Jr. Sequential and parallel dual labeling of nanoparticles using click chemistry. Bioorg. Med. Chem. 2014, 22, 6288–6296. [Google Scholar] [CrossRef] [PubMed]
- Haldón, E.; Nicasio, M.C.; Perez, P.J. Copper-catalysed azide-alkyne cycloadditions (CuAAC): An update. Org. Biomol. Chem. 2015, 13, 9528–9550. [Google Scholar] [CrossRef]
- Meldal, M.; Diness, F. Recent Fascinating Aspects of the CuAAC Click Reaction. Trends Chem. 2020, 2, 569–584. [Google Scholar] [CrossRef]
- Fantoni, N.Z.; El-Sagheer, A.H.; Brown, T. A Hitchhiker’s Guide to Click-Chemistry with Nucleic Acids. Chem. Rev. 2021. [Google Scholar] [CrossRef] [PubMed]
- Gavrilyuk, J.I.; Wuellner, U.; Salahuddin, S.; Goswami, R.K.; Sinha, S.C.; Barbas, C.F., 3rd. An efficient chemical approach to bispecific antibodies and antibodies of high valency. Bioorg. Med. Chem. Lett. 2009, 19, 3716–3720. [Google Scholar] [CrossRef] [PubMed]
- Gnjatic, S.; Sawhney, N.B.; Bhardwaj, N. Toll-like receptor agonists: Are they good adjuvants? Cancer J. 2010, 16, 382–391. [Google Scholar] [CrossRef] [PubMed]
- Kirtland, M.E.; Tsitoura, D.C.; Durham, S.R.; Shamji, M.H. Toll-Like Receptor Agonists as Adjuvants for Allergen Immunotherapy. Front. Immunol. 2020, 11, 599083. [Google Scholar] [CrossRef] [PubMed]
- Tom, J.K.; Dotsey, E.Y.; Wong, H.Y.; Stutts, L.; Moore, T.; Davies, D.H.; Felgner, P.L.; Esser-Kahn, A.P. Modulation of Innate Immune Responses via Covalently Linked TLR Agonists. ACS Cent. Sci. 2015, 1, 439–448. [Google Scholar] [CrossRef] [PubMed]
- Kuhri, S.; Charalambidis, G.; Angaridis, P.A.; Lazarides, T.; Pagona, G.; Tagmatarchis, N.; Coutsolelos, A.G.; Guldi, D.M. A new approach for the photosynthetic antenna-reaction center complex with a model organized around an s-triazine linker. Chem. Eur. J. 2014, 20, 2049–2057. [Google Scholar] [CrossRef]
- Wang, T.; Vineberg, J.G.; Honda, T.; Ojima, I. Design and synthesis of tumor-targeting theranostic drug conjugates for SPECT and PET imaging studies. Bioorg. Chem. 2018, 76, 458–467. [Google Scholar] [CrossRef]
- Vineberg, J.G.; Wang, T.; Zuniga, E.S.; Ojima, I. Design, synthesis, and biological evaluation of theranostic vitamin-linker-taxoid conjugates. J. Med. Chem. 2015, 58, 2406–2416. [Google Scholar] [CrossRef]
- Vineberg, J.G.; Zuniga, E.S.; Kamath, A.; Chen, Y.J.; Seitz, J.D.; Ojima, I. Design, synthesis, and biological evaluations of tumor-targeting dual-warhead conjugates for a taxoid-camptothecin combination chemotherapy. J. Med. Chem. 2014, 57, 5777–5791. [Google Scholar] [CrossRef]
- Lee, C.; Ji, K.; Simanek, E.E. Functionalization of a Triazine Dendrimer Presenting Four Maleimides on the Periphery and a DOTA Group at the Core. Molecules 2016, 21, 335. [Google Scholar] [CrossRef]
- Albin, T.J.; Tom, J.K.; Manna, S.; Gilkes, A.P.; Stetkevich, S.A.; Katz, B.B.; Supnet, M.; Felgner, J.; Jain, A.; Nakajima, R.; et al. Linked Toll-Like Receptor Triagonists Stimulate Distinct, Combination-Dependent Innate Immune Responses. ACS Cent. Sci 2019, 5, 1137–1145. [Google Scholar] [CrossRef] [PubMed]
- Gilkes, A.P.; Albin, T.J.; Manna, S.; Supnet, M.; Ruiz, S.; Tom, J.; Badten, A.J.; Jain, A.; Nakajima, R.; Felgner, J.; et al. Tuning Subunit Vaccines with Novel TLR Triagonist Adjuvants to Generate Protective Immune Responses against Coxiella burnetii. J. Immunol. 2020, 204, 611–621. [Google Scholar] [CrossRef] [PubMed]
- Walsh, S.J.; Omarjee, S.; Galloway, W.; Kwan, T.T.; Sore, H.F.; Parker, J.S.; Hyvonen, M.; Carroll, J.S.; Spring, D.R. A general approach for the site-selective modification of native proteins, enabling the generation of stable and functional antibody-drug conjugates. Chem. Sci. 2019, 10, 694–700. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.G.; Huangfu, S.Y.; Patil, S.; Tang, Q.; Sun, W.; Li, Y.; Lu, Z.L.; Qian, A. [12]aneN3-based multifunctional compounds as fluorescent probes and nucleic acids delivering agents. Drug Deliv. 2020, 27, 66–80. [Google Scholar] [CrossRef] [PubMed]
- Khongorzul, P.; Ling, C.J.; Khan, F.U.; Ihsan, A.U.; Zhang, J. Antibody-Drug Conjugates: A Comprehensive Review. Mol. Cancer Res. 2020, 18, 3–19. [Google Scholar] [CrossRef] [PubMed]
- Tsuchikama, K.; An, Z. Antibody-drug conjugates: Recent advances in conjugation and linker chemistries. Protein Cell 2016, 9, 33–46. [Google Scholar] [CrossRef]
- Tiberghien, A.C.; Gregson, S.J.; Masterson, L.A.; Levy, J.-N.; Kemp, G.C.; Adams, L.R.; Patel, N.V.; Howard, P.W. An optimised synthesis of SG3376, a non-cleavable antibody-drug conjugate pyrrolobenzodiazepine drug-linker. Tetrahedron Lett. 2017, 58, 4363–4366. [Google Scholar] [CrossRef]
- Baker, M.B.; Ghiviriga, I.; Castellano, R.K. Molecular multifunctionalization via electronically coupled lactones. Chem. Sci. 2012, 3, 1095–1099. [Google Scholar] [CrossRef]
- Baker, M.B.; Ferreira, R.B.; Tasseroul, J.; Lampkins, A.J.; Al Abbas, A.; Abboud, K.A.; Castellano, R.K. Selective and Sequential Aminolysis of Benzotrifuranone: Synergism of Electronic Effects and Ring Strain Gradient. J. Org. Chem. 2016, 81, 9279–9288. [Google Scholar] [CrossRef]
- Li, H.; Homan, E.A.; Lampkins, A.J.; Ghiviriga, I.; Castellano, R.K. Synthesis and self-assembly of functionalized donor-sigma-acceptor molecules. Org. Lett. 2005, 7, 443–446. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Lampkins, A.J.; Baker, M.B.; Sumpter, B.G.; Huang, J.; Abboud, K.A.; Castellano, R.K. Benzotrifuranone: Synthesis, structure, and access to polycyclic heteroaromatics. Org. Lett. 2009, 11, 4314–4317. [Google Scholar] [CrossRef] [PubMed]
- Longin, O.; van de Langemheen, H.; Gannon, S.; Ward, D.; Liskamp, R.M.J. Synthesis of tris-tertiary amine CycloTriVeratrilene (TACTV) derivatives as water soluble pre-organized three aromatic ring containing molecular scaffolds for the construction of protein mimics. Tetrahedron Lett. 2019, 60, 151245–151249. [Google Scholar] [CrossRef]
- Longin, O.; Hezwani, M.; van de Langemheen, H.; Liskamp, R.M.J. Synthetic antibody protein mimics of infliximab by molecular scaffolding on novel CycloTriVeratrilene (CTV) derivatives. Org. Biomol. Chem. 2018, 16, 5254–5274. [Google Scholar] [CrossRef] [PubMed]
- Pícha, J.; Fabre, B.; Buděšínský, M.; Hajduch, J.; Abdellaoui, M.; Jiráček, J. Tri-Orthogonal Scaffolds for the Solid-Phase Synthesis of Peptides. Eur. J. Org. Chem. 2018, 2018, 5180–5192. [Google Scholar] [CrossRef]
- Watzke, A.; Gutierrez-Rodriguez, M.; Kohn, M.; Wacker, R.; Schroeder, H.; Breinbauer, R.; Kuhlmann, J.; Alexandrov, K.; Niemeyer, C.M.; Goody, R.S.; et al. A generic building block for C- and N-terminal protein-labeling and protein-immobilization. Bioorg. Med. Chem. 2006, 14, 6288–6306. [Google Scholar] [CrossRef]
- Viault, G.; Dautrey, S.; Maindron, N.; Hardouin, J.; Renard, P.Y.; Romieu, A. The first “ready-to-use” benzene-based heterotrifunctional cross-linker for multiple bioconjugation. Org. Biomol. Chem. 2013, 11, 2693–2705. [Google Scholar] [CrossRef]
- Yoshida, S.; Kanno, K.; Kii, I.; Misawa, Y.; Hagiwara, M.; Hosoya, T. Convergent synthesis of trifunctional molecules by three sequential azido-type-selective cycloadditions. Chem. Commun. 2018, 54, 3705–3708. [Google Scholar] [CrossRef]
- Hospital, A.; Gibard, C.; Gaulier, C.; Nauton, L.; Thery, V.; El-Ghozzi, M.; Avignant, D.; Cisnetti, F.; Gautier, A. Access to functionalised silver(I) and gold(I) N-heterocyclic carbenes by [2 + 3] dipolar cycloadditions. Dalton Trans. 2012, 41, 6803–6812. [Google Scholar] [CrossRef]
- Yoshida, S.; Misawa, Y.; Hosoya, T. Formal C-H-Azidation—Based Shortcut to Diazido Building Blocks for the Versatile Preparation of Photoaffinity Labeling Probes. Eur. J. Org. Chem. 2014, 2014, 3991–3995. [Google Scholar] [CrossRef]
- Wagner, R.W.; Lindsey, J.S. Boron-Dipyrromethene Dyes for Incorporation in Synthetic Multi-Pigment Light-Harvesting Arrays. Pure Appl. Chem. 1996, 68, 1373–1380. [Google Scholar] [CrossRef]
- Azov, V.A.; Schlegel, A.; Diederich, F. Functionalized Calix[4]resorcinarene Cavitands. Versatile Platforms for the Modular Construction of Extended Molecular Switches. Bull. Chem. Soc. Jpn. 2006, 79, 1926–1940. [Google Scholar] [CrossRef]
- Yoshida, S.; Sakata, Y.; Misawa, Y.; Morita, T.; Kuribara, T.; Ito, H.; Koike, Y.; Kii, I.; Hosoya, T. Assembly of four modules onto a tetraazide platform by consecutive 1,2,3-triazole formations. Chem. Commun. 2021, 57, 899–902. [Google Scholar] [CrossRef] [PubMed]
- Ballell, L.; van Scherpenzeel, M.; Buchalova, K.; Liskamp, R.M.; Pieters, R.J. A new chemical probe for the detection of the cancer-linked galectin-3. Org. Biomol. Chem. 2006, 4, 4387–4394. [Google Scholar] [CrossRef]
- Rashidian, M.; Kumarapperuma, S.C.; Gabrielse, K.; Fegan, A.; Wagner, C.R.; Distefano, M.D. Simultaneous dual protein labeling using a triorthogonal reagent. J. Am. Chem. Soc. 2013, 135, 16388–16396. [Google Scholar] [CrossRef]
- Rashidian, M.; Song, J.M.; Pricer, R.E.; Distefano, M.D. Chemoenzymatic reversible immobilization and labeling of proteins without prior purification. J. Am. Chem. Soc. 2012, 134, 8455–8467. [Google Scholar] [CrossRef]
- Gaon, I.; Turek, T.C.; Distefano, M.D. Farnesyl and geranylgeranyl pyrophosphate analogs incorporating benzoylbenzyl ethers: Synthesis and inhibition of yeast protein farnesyltransferase. Tetrahedron Lett. 1996, 37, 8833–8836. [Google Scholar] [CrossRef]
- Fegan, A.; Kumarapperuma, S.C.; Wagner, C.R. Chemically self-assembled antibody nanostructures as potential drug carriers. Mol. Pharm. 2012, 9, 3218–3227. [Google Scholar] [CrossRef]
- Wu, Y.X.; Zhang, X.B.; Li, J.B.; Zhang, C.C.; Liang, H.; Mao, G.J.; Zhou, L.Y.; Tan, W.; Yu, R.Q. Bispyrene-fluorescein hybrid based FRET cassette: A convenient platform toward ratiometric time-resolved probe for bioanalytical applications. Anal. Chem. 2014, 86, 10389–10396. [Google Scholar] [CrossRef]
- Lamos, S.M.; Krusemark, C.J.; McGee, C.J.; Scalf, M.; Smith, L.M.; Belshaw, P.J. Mixed isotope photoaffinity reagents for identification of small-molecule targets by mass spectrometry. Angew. Chem. Int. Ed. 2006, 45, 4329–4333. [Google Scholar] [CrossRef]
- Vall-Sagarra, A.; Litau, S.; Decristoforo, C.; Wangler, B.; Schirrmacher, R.; Fricker, G.; Wangler, C. Design, Synthesis, In Vitro, and Initial In Vivo Evaluation of Heterobivalent Peptidic Ligands Targeting Both NPY(Y(1))- and GRP-Receptors–An Improvement for Breast Cancer Imaging? Pharmaceuticals 2018, 11, 65. [Google Scholar] [CrossRef] [PubMed]
- Yokoi, T.; Ueda, T.; Tanimoto, H.; Morimoto, T.; Kakiuchi, K. Site-selective conversion of azido groups at carbonyl alpha-positions into oxime groups leading triazide to a triple click conjugation scaffold. Chem. Commun. 2019, 55, 1891–1894. [Google Scholar] [CrossRef]
- Yokoi, T.; Tanimoto, H.; Ueda, T.; Morimoto, T.; Kakiuchi, K. Site-Selective Conversion of Azido Groups at Carbonyl alpha-Positions to Diazo Groups in Diazido and Triazido Compounds. J. Org. Chem. 2018, 83, 12103–12121. [Google Scholar] [CrossRef]
- Daghish, M.; Hennig, L.; Findeisen, M.; Giesa, S.; Schumer, F.; Hennig, H.; Beck-Sickinger, A.G.; Welzel, P. Tetrafunctional photoaffinity labels based on Nakanish’s m-nitroalkoxy-substituted phenyltrifluoromethyldiazirine. Angew. Chem. Int. Ed. 2002, 41, 2293–2297. [Google Scholar] [CrossRef]
- Sorkin, M.R.; Walker, J.A.; Brown, J.S.; Alabi, C.A. Versatile Platform for the Synthesis of Orthogonally Cleavable Heteromultifunctional Cross-Linkers. Bioconjug. Chem. 2017, 28, 907–912. [Google Scholar] [CrossRef]
- Cline, L.L.; Janout, V.; Fisher, M.; Juliano, R.L.; Regen, S.L. A molecular umbrella approach to the intracellular delivery of small interfering RNA. Bioconjug. Chem. 2011, 22, 2210–2216. [Google Scholar] [CrossRef]
- Werkhoven, P.R.; van de Langemheen, H.; van der Wal, S.; Kruijtzer, J.A.; Liskamp, R.M. Versatile convergent synthesis of a three peptide loop containing protein mimic of whooping cough pertactin by successive Cu(I)-catalyzed azide alkyne cycloaddition on an orthogonal alkyne functionalized TAC-scaffold. J. Pept. Sci. 2014, 20, 235–239. [Google Scholar] [CrossRef]
- Dai, Y.; Weng, J.; George, J.; Chen, H.; Lin, Q.; Wang, J.; Royzen, M.; Zhang, Q. Three-Component Protein Modification Using Mercaptobenzaldehyde Derivatives. Org. Lett. 2019, 21, 3828–3833. [Google Scholar] [CrossRef]
- Haley, C.A.C.; Maitland, P. Organic Reactions in Aqueous Solution at Room Temperature. Part 1. The Influence of pH on Condensations Involving the Linking of Carbon to Nitrogen and of Carbon to Carbon. J. Chem. Soc. 1951, 3155–3174. [Google Scholar] [CrossRef]
- Medley, J.W.; Movassaghi, M. Robinson’s landmark synthesis of tropinone. Chem. Commun. 2013, 49, 10775–10777. [Google Scholar] [CrossRef] [PubMed]
- Boutureira, O.; Bernardes, G.J. Advances in chemical protein modification. Chem. Rev. 2015, 115, 2174–2195. [Google Scholar] [CrossRef]
- Shadish, J.A.; DeForest, C.A. Site-Selective Protein Modification: From Functionalized Proteins to Functional Biomaterials. Matter 2020, 2, 50–77. [Google Scholar] [CrossRef]
- Maruani, A.; Richards, D.A.; Chudasama, V. Dual modification of biomolecules. Org. Biomol. Chem. 2016, 14, 6165–6178. [Google Scholar] [CrossRef]
- Tessier, R.; Ceballos, J.; Guidotti, N.; Simonet-Davin, R.; Fierz, B.; Waser, J. “Doubly Orthogonal” Labeling of Peptides and Proteins. Chem 2019, 5, 2243–2263. [Google Scholar] [CrossRef]
- Baslé, E.; Joubert, N.; Pucheault, M. Protein chemical modification on endogenous amino acids. Chem. Biol. 2010, 17, 213–227. [Google Scholar] [CrossRef] [PubMed]
- Spicer, C.D.; Davis, B.G. Selective chemical protein modification. Nat. Commun. 2014, 5, 4740–4753. [Google Scholar] [CrossRef] [PubMed]
- Ramesh, S.; Cherkupally, P.; Govender, T.; Kruger, H.G.; Albericio, F.; de la Torre, B.G. Highly chemoselective ligation of thiol- and amino-peptides on a bromomaleimide core. Chem. Commun. 2016, 52, 2334–2337. [Google Scholar] [CrossRef]
- Gil de Montes, E.; Jimenez-Moreno, E.; Oliveira, B.L.; Navo, C.D.; Cal, P.; Jimenez-Oses, G.; Robina, I.; Moreno-Vargas, A.J.; Bernardes, G.J.L. Azabicyclic vinyl sulfones for residue-specific dual protein labelling. Chem. Sci. 2019, 10, 4515–4522. [Google Scholar] [CrossRef]
- Morales-Sanfrutos, J.; Lopez-Jaramillo, J.; Ortega-Munoz, M.; Megia-Fernandez, A.; Perez-Balderas, F.; Hernandez-Mateo, F.; Santoyo-Gonzalez, F. Vinyl sulfone: A versatile function for simple bioconjugation and immobilization. Org. Biomol. Chem. 2010, 8, 667–675. [Google Scholar] [CrossRef]
- Lang, K.; Davis, L.; Torres-Kolbus, J.; Chou, C.; Deiters, A.; Chin, J.W. Genetically encoded norbornene directs site-specific cellular protein labelling via a rapid bioorthogonal reaction. Nat. Chem. 2012, 4, 298–304. [Google Scholar] [CrossRef] [PubMed]
- Mushtaq, S.; Yun, S.J.; Jeon, J. Recent Advances in Bioorthogonal Click Chemistry for Efficient Synthesis of Radiotracers and Radiopharmaceuticals. Molecules 2019, 24, 3567. [Google Scholar] [CrossRef]
- Koniev, O.; Wagner, A. Developments and recent advancements in the field of endogenous amino acid selective bond forming reactions for bioconjugation. Chem. Soc. Rev. 2015, 44, 5495–5551. [Google Scholar] [CrossRef]
- Gai, Y.; Xiang, G.; Ma, X.; Hui, W.; Ouyang, Q.; Sun, L.; Ding, J.; Sheng, J.; Zeng, D. Universal Molecular Scaffold for Facile Construction of Multivalent and Multimodal Imaging Probes. Bioconjug. Chem. 2016, 27, 515–520. [Google Scholar] [CrossRef]
- Hashimoto, M.; Hatanaka, Y.; Yang, J.; Dhesi, J.; Holman, G.D. Synthesis of biotinylated bis(D-glucose) derivatives for glucose transporter photoaffinity labelling. Carbohydr. Res. 2001, 331, 119–127. [Google Scholar] [CrossRef]
- Ji, C.; Miller, P.A.; Miller, M.J. Iron transport-mediated drug delivery: Practical syntheses and in vitro antibacterial studies of tris-catecholate siderophore-aminopenicillin conjugates reveals selectively potent antipseudomonal activity. J. Am. Chem. Soc. 2012, 134, 9898–9901. [Google Scholar] [CrossRef]
- Knall, A.C.; Hollauf, M.; Saf, R.; Slugovc, C. A trifunctional linker suitable for conducting three orthogonal click chemistries in one pot. Org. Biomol. Chem. 2016, 14, 10576–10580. [Google Scholar] [CrossRef] [PubMed]
- Knall, A.C.; Slugovc, C. Inverse electron demand Diels-Alder (iEDDA)-initiated conjugation: A (high) potential click chemistry scheme. Chem. Soc. Rev. 2013, 42, 5131–5142. [Google Scholar] [CrossRef]
- Pagel, M. Inverse electron demand Diels-Alder (IEDDA) reactions in peptide chemistry. J. Pept. Sci. 2019, 25, e3141. [Google Scholar] [CrossRef]
- Citron, C.A.; Wickel, S.M.; Schulz, B.; Draeger, S.; Dickschat, J.S. A Diels-Alder/Retro-Diels-Alder Approach for the Enantioselective Synthesis of Microbial Butenolides. Eur. J. Org. Chem. 2012, 6636–6646. [Google Scholar] [CrossRef]
- Gramlich, W.M.; Robertson, M.L.; Hillmyer, M.A. Reactive Compatibilization of Poly(l-lactide) and Conjugated Soybean Oil. Macromolecules 2010, 43, 2313–2321. [Google Scholar] [CrossRef]
- Ho, H.T.; Levere, M.E.; Fournier, D.; Montembault, V.; Pascual, S.; Fontaine, L. Introducing the Azlactone Functionality into Polymers through Controlled Radical Polymerization: Strategies and Recent Developments. Aust. J. Chem. 2012, 65, 970–977. [Google Scholar] [CrossRef]
- de Castro, P.P.; Carpanez, A.G.; Amarante, G.W. Azlactone Reaction Developments. Chem. Eur. J. 2016, 22, 10294–10318. [Google Scholar] [CrossRef]
- Yu, X.; Herberg, A.; Kuckling, D. Micellar Organocatalysis Using Smart Polymer Supports: Influence of Thermoresponsive Self-Assembly on Catalytic Activity. Polymers 2020, 12, 2265. [Google Scholar] [CrossRef]
- Pantin, M.; Caillé, J.; Boeda, F.; Fontaine, L.; Pearson-Long, M.S.M.; Bertus, P. Heteromultifunctional Oxazolones as Versatile Linkers for Click Chemistry Reactions. Eur. J. Org. Chem. 2019, 2019, 7359–7366. [Google Scholar] [CrossRef]
- Moss, J.A.; Stokols, S.; Hixon, M.S.; Ashley, F.T.; Chang, J.Y.; Janda, K.D. Solid-phase synthesis and kinetic characterization of fluorogenic enzyme-degradable hydrogel cross-linkers. Biomacromolecules 2006, 7, 1011–1016. [Google Scholar] [CrossRef] [PubMed]
- Masselin, A.; Petrelli, A.; Donzel, M.; Armand, S.; Cottaz, S.; Fort, S. Unprecedented Affinity Labeling of Carbohydrate-Binding Proteins with s-Triazinyl Glycosides. Bioconjug. Chem. 2019, 30, 2332–2339. [Google Scholar] [CrossRef]
- Blencowe, C.A.; Russell, A.T.; Greco, F.; Hayes, W.; Thornthwaite, D.W. Self-immolative linkers in polymeric delivery systems. Polym. Chem. 2011, 2, 773–790. [Google Scholar] [CrossRef]
- Gonzaga, R.V.; do Nascimento, L.A.; Santos, S.S.; Machado Sanches, B.A.; Giarolla, J.; Ferreira, E.I. Perspectives About Self-Immolative Drug Delivery Systems. J. Pharm. Sci. 2020, 109, 3262–3281. [Google Scholar] [CrossRef] [PubMed]
- Jeffrey, S.C.; De Brabander, J.; Miyamoto, J.; Senter, P.D. Expanded Utility of the beta-Glucuronide Linker: ADCs That Deliver Phenolic Cytotoxic Agents. ACS Med. Chem. Lett. 2010, 1, 277–280. [Google Scholar] [CrossRef]
- Kumar, R.; Han, J.; Lim, H.J.; Ren, W.X.; Lim, J.Y.; Kim, J.H.; Kim, J.S. Mitochondrial induced and self-monitored intrinsic apoptosis by antitumor theranostic prodrug: In vivo imaging and precise cancer treatment. J. Am. Chem. Soc. 2014, 136, 17836–17843. [Google Scholar] [CrossRef] [PubMed]
- Alsarraf, J.; Peraudeau, E.; Poinot, P.; Tranoy-Opalinski, I.; Clarhaut, J.; Renoux, B.; Papot, S. A dendritic beta-galactosidase-responsive folate-monomethylauristatin E conjugate. Chem. Commun. 2015, 51, 15792–15795. [Google Scholar] [CrossRef]
- Legigan, T.; Clarhaut, J.; Tranoy-Opalinski, I.; Monvoisin, A.; Renoux, B.; Thomas, M.; Le Pape, A.; Lerondel, S.; Papot, S. The first generation of beta-galactosidase-responsive prodrugs designed for the selective treatment of solid tumors in prodrug monotherapy. Angew. Chem. Int. Ed. 2012, 51, 11606–11610. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Zhou, X.-Y.; Wu, H.; Lei, Y.-Z.; Li, J.-H. Highly efficient reduction of nitro compounds: Recyclable Pd/C-catalyzed transfer hydrogenation with ammonium formate or hydrazine hydrate as hydrogen source. Synth. Commun. 2018, 48, 2475–2484. [Google Scholar] [CrossRef]
- Wiener, H.; Blum, J.; Sasson, Y. Studies on the Mechanism of Transfer Hydrogenation of Nitroarenes by Formate Salts Catalyzed by Pd/C. J. Org. Chem. 1991, 56, 4481–4486. [Google Scholar] [CrossRef]
- Erez, R.; Shabat, D. The azaquinone-methide elimination: Comparison study of 1,6- and 1,4-eliminations under physiological conditions. Org. Biomol. Chem. 2008, 6, 2669–2672. [Google Scholar] [CrossRef] [PubMed]
- Grinda, M.; Legigan, T.; Clarhaut, J.; Peraudeau, E.; Tranoy-Opalinski, I.; Renoux, B.; Thomas, M.; Guilhot, F.; Papot, S. An enzyme-responsive system programmed for the double release of bioactive molecules through an intracellular chemical amplification process. Org. Biomol. Chem. 2013, 11, 7129–7133. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, H.; Ogawa, M.; Alford, R.; Choyke, P.L.; Urano, Y. New strategies for fluorescent probe design in medical diagnostic imaging. Chem. Rev. 2010, 110, 2620–2640. [Google Scholar] [CrossRef]
- Peterson, J.D. Paradigms in Fluorescence Molecular Imaging: Maximizing Measurement of Biological Changes in Disease, Therapeutic Efficacy, and Toxicology/Safety. Mol. Imaging Biol. 2019, 21, 599–611. [Google Scholar] [CrossRef]
- Yan, R.; Ye, D. Molecular imaging of enzyme activity in vivo using activatable probes. Sci. Bull. 2016, 61, 1672–1679. [Google Scholar] [CrossRef]
- Liu, H.W.; Chen, L.; Xu, C.; Li, Z.; Zhang, H.; Zhang, X.B.; Tan, W. Recent progresses in small-molecule enzymatic fluorescent probes for cancer imaging. Chem. Soc. Rev. 2018, 47, 7140–7180. [Google Scholar] [CrossRef]
- Singh, H.; Tiwari, K.; Tiwari, R.; Pramanik, S.K.; Das, A. Small Molecule as Fluorescent Probes for Monitoring Intracellular Enzymatic Transformations. Chem. Rev. 2019, 119, 11718–11760. [Google Scholar] [CrossRef]
- Baruch, A.; Jeffery, D.A.; Bogyo, M. Enzyme activity—it’s all about image. Trends Cell Biol. 2004, 14, 29–35. [Google Scholar] [CrossRef] [PubMed]
- Martinon, F.; Tschopp, J. Inflammatory caspases: Linking an intracellular innate immune system to autoinflammatory diseases. Cell 2004, 117, 561–574. [Google Scholar] [CrossRef] [PubMed]
- Van Opdenbosch, N.; Lamkanfi, M. Caspases in Cell Death, Inflammation, and Disease. Immunity 2019, 50, 1352–1364. [Google Scholar] [CrossRef] [PubMed]
- Hu, M.; Li, L.; Wu, H.; Su, Y.; Yang, P.Y.; Uttamchandani, M.; Xu, Q.H.; Yao, S.Q. Multicolor, one- and two-photon imaging of enzymatic activities in live cells with fluorescently Quenched Activity-Based Probes (qABPs). J. Am. Chem. Soc. 2011, 133, 12009–12020. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Q.; Girish, A.; Chattopadhaya, S.; Yao, S.Q. Developing novel activity-based fluorescent probes that target different classes of proteases. Chem. Commun. 2004, 1512–1513. [Google Scholar] [CrossRef] [PubMed]
- Sellars, J.D.; Landrum, M.; Congreve, A.; Dixon, D.P.; Mosely, J.A.; Beeby, A.; Edwards, R.; Steel, P.G. Fluorescence quenched quinone methide based activity probes—A cautionary tale. Org. Biomol. Chem. 2010, 8, 1610–1618. [Google Scholar] [CrossRef]
- Gnaim, S.; Shabat, D. Quinone-methide species, a gateway to functional molecular systems: From self-immolative dendrimers to long-wavelength fluorescent dyes. Acc. Chem. Res. 2014, 47, 2970–2984. [Google Scholar] [CrossRef]
- Gnaim, S.; Shabat, D. Activity-Based Optical Sensing Enabled by Self-Immolative Scaffolds: Monitoring of Release Events by Fluorescence or Chemiluminescence Output. Acc. Chem. Res. 2019, 52, 2806–2817. [Google Scholar] [CrossRef]
- Chevalier, A.; Hardouin, J.; Renard, P.Y.; Romieu, A. Universal dark quencher based on “clicked” spectrally distinct azo dyes. Org. Lett. 2013, 15, 6082–6085. [Google Scholar] [CrossRef]
- Huang, J.; Li, J.; Lyu, Y.; Miao, Q.; Pu, K. Molecular optical imaging probes for early diagnosis of drug-induced acute kidney injury. Nat. Mater. 2019, 18, 1133–1143. [Google Scholar] [CrossRef] [PubMed]
- Arja, K.; Elgland, M.; Appelqvist, H.; Konradsson, P.; Lindgren, M.; Nilsson, K.P.R. Synthesis and Characterization of Novel Fluoro-glycosylated Porphyrins that can be Utilized as Theranostic Agents. ChemistryOpen 2018, 7, 495–503. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Deng, J.; Guo, D.; Sun, Q.; Wang, Z.; Wang, K.; Wu, F. Mitochondria-targeting Pt/Mn porphyrins as efficient photosensitizers for magnetic resonance imaging and photodynamic therapy. Dyes Pigment. 2019, 166, 189–195. [Google Scholar] [CrossRef]
- Lindsey, J.S. Synthesis of meso-Substituted Porphyrins. In The Porphyrin Handbook; Kadish, K.M., Smith, K.M., Guilard, R., Eds.; Academic Press: San Diego, CA, USA, 2000; Volume 1, pp. 45–118. [Google Scholar]
- Taniguchi, M.; Lindsey, J.S. Enumeration of Isomers of Substituted Tetrapyrrole Macrocycles: From Classical Problems in Biology to Modern Combinatorial Libraries. In Handbook of Porphyrin Science; Kadish, K.M., Smith, K.M., Guilard, R., Eds.; World Scientific: Singapore, 2012; Volume 23, pp. 1–80. [Google Scholar]
- Yang, J.; Yang, Y.W. Metal-Organic Frameworks for Biomedical Applications. Small 2020, 16, e1906846. [Google Scholar] [CrossRef] [PubMed]
- Pliquett, J.; Amor, S.; Ponce-Vargas, M.; Laly, M.; Racoeur, C.; Rousselin, Y.; Denat, F.; Bettaieb, A.; Fleurat-Lessard, P.; Paul, C.; et al. Design of a multifunctionalizable BODIPY platform for the facile elaboration of a large series of gold(I)-based optical theranostics. Dalton Trans. 2018, 47, 11203–11218. [Google Scholar] [CrossRef] [PubMed]
- Laine, M.; Barbosa, N.A.; Kochel, A.; Osiecka, B.; Szewczyk, G.; Sarna, T.; Ziółkowski, P.; Wieczorek, R.; Filarowski, A. Synthesis, structural, spectroscopic, computational and cytotoxic studies of BODIPY dyes. Sens. Actuators B Chem. 2017, 238, 548–555. [Google Scholar] [CrossRef]
- Gryko, D.; Li, J.; Diers, J.R.; Roth, K.M.; Bocian, D.F.; Kuhr, W.G.; Lindsey, J.S. Studies Related to the Design and Synthesis of a Molecular Octal Counter. J. Mater. Chem. 2001, 11, 1162–1180. [Google Scholar] [CrossRef]
- Li, J.; Ambroise, A.; Yang, S.I.; Diers, J.R.; Seth, J.; Wack, C.R.; Bocian, D.F.; Holten, D.; Lindsey, J.S. Template-Directed Synthesis, Excited-State Photodynamics, and Electronic Communication in a Hexameric Wheel of Porphyrins. J. Am. Chem. Soc. 1999, 121, 8927–8940. [Google Scholar] [CrossRef]
- Ambroise, A.; Li, J.; Yu, L.; Lindsey, J.S. A Self-Assembled Light-Harvesting Array of Seven Porphyrins in a Wheel and Spoke Architecture. Org. Lett. 2000, 2, 2563–2566. [Google Scholar] [CrossRef]
- Yu, L.; Lindsey, J.S. Rational Syntheses of Cyclic Hexameric Porphyrin Arrays for Studies of Self-Assembling Light-Harvesting Systems. J. Org. Chem. 2001, 66, 7402–7419. [Google Scholar] [CrossRef]
- Tomizaki, K.-y.; Yu, L.; Wei, L.; Bocian, D.F.; Lindsey, J.S. Synthesis of Cyclic Hexameric Porphyrin Arrays. Anchors for Surface Immobilization and Columnar Self-Assembly. J. Org. Chem. 2003, 68, 8199–8207. [Google Scholar] [CrossRef]
- Lindsey, J.S. Synthetic Routes to meso-Patterned Porphyrins. Acc. Chem. Res. 2010, 43, 300–311. [Google Scholar] [CrossRef]
- Holten, D.; Bocian, D.F.; Lindsey, J.S. Probing Electronic Communication in Covalently Linked Multiporphyrin Arrays. A Guide to the Rational Design of Molecular Photonic Devices. Acc. Chem. Res. 2002, 35, 57–69. [Google Scholar] [CrossRef]
- Song, H.-e.; Kirmaier, C.; Schwartz, J.K.; Hindin, E.; Yu, L.; Bocian, D.F.; Lindsey, J.S.; Holten, D. Mechanisms, Pathways, and Dynamics of Excited-State Energy Flow in Self-Assembled Wheel-and-Spoke Light-Harvesting Architectures. J. Phys. Chem. B 2006, 110, 19121–19130. [Google Scholar] [CrossRef]
- Song, H.-e.; Kirmaier, C.; Schwartz, J.K.; Hindin, E.; Yu, L.H.; Bocian, D.F.; Lindsey, J.S.; Holten, D. Effects of Multiple Pathways on Excited-State Energy Flow in Self-Assembled Wheel-and-Spoke Light-Harvesting Architectures. J. Phys. Chem. B 2006, 110, 19131–19139. [Google Scholar] [CrossRef]
- Tiede, D.M.; Zhang, R.; Chen, L.X.; Yu, L.; Lindsey, J.S. Structural Characterization of Modular Supramolecular Architectures in Solution. J. Am. Chem. Soc. 2004, 126, 14054–14062. [Google Scholar] [CrossRef]
- Mardis, K.L.; Sutton, H.M.; Zuo, X.; Lindsey, J.S.; Tiede, D.M. Solution-State Conformational Ensemble of a Hexameric Porphyrin Array Characterized Using Molecular Dynamics and X-Ray Scattering. J. Phys. Chem. A 2009, 113, 2516–2523. [Google Scholar] [CrossRef][Green Version]
- Geier, G.R., III; Callinan, J.B.; Rao, P.D.; Lindsey, J.S. A Survey of Acid Catalysts in Dipyrromethanecarbinol Condensations Leading to meso-Substituted Porphyrins. J. Porphyr. Phthalocyanines 2001, 5, 810–823. [Google Scholar] [CrossRef]
- Senge, M.O. Stirring the porphyrin alphabet soup–functionalization reactions for porphyrins. Chem. Commun. 2011, 47, 1943–1960. [Google Scholar] [CrossRef] [PubMed]
- Laha, J.K.; Dhanalekshmi, S.; Taniguchi, M.; Ambroise, A.; Lindsey, J.S. A Scalable Synthesis of Meso-Substituted Dipyrromethanes. Org. Process Res. Dev. 2003, 7, 799–812. [Google Scholar] [CrossRef]













































































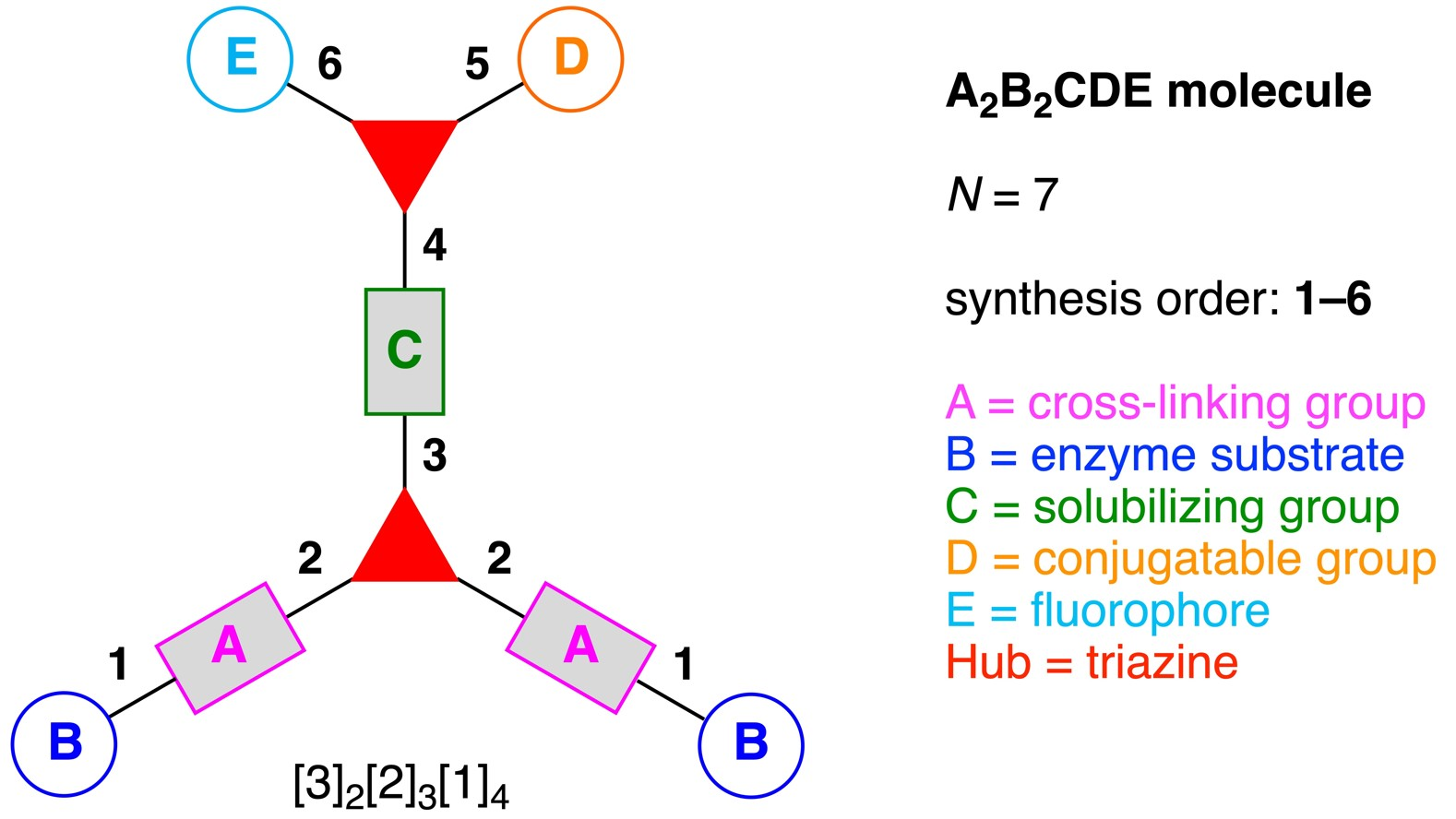{kind=link}
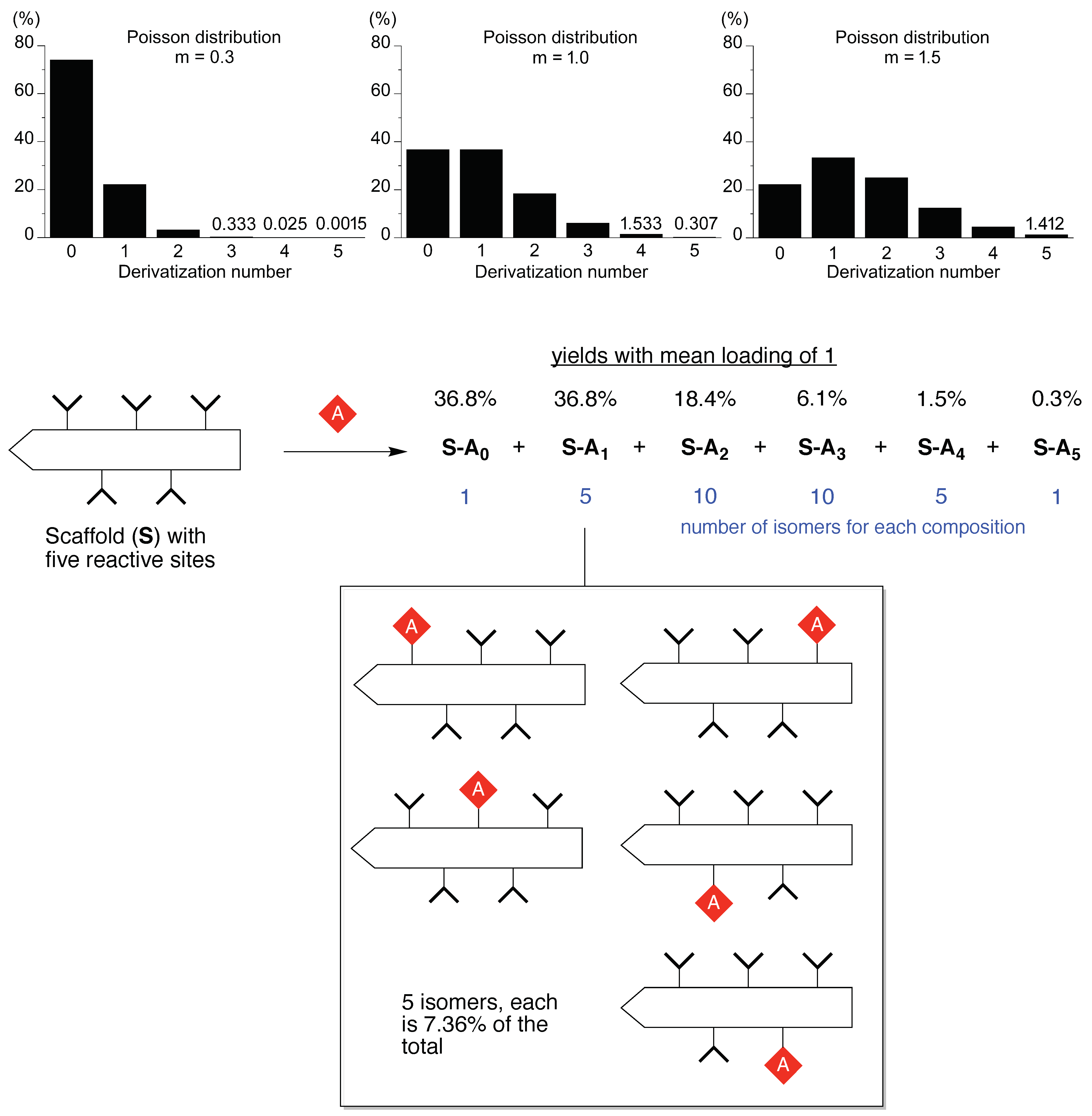{kind=link}
{kind=link}
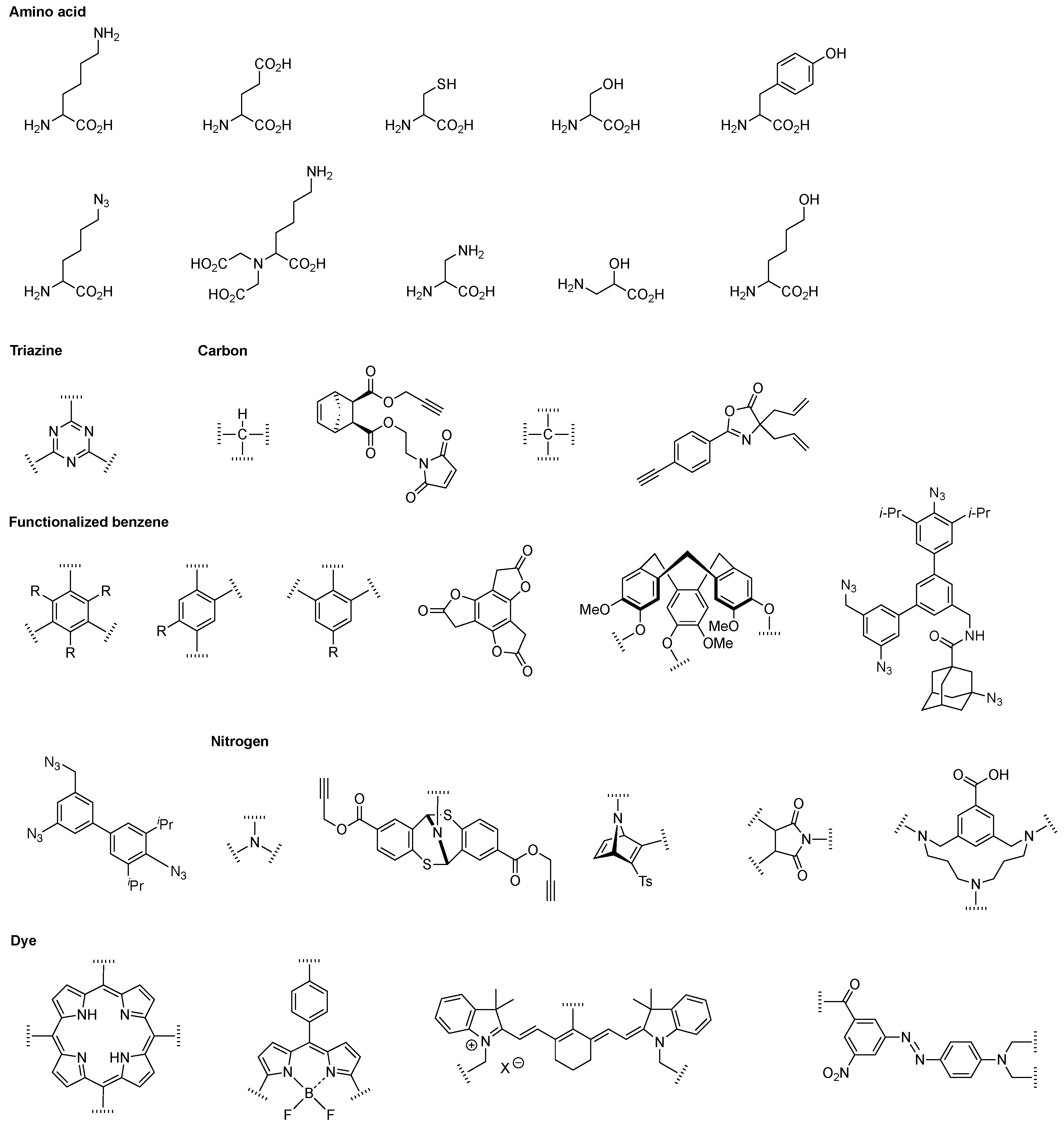{kind=link}
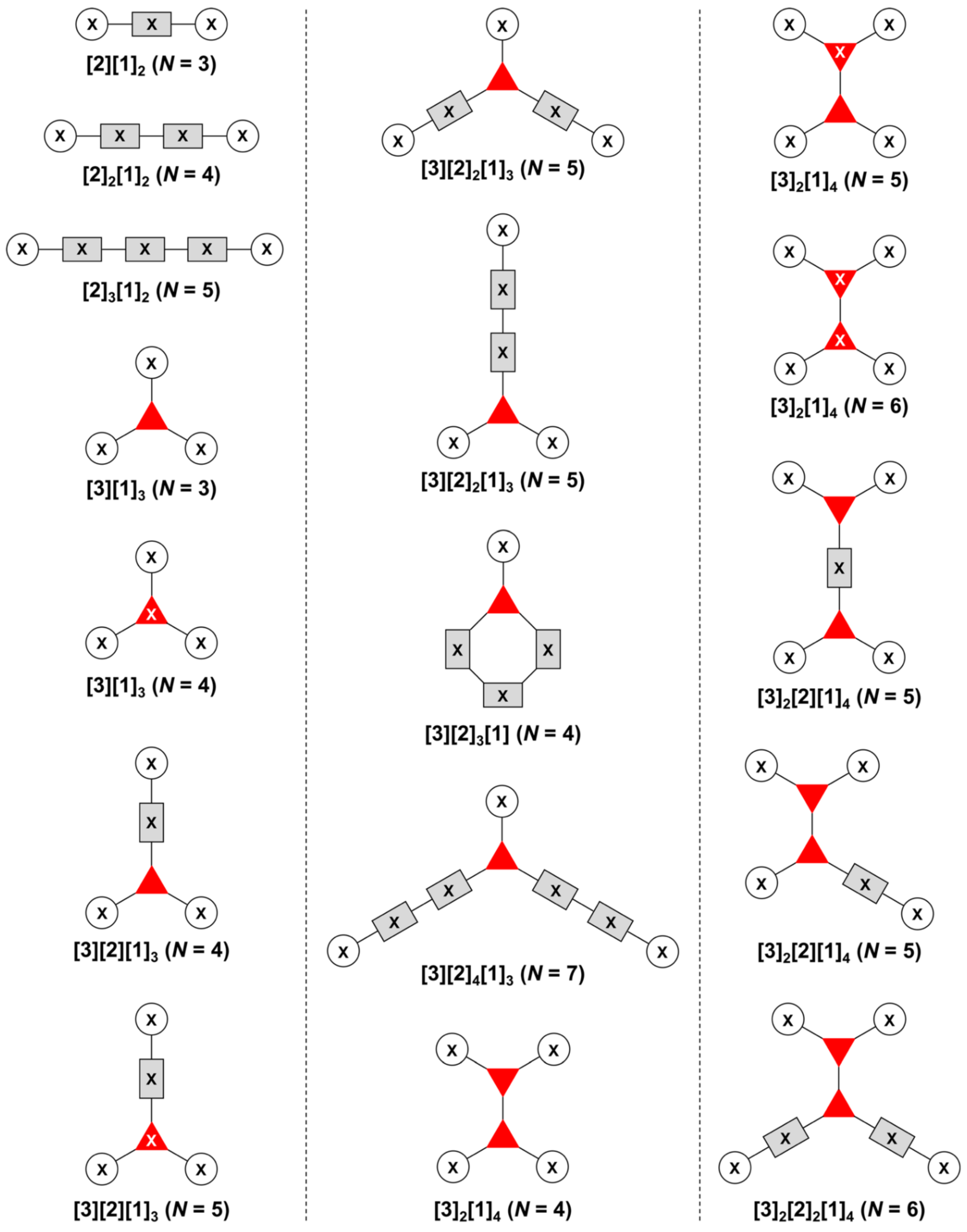{kind=link}
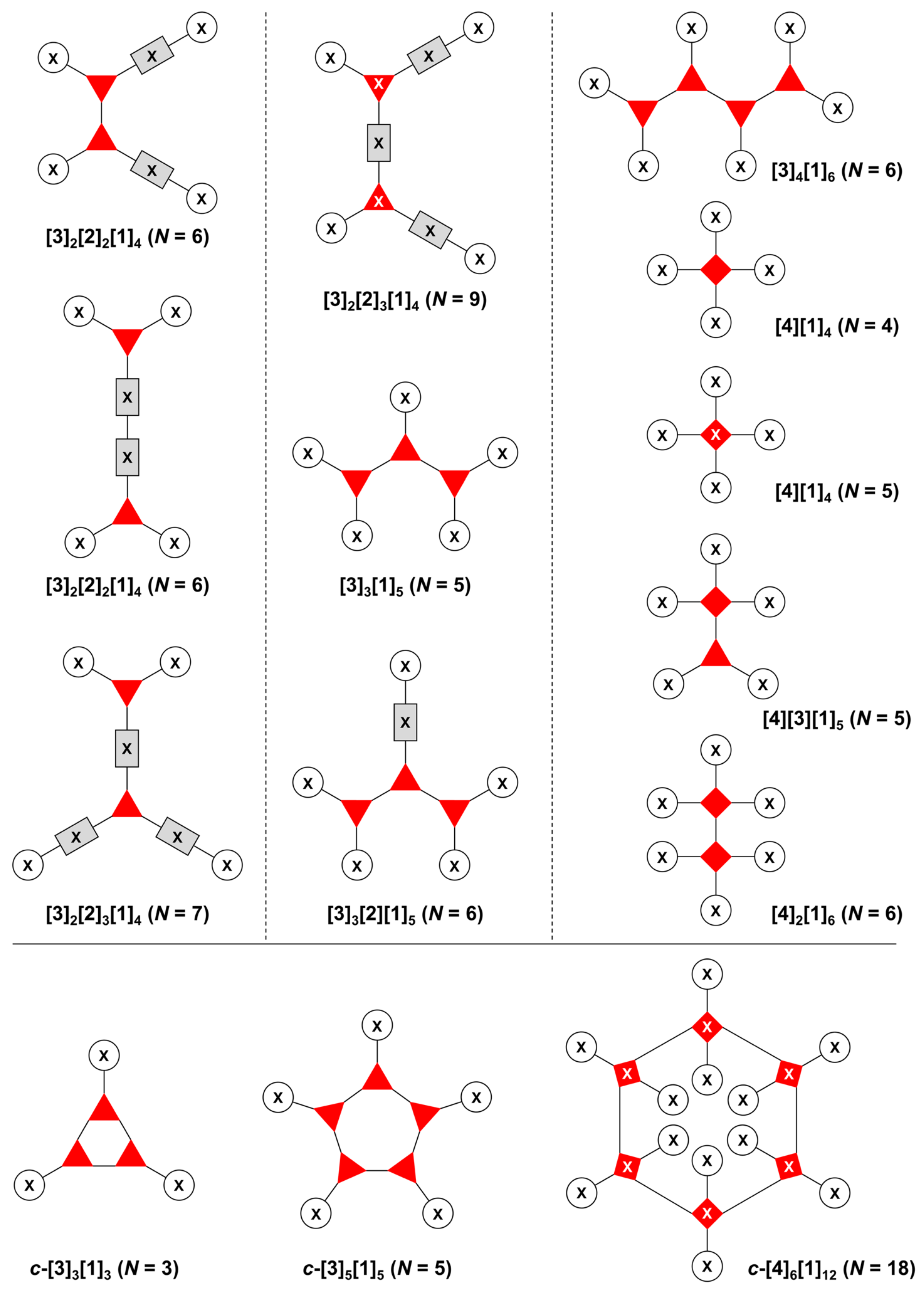{kind=link}
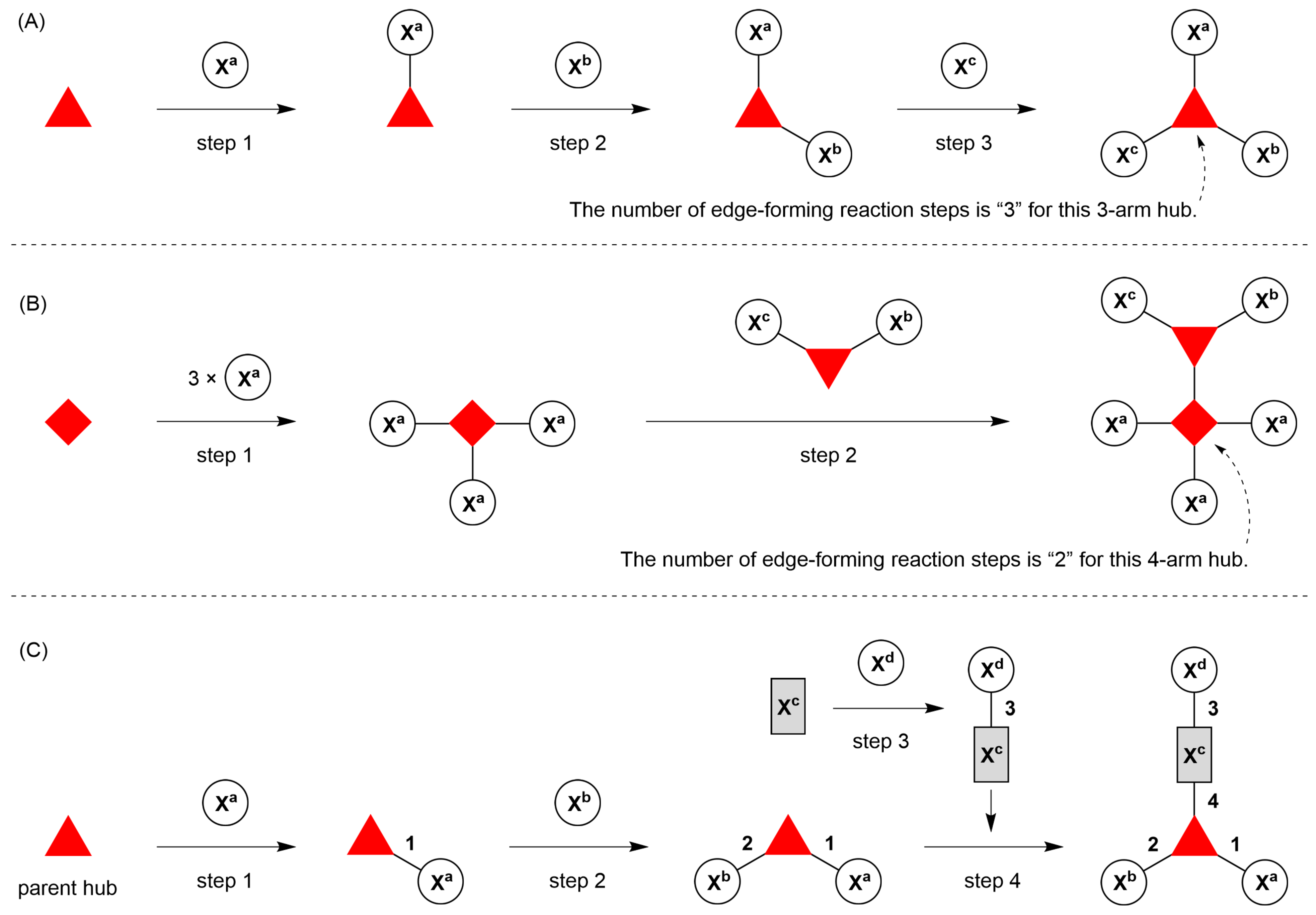{kind=link}
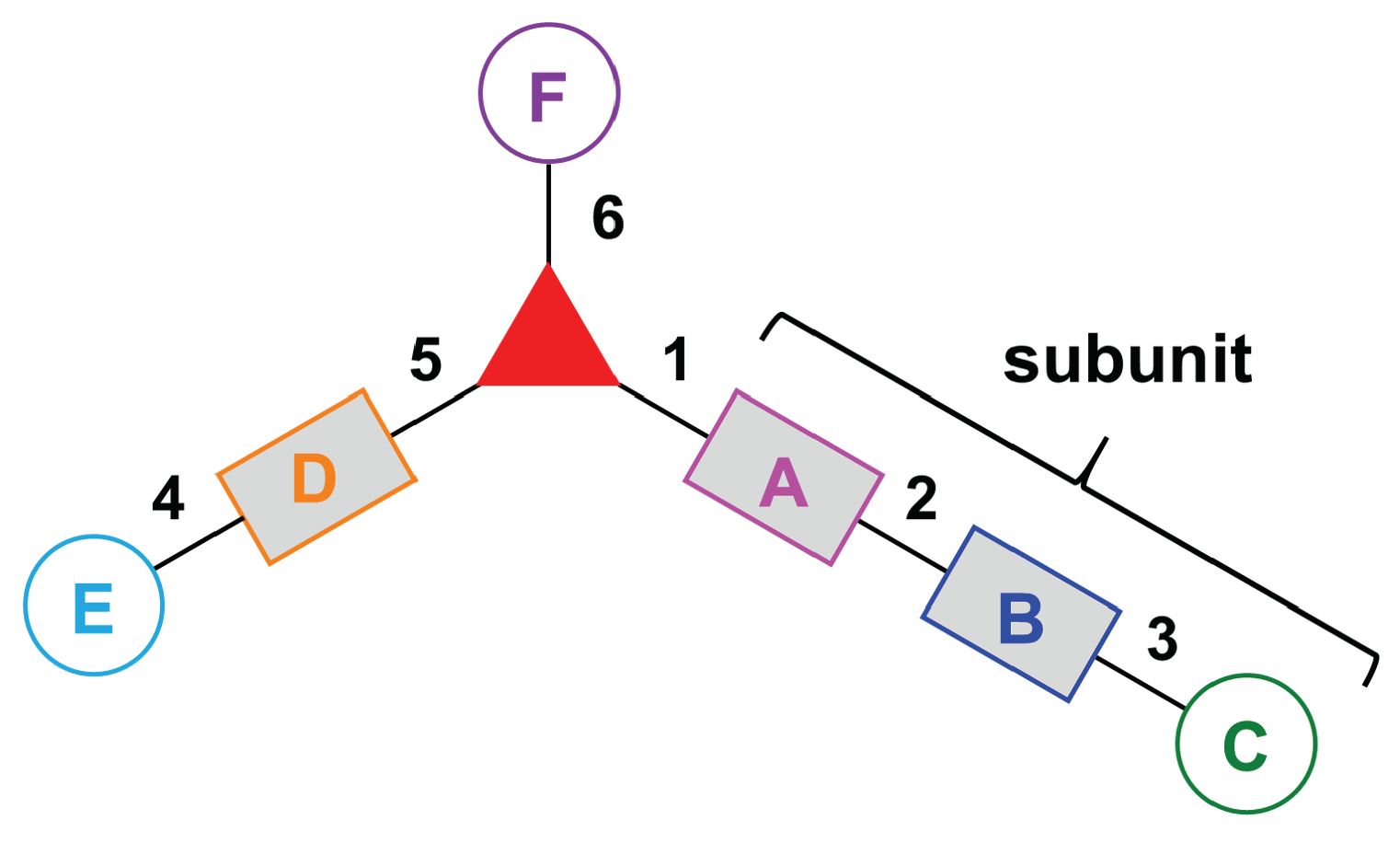{kind=link}
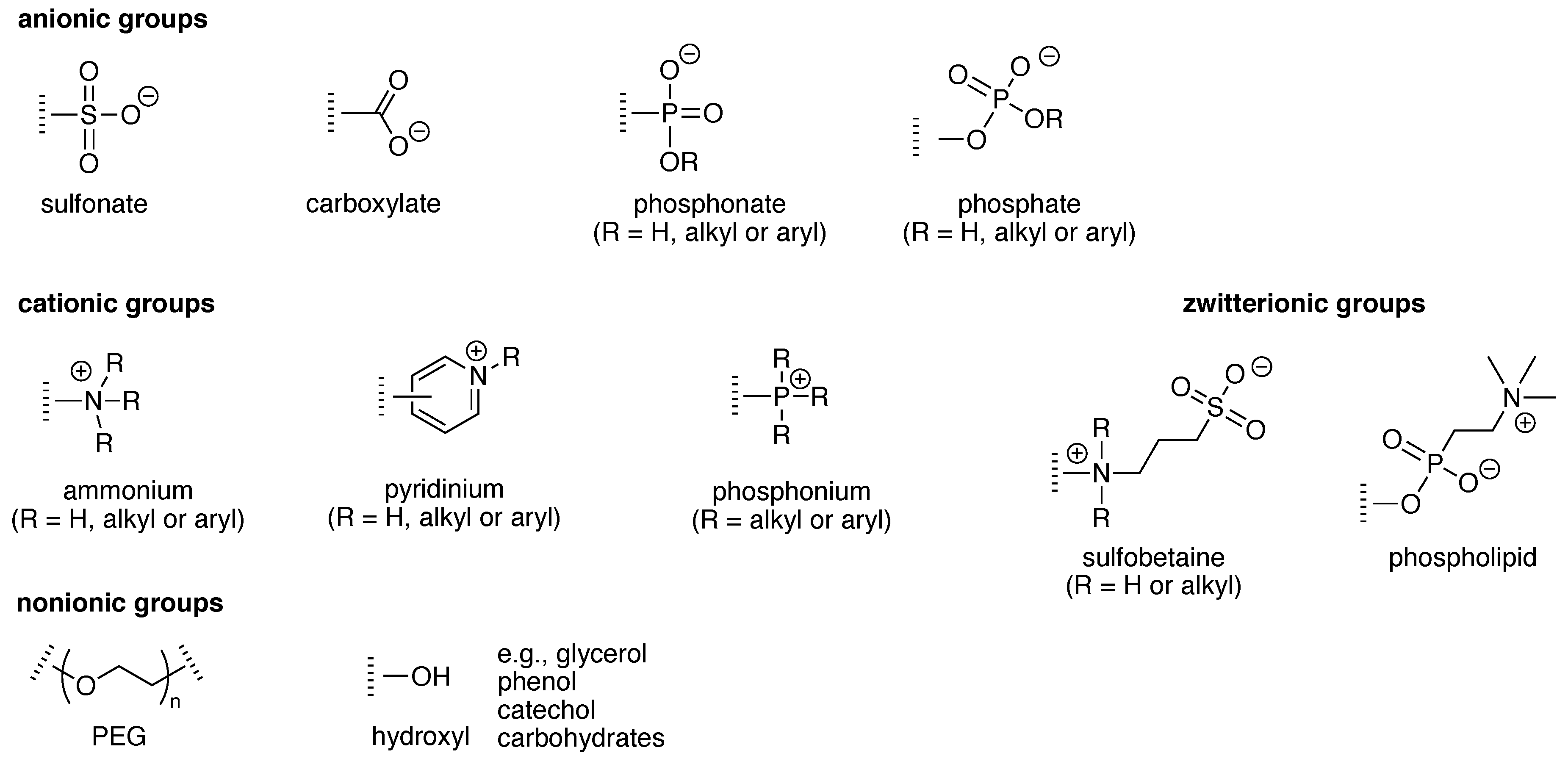{kind=link}
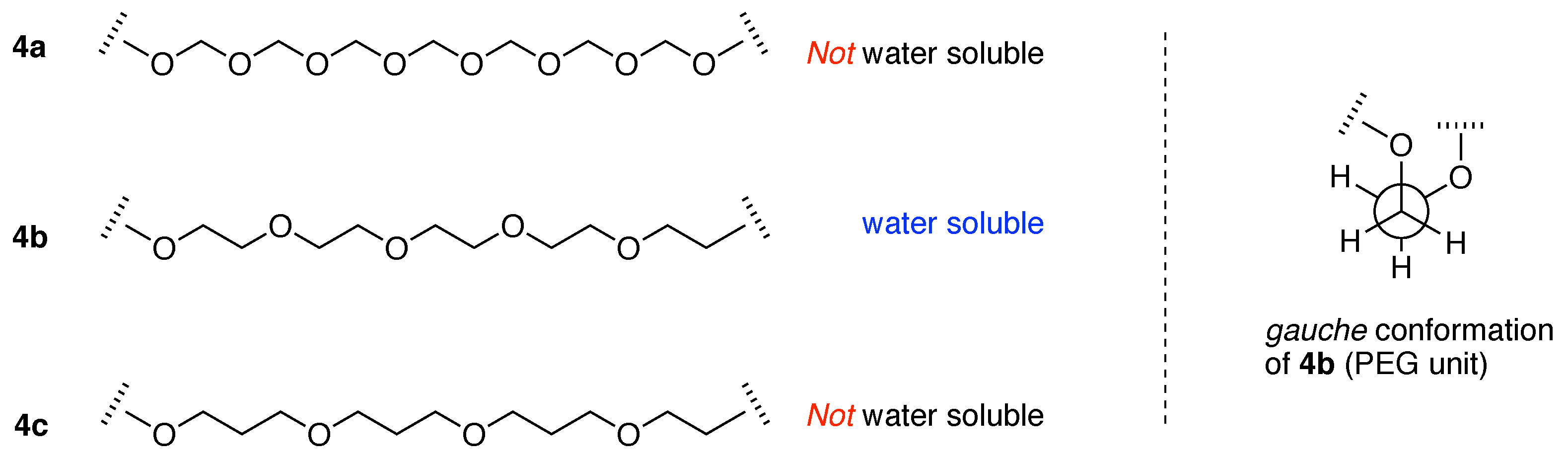{kind=link}
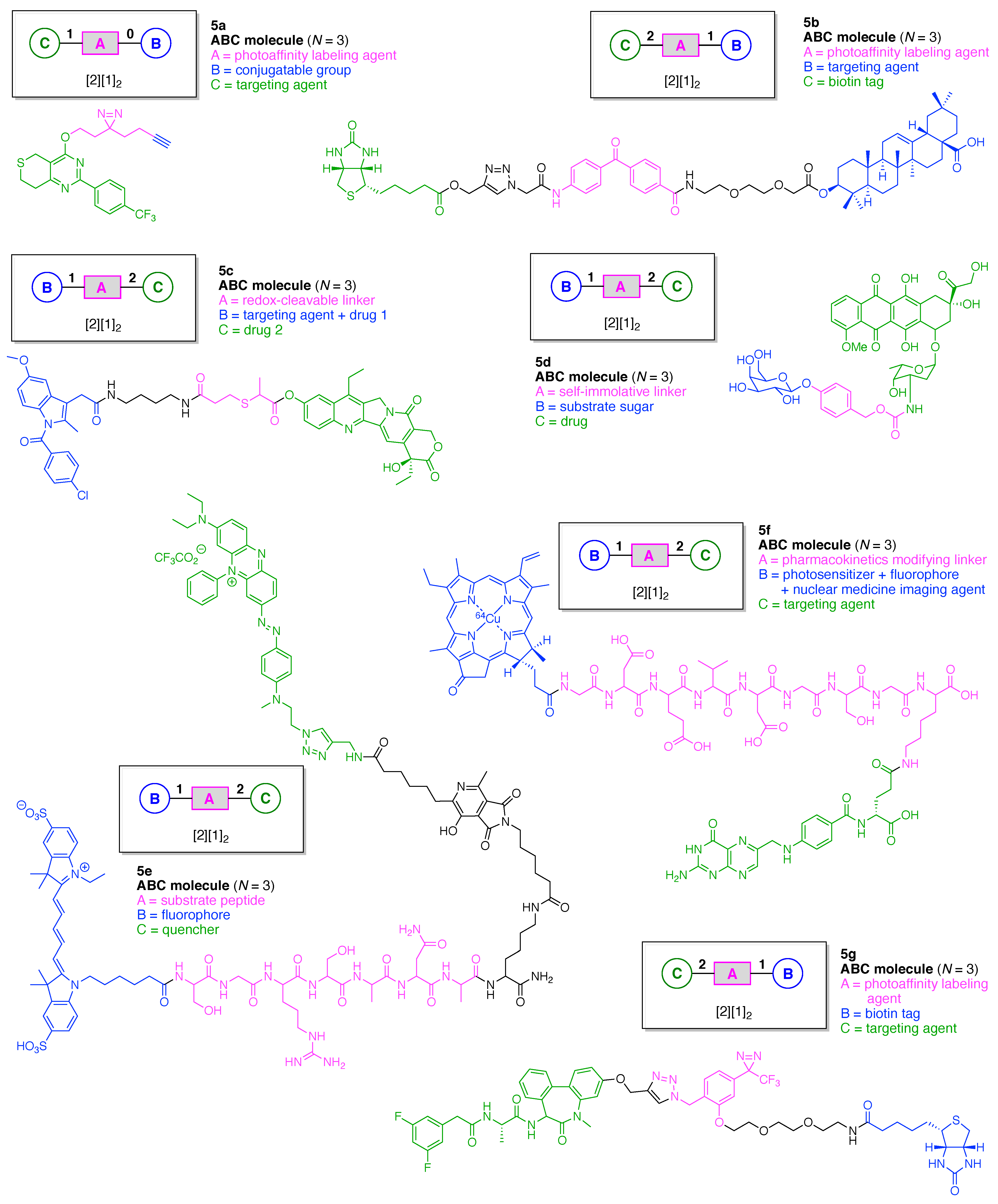{kind=link}
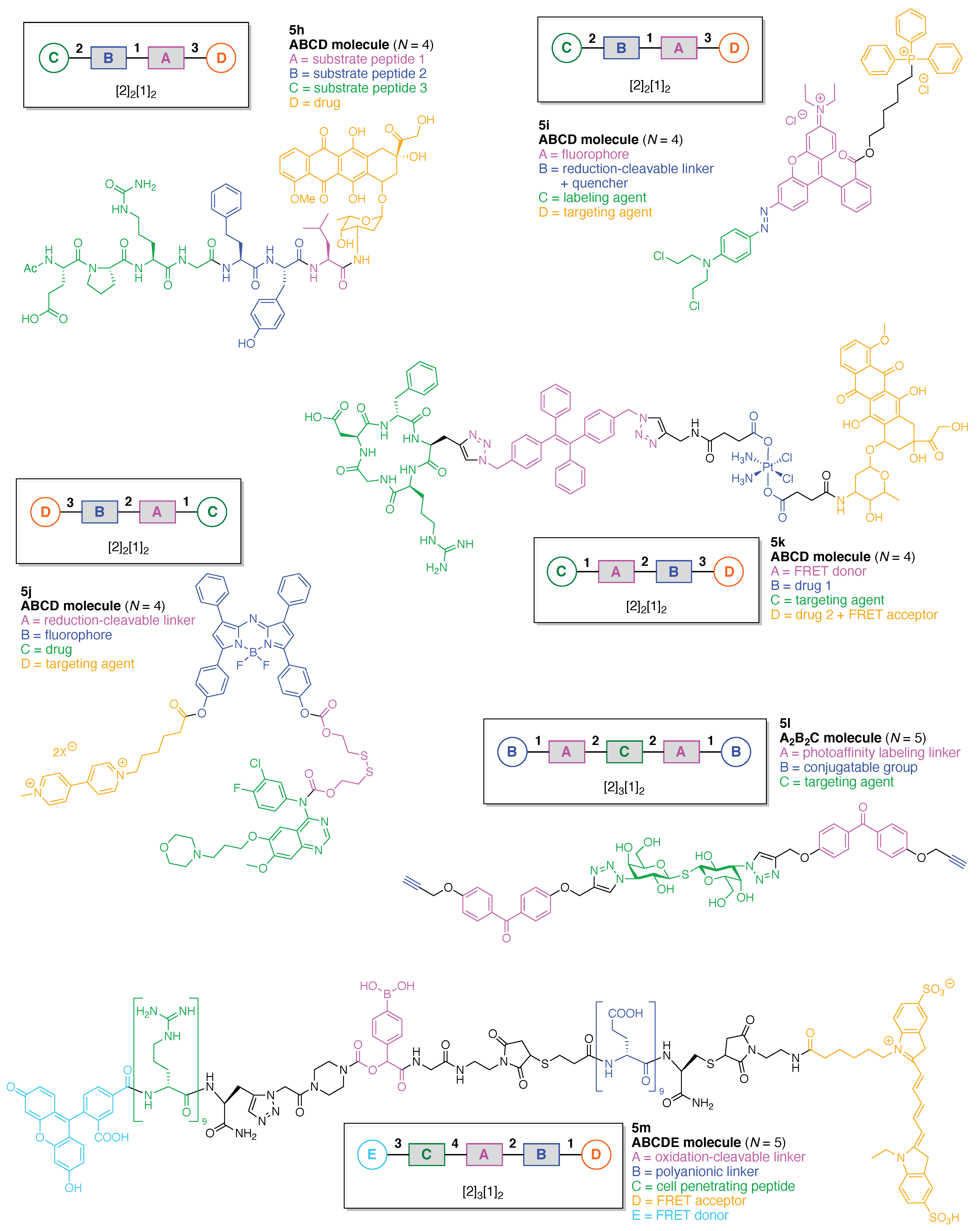{kind=link}
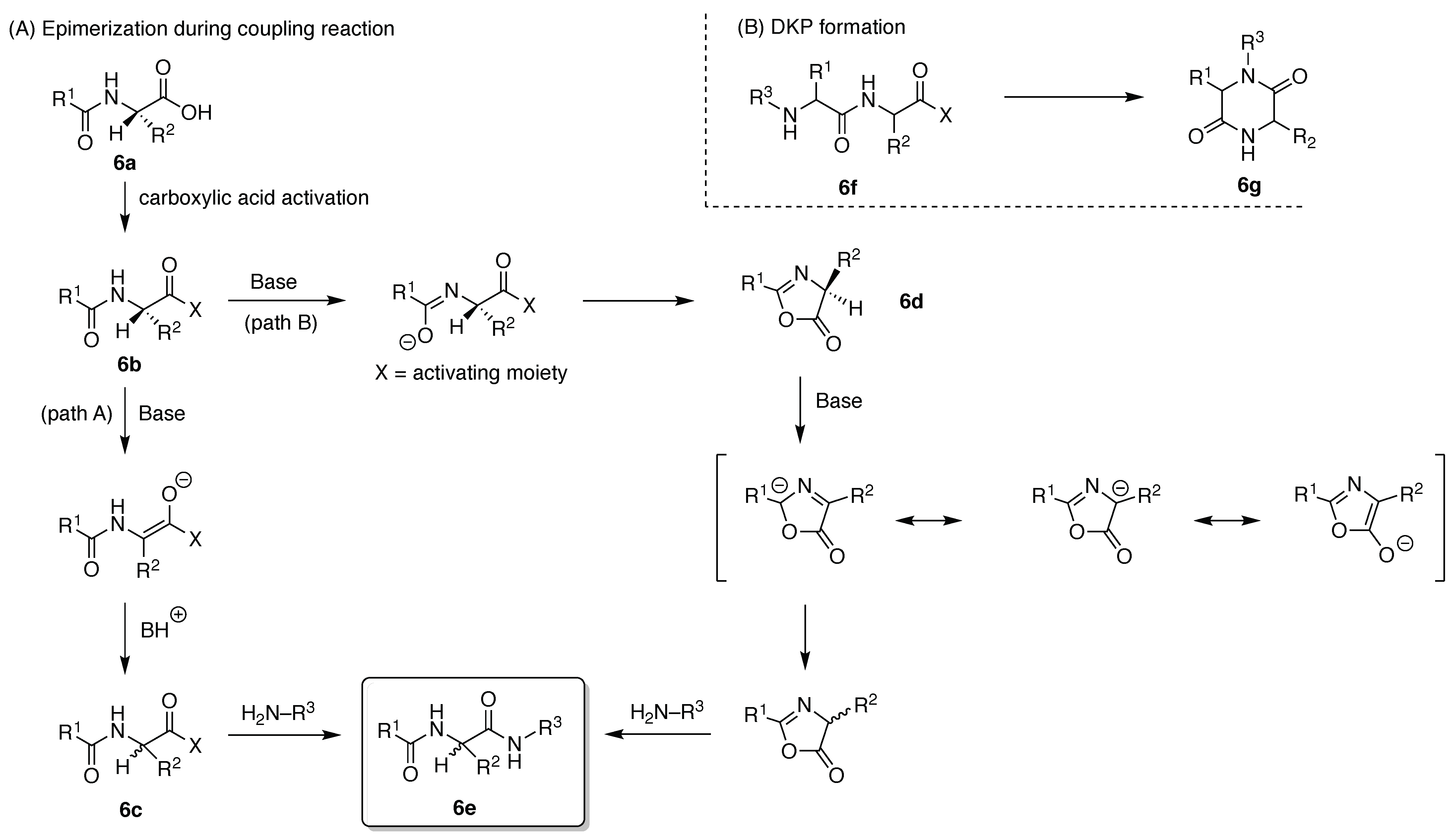{kind=link}
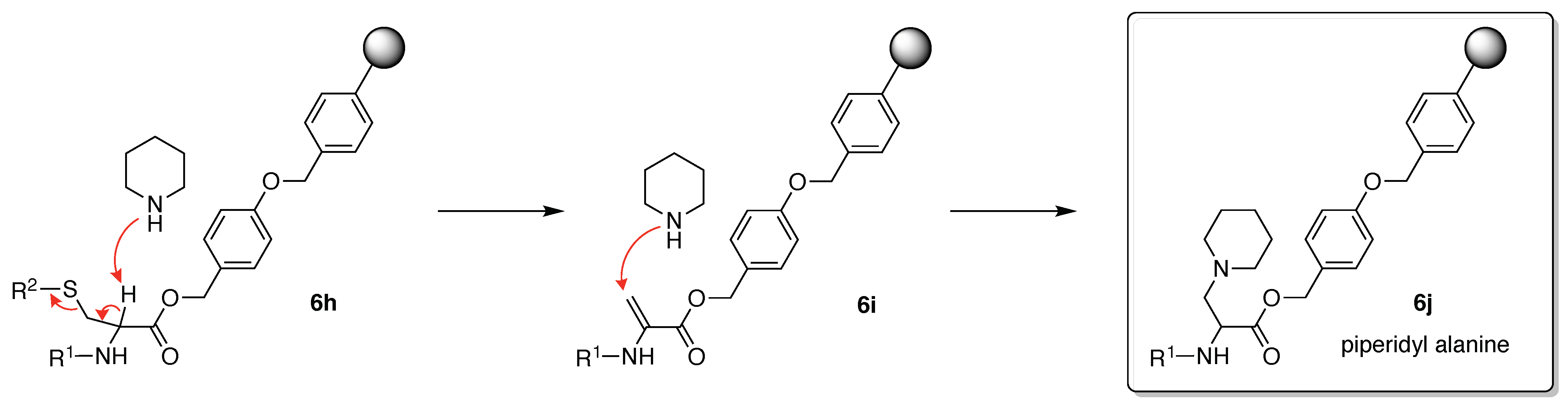{kind=link}
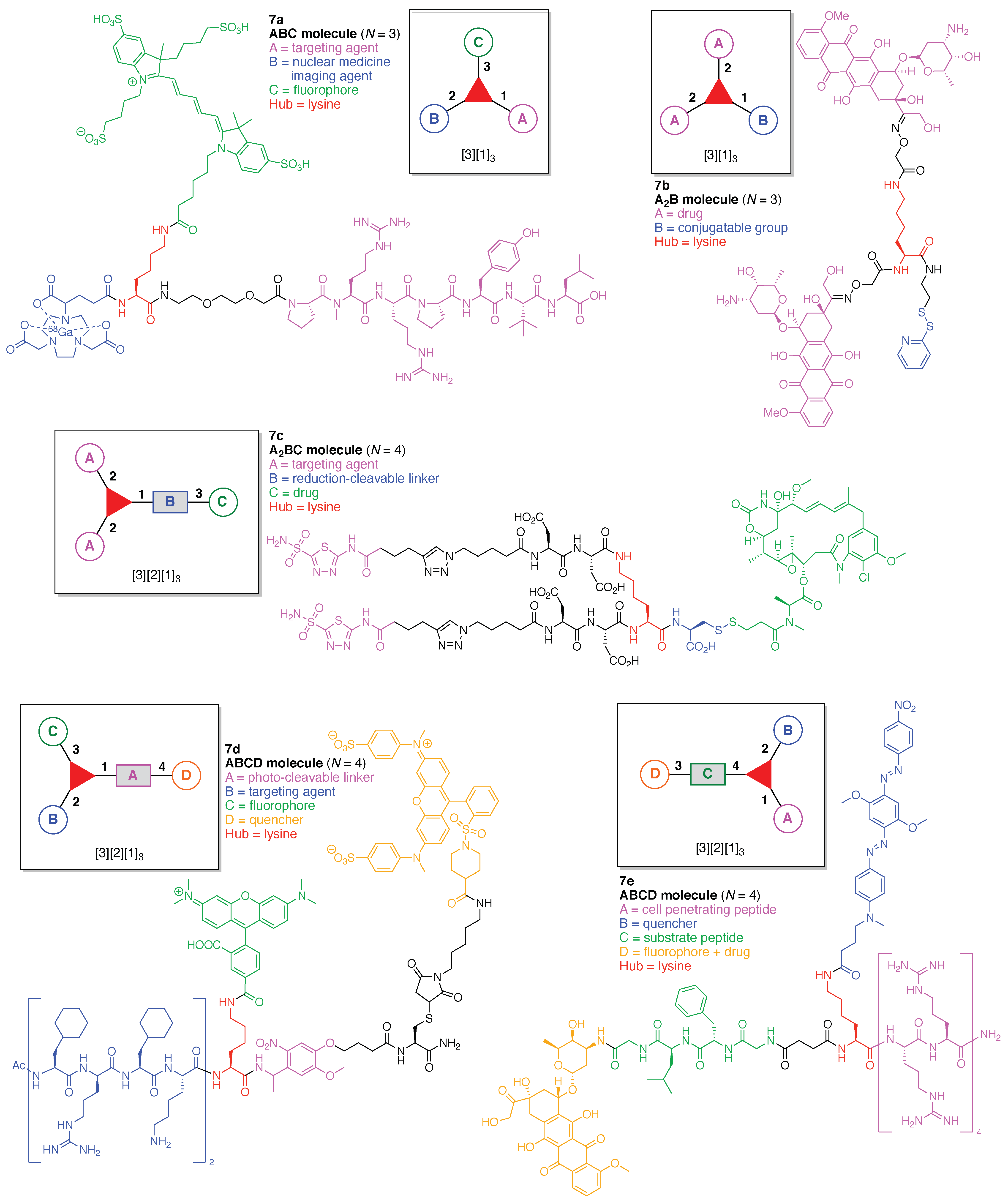{kind=link}
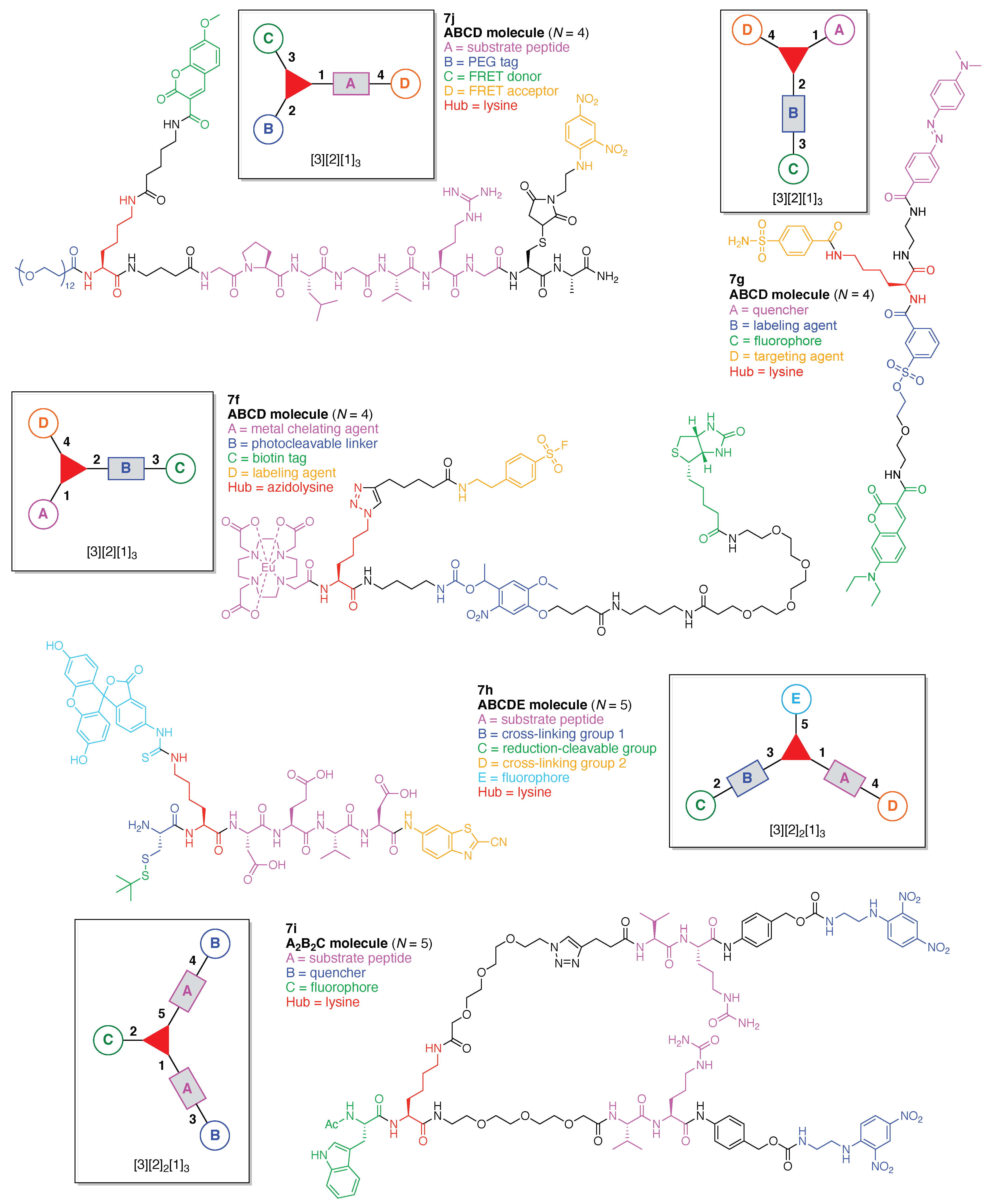{kind=link}
{kind=link}
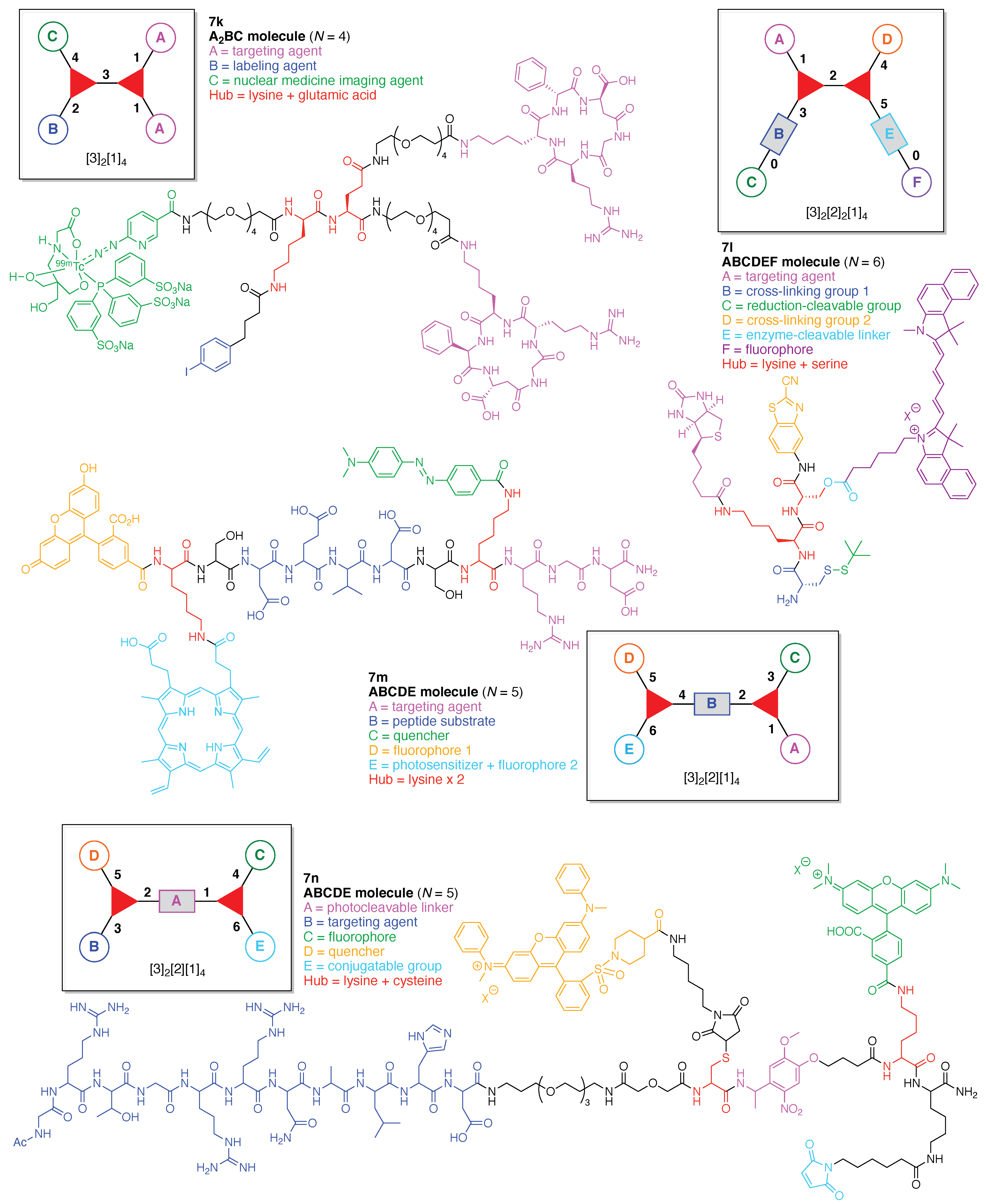{kind=link}
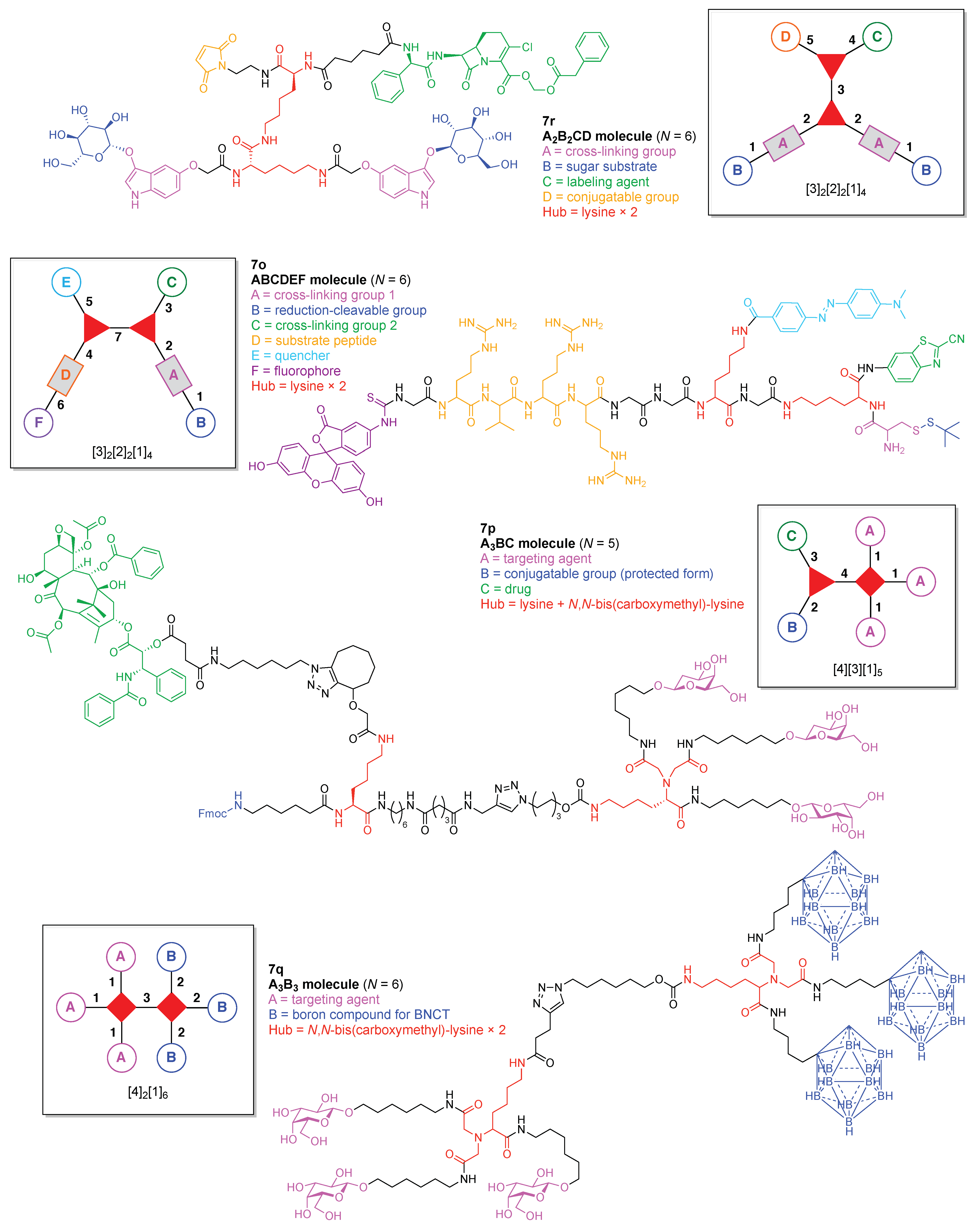{kind=link}
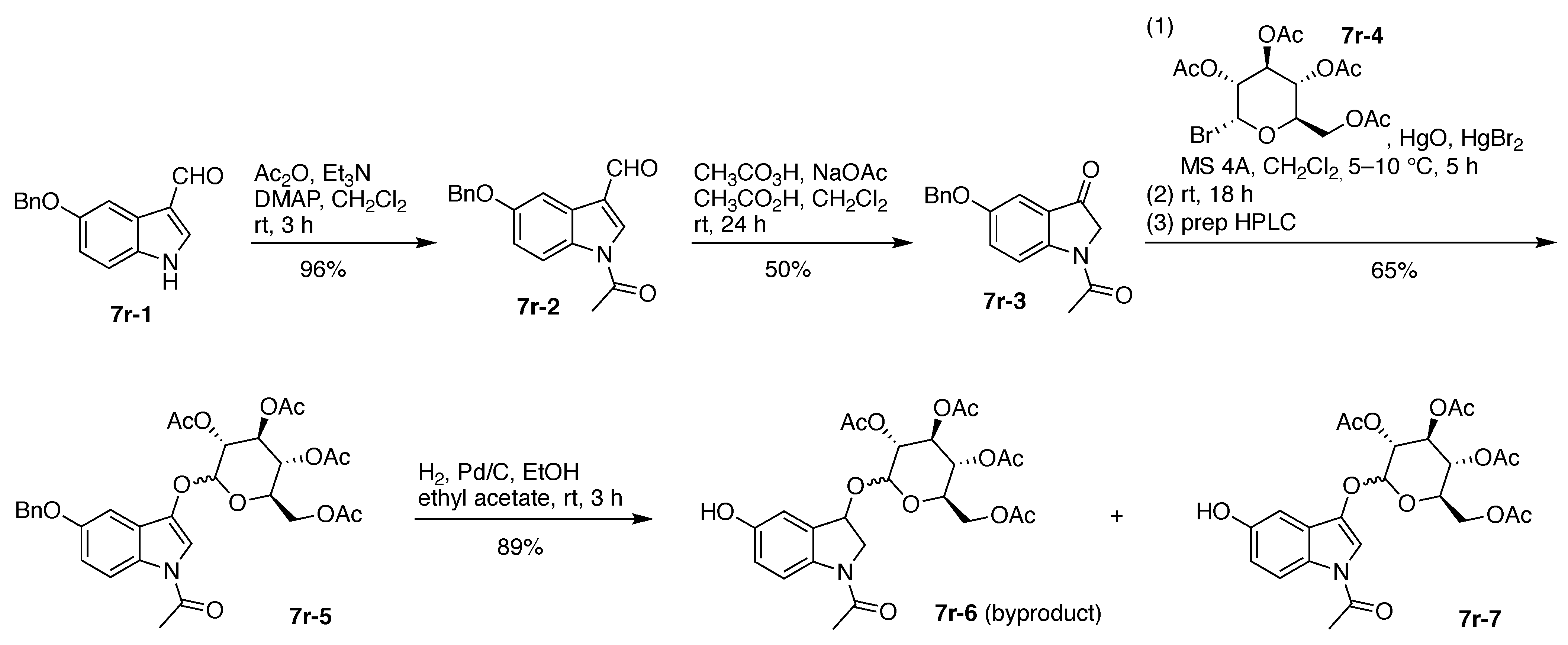{kind=link}
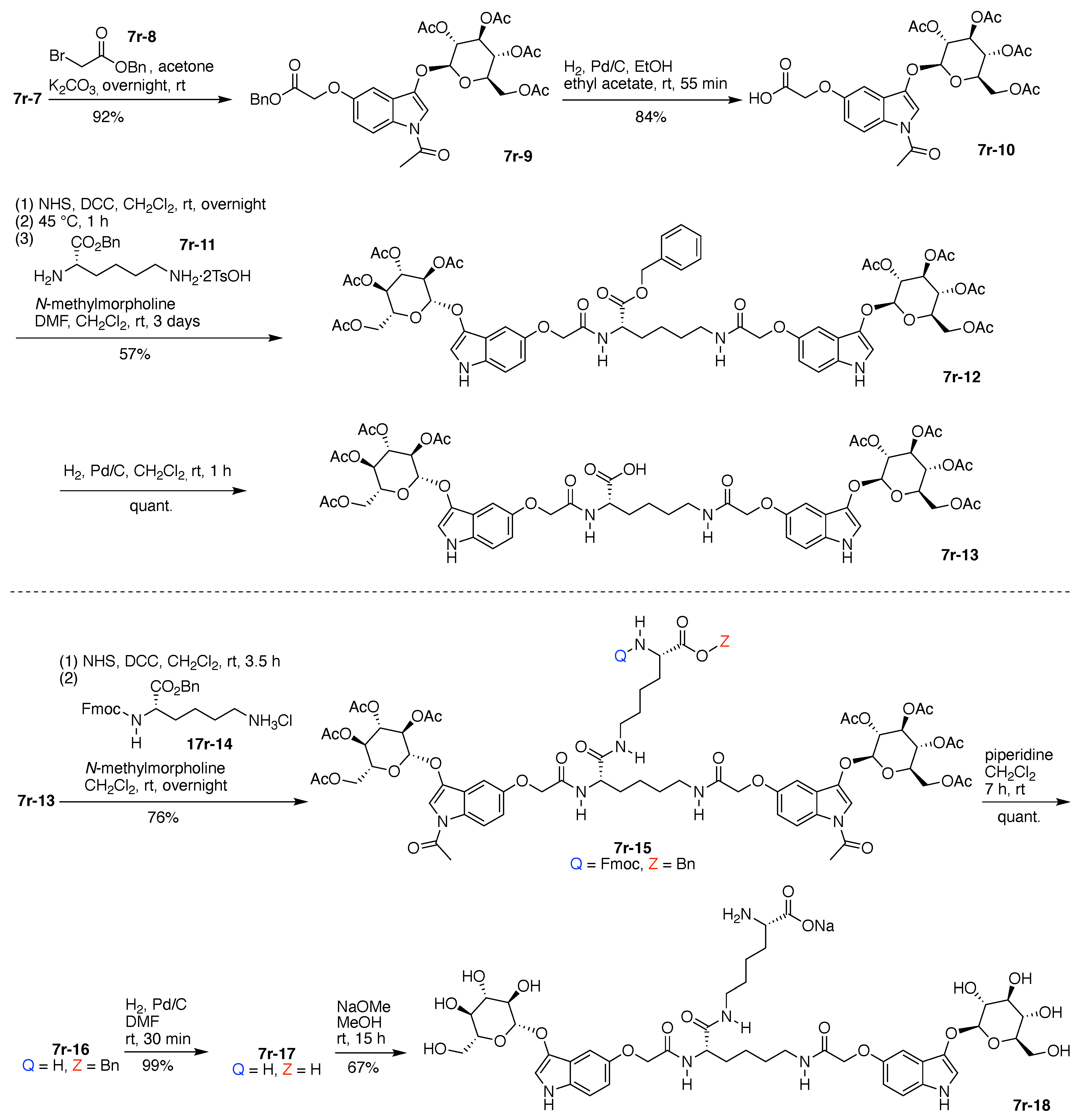{kind=link}
{kind=link}
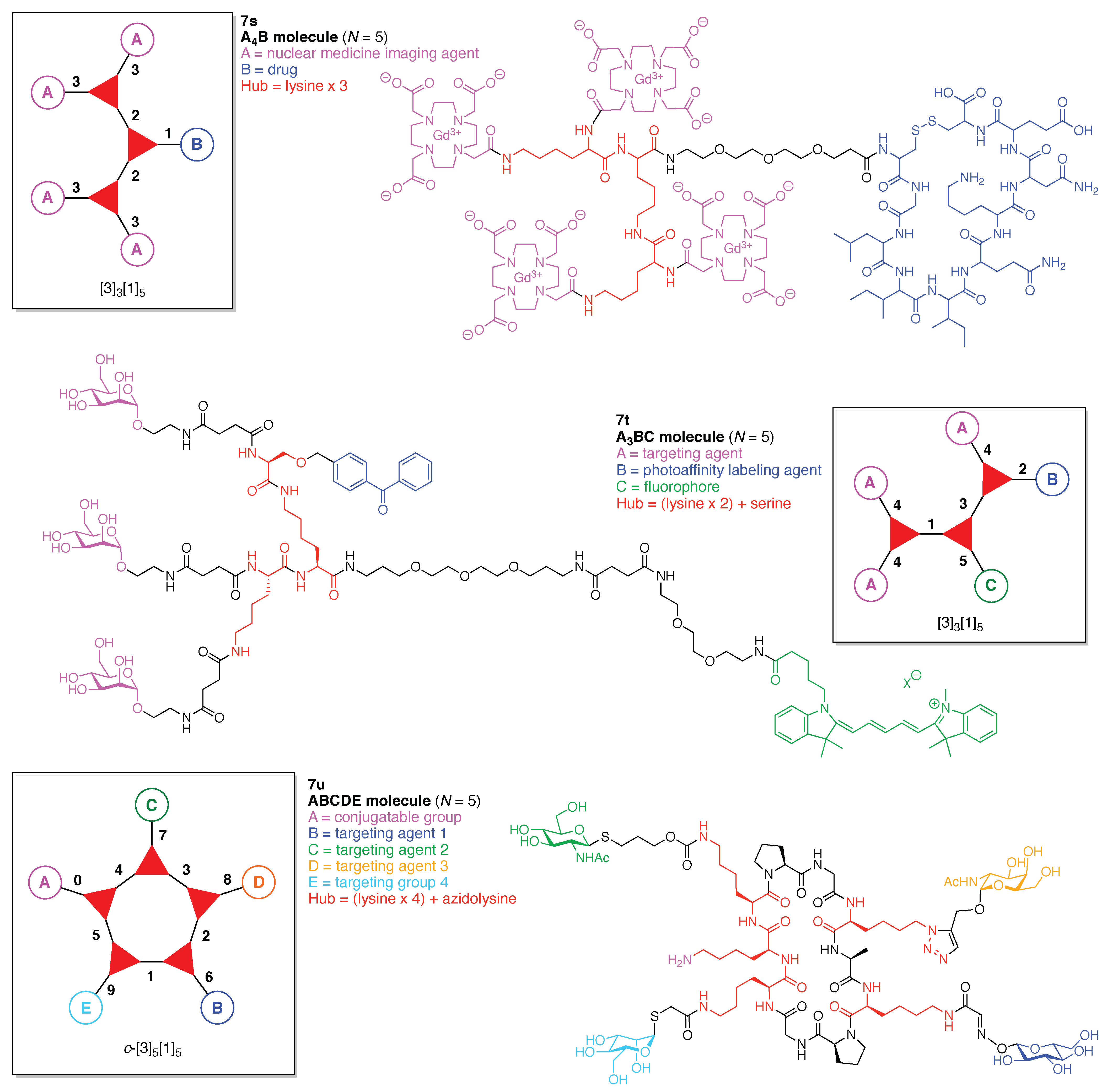{kind=link}
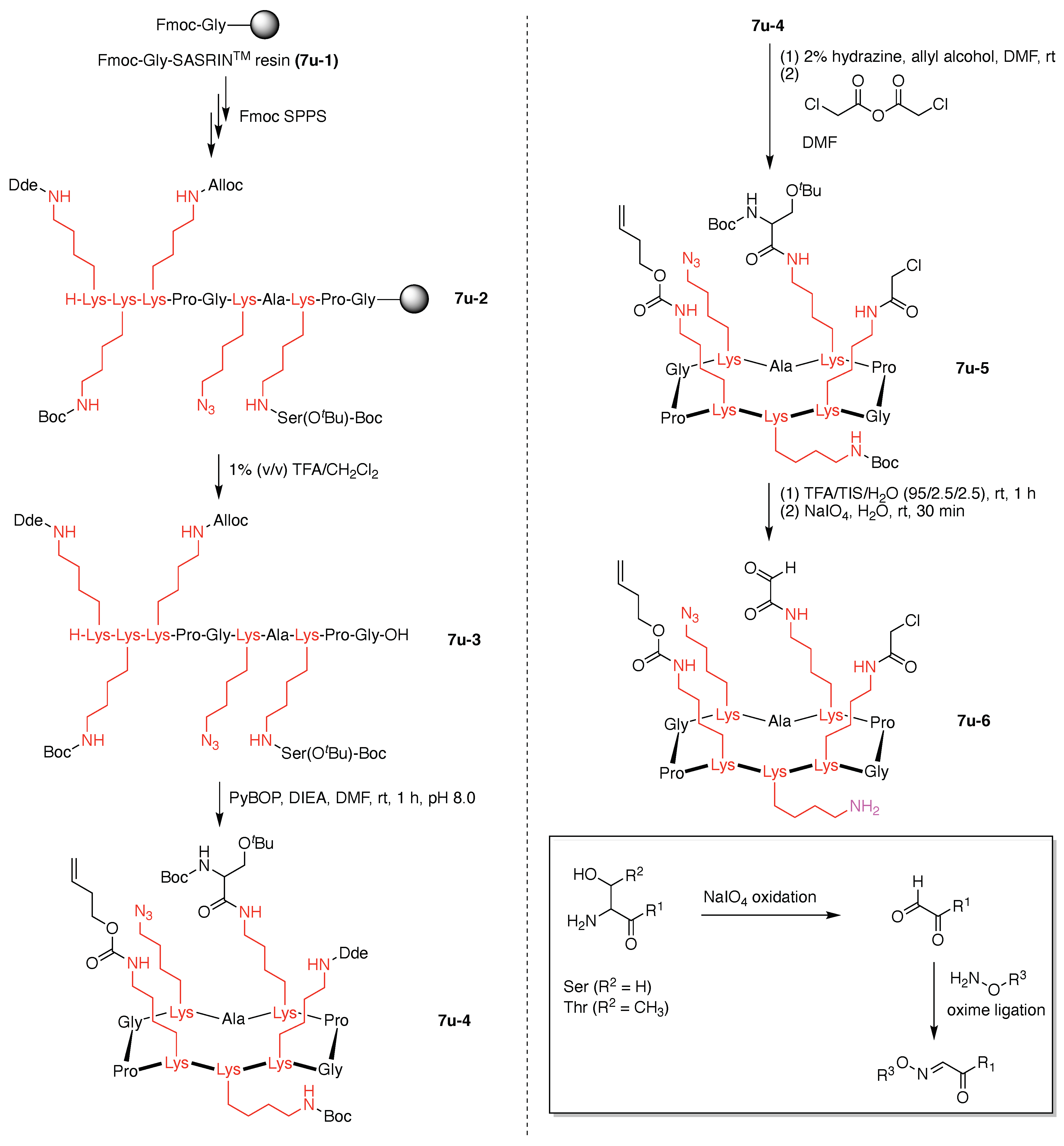{kind=link}
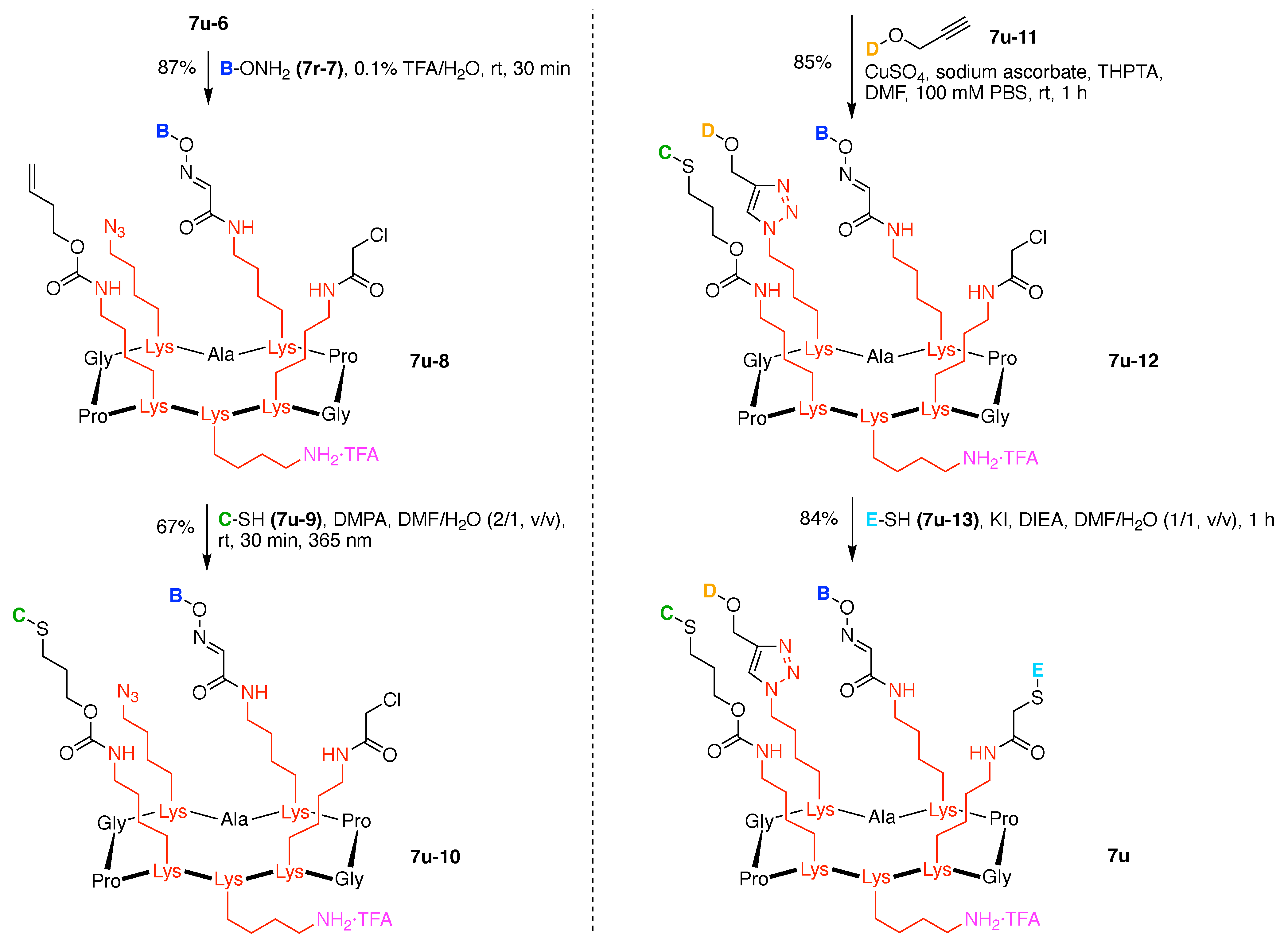{kind=link}
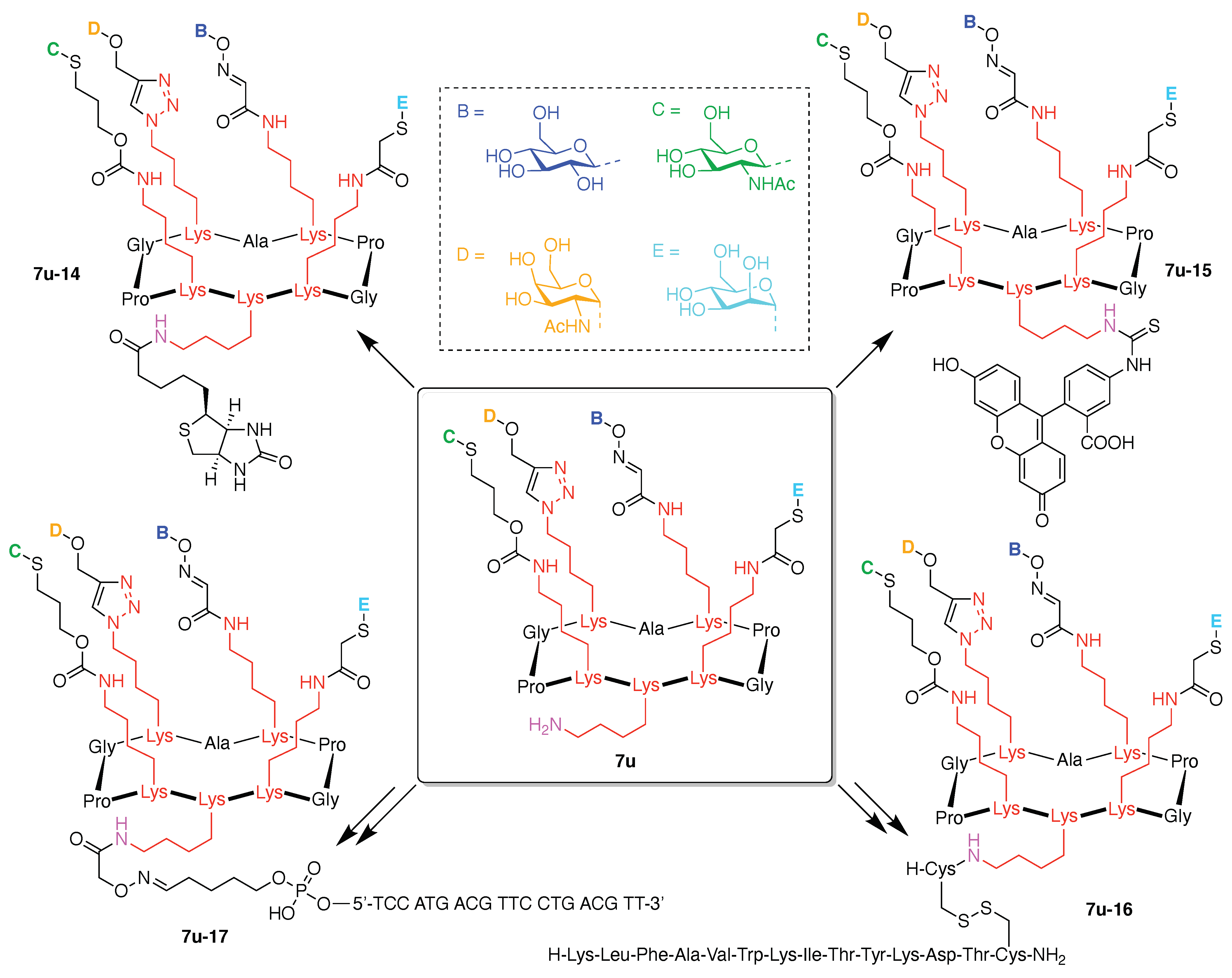{kind=link}
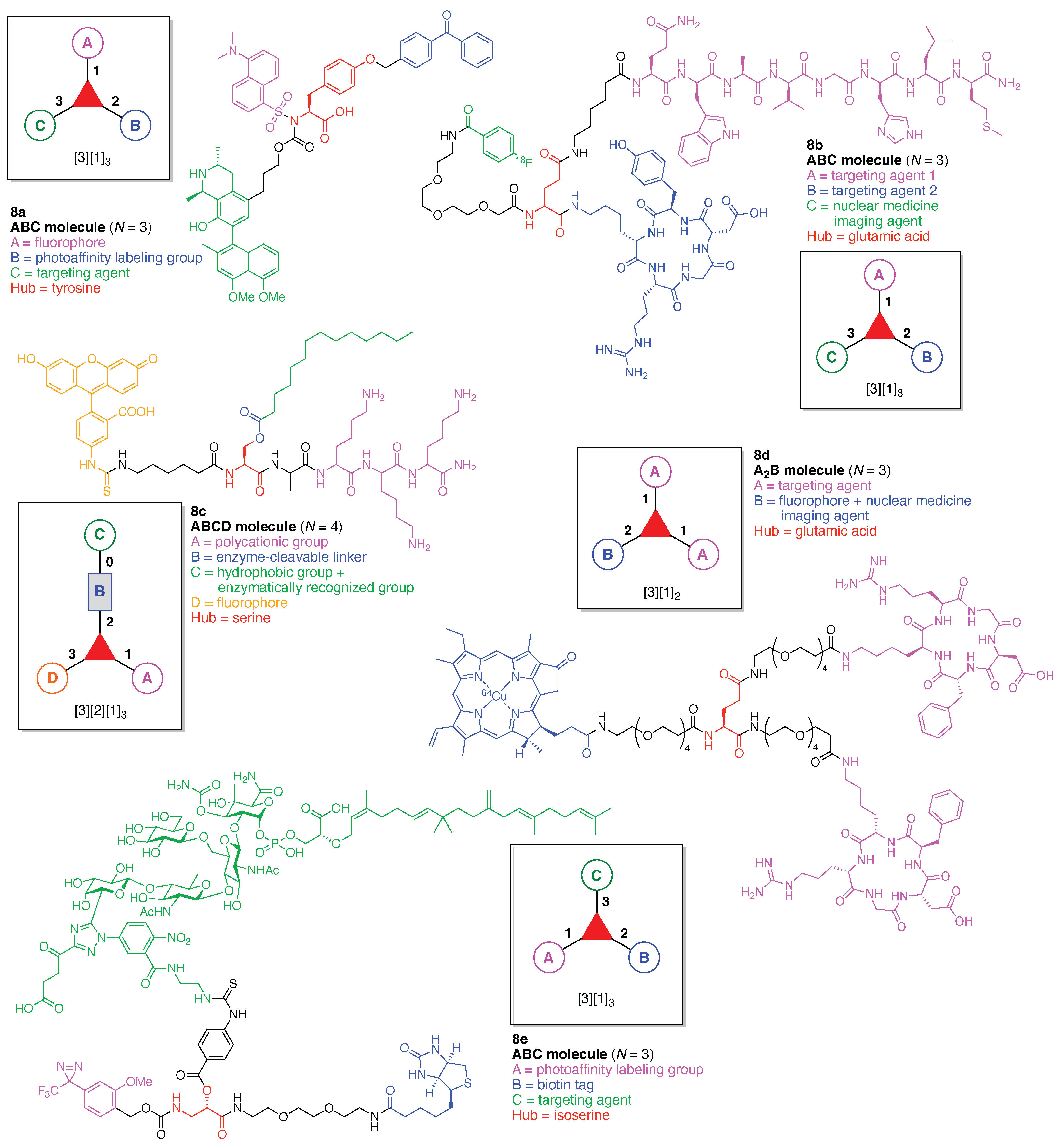{kind=link}
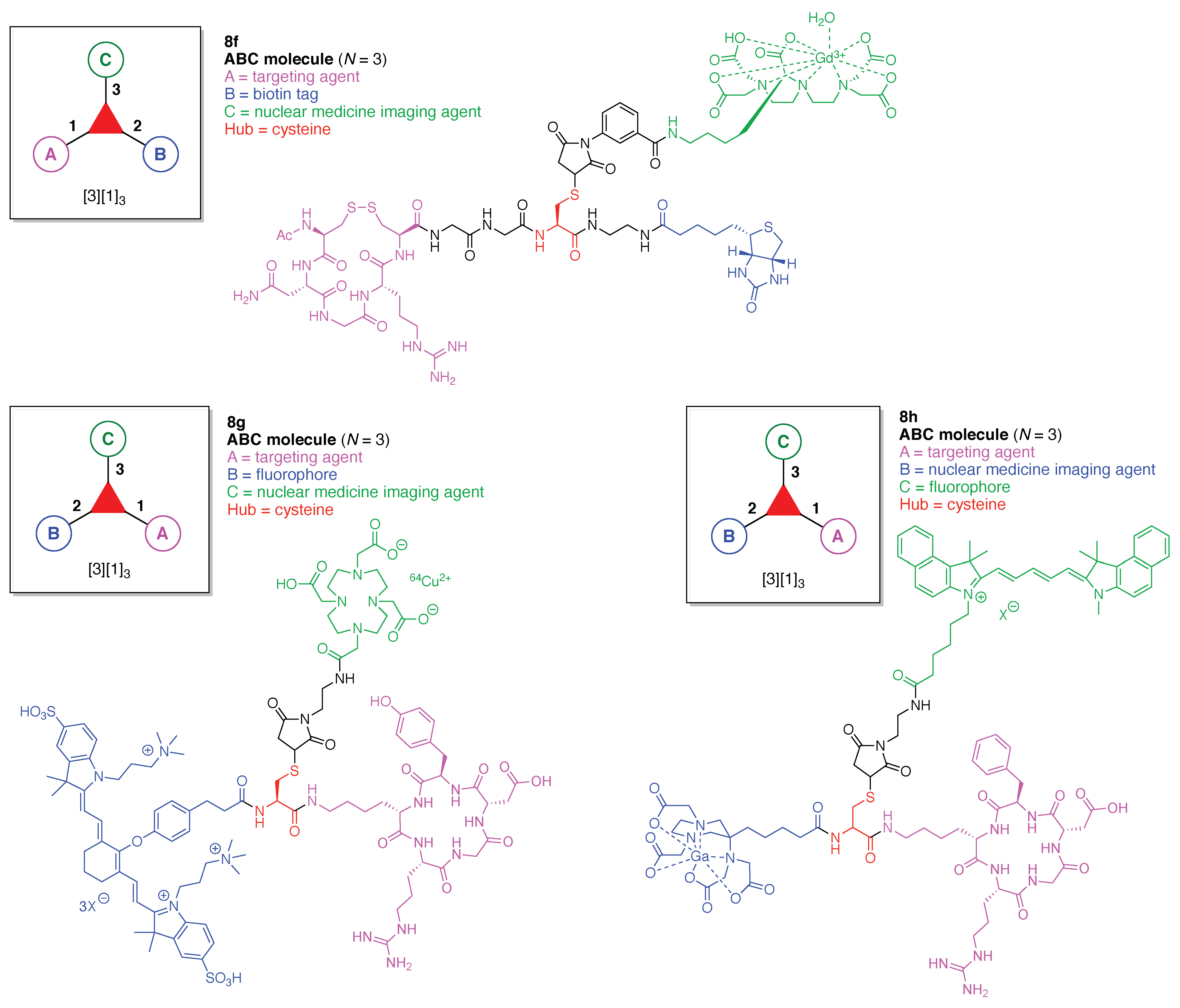{kind=link}
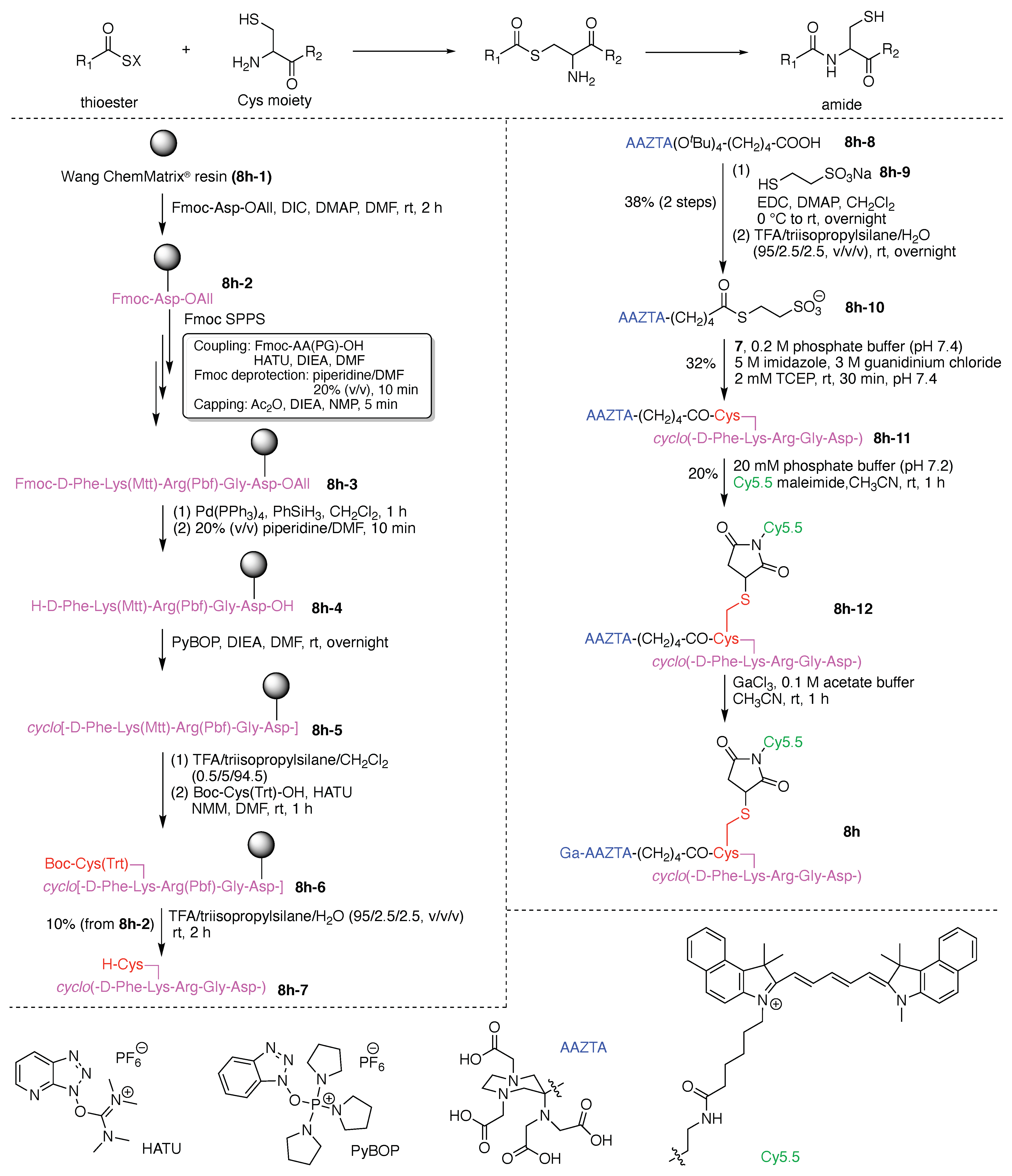{kind=link}
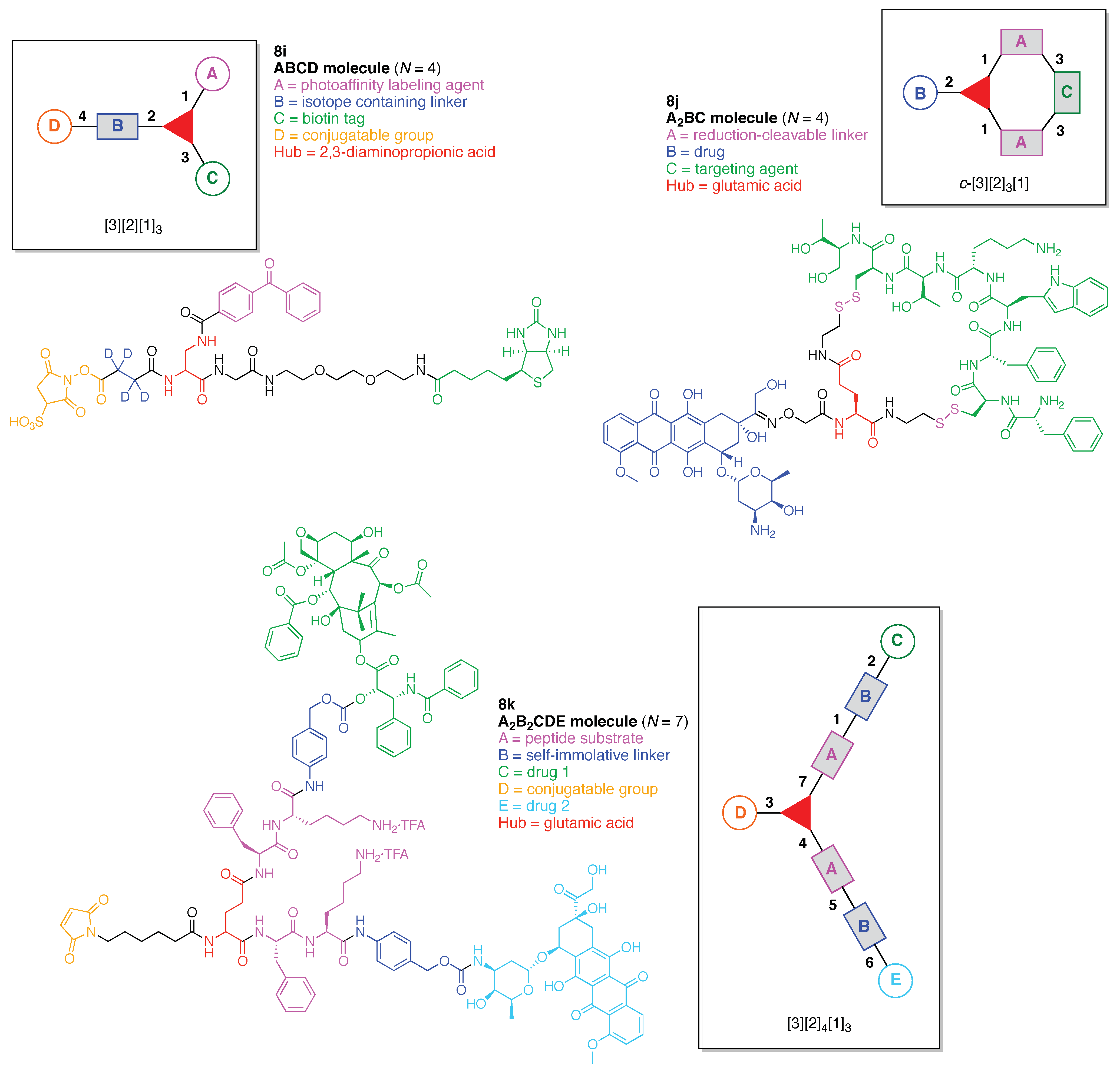{kind=link}
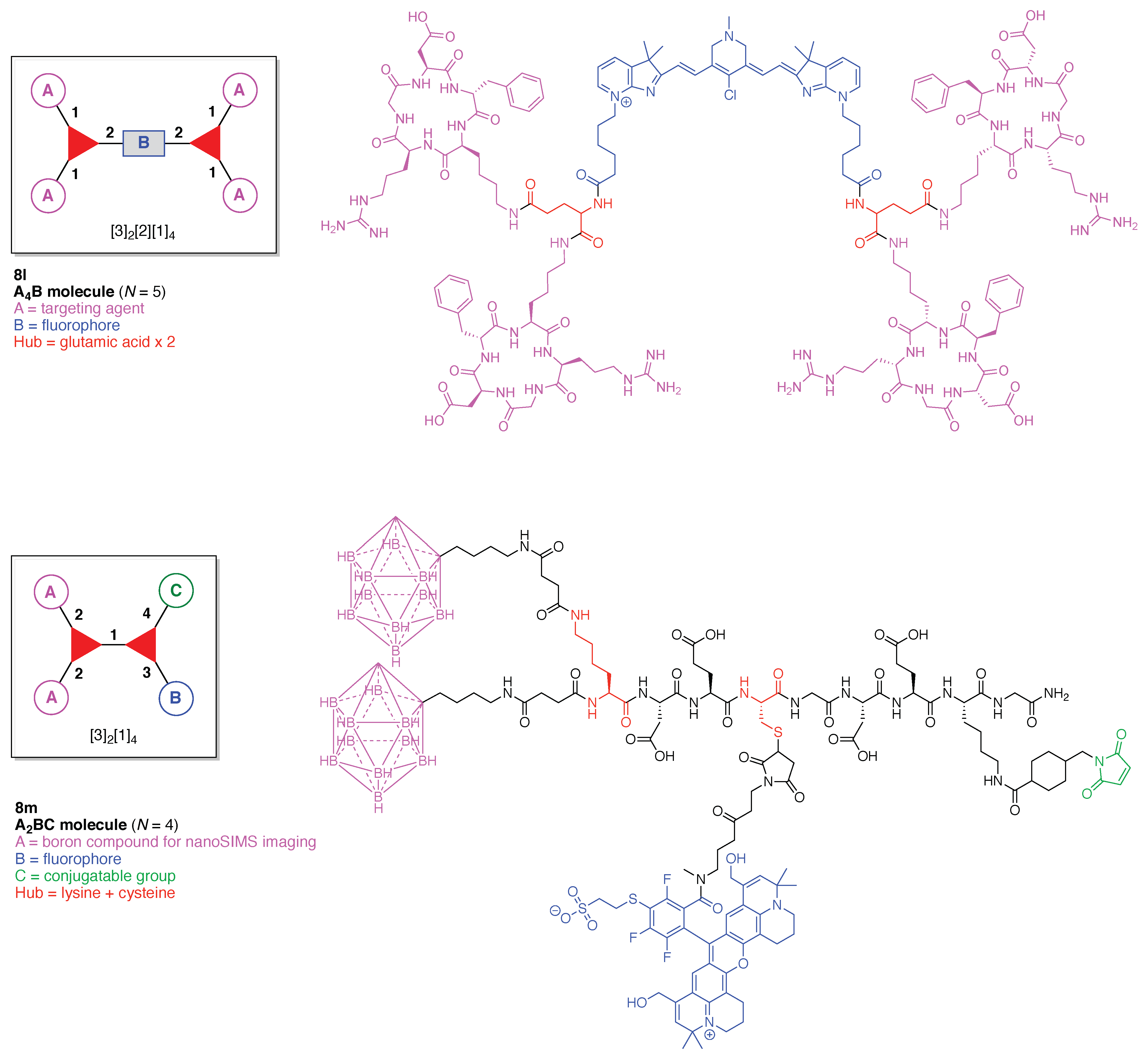{kind=link}
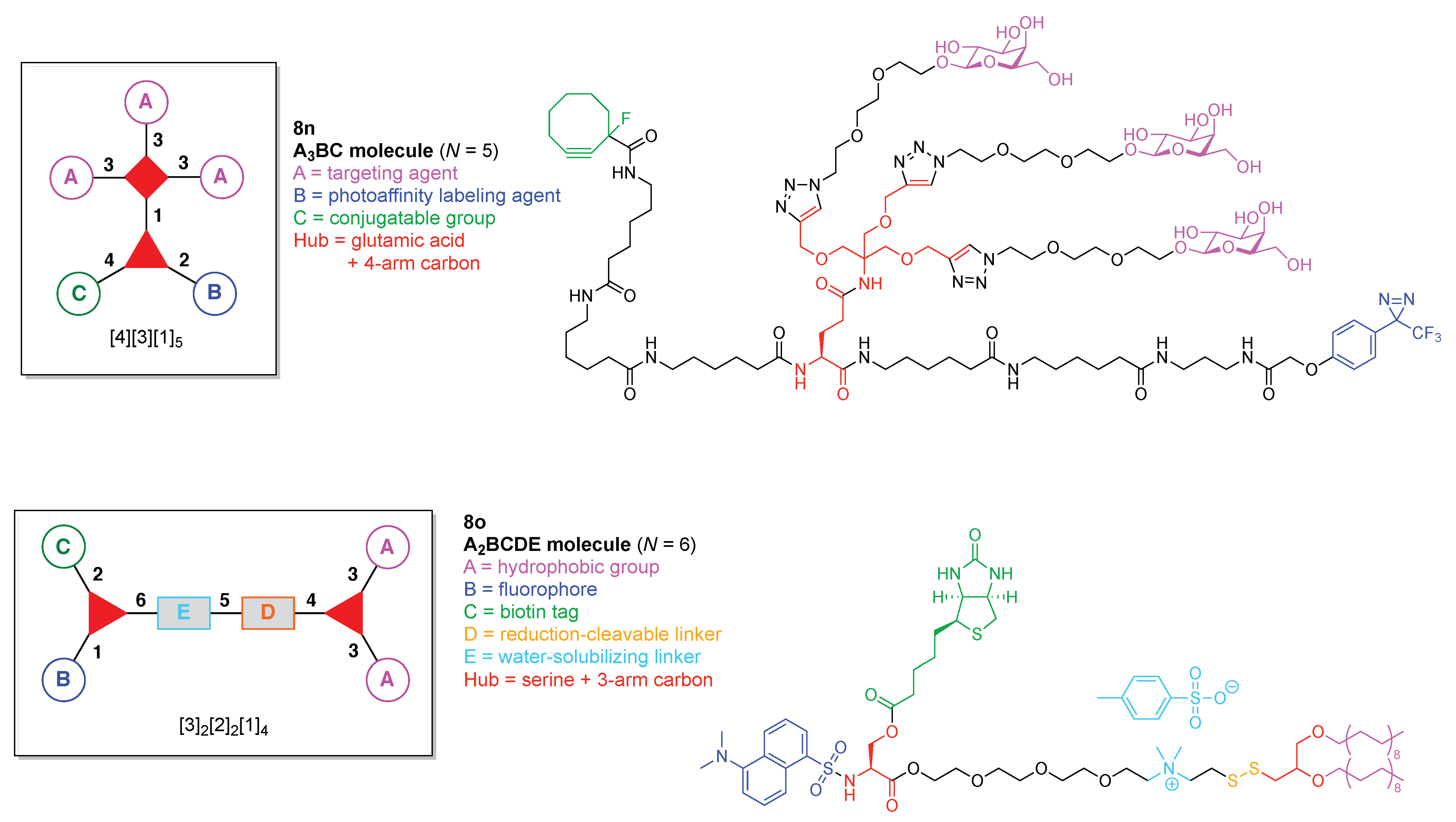{kind=link}
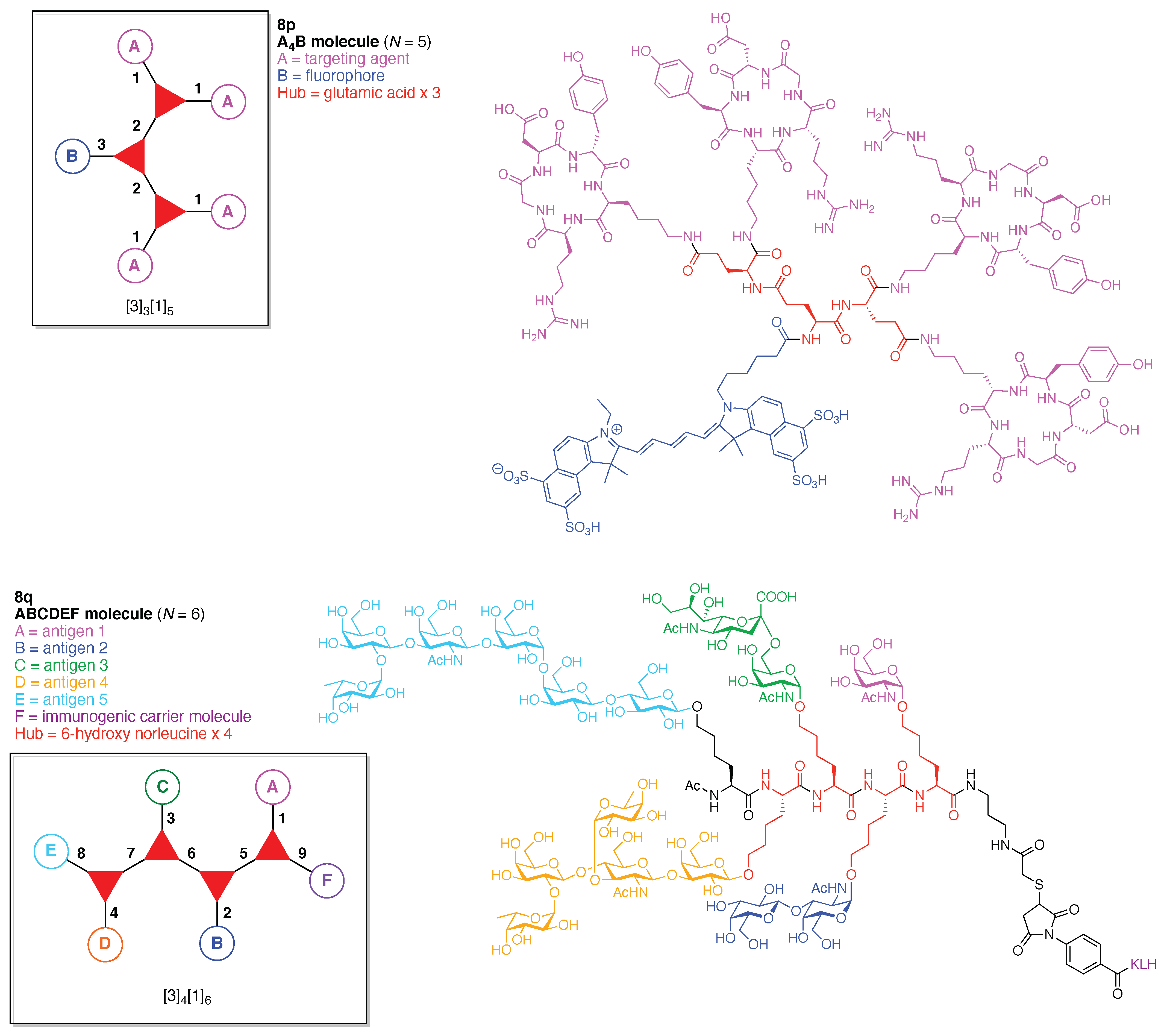{kind=link}
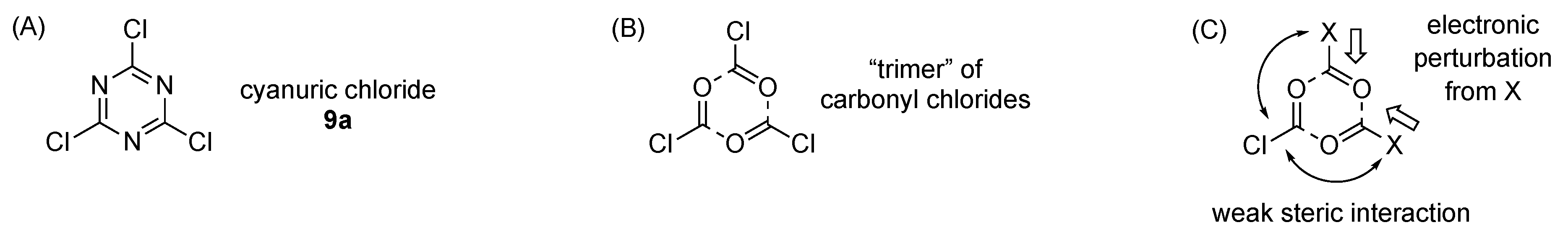{kind=link}
{kind=link}
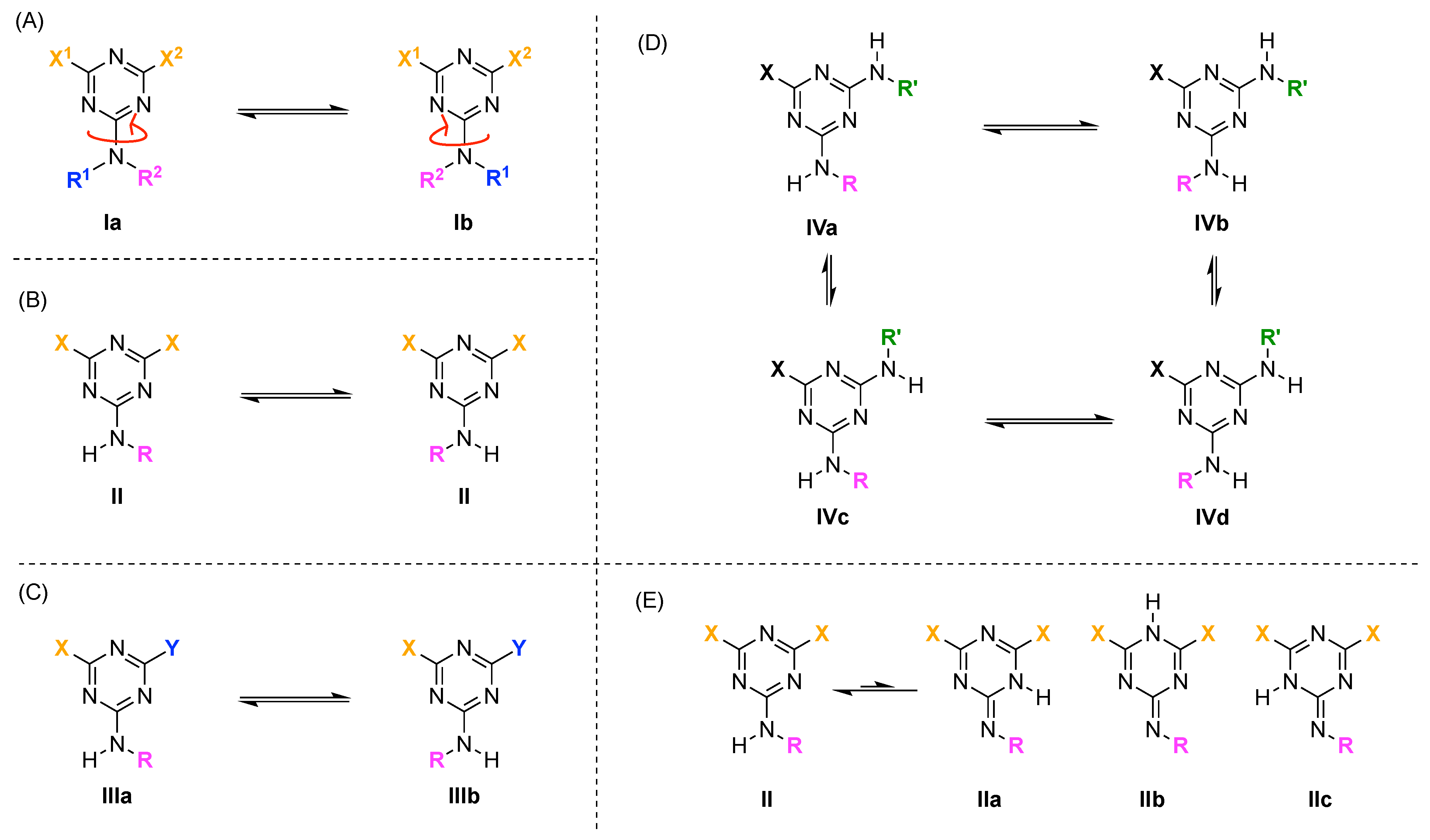{kind=link}
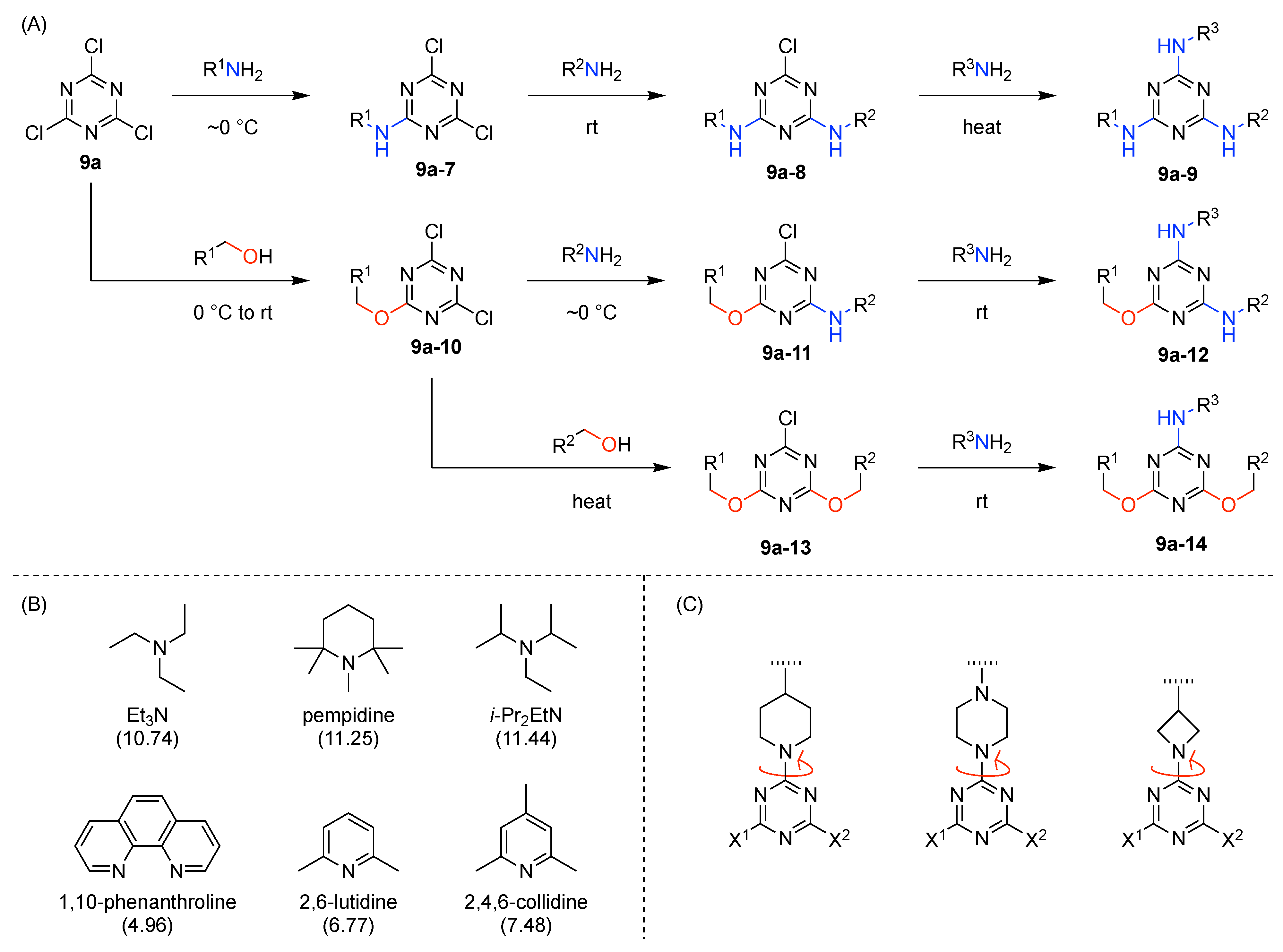{kind=link}
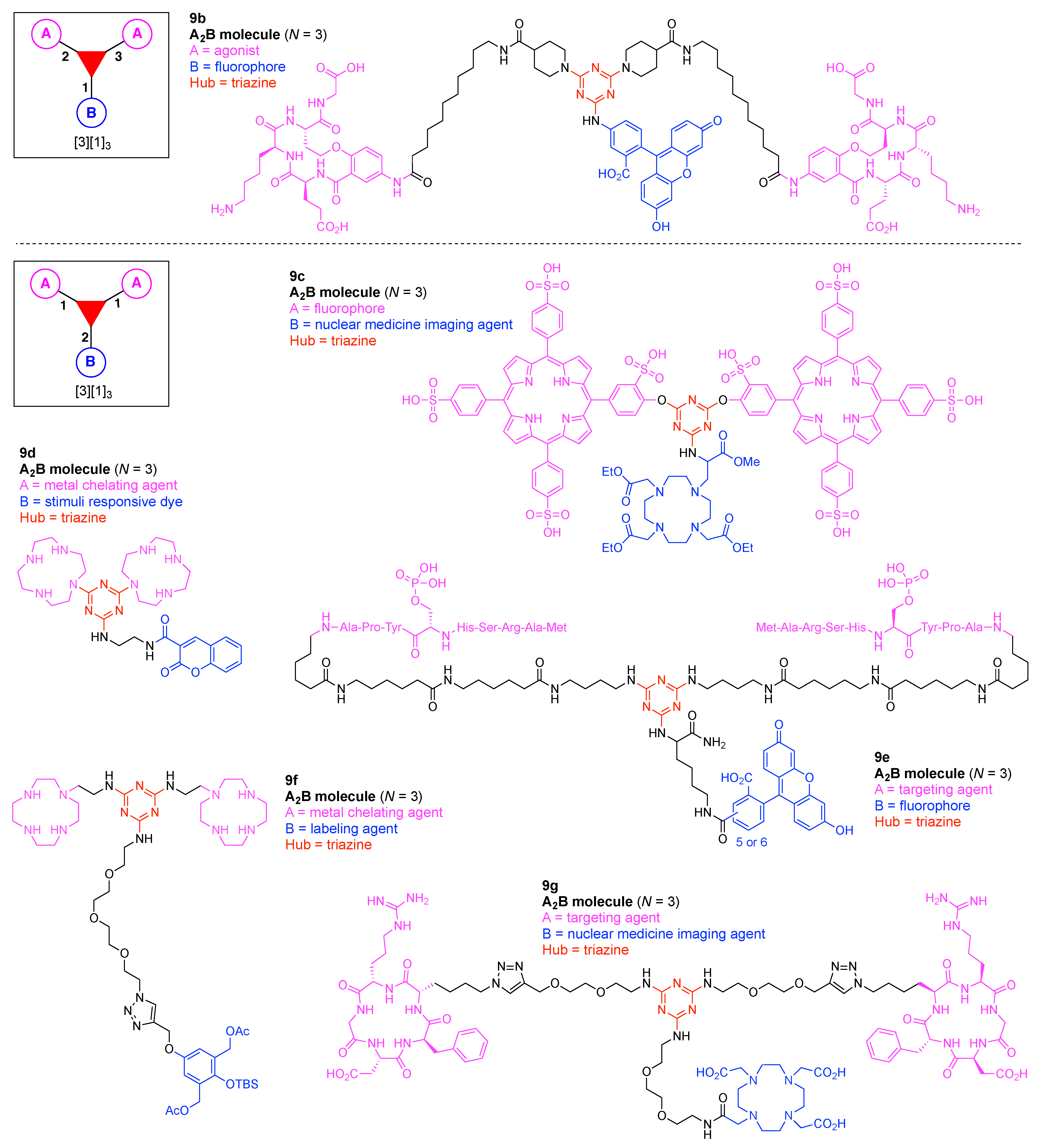{kind=link}
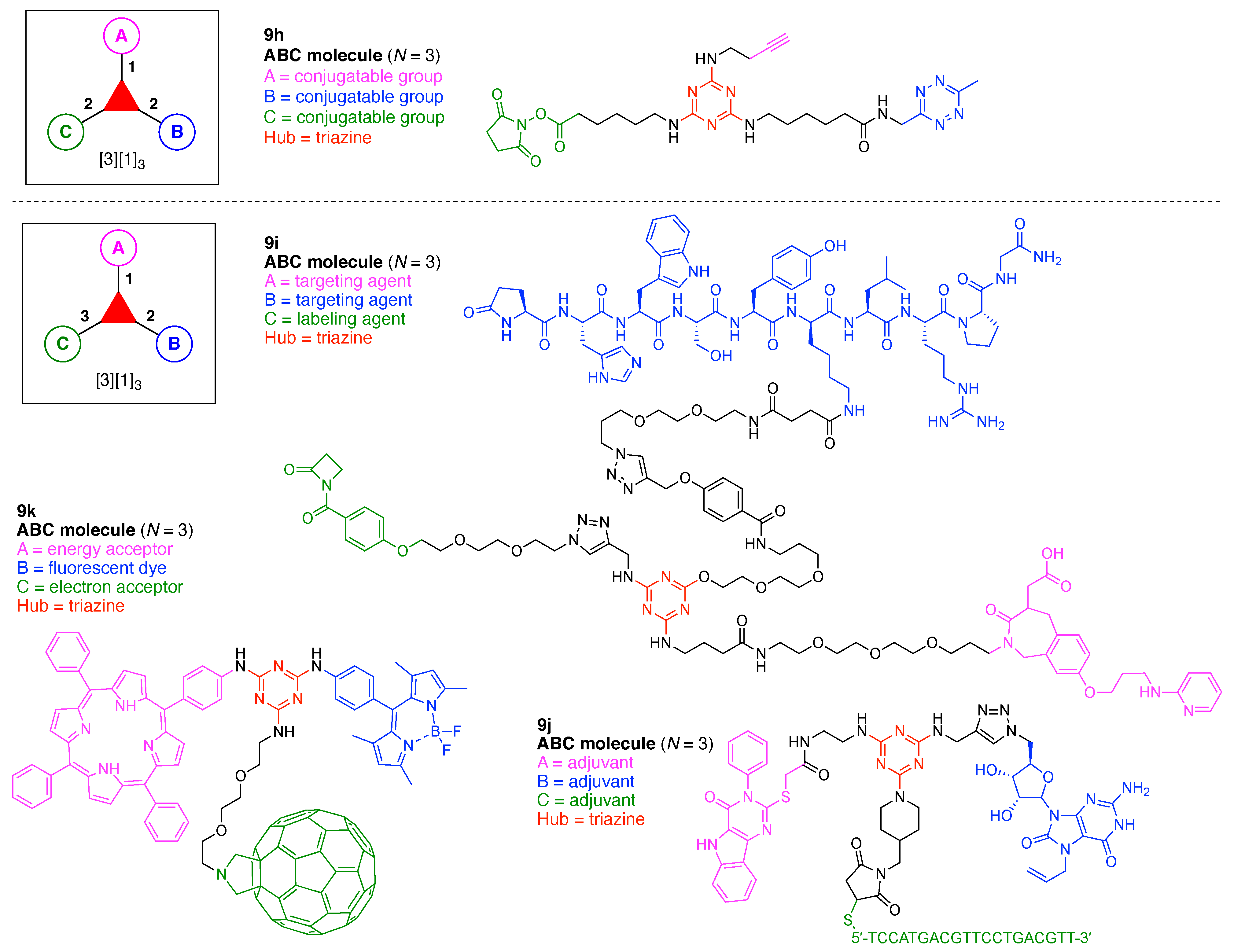{kind=link}
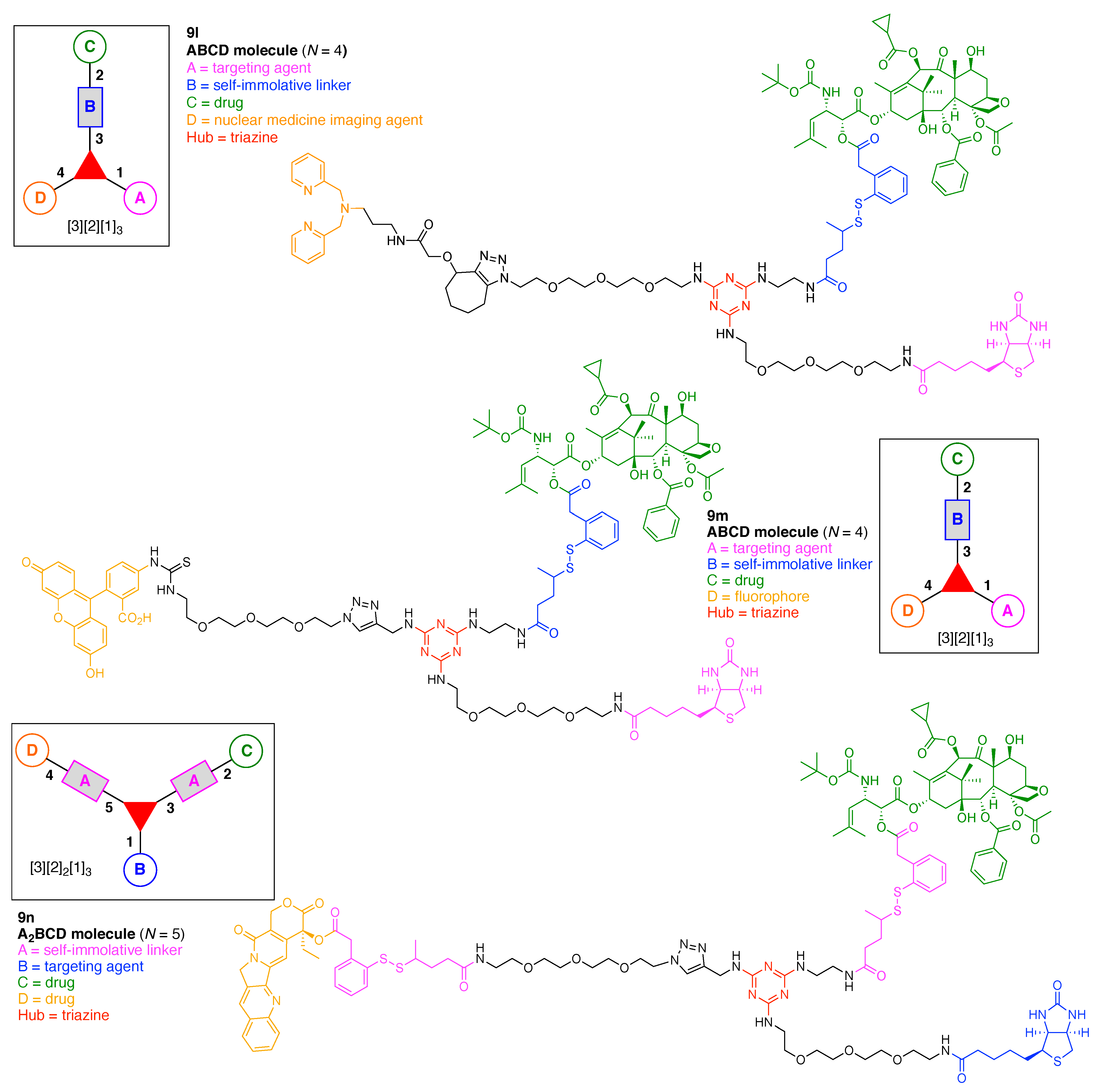{kind=link}
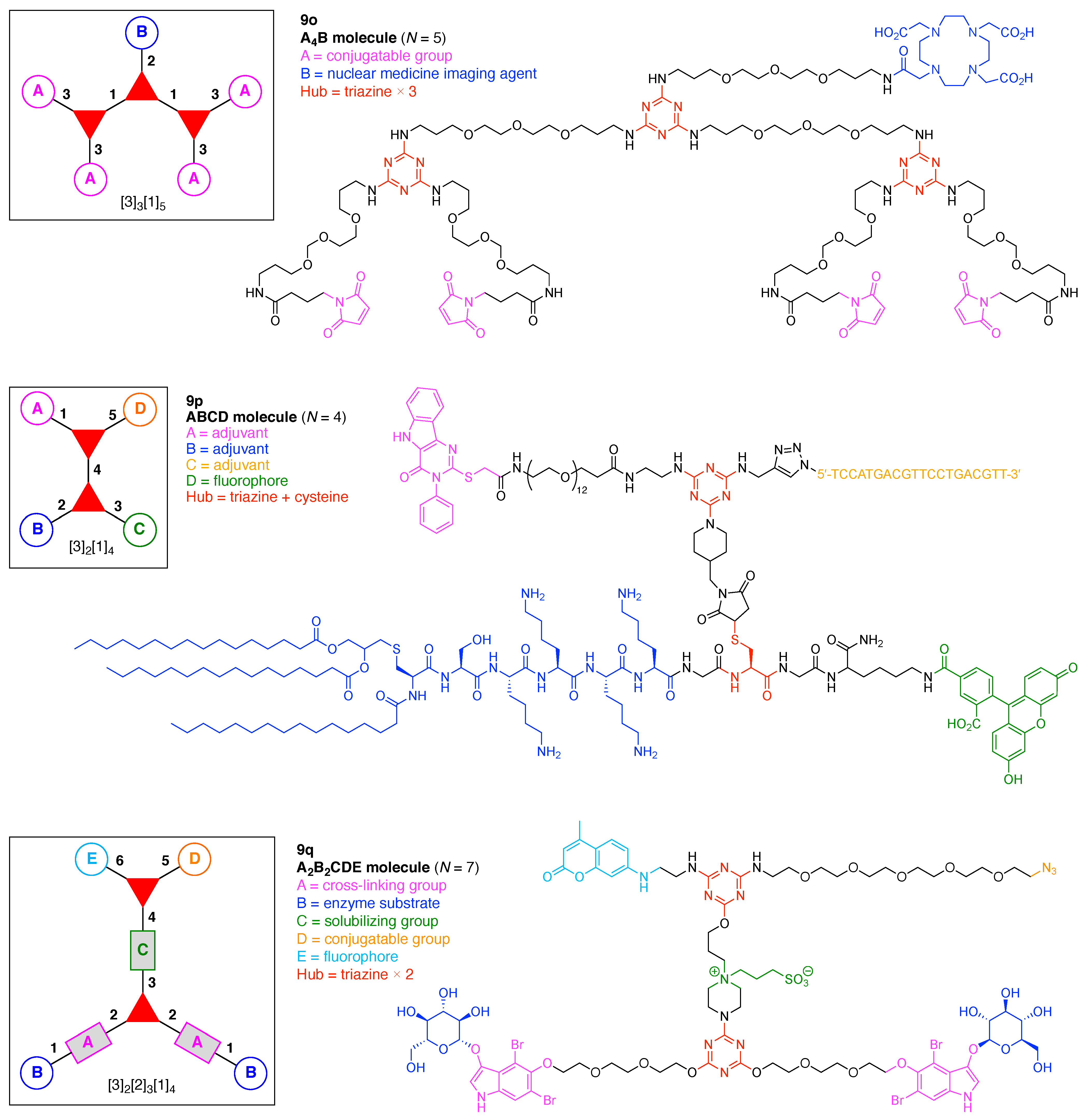{kind=link}
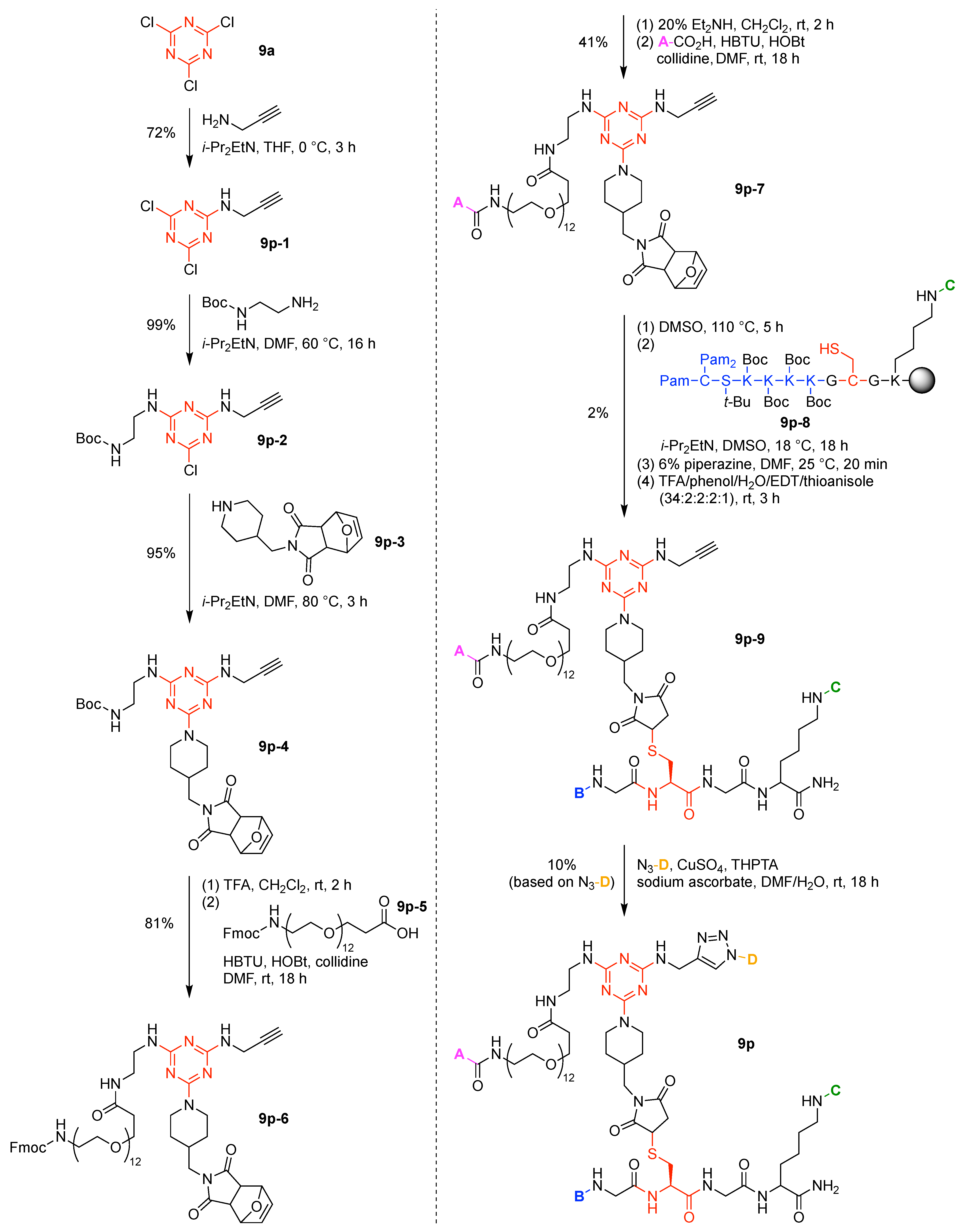{kind=link}
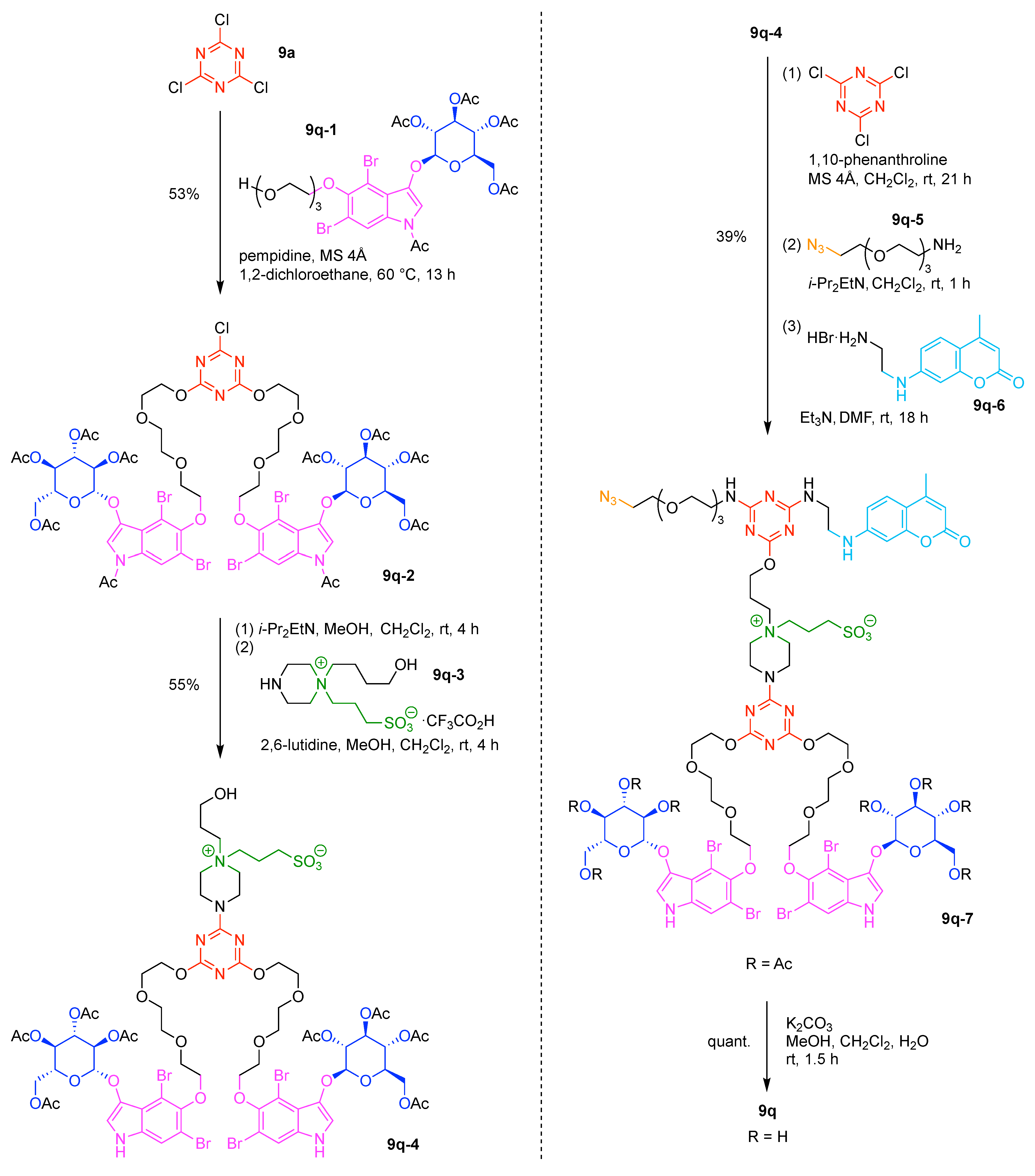{kind=link}
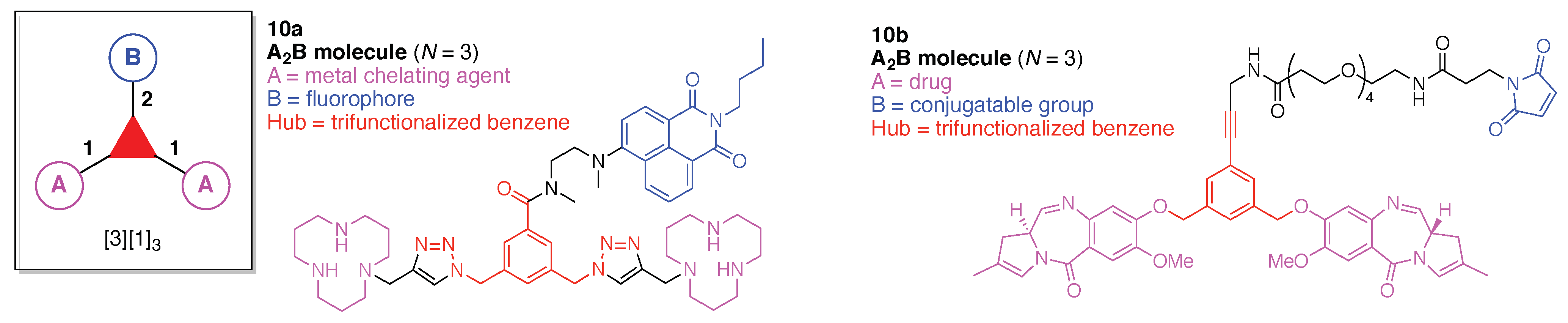{kind=link}
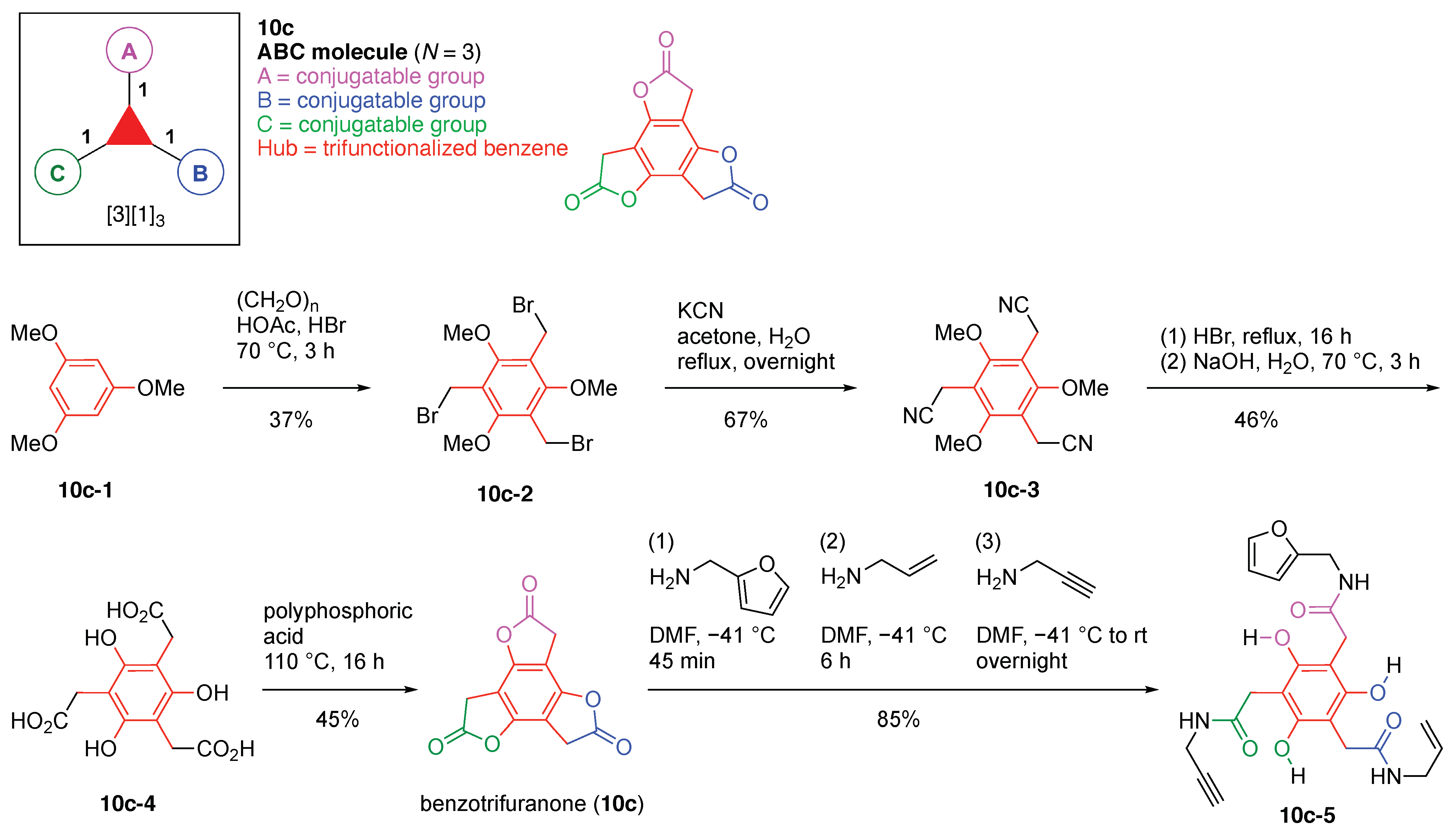{kind=link}
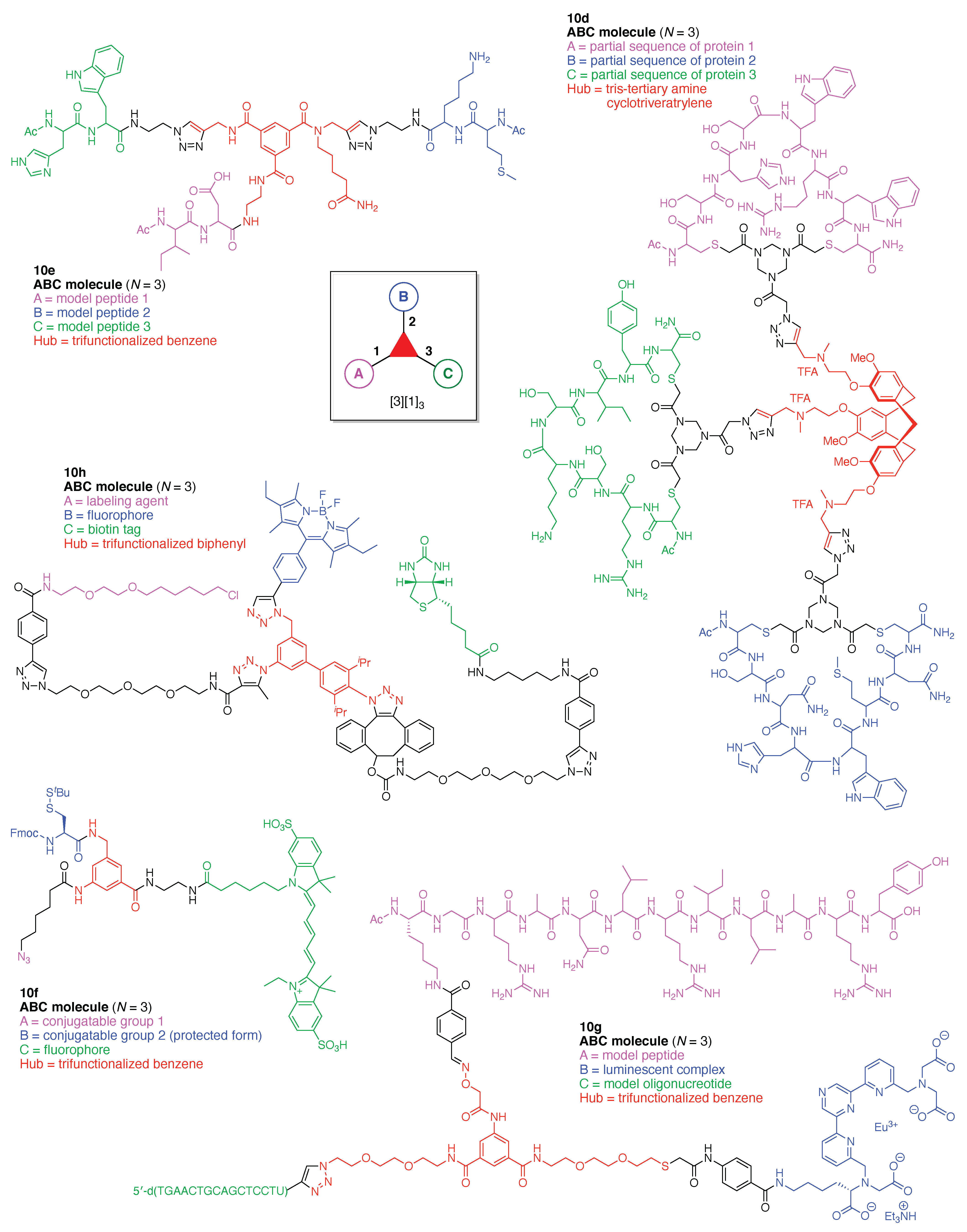{kind=link}
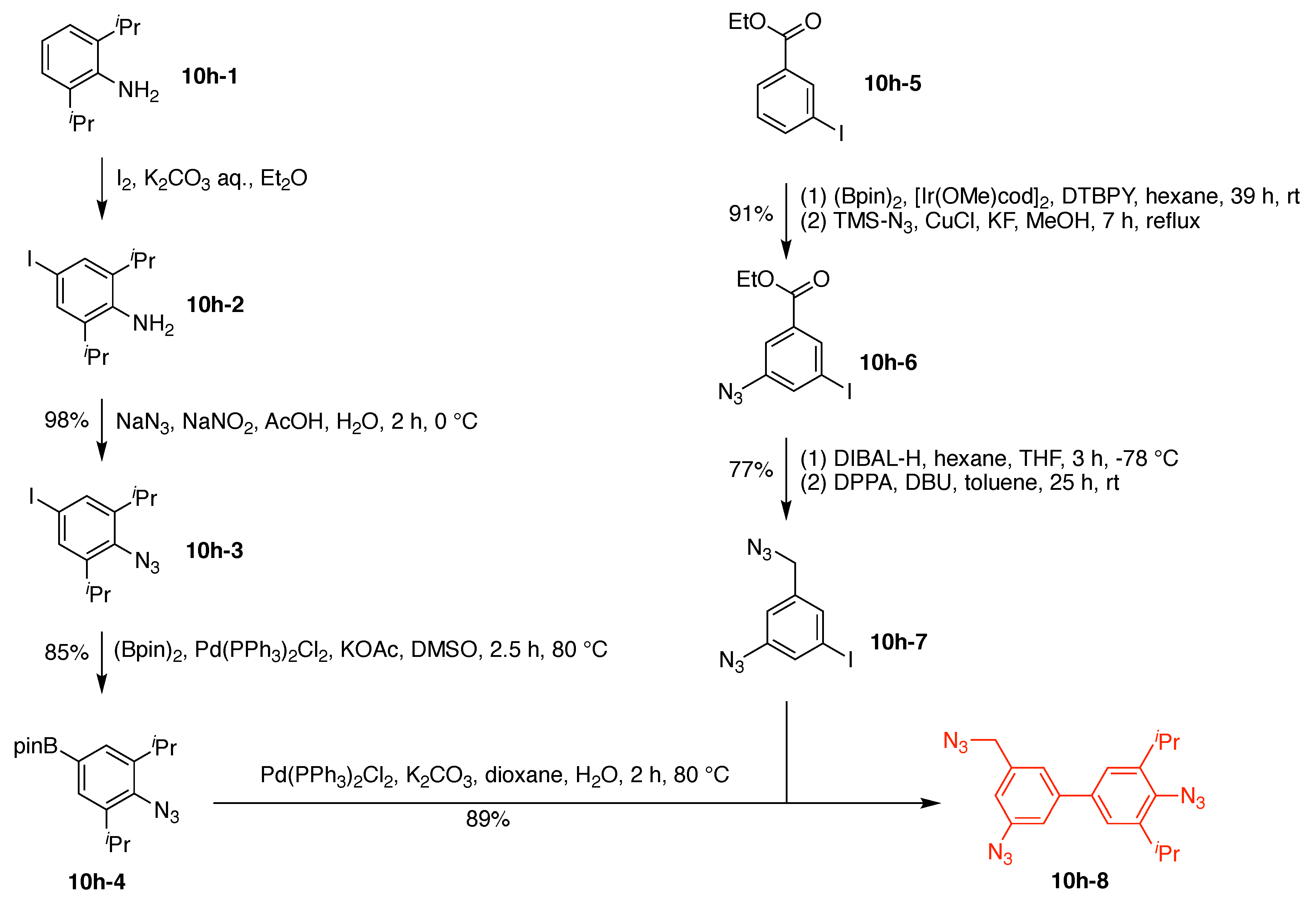{kind=link}
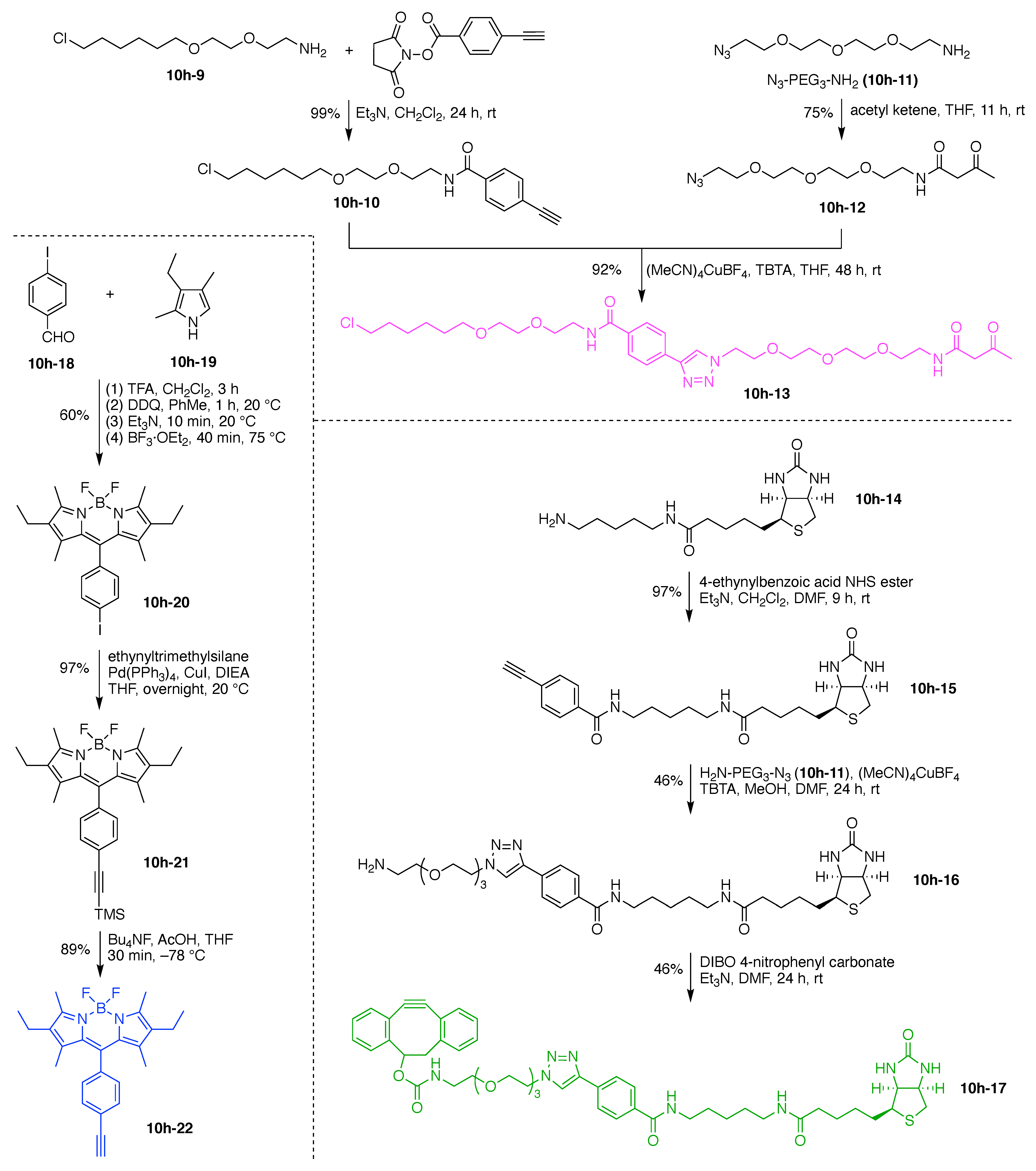{kind=link}
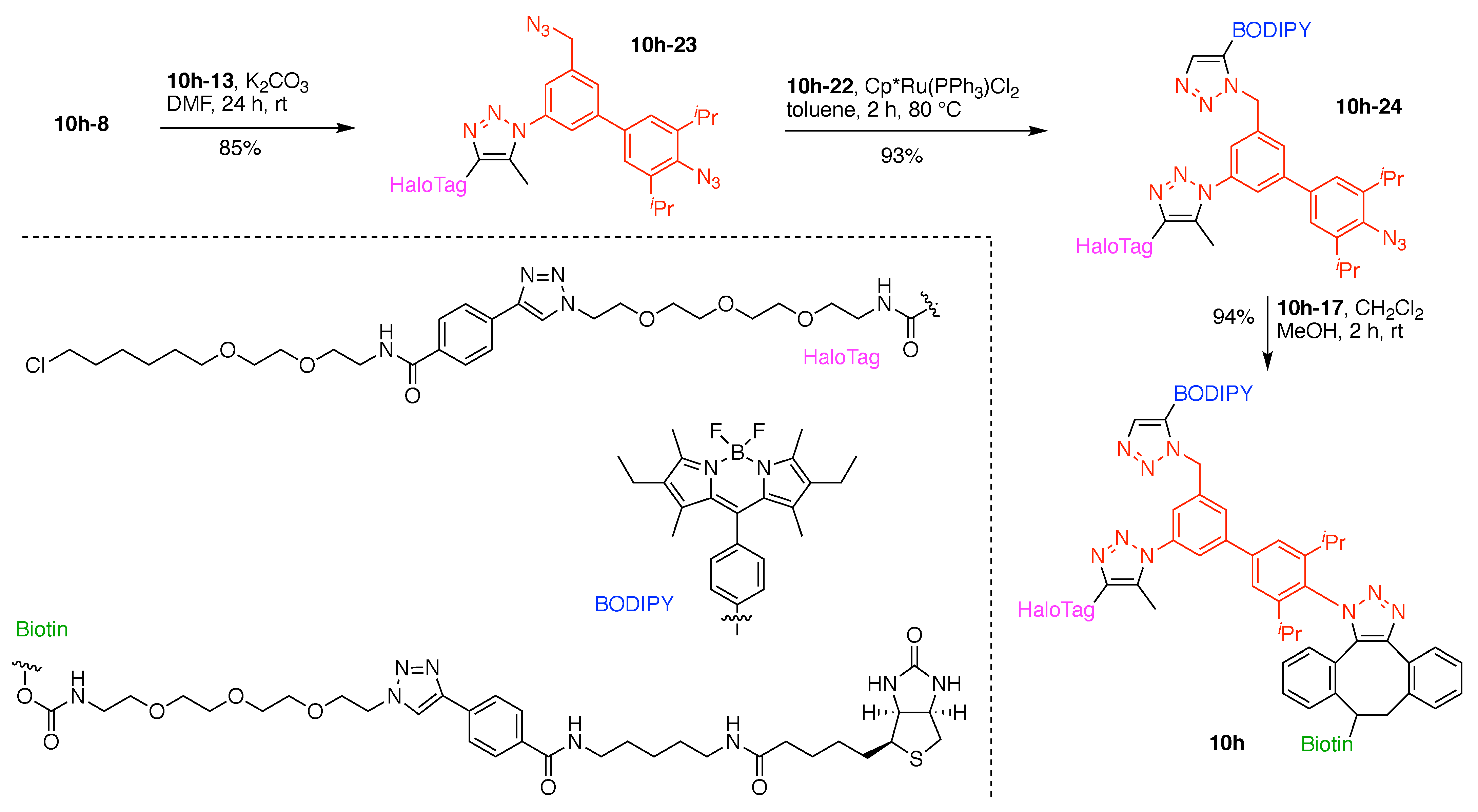{kind=link}
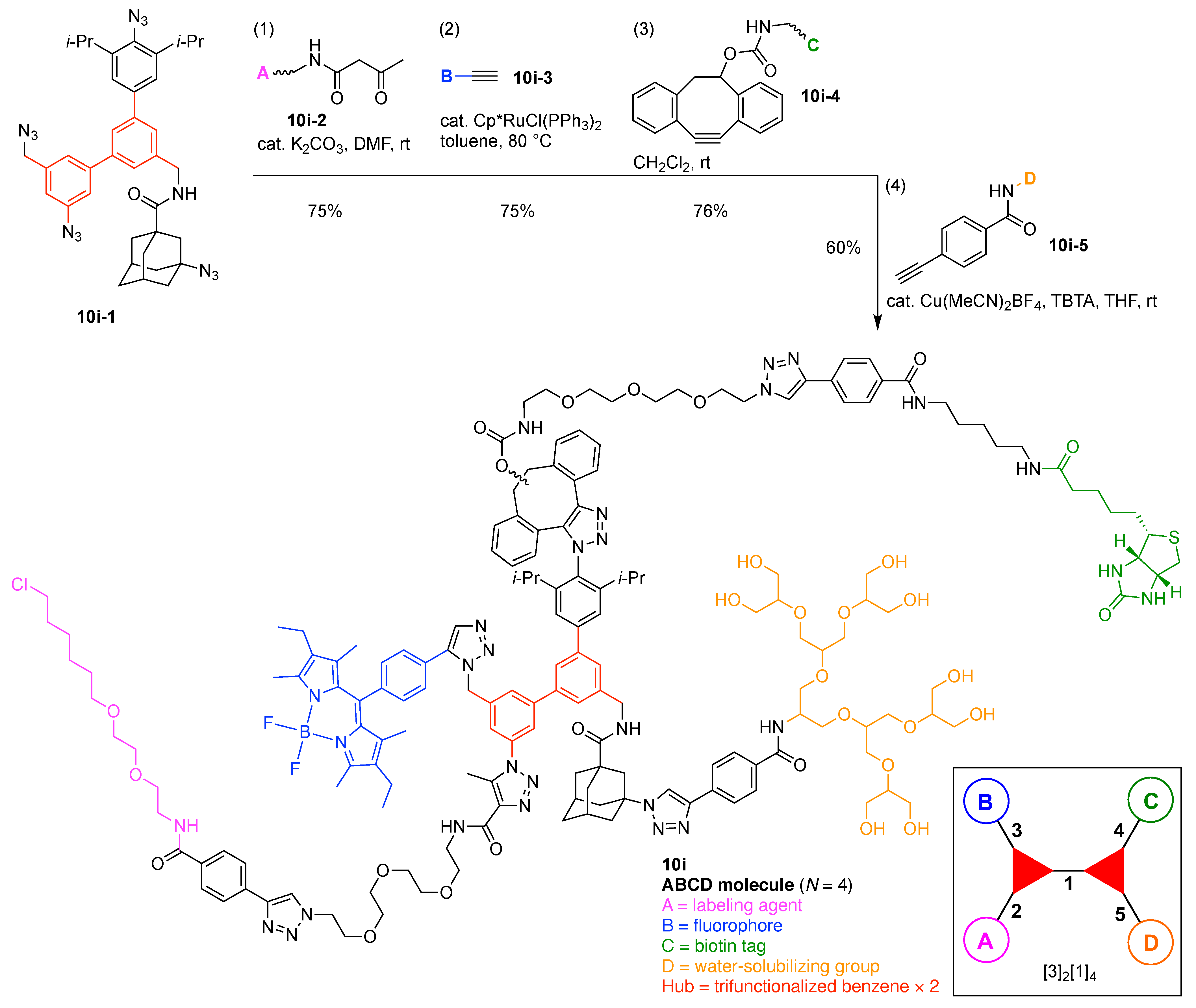{kind=link}
{kind=link}
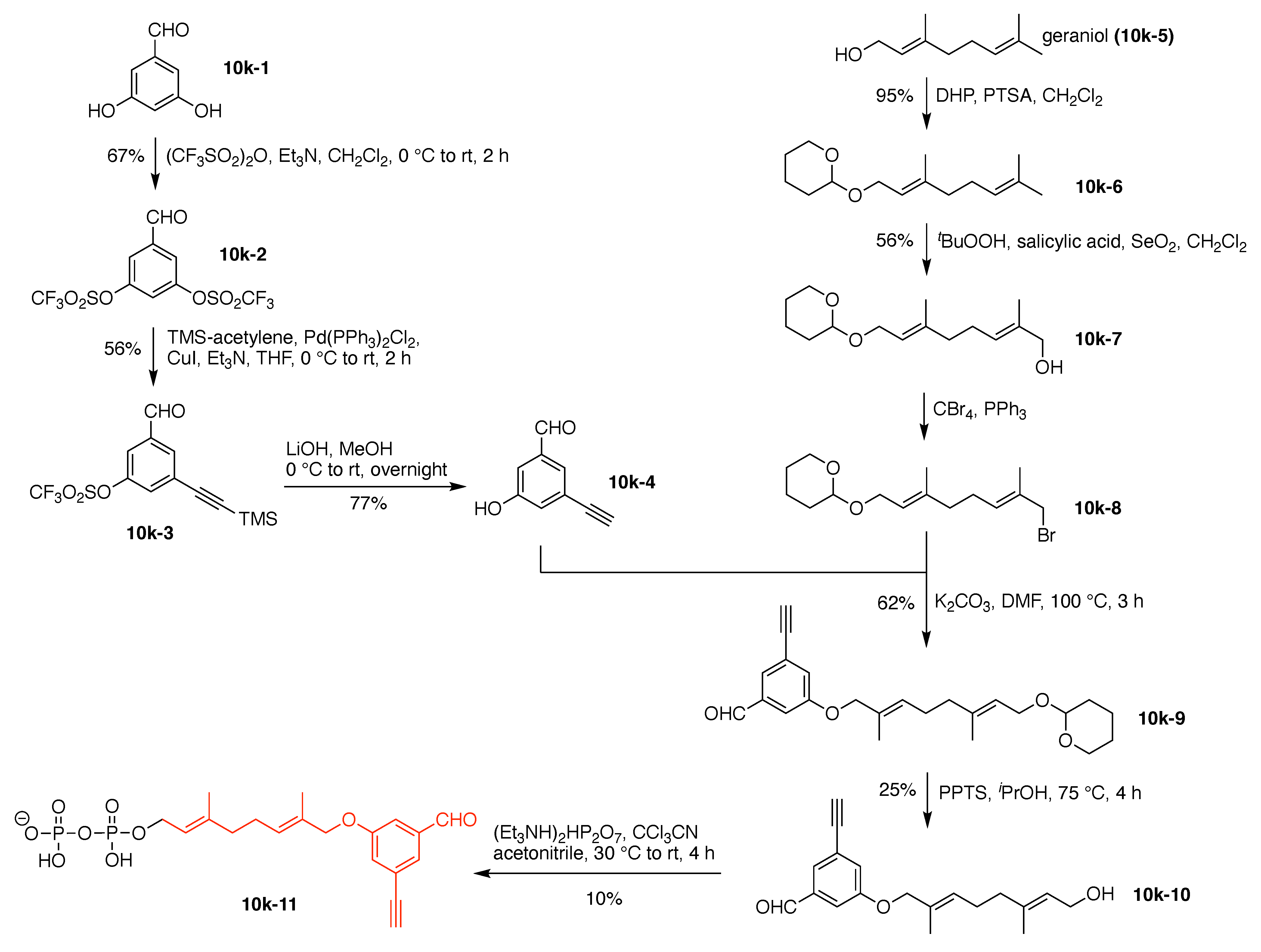{kind=link}
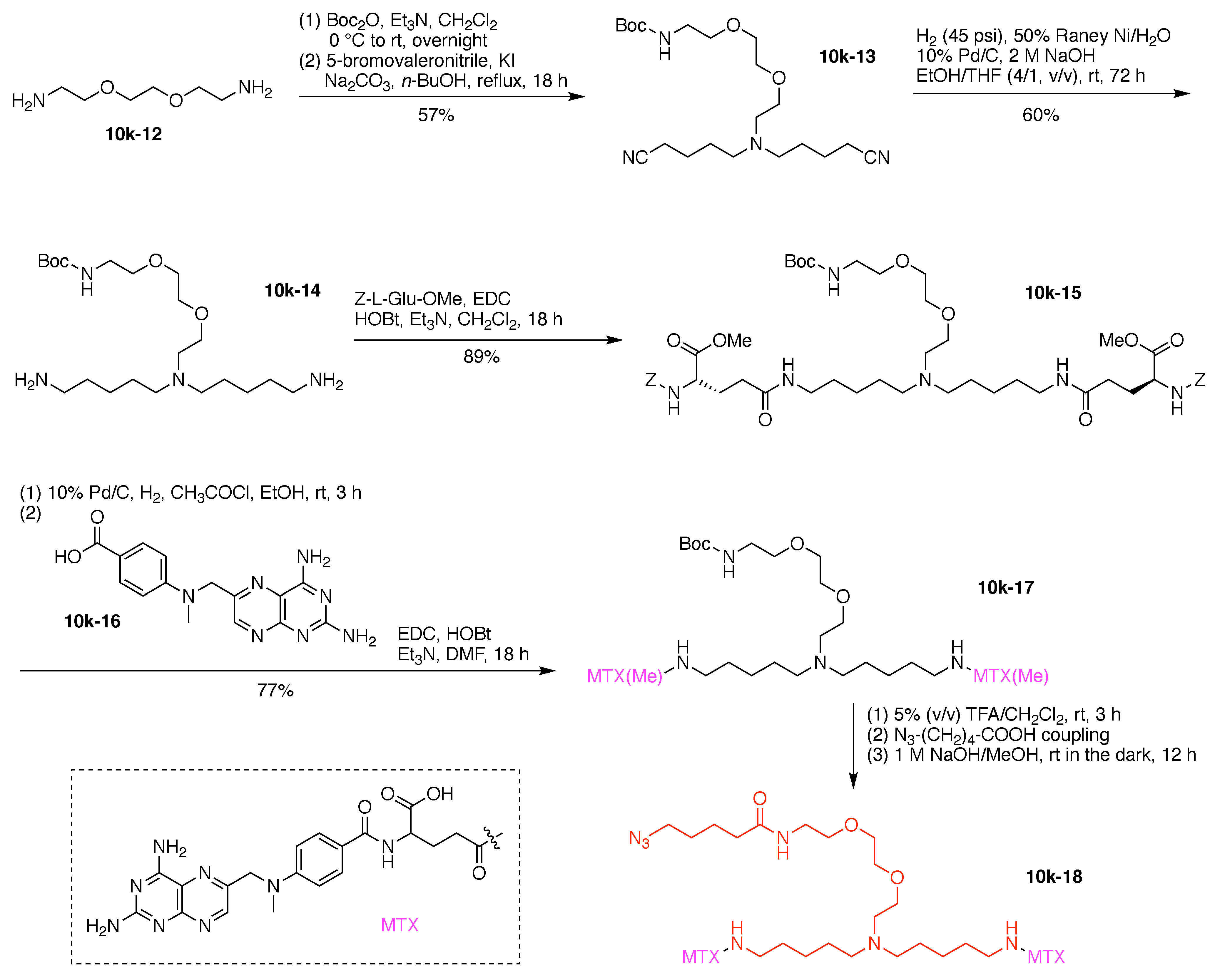{kind=link}
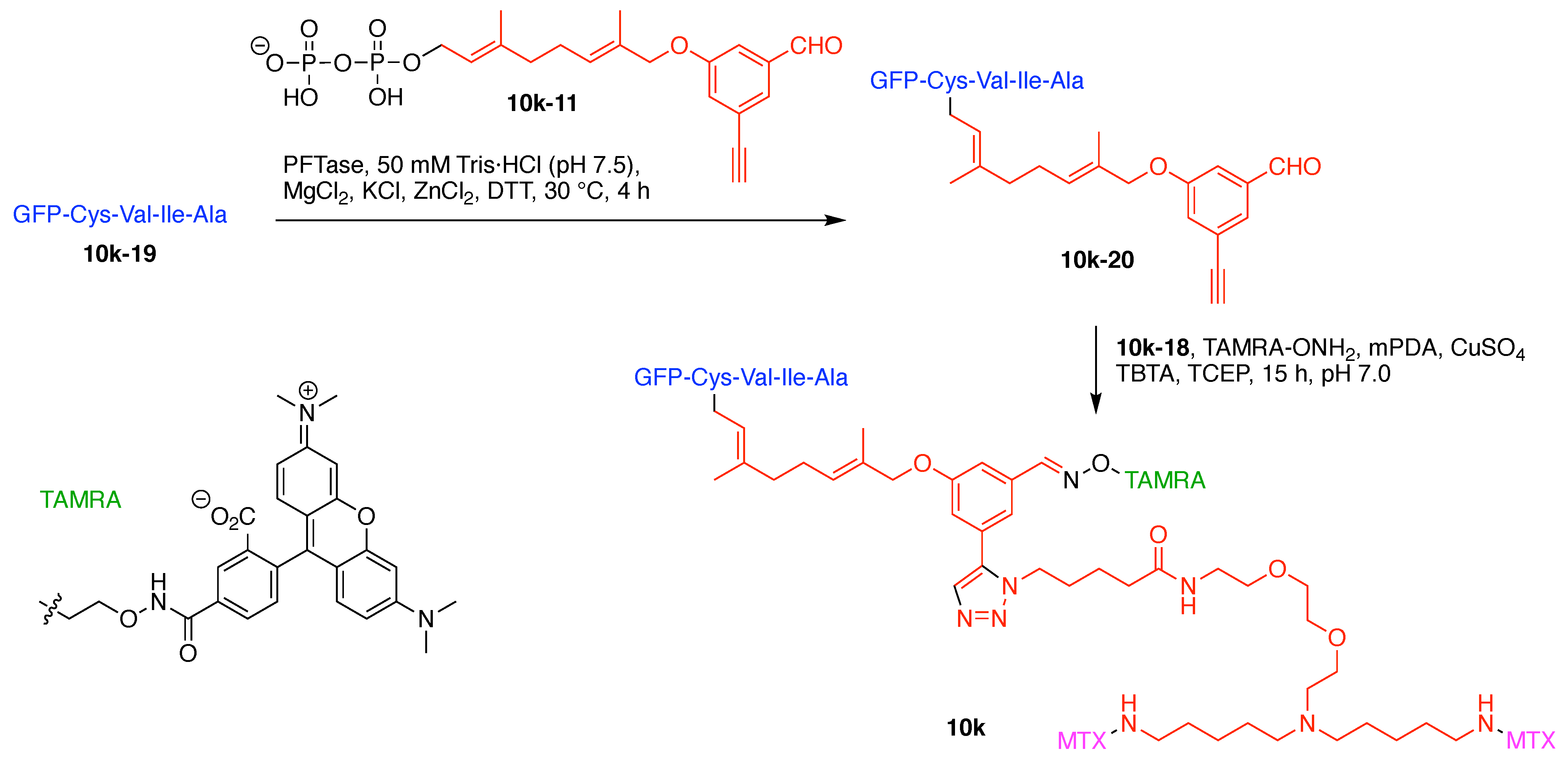{kind=link}
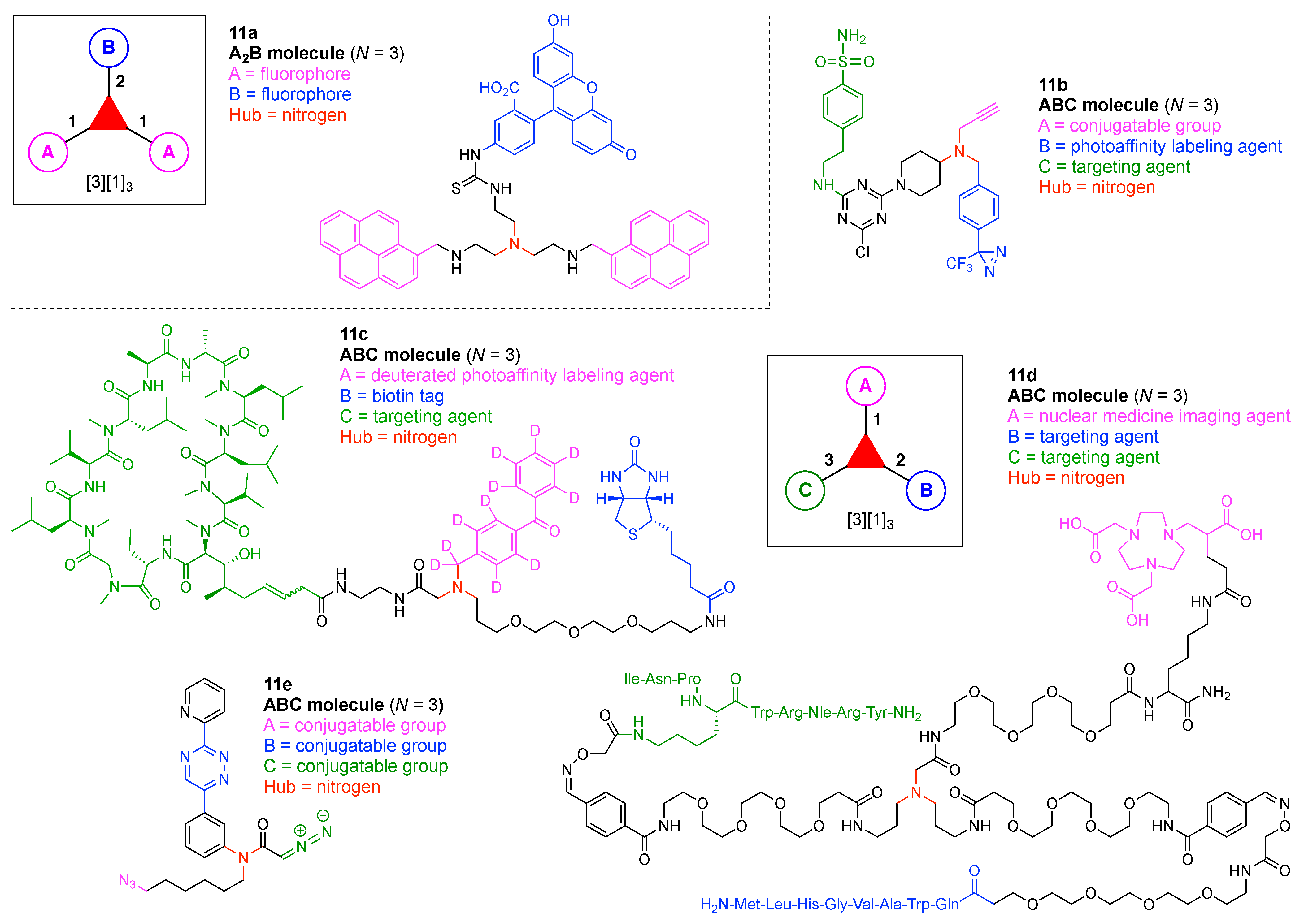{kind=link}
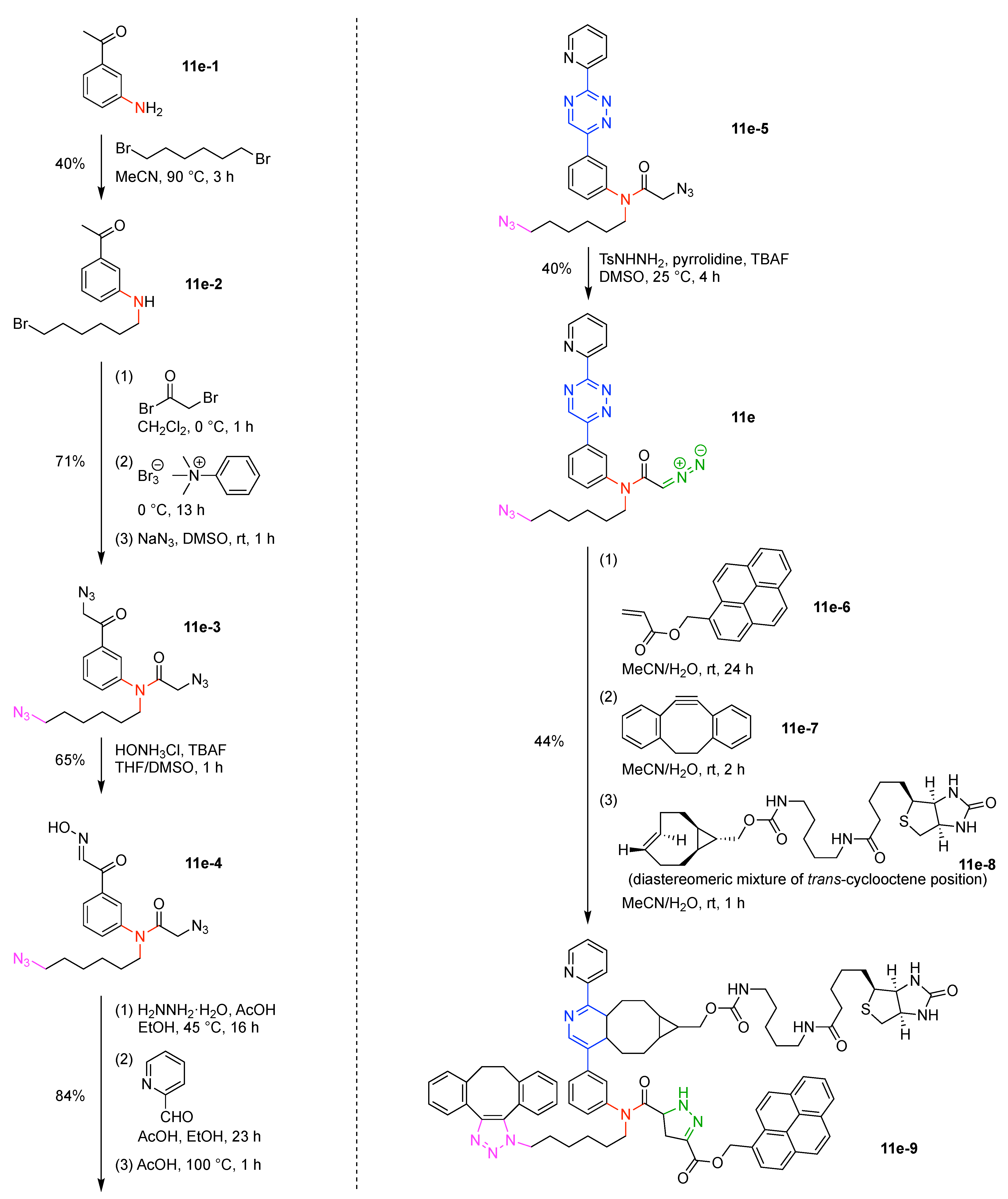{kind=link}
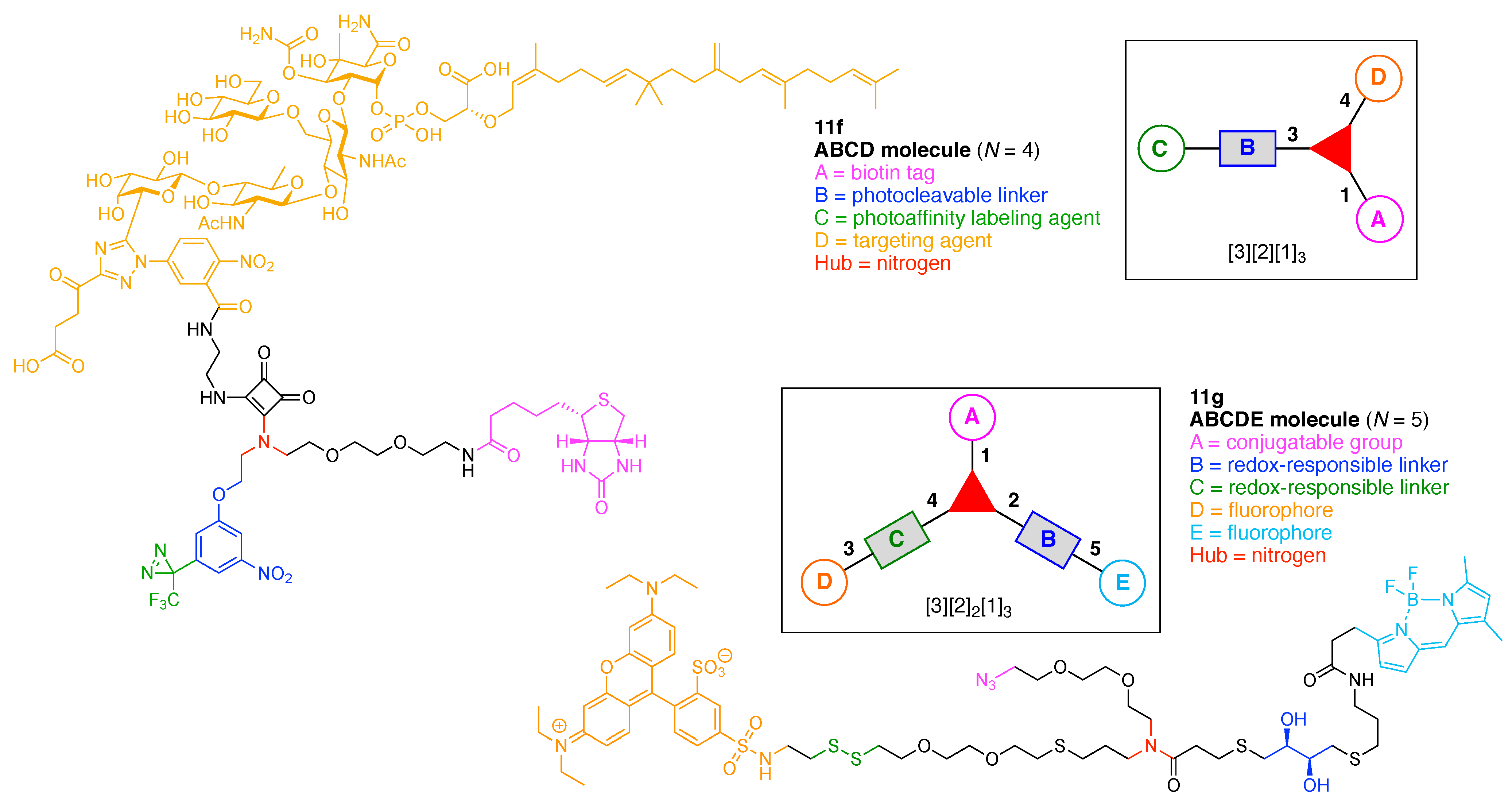{kind=link}
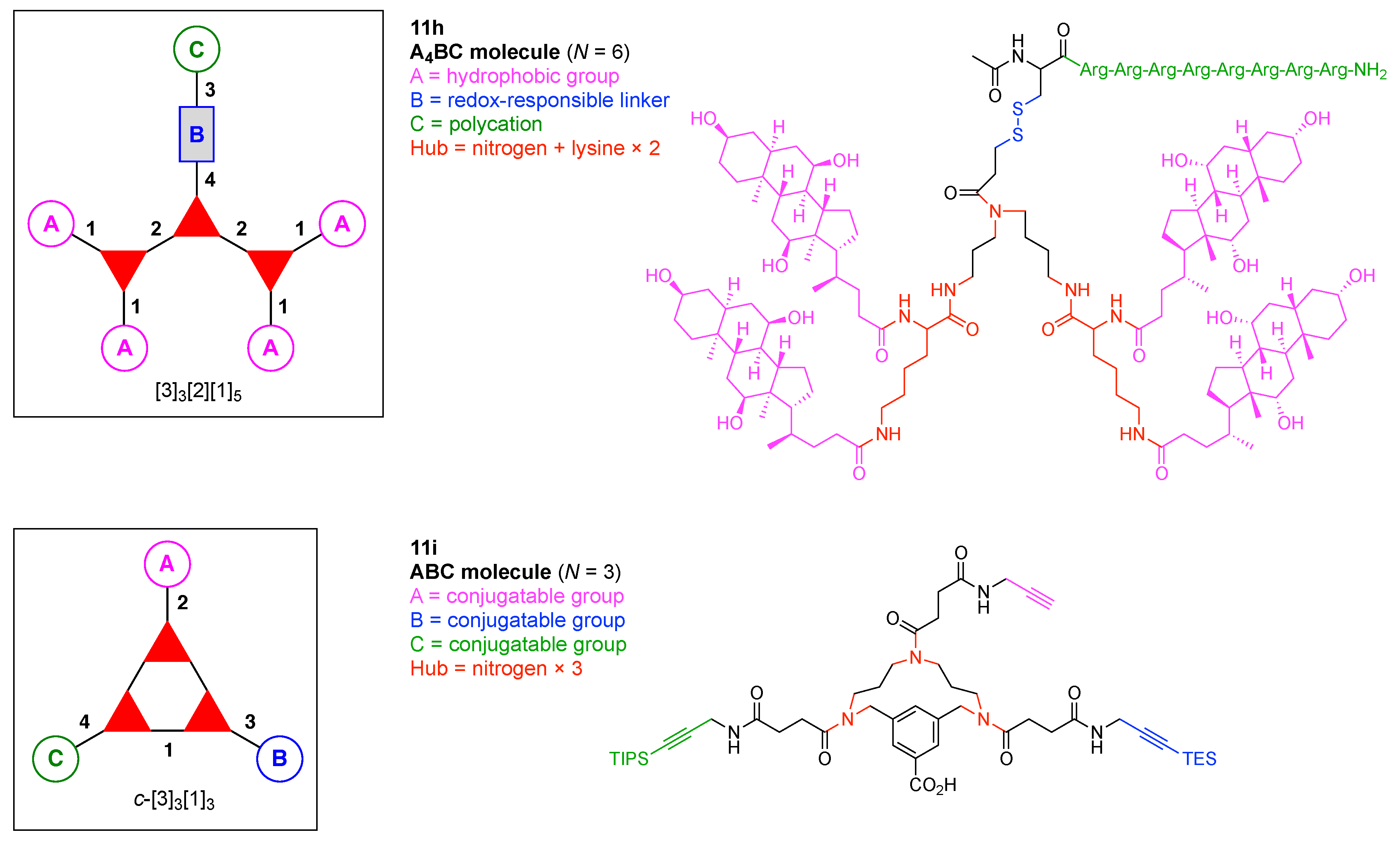{kind=link}
{kind=link}
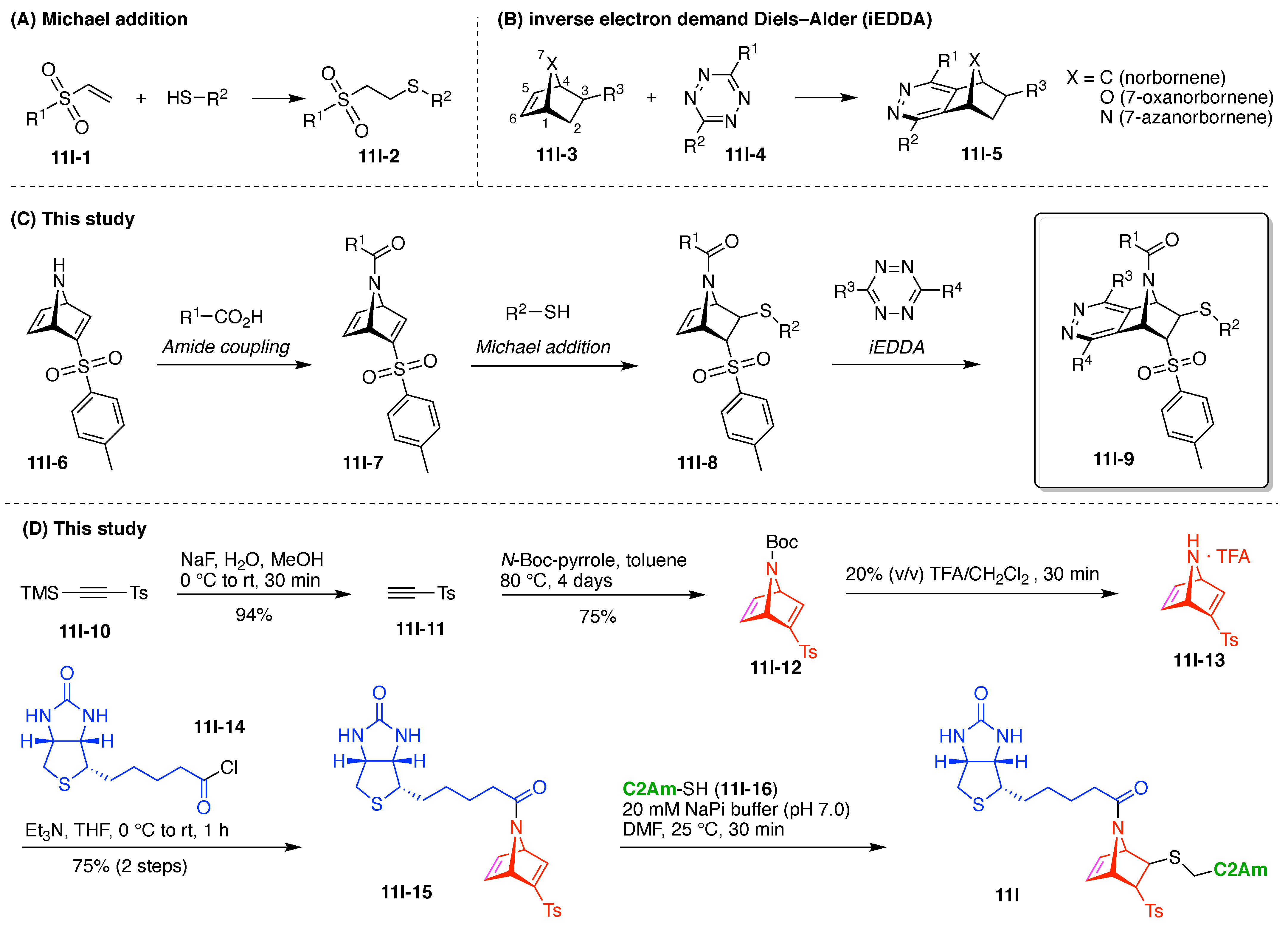{kind=link}
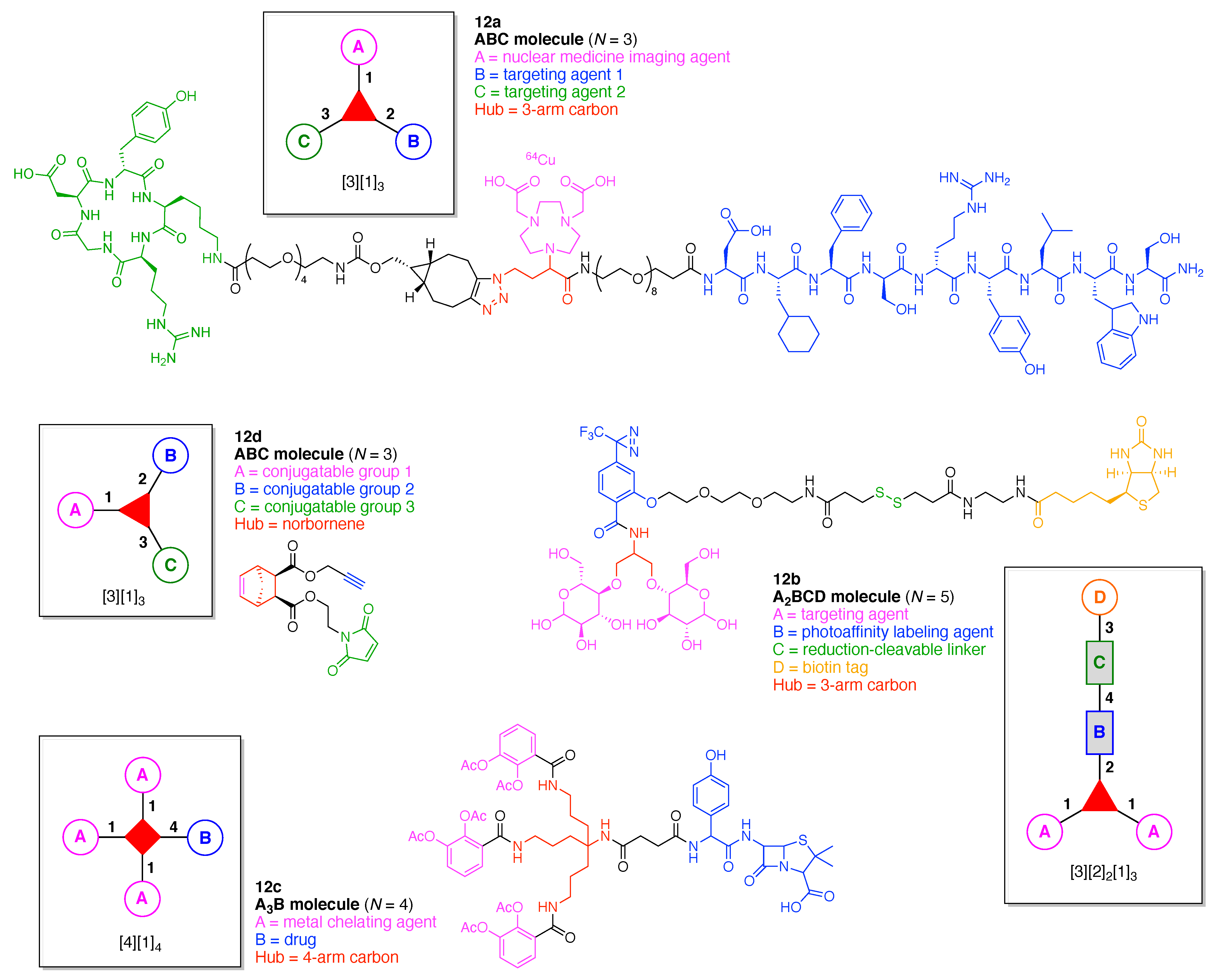{kind=link}
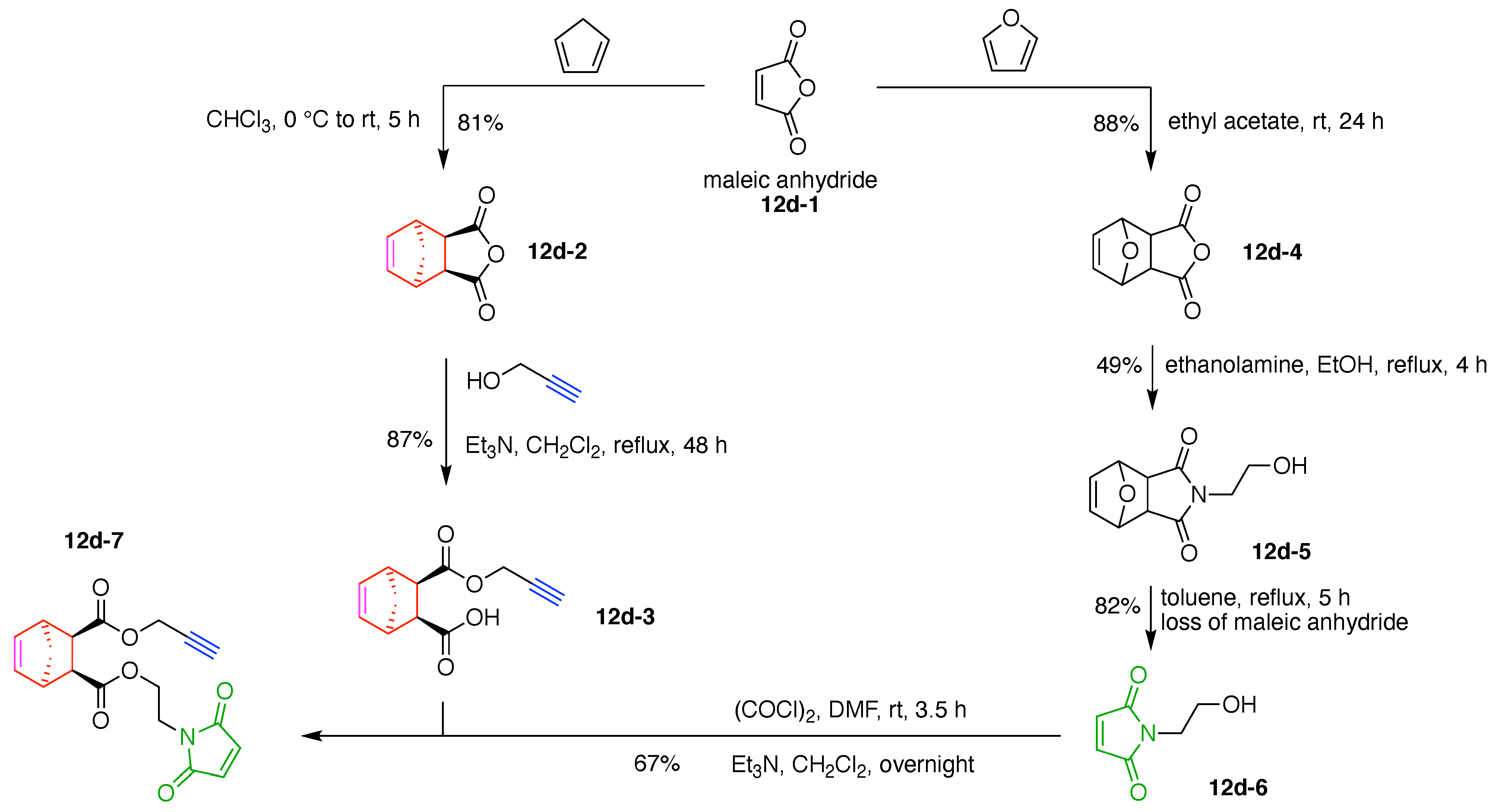{kind=link}
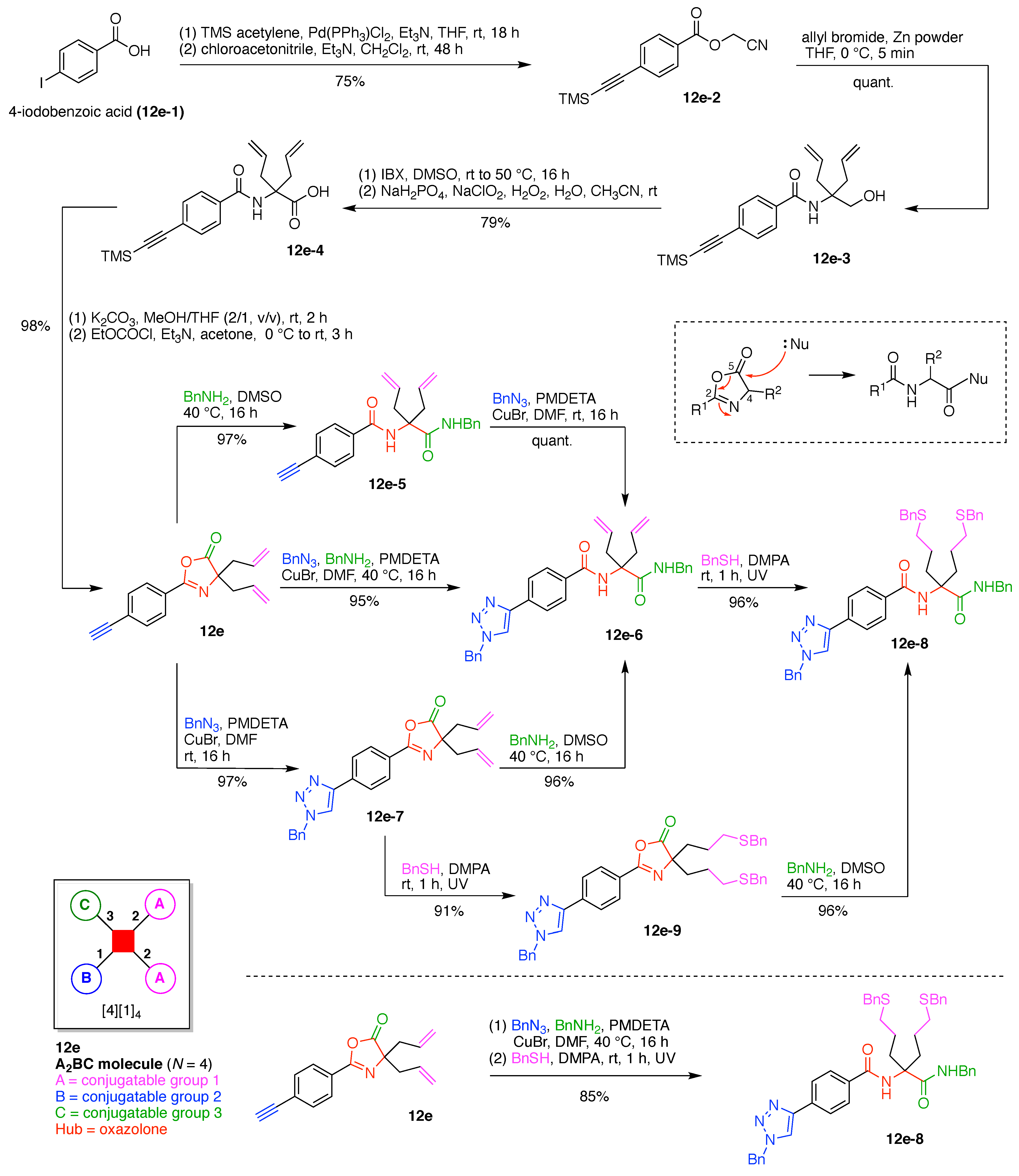{kind=link}
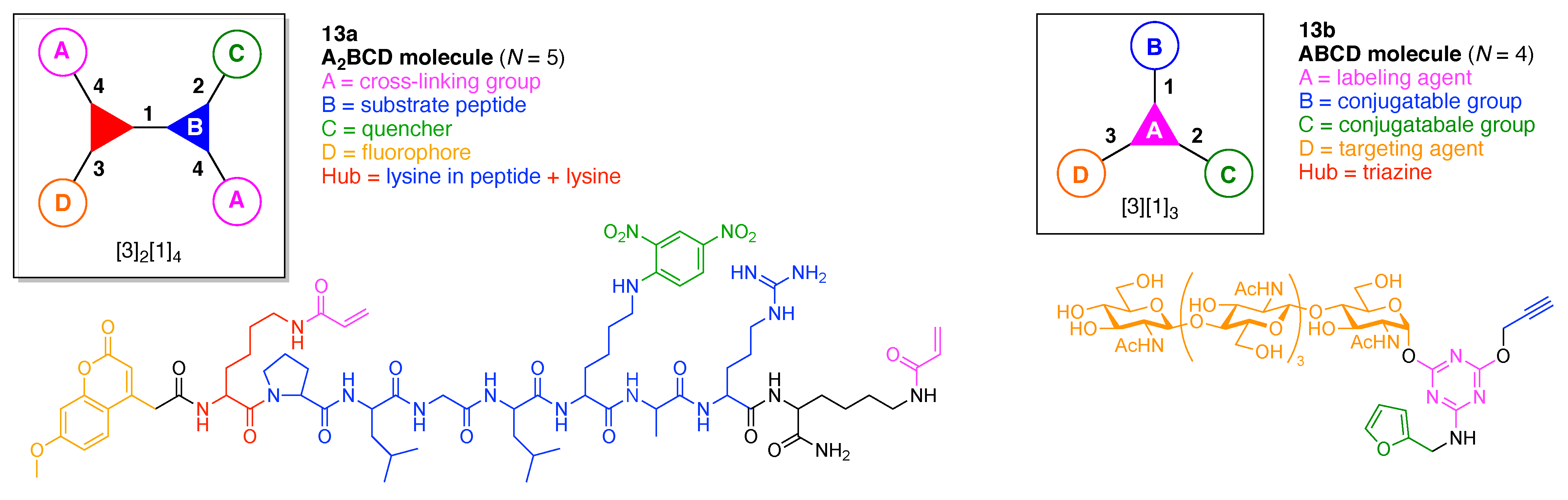{kind=link}
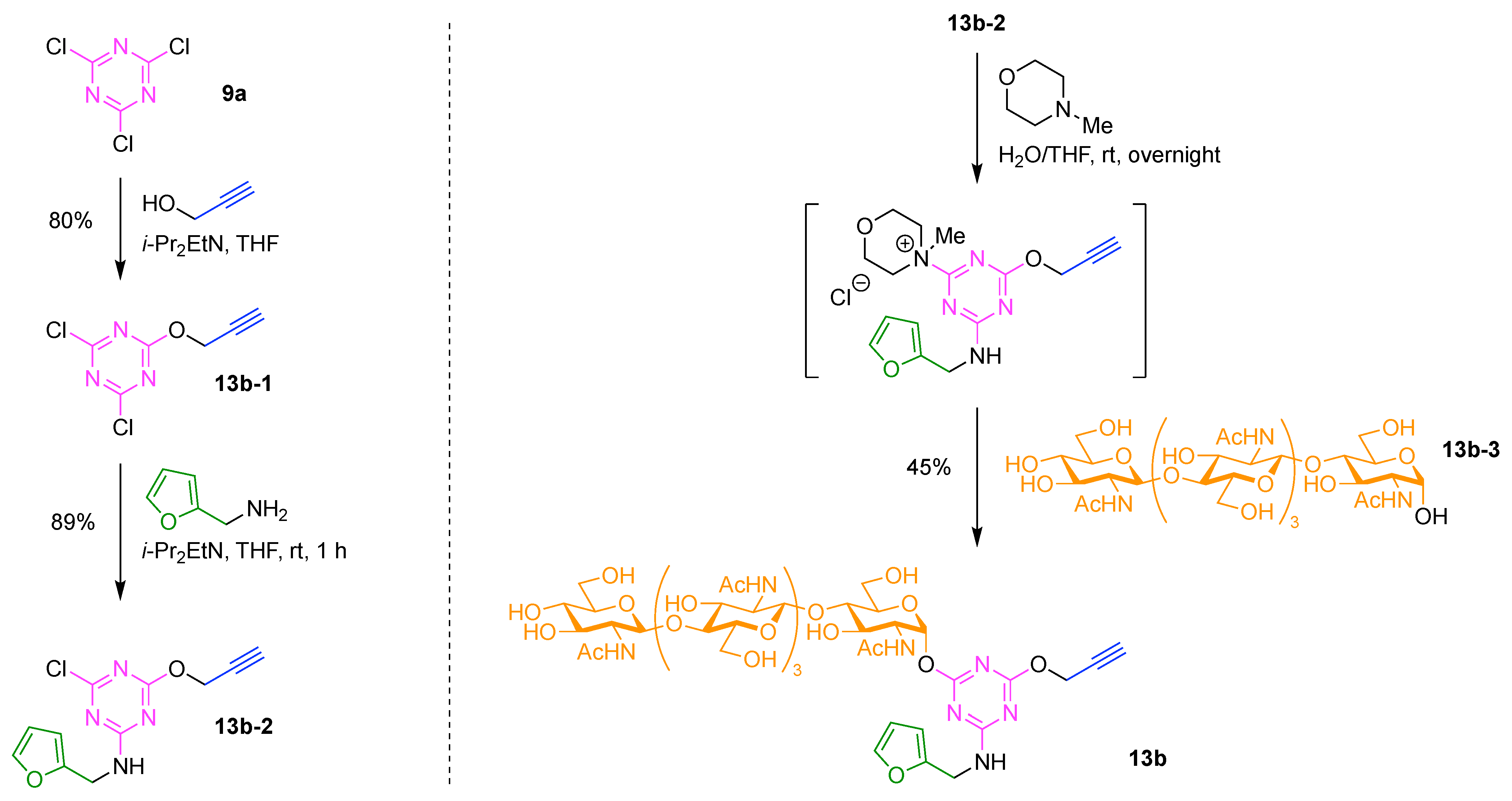{kind=link}
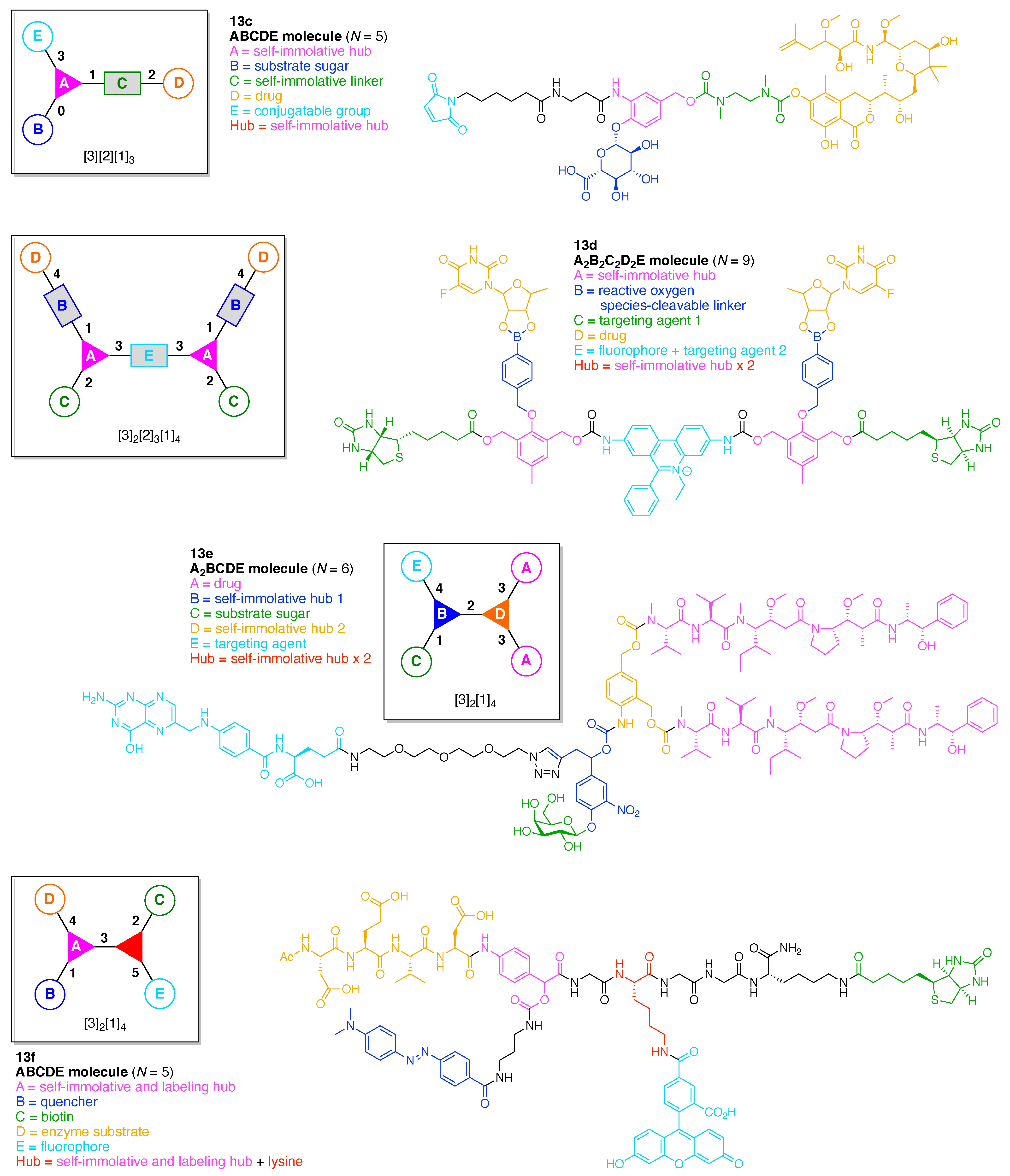{kind=link}
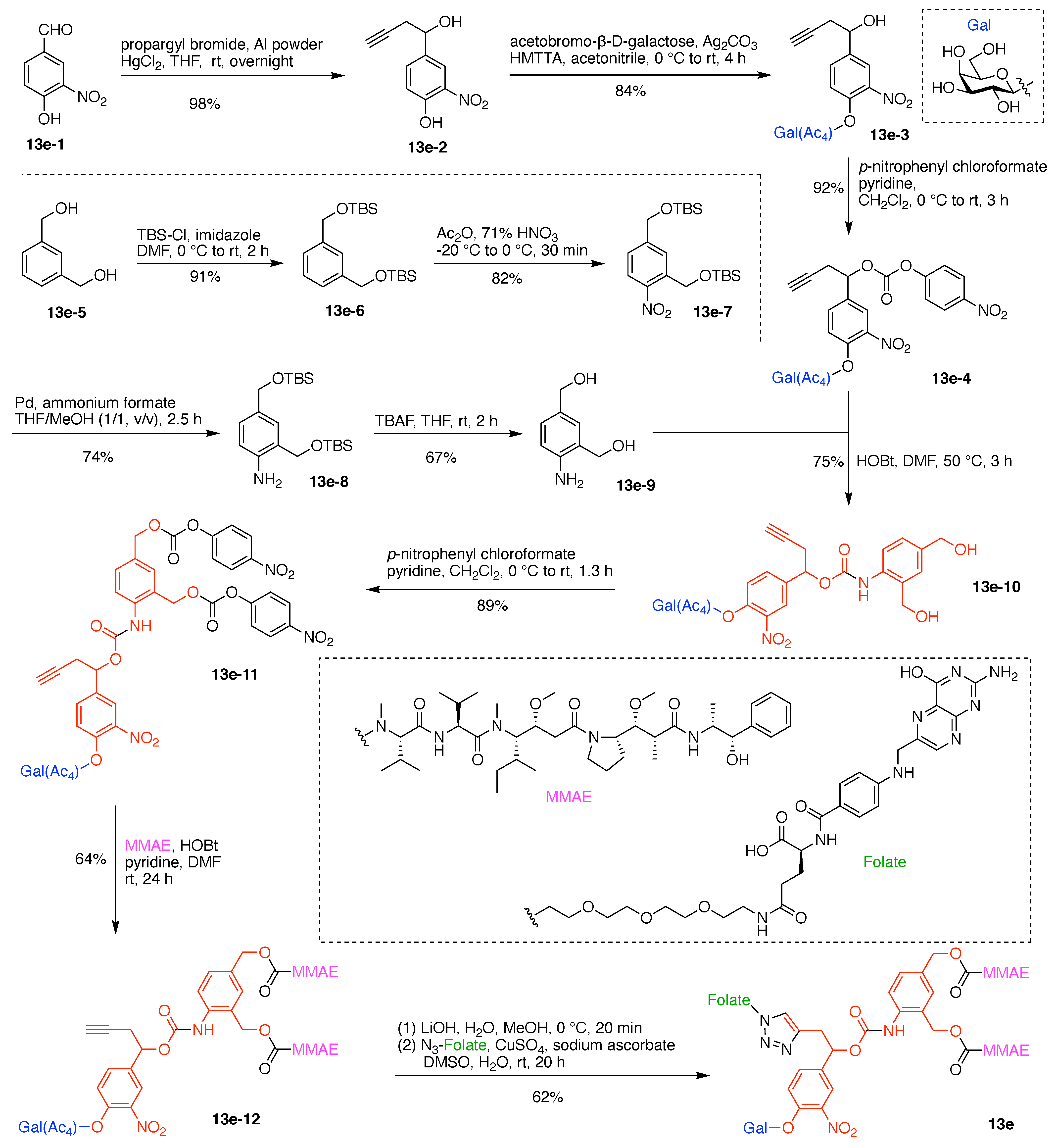{kind=link}
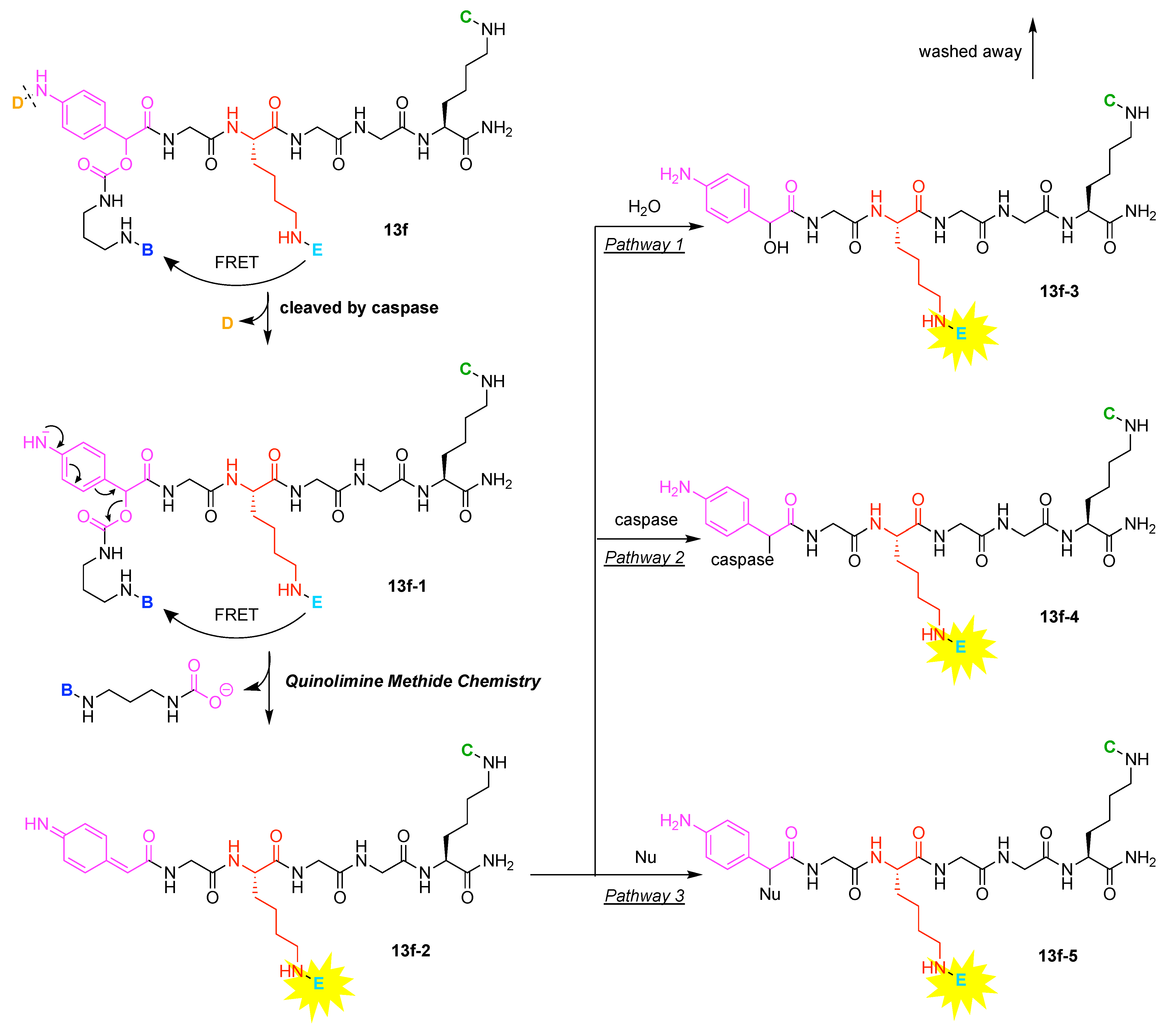{kind=link}
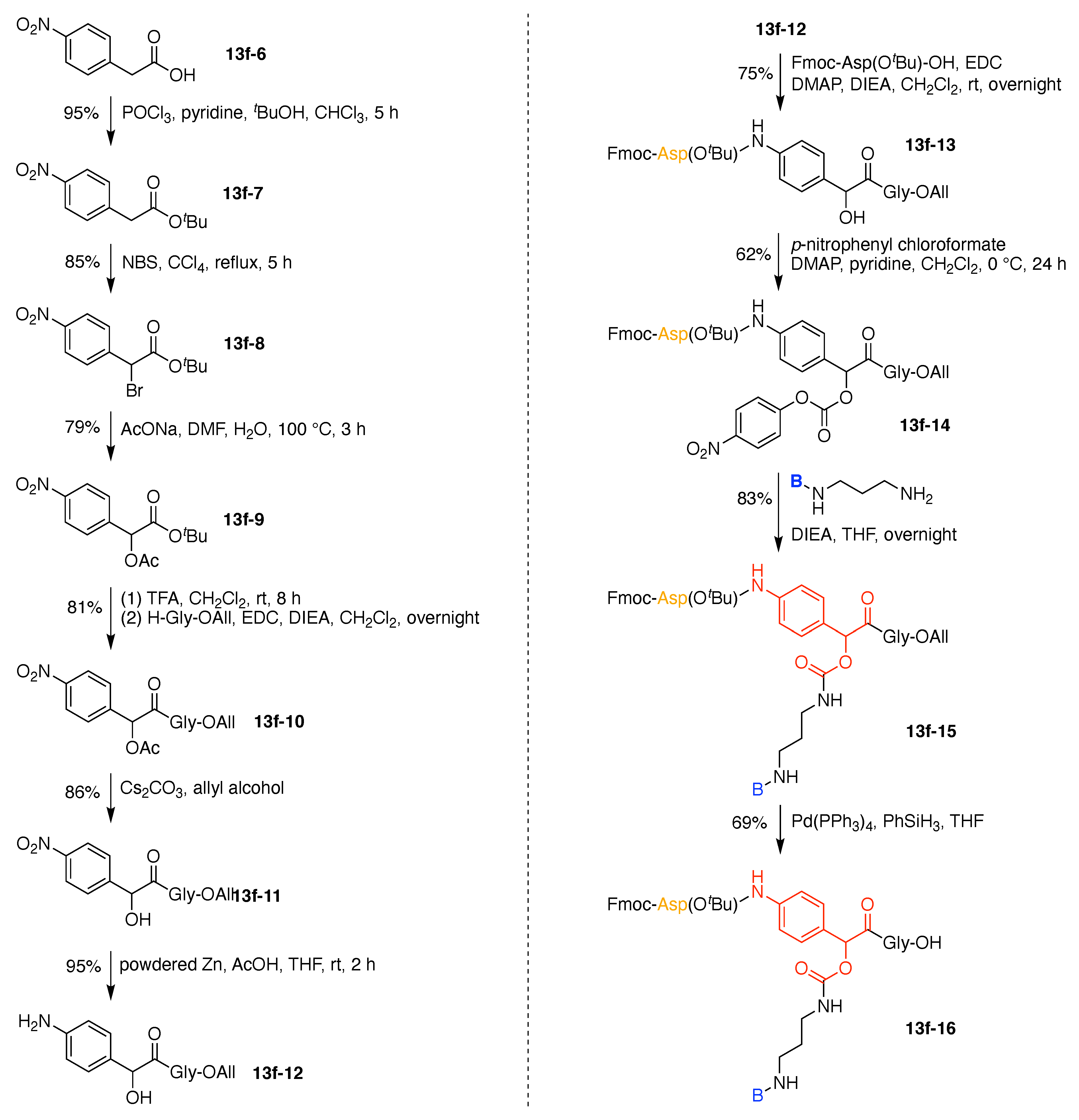{kind=link}
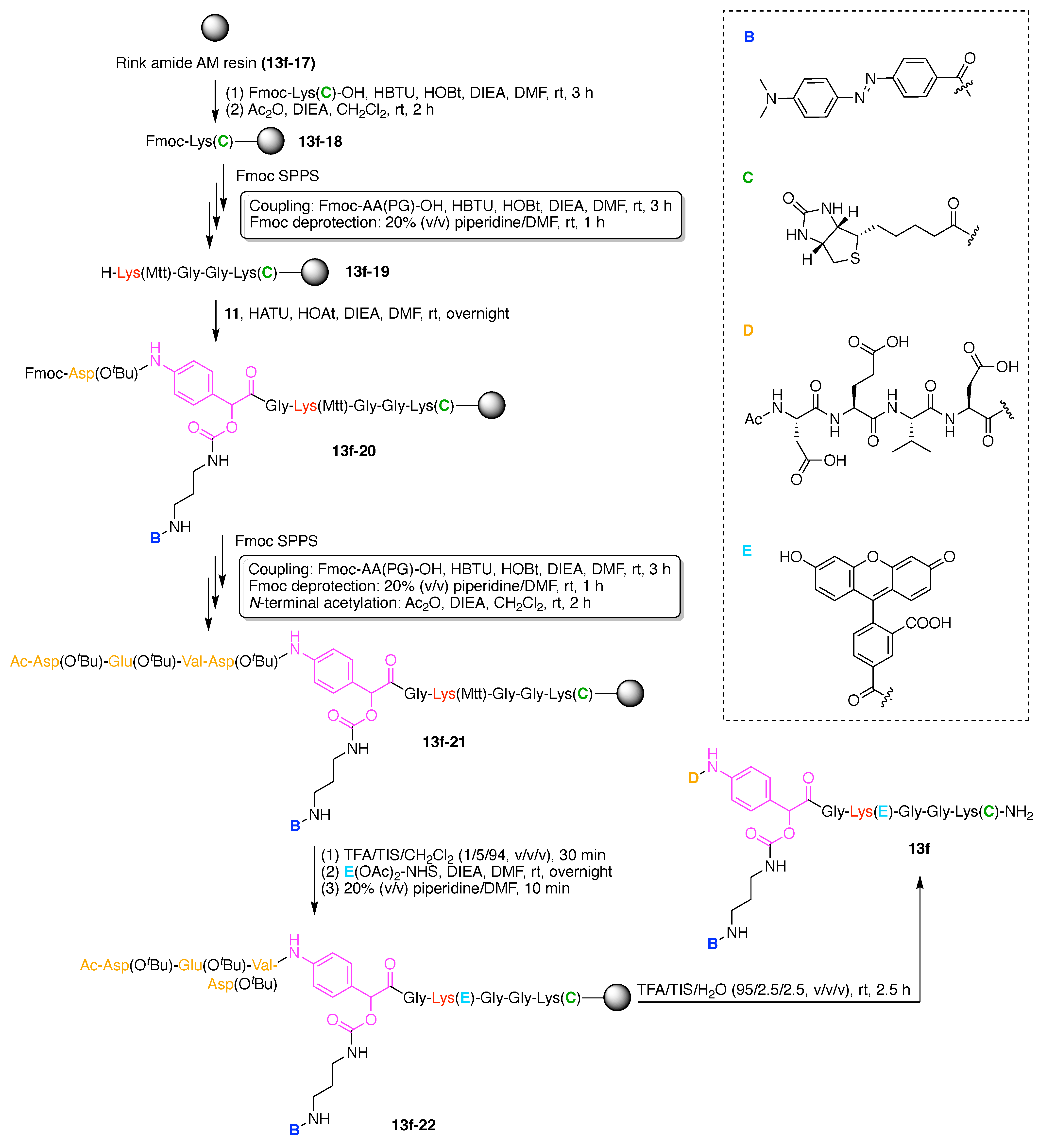{kind=link}
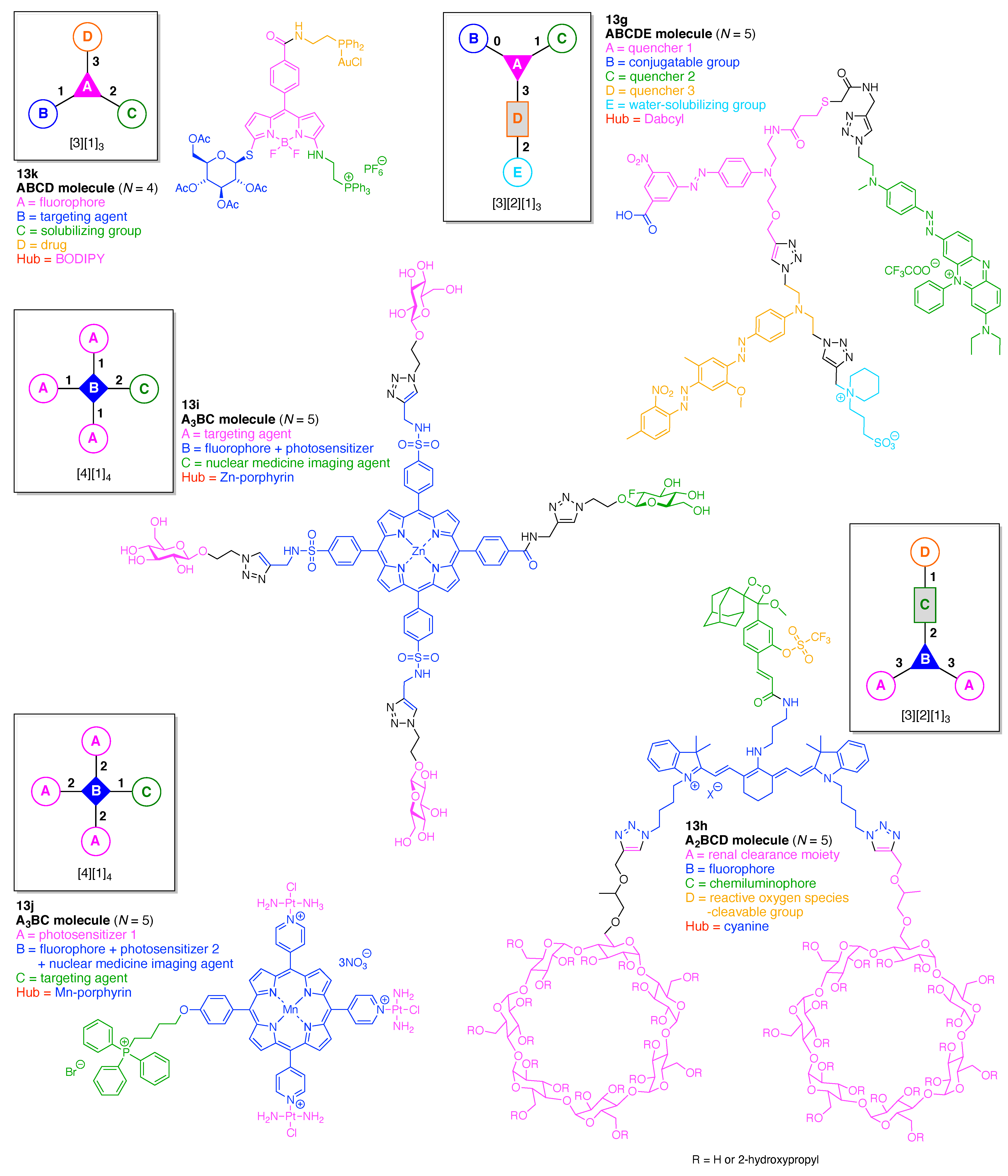{kind=link}
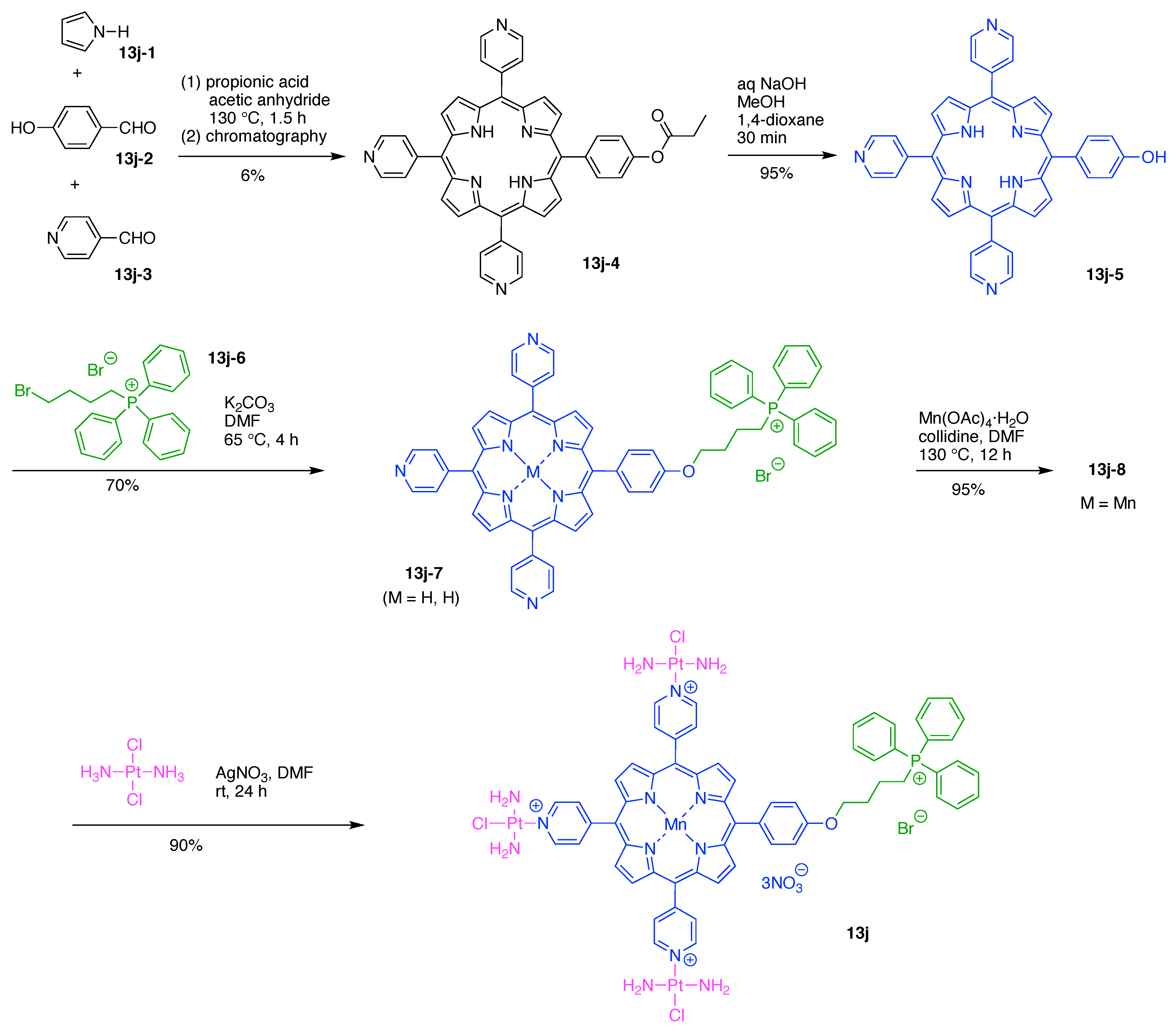{kind=link}
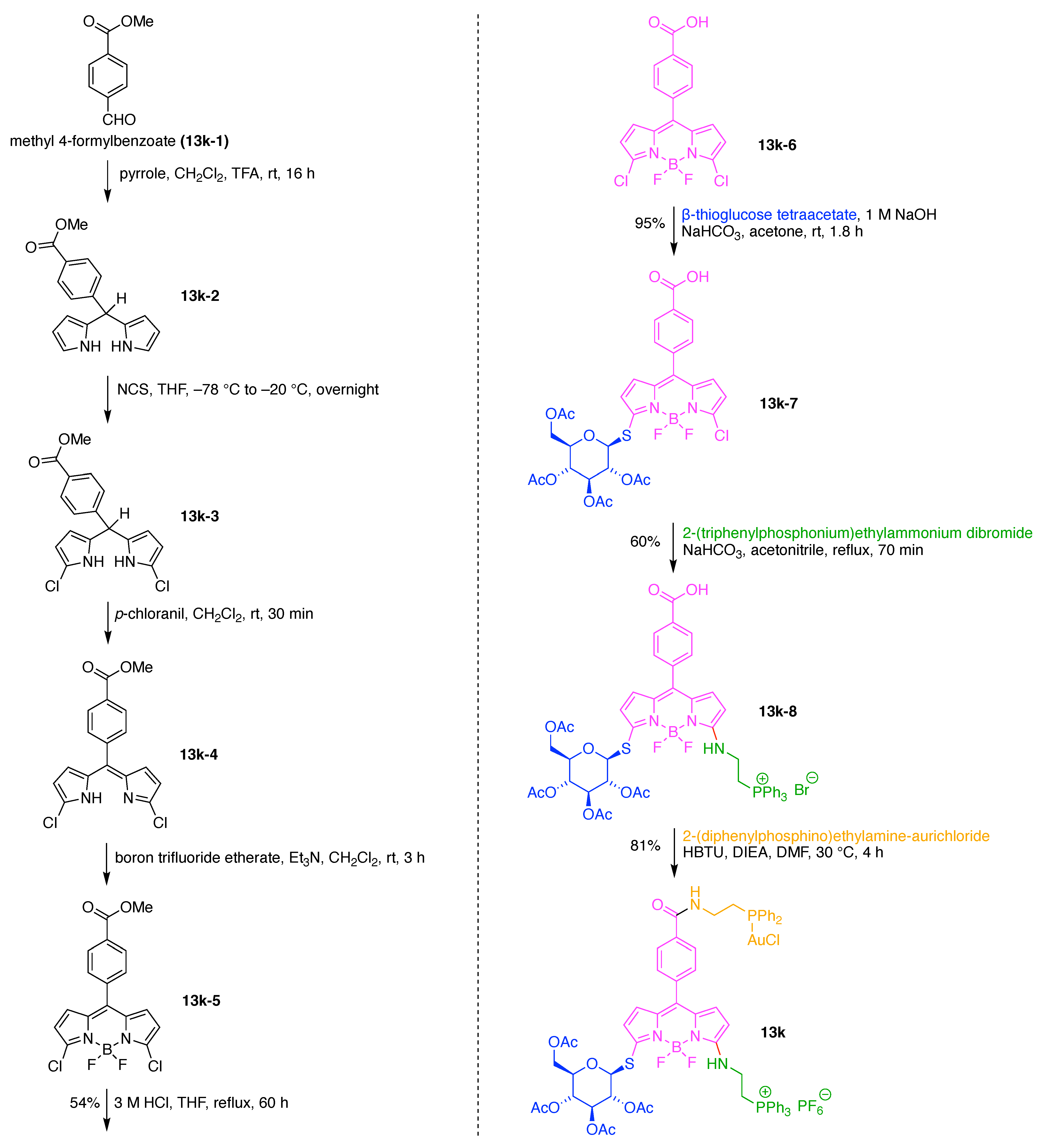{kind=link}
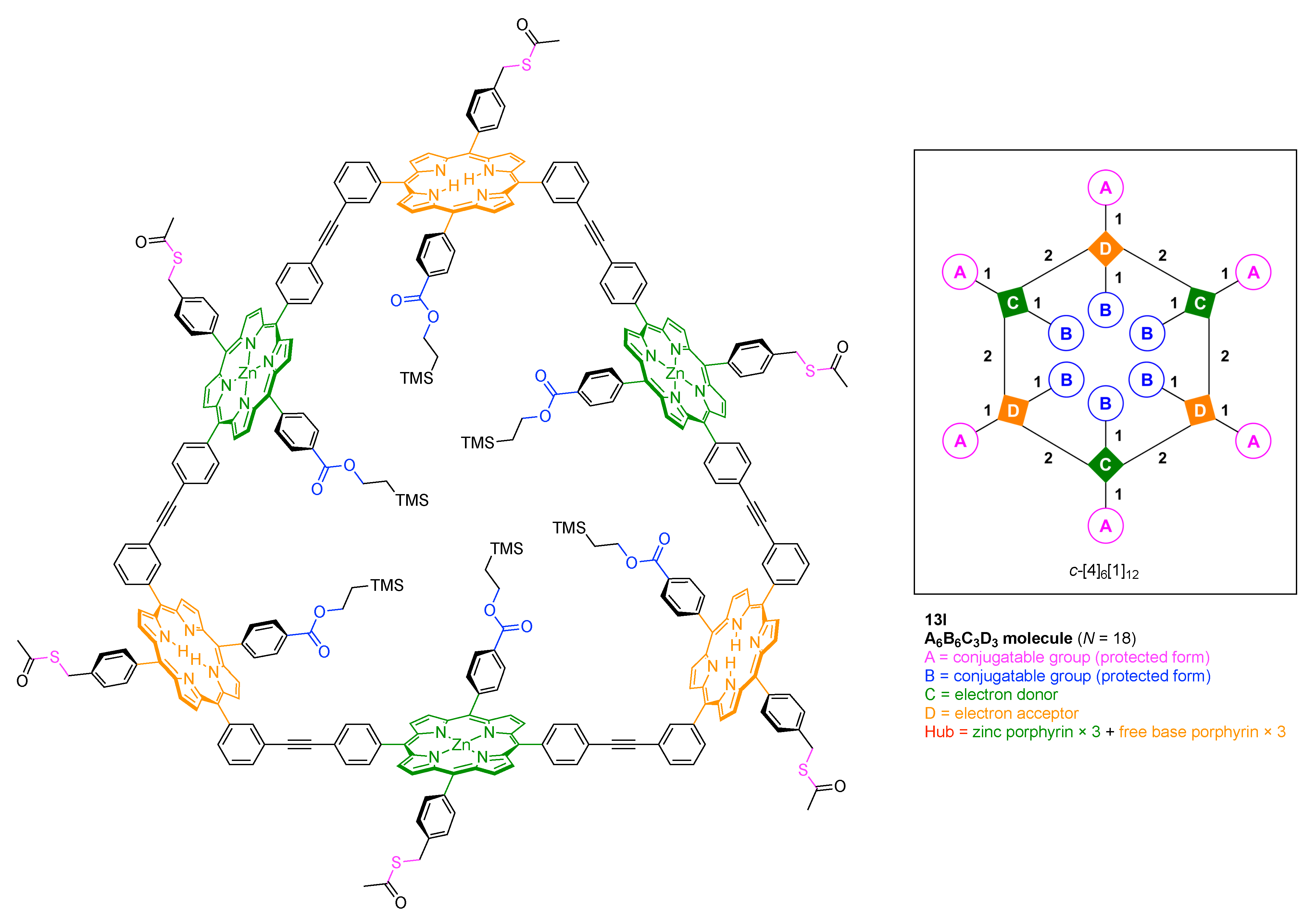{kind=link}
{kind=link}
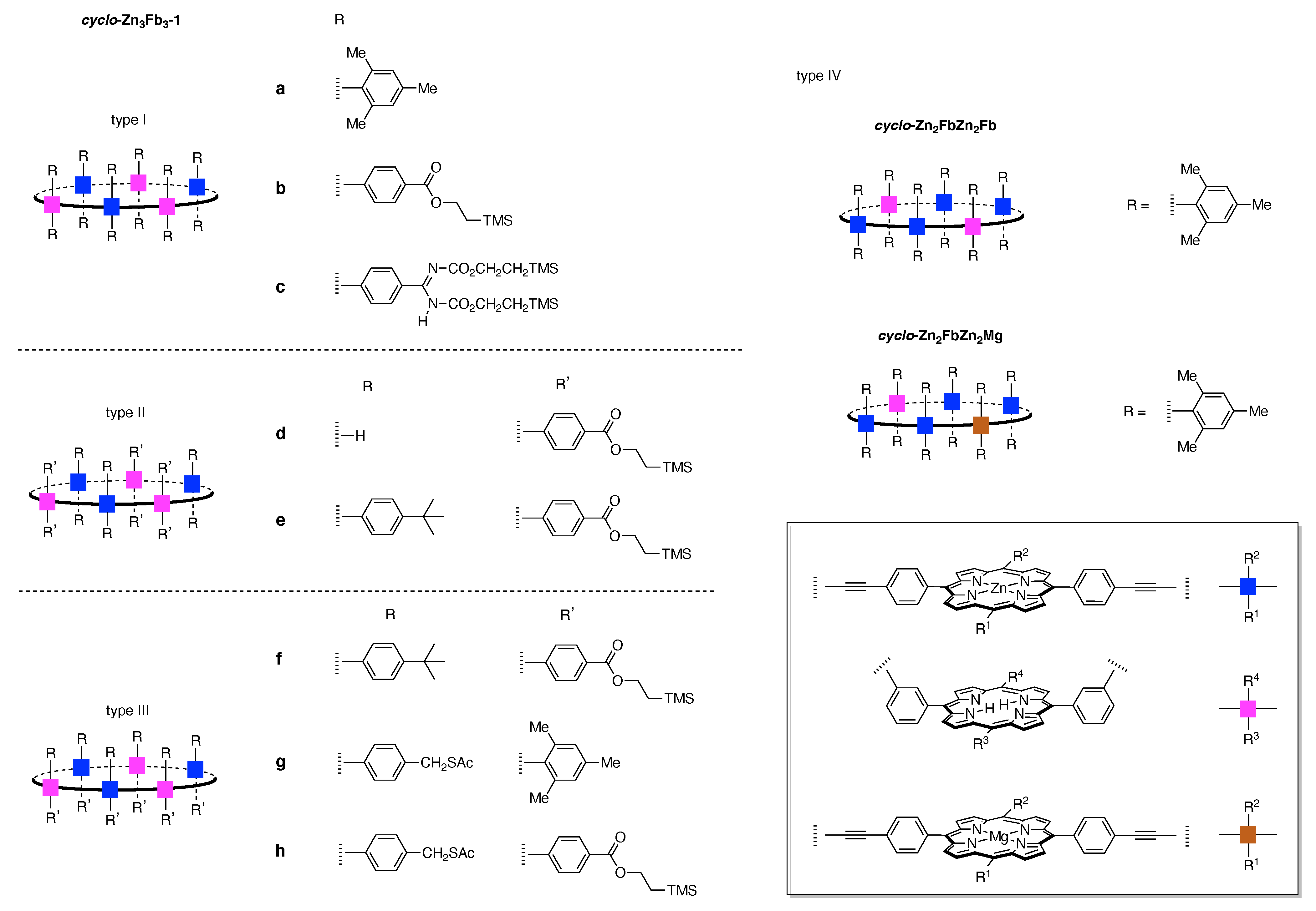{kind=link}
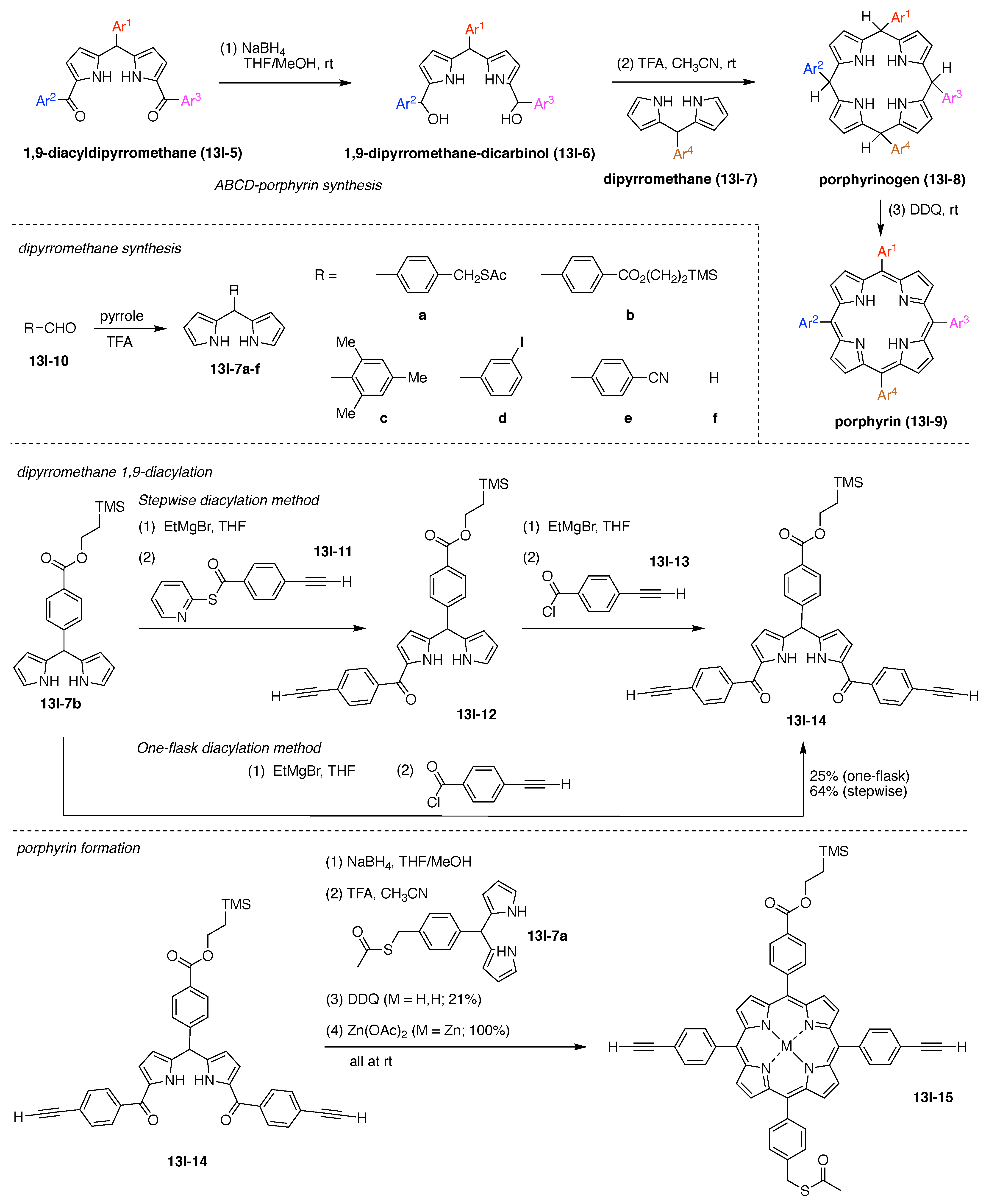{kind=link}

| Entry | X | σma | Relative Rate Constant | |
|---|---|---|---|---|
| Reaction A | Reaction B | |||
| 1 | –Cl | 0.37 | 1.0 b | 1.0 b |
| 2 | –OPh | 0.25 | 1/52 | |
| 3 | –SMe | 0.15 | 1/130 | |
| 4 | –OMe | 0.12 | 1/150 | 1/190 |
| 5 | –Ph | 0.06 | 1/290 | |
| 6 | –NHPh | −0.02 | 1/2900 | |
| 7 | –N-morpholino | 1/6000 | ||
| 8 | –NMe2 | −0.16 | 1/17,000 | 1/75,000 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sato, D.; Wu, Z.; Fujita, H.; Lindsey, J.S. Design, Synthesis, and Utility of Defined Molecular Scaffolds. Organics 2021, 2, 161-273. https://doi.org/10.3390/org2030013
Sato D, Wu Z, Fujita H, Lindsey JS. Design, Synthesis, and Utility of Defined Molecular Scaffolds. Organics. 2021; 2(3):161-273. https://doi.org/10.3390/org2030013
Chicago/Turabian StyleSato, Daisuke, Zhiyuan Wu, Hikaru Fujita, and Jonathan S. Lindsey. 2021. "Design, Synthesis, and Utility of Defined Molecular Scaffolds" Organics 2, no. 3: 161-273. https://doi.org/10.3390/org2030013
APA StyleSato, D., Wu, Z., Fujita, H., & Lindsey, J. S. (2021). Design, Synthesis, and Utility of Defined Molecular Scaffolds. Organics, 2(3), 161-273. https://doi.org/10.3390/org2030013