
A Sustainable Improvement of ω-Bromoalkylphosphonates Synthesis to Access Novel KuQuinones

 , ,
, ,  ,
,  and
and
Abstract
:1. Introduction
2. Materials and Methods
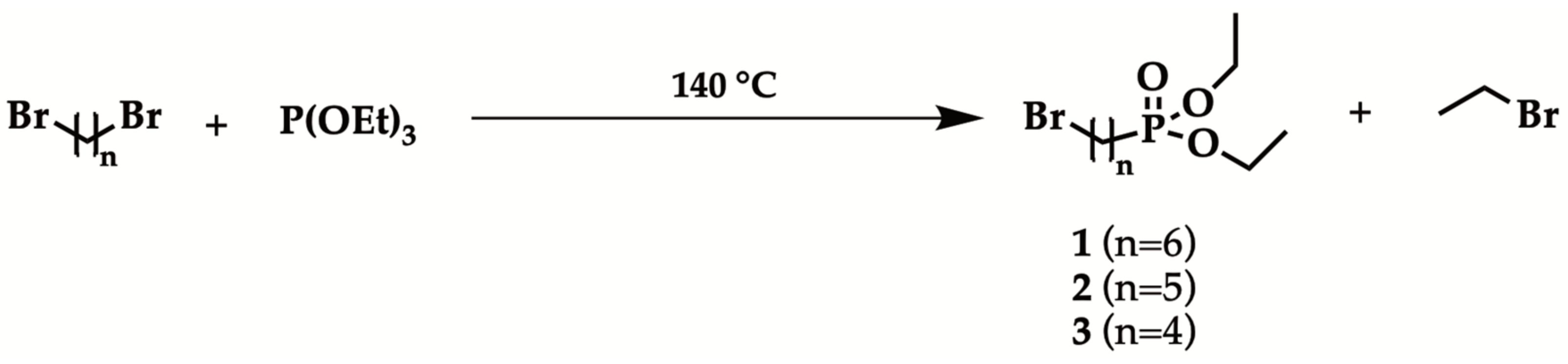
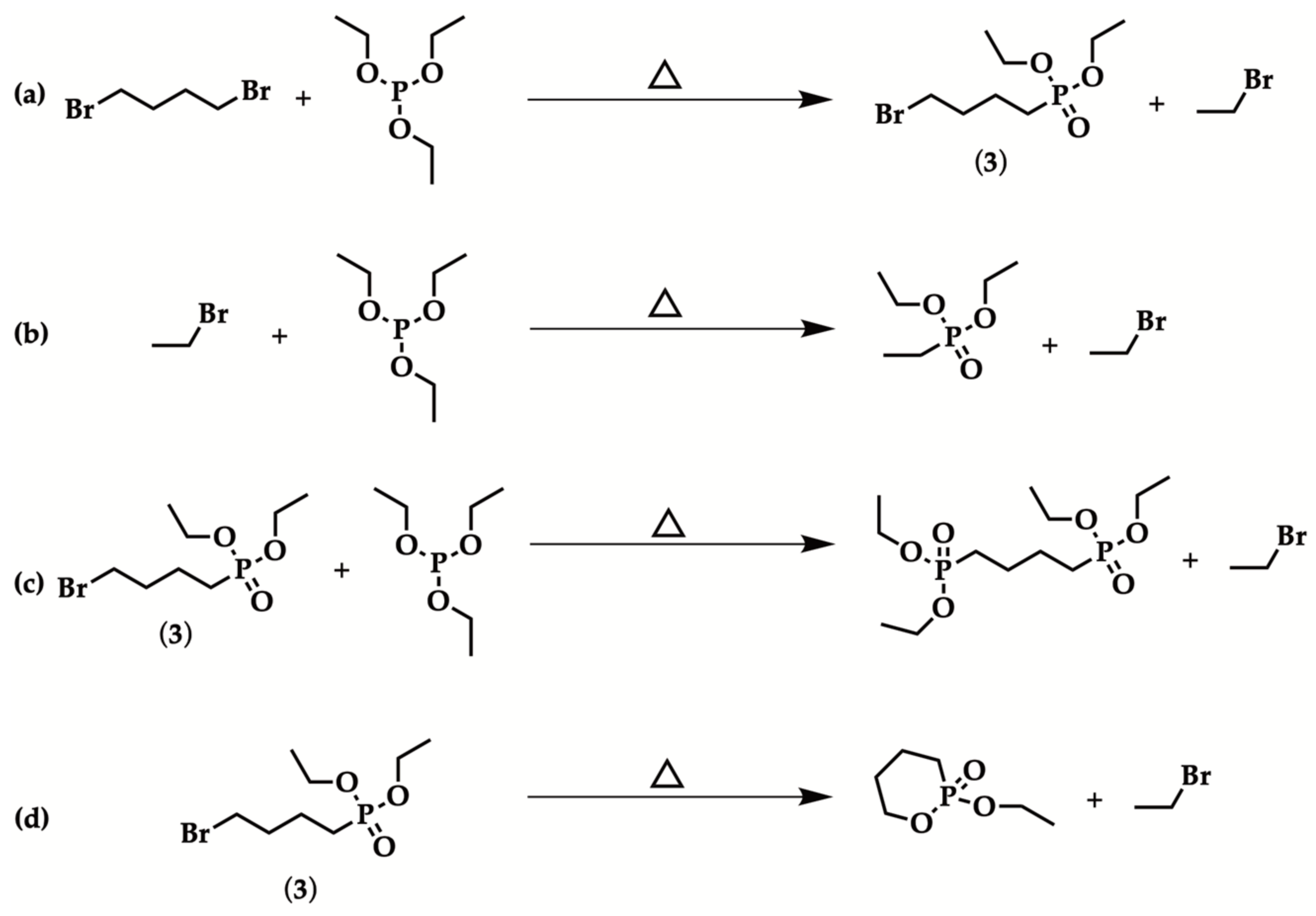
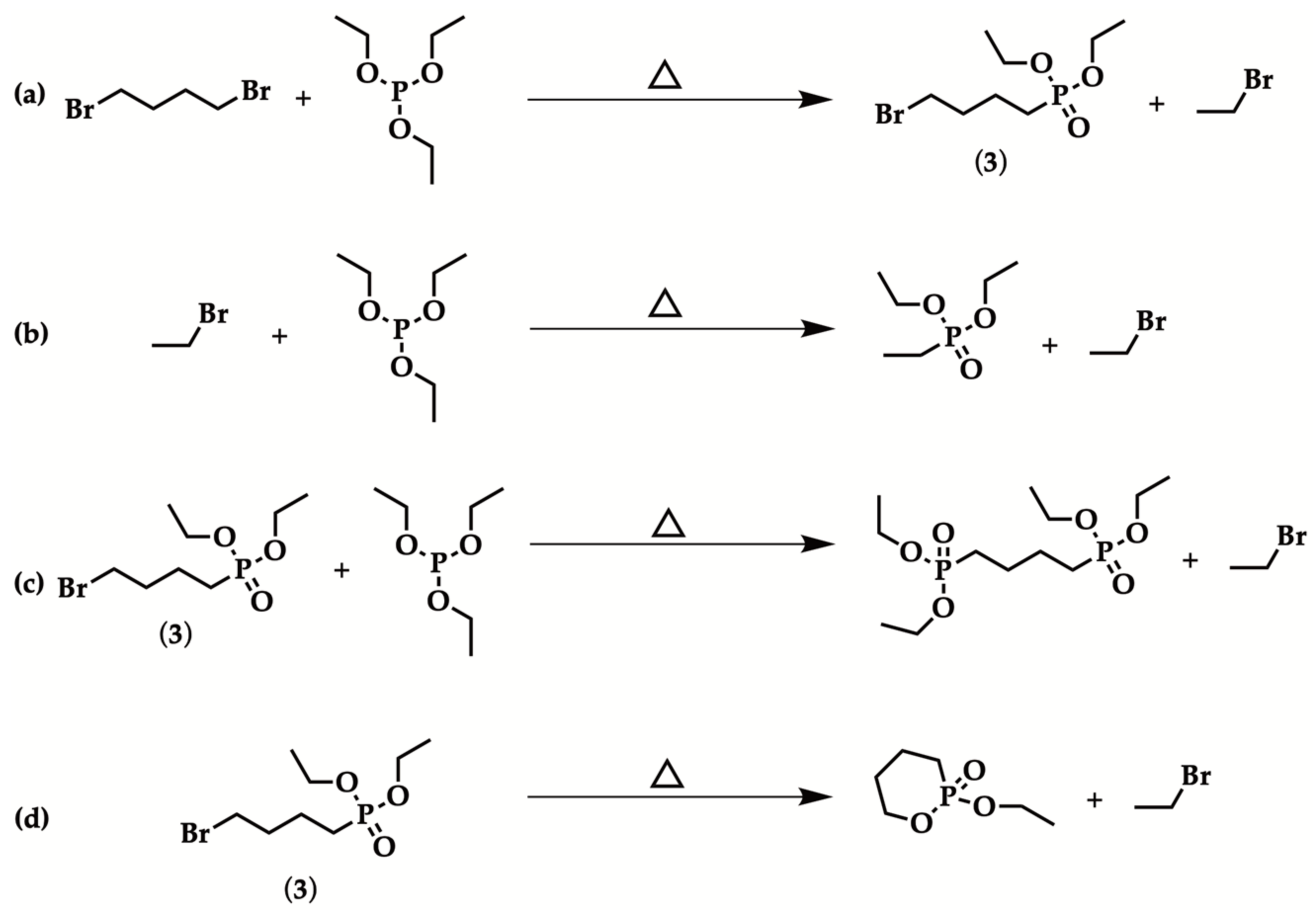
2.1. Optimized Procedure for the Synthesis of Diethyl ω-Bromoalkylphosphonate
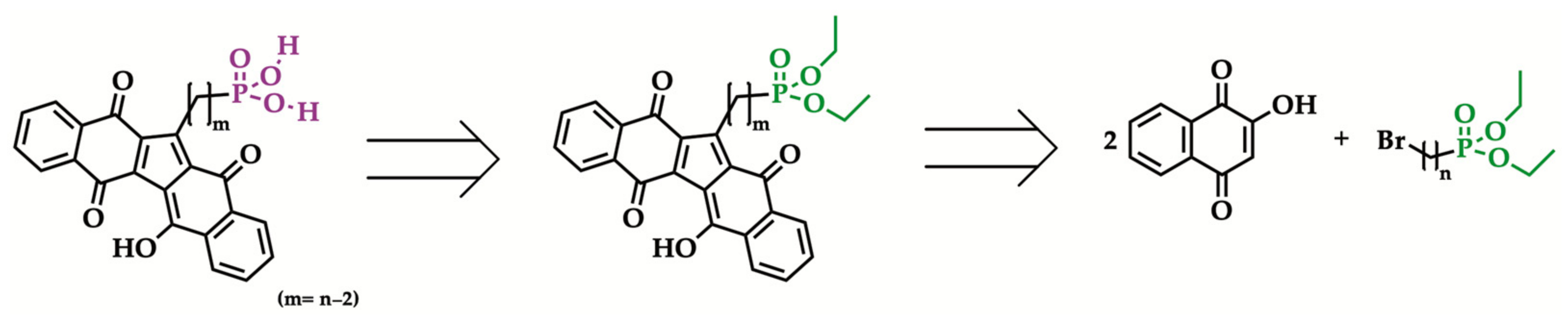
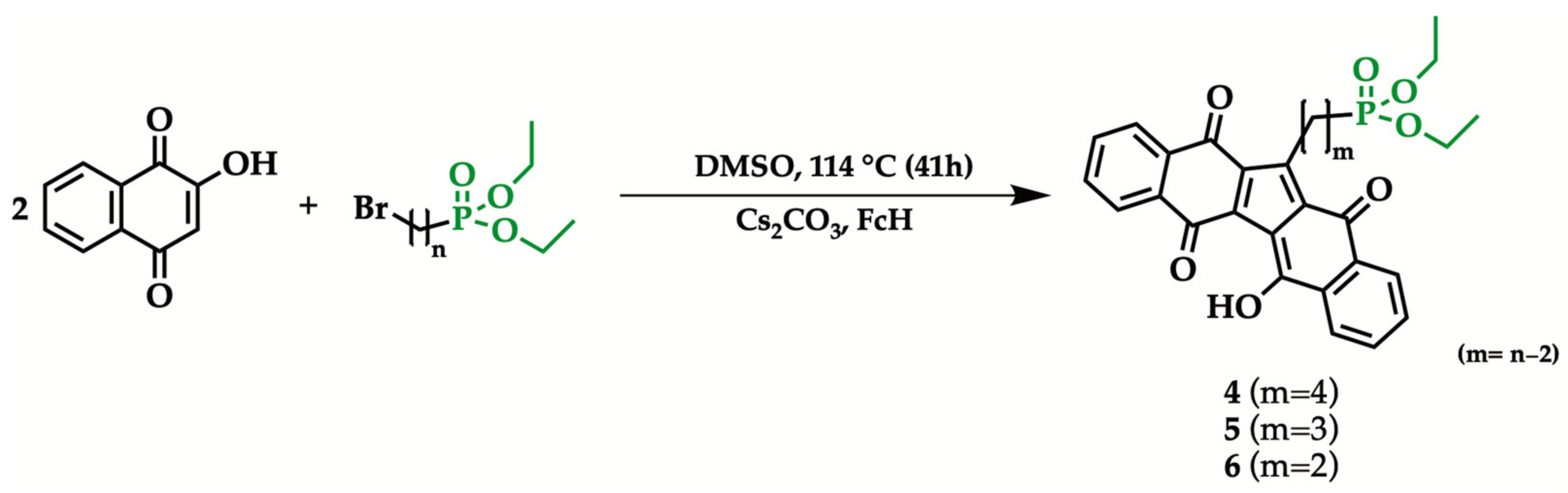
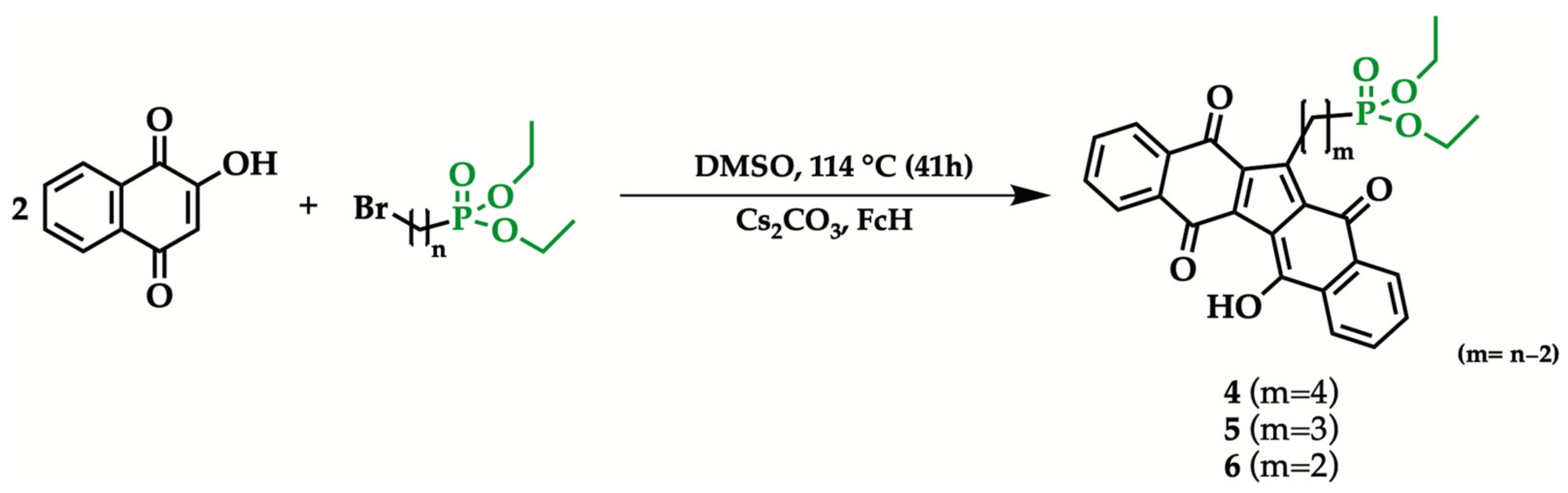
2.2. General Procedure for the Synthesis of 1-[n-(Diethyl phosphonyl) alkyl]KuQuinone
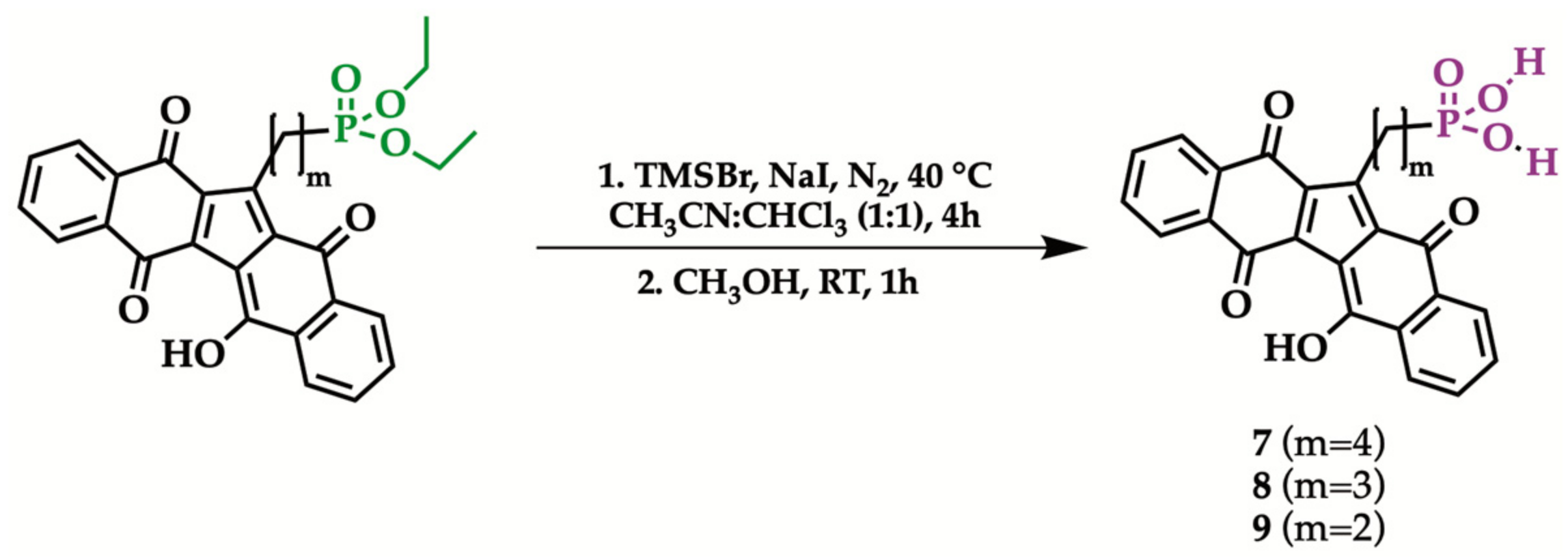
2.3. Hydrolysis of Diethyl Phosphonate KuQuinone Derivatives
3. Results and Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lyagin, I.; Efremenko, E. Enzymes, Reacting with Organophosphorus Compounds as Detoxifiers: Diversity and Functions. Int. J. Mol. Sci. 2021, 22, 1761. [Google Scholar] [CrossRef]
- Wiemer, A.J.; Wiemer, D.F. Prodrugs of phosphonates and phosphates: Crossing the membrane barrier. Top. Curr. Chem. 2015, 360, 115–160. [Google Scholar] [CrossRef] [Green Version]
- Tan, E.L.; Ooi, E.E.; Lin, C.Y.; Tan, H.C.; Ling, A.E.; Lim, B.; Stanton, L.W. Inhibition of SARS coronavirus infection in vitro with clinically approved antiviral drugs. Emerg. Infect. Dis. 2004, 10, 581–586. [Google Scholar] [CrossRef] [Green Version]
- De Clercq, E. Clinical Potential of the Acyclic Nucleoside Phosphonates Cidofovir, Adefovir, and Tenofovir in Treatment of DNA Virus and Retrovirus Infections. E. Clin. Microbiol. Rev. 2003, 16, 569–596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bray, W. Covid-19 Drug Design via Quantum Mechanical Principles Leads to a New Corona-SARS Anti- Viral Candidate 2-Phosphono-Benzoic-Acid. J. Theor. Comput. Sci. 2020. [Google Scholar] [CrossRef]
- Verbrugghen, T.; Vandurm, P.; Pouyez, J.; Maes, L.; Wouters, J.; Van Calenbergh, S.J. Alpha-Heteroatom Derivatized Analogues of 3-(Acetylhydroxyamino)propyl Phosphonic Acid (FR900098) as Antimalarials. Med. Chem. 2013, 56, 376–380. [Google Scholar] [CrossRef] [PubMed]
- Falagas, M.E.; Vouloumanou, E.K.; Samonis, G.; Vardakas, K.Z. Fosfomycin. Clin. Microbiol. Rev. 2016, 29, 321–347. [Google Scholar] [CrossRef] [Green Version]
- Savjani, K.T.; Gajjar, A.K.; Savjani, J.K. Drug solubility: Importance and enhancement techniques. ISRN Pharm. 2012, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, X.; Hu, J.Y.; Luo, J.; Liao, W.M.; He, J. Conductive Metal–Organic Frameworks: Mechanisms, Design Strategies and Recent Advances. Top. Curr. Chem. 2020, 378, 27. [Google Scholar] [CrossRef]
- Hix, G.B.; Caignaert, V.; Rueff, J.M.; Le Pluart, L.; Warren, J.E.; Jaffrès, P.A. A Supramolecular Ladderlike Structure Formed by the Auto-Assembly of Benzene-1,3,5-triphosphonic Acid. Cryst. Growth Des. 2007, 7, 208–211. [Google Scholar] [CrossRef]
- Sopkova-de Oliveira Santos, J.; Montouillout, V.; Fayon, F.; Fernandez, C.; Delain-Bioton, L.; Villemin, D.; Jaffrès, P.A. Assembly of benzene-1,3,5-tris(methylenephosphonic acid) and guanidinium salt: Single crystal-X-ray characterisation and 31P solid state NMR investigations. New J. Chem. 2004, 28, 1244–1249. [Google Scholar] [CrossRef]
- Jiménez-García, L.; Kaltbeitzel, A.; Enkelmann, V.; Gutmann, J.S.; Klapper, M.; Müllen, K. Organic Proton-Conducting Molecules as Solid-State Separator Materials for Fuel Cell Applications. Adv. Funct. Mater. 2011, 21, 2216–2224. [Google Scholar] [CrossRef]
- Hotchkiss, P.J.; Jones, S.C.; Paniagua, S.A.; Sharma, A.; Kippelen, B.; Armstrong, N.R.; Marder, S.R. The Modification of Indium Tin Oxide with Phosphonic Acids: Mechanism of Binding, Tuning of Surface Properties, and Potential for Use in Organic Electronic Applications. Acc. Chem. Res. 2012, 45, 337–346. [Google Scholar] [CrossRef]
- Queffélec, C.; Petit, M.; Janvier, P.; Knight, D.A.; Bujoli, B. Surface Modification Using Phosphonic Acids and Esters. Chem. Rev. 2012, 112, 3777–3807. [Google Scholar] [CrossRef]
- Pujari, S.P.; Scheres, L.; Marcelis, A.T.M.; Zuilhof, H. Covalent Surface Modification of Oxide Surfaces. Angew. Chem. Int. Ed. 2014, 53, 6322–6356. [Google Scholar] [CrossRef]
- Boissezon, R.; Muller, J.; Beaugeard, V.; Monge, S.; Robin, J.J. Organophosphonates as anchoring agents onto metal oxide-based materials: Synthesis and applications. RSC Adv. 2014, 4, 35690–35707. [Google Scholar] [CrossRef]
- Villemin, D.; Moreau, B.; Siméon, F.; Maheut, G.; Fernandez, C.; Montouillout, V.; Caignaert, V.; Jaffrès, P.A. A one step process for grafting organic pendants on alumina via the reaction of alumina and phosphonate under microwave irradiation. Chem. Commun. 2001, 2060–2061. [Google Scholar] [CrossRef]
- Zeininger, L.; Portilla, L.; Halik, M.; Hirsch, A. Quantitative Determination and Comparison of the Surface Binding of Phosphonic Acid, Carboxylic Acid, and Catechol Ligands on TiO2 Nanoparticles. Chem. Eur. J. 2016, 22, 13506–13512. [Google Scholar] [CrossRef] [PubMed]
- Lafond, V.; Gervais, C.; Maquet, J.; Prochnow, D.; Babonneau, F.; Mutin, P.H. 17O MAS NMR Study of the Bonding Mode of Phosphonate Coupling Molecules in a Titanium Oxo-Alkoxo-Phosphonate and in Titania-Based Hybrid Materials. Chem. Mater. 2003, 15, 4098–4103. [Google Scholar] [CrossRef]
- Holland, G.P.; Sharma, R.; Agola, J.O.; Amin, S.; Solomon, V.C.; Singh, P.; Buttry, D.A.; Yarger, J.L. NMR Characterization of Phosphonic Acid Capped SnO2 Nanoparticles. Chem. Mater. 2007, 19, 2519–2526. [Google Scholar] [CrossRef]
- Tudisco, C.; Oliveri, V.; Cantarella, M.; Vecchio, G.; Condorelli, G.G. Cyclodextrin Anchoring on Magnetic Fe3O4 Nanoparticles Modified with Phosphonic Linkers. Eur. J. Inorg. Chem. 2012, 32, 5323–5331. [Google Scholar] [CrossRef]
- Zhang, L.; Cole, J.M. Anchoring Groups for Dye-Sensitized Solar Cells. ACS Appl. Mater. Interfaces 2015, 7, 3427–3455. [Google Scholar] [CrossRef]
- Büttner, A.; Brauchli, S.Y.; Vogt, R.; Constable, E.C.; Housecroft, C.E. Combining phosphonic acid-functionalized anchoring ligands with asymmetric ancillary ligands in bis(diimine)copper(I) dyes for dye sensitized solar cells. RSC Adv. 2016, 6, 5205–5213. [Google Scholar] [CrossRef] [Green Version]
- De Tovar, J.; Romero, N.; Denisov, S.A.; Bofill, R.; Gimbert-Surinach, C.; Ciuculescu-Pradines, D.; Drouet, S.; Llobet, A.; Lecante, P.; Colliere, V.; et al. Light-driven water oxidation using hybrid photosensitizer-decorated Co3O4 nanoparticles. Mat. Today Energy 2018, 9, 506–515. [Google Scholar] [CrossRef]
- Xie, G.; Feng, D.; Ma, X. 9-Amino(9-deoxy)epi-cinchona alkaloid-tethered aluminium phosphonate architectures for heterogeneous cooperative catalysis: Asymmetric aldol and double-Michael cascade reaction. Mol. Cat. 2017, 434, 86–95. [Google Scholar] [CrossRef]
- Angeloni, M.; Piermatti, O.; Pizzo, F.; Vaccaro, L. Synthesis of Zirconium Phosphonate Supported L-Proline as an Effective Organocatalyst for Direct Asymmetric Aldol Addition. Eur. J. Org. Chem. 2014, 8, 1716–1726. [Google Scholar] [CrossRef]
- Wan, J.; Ding, L.; Wu, T.; Ma, X.; Tang, Q. Facile one-pot fabrication of magnetic nanoparticles (MNPs)-supported organocatalysts using phosphonate as an anchor point through direct co-precipitation method. RSC Adv. 2014, 4, 38323–38333. [Google Scholar] [CrossRef]
- Sabuzi, F.; Armuzza, V.; Conte, V.; Floris, B.; Venanzi, M.; Galloni, P.; Gatto, M. KuQuinones: A new class of quinoid compounds as photoactive species on ITO. J. Mater. Chem. C 2016, 4, 622–629. [Google Scholar] [CrossRef]
- Bonomo, M.; Sabuzi, F.; Di Carlo, A.; Conte, V.; Dini, D.; Galloni, P. KuQuinones as sensitizers of NiO based p-type dye sensitized solar cells. New J. Chem. 2017, 41, 2769–2779. [Google Scholar] [CrossRef]
- Volpato, G.A.; Marasi, M.; Gobbato, T.; Valentini, F.; Sabuzi, F.; Gagliardi, V.; Bonetto, A.; Marcomini, A.; Berardi, S.; Conte, V.; et al. Photoanodes for water oxidation with visible light based on a pentacyclic quinoid organic dye enabling proton-coupled electron transfer. Chem. Commun. 2020, 56, 2248–2251. [Google Scholar] [CrossRef] [PubMed]
- Valentini, F.; Sabuzi, F.; Conte, V.; Nemykin, V.N.; Galloni, P. Unveiling KuQuinone redox species: An electrochemical and computational cross study. J. Org. Chem. 2021, 86, 5680–5689. [Google Scholar] [CrossRef]
- Coletti, A.; Lentini, S.; Conte, V.; Floris, B.; Bortolini, O.; Sforza, F.; Grepioni, F.; Galloni, P. Unexpected One-Pot Synthesis of Highly Conjugated Pentacyclic Diquinoid Compounds. J. Org. Chem. 2012, 77, 6873–6879. [Google Scholar] [CrossRef] [Green Version]
- Sabuzi, F.; Lentini, S.; Sforza, F.; Pezzola, S.; Fratelli, S.; Bortolini, O.; Floris, B.; Conte, V.; Galloni, P. KuQuinones Equilibria Assessment for Biomedical Applications. J. Org. Chem. 2017, 82, 10129–10138. [Google Scholar] [CrossRef] [PubMed]
- Guerrero, G.; Alauzun, J.G.; Granier, M.; Laurencin, D.; Mutin, P.H. Phosphonate Coupling Molecules for the Control of Surface/Interface Properties and the Synthesis of Nanomaterials. Dalton Trans. 2013, 42, 12569–12585. [Google Scholar] [CrossRef] [PubMed]
- Hagfeldt, A.; Boschloo, G.; Sun, L.; Kloo, L.; Pettersson, H. Dye-Sensitized Solar Cells. Chem. Rev. 2010, 110, 6595–6663. [Google Scholar] [CrossRef]
- Gardner, T.J.; Frisbie, C.D.; Wrighton, M.S. Systems for Orthogonal Self-Assembly of Electroactive Monolayers on Au and ITO: An Approach to Molecular Electronics. J. Am. Chem. Soc. 1995, 117, 6927–6933. [Google Scholar] [CrossRef]
- Lalatonne, Y.; Paris, C.; Serfaty, J.M.; Weinmann, P.; Lecouvey, M.; Motte, L. Bis-Phosphonates-Ultra Small Superparamagnetic Iron Oxide Nanoparticles: A Platform Towards Diagnosis and Therapy. Chem. Commun. 2008, 2553–2555. [Google Scholar] [CrossRef] [PubMed]
- Péchy, P.; Rotzinger, F.P.; Nazeeruddin, M.K.; Kohle, O.; Zakeeruddin, S.M.; Humphry-Baker, R.; Grätzel, M. Preparation of Phosphonated Polypyridyl Ligands to Anchor Transition-Metal Complexes on Oxide Surfaces: Application for the Conversion of Light to Electricity with Nanocrystalline TiO2 Films. J. Chem. Soc. Chem. Commun. 1995, 65–66. [Google Scholar] [CrossRef]
- Koh, S.E.; McDonald, K.D.; Holt, D.H.; Dulcey, C.S. Phenylphosphonic Acid Functionalization of Indium Tin Oxide: Surface Chemistry and Work functions. Langmuir 2006, 22, 6249–6255. [Google Scholar] [CrossRef]
- Michaelis, A.; Kaehne, R. Ueber das Verhalten der Jodalkyle gegen die sogen. Phosphorigsäureester oder O-Phosphine. Ber. Dtsch. Chem. Ges. 1898, 31, 1048–1055. [Google Scholar] [CrossRef] [Green Version]
- Bhattacharya, A.K.; Thyagarajan, G. The Michaelis-Arbuzov Rearrangement. Chem. Rev. 1981, 81, 415. [Google Scholar] [CrossRef]
- Catel, Y.; Degrange, M.; Le Pluart, L.; Madec, P.J.; Pham, T.N.; Picton, L. Synthesis, Photopolymerization and Adhesive Properties of New Hydrolytically Stable Phosphonic Acids for Dental Applications. J. Polym. Sci. A Polym. Chem. 2008, 46, 7074–7090. [Google Scholar] [CrossRef]
- Ohashi, K.; Kosai, S.; Arizuka, M.; Watanabe, T.; Yamagiwa, Y.; Kamikawa, T. Syntheses of D-erythro-1-deoxydihydroceramide-1-sulfonic acid and phosphonosphingoglycolipid found in marine organisms via a common precursor. Tetrahedron 1989, 45, 2557–2570. [Google Scholar] [CrossRef]
- Ragulin, V.V. ω-Haloalkylphosphoryl Compounds: Synthesis and Properties. Russian, J. Gen. Chem. 2012, 82, 1928–1937. [Google Scholar] [CrossRef]
- Hewitt, D.G.; Newland, G.L. Organophosphorus compounds. P-arylated perhydro-1,2-azaphosphorines. Aust. J. Chem 1977, 30, 579–587. [Google Scholar] [CrossRef]
- Matoba, K.; Yonemoto, H.; Fukui, M.; Yamazaki, T. Structural modification of bioactive compounds. Syntheses of aminophosphonic acids. Chem. Pharm. Bull. 1984, 32, 3918–3925. [Google Scholar] [CrossRef] [Green Version]
- Groenewold, G.S.; Scott, J.R.; Lee, E.D.; Lammert, S.A. Rapid analysis of organophosphonate compounds recovered from vinyl floor tile using vacuum extraction coupled with a fast-duty cycle GC/MS. Anal. Methods 2013, 5, 2227–2236. [Google Scholar] [CrossRef]
- McLafferty, F.W. Mass spectrometric analysis. Molecular rearrangements. Anal. Chem. 1959, 31, 82–87. [Google Scholar] [CrossRef]
- Li, F.; Shishkin, E.; Mastro, M.A.; Hite, J.K.; Eddy, C.R.; Edgar, J.H.; Ito, T. Photopolymerization of Self-Assembled Monolayers of Diacetylenic Alkylphosphonic Acids on Group-III Nitride Substrates. Langmuir 2010, 26, 10725–10730. [Google Scholar] [CrossRef]
- Subedi, Y.P.; Alfindee, M.N.; Shrestha, J.P.; Becker, G.; Grilley, M.; Takemoto, J.Y.; Chang, C.W.T. Synthesis and biological activity investigation of azole and quinone hybridized phosphonates. Bioorg. Med. Chem. Lett. 2018, 28, 3034–3037. [Google Scholar] [CrossRef]
- Villemin, D.; Simeon, F.; Decreus, H.; Jaffres, P.A. Rapid and efficient arbuzov reaction under microwave irradiation. Phosphorus Sulfur Silicon Relat. Elem. 1998, 133, 209–213. [Google Scholar] [CrossRef]
- Cherevatskaya, M.; Neumann, M.; Füldner, S.; Harlander, C.; Kümmel, S.; Dankesreiter, S.; Pfitzner, A.; Zeitler, K.; König, B. Visible-Light-Promoted Stereoselective Alkylation by Combining Heterogeneous Photocatalysis with Organocatalysis. Angew. Chem. Int. Ed. 2012, 51, 4062–4066. [Google Scholar] [CrossRef]
- Sevrain, C.M.; Berchel, M.; Couthon, H.; Jaffrès, P.A. Phosphonic acid: Preparation and applications. Beilstein J. Org. Chem. 2017, 13, 2186–2213. [Google Scholar] [CrossRef] [PubMed]
- Ikenberry, M.; Peña, L.; Wei, D.; Wang, H.; Bossmann, S.H.; Wilke, T.; Wang, D.; Komreddy, V.R.; Rillema, D.P.; Hohn, K.L. Acid monolayer functionalized iron oxide nanoparticles as catalysts for carbohydrate hydrolysis. Green Chem. 2014, 16, 836–843. [Google Scholar] [CrossRef] [Green Version]
- Jansa, P.; Baszczyňski, O.; Procházková, E.; Dračìnský, M.; Janeba, Z. Microwave-assisted hydrolysis of phosphonate diesters: An efficient protocol for the preparation of phosphonic acids. Green Chem. 2012, 14, 2282–2288. [Google Scholar] [CrossRef]
- Besse, V.; Le Pluart, L.; Cook, W.D.; Pham, T.N.; Madec, P.J. Synthesis and polymerization kinetics of acrylamide phosphonic acids and esters as new dentine adhesives. J. Polym. Sci. A Polym. Chem. 2013, 51, 149–157. [Google Scholar] [CrossRef]
- Norris, M.R.; Concepcion, J.J.; Glasson, C.R.K.; Fang, Z.; Lapides, A.M.; Ashford, D.L.; Templeton, J.L.; Meyer, T.J. Synthesis of Phosphonic Acid Derivatized Bipyridine Ligands and Their Ruthenium Complexes. Inorg. Chem. 2013, 52, 12492–12501. [Google Scholar] [CrossRef]
- Zhou, Y.; Ayad, S.; Ruchlin, C.; Posey, V.; Hill, S.P.; Wu, Q.; Hanson, K. Examining the Role of Acceptor Molecule Structure in Self-Assembled Bilayers: Surface Loading, Stability, Energy Transfer, and Upconverted Emission. Phys. Chem. Chem. Phys. 2018, 20, 20513–20524. [Google Scholar] [CrossRef]
- Morita, T.; Okamoto, Y.; Sakurai, H. A convenient dealkylation of dialkyl phosphonates by chlorotrimethylsilane in the presence of sodium iodide. Tetrahedron Lett. 1978, 28, 2523–2526. [Google Scholar] [CrossRef]
- Franz, R.G. Comparisons of pKa and log P values of some carboxylic and phosphonic acids: Synthesis and measurement. AAPS Pharm. Sci. 2001, 3, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Papadimitriou, K.D.; Andreopoulou, A.K.; Kallitsis, J.K. Phosphonated fully aromatic polyethers for PEMFCs applications. J. Polym. Sci. A Polym. Chem. 2010, 48, 2817–2827. [Google Scholar] [CrossRef]
- Kahraman, G.; Wang, D.Y.; Von Irmer, J.; Gallei, M.; Hey-Hawkins, E.; Eren, T. Synthesis and Characterization of Phosphorus- and Carborane-Containing Polyoxanorbornene Block Copolymers. Polymers 2019, 11, 613–629. [Google Scholar] [CrossRef] [PubMed] [Green Version]

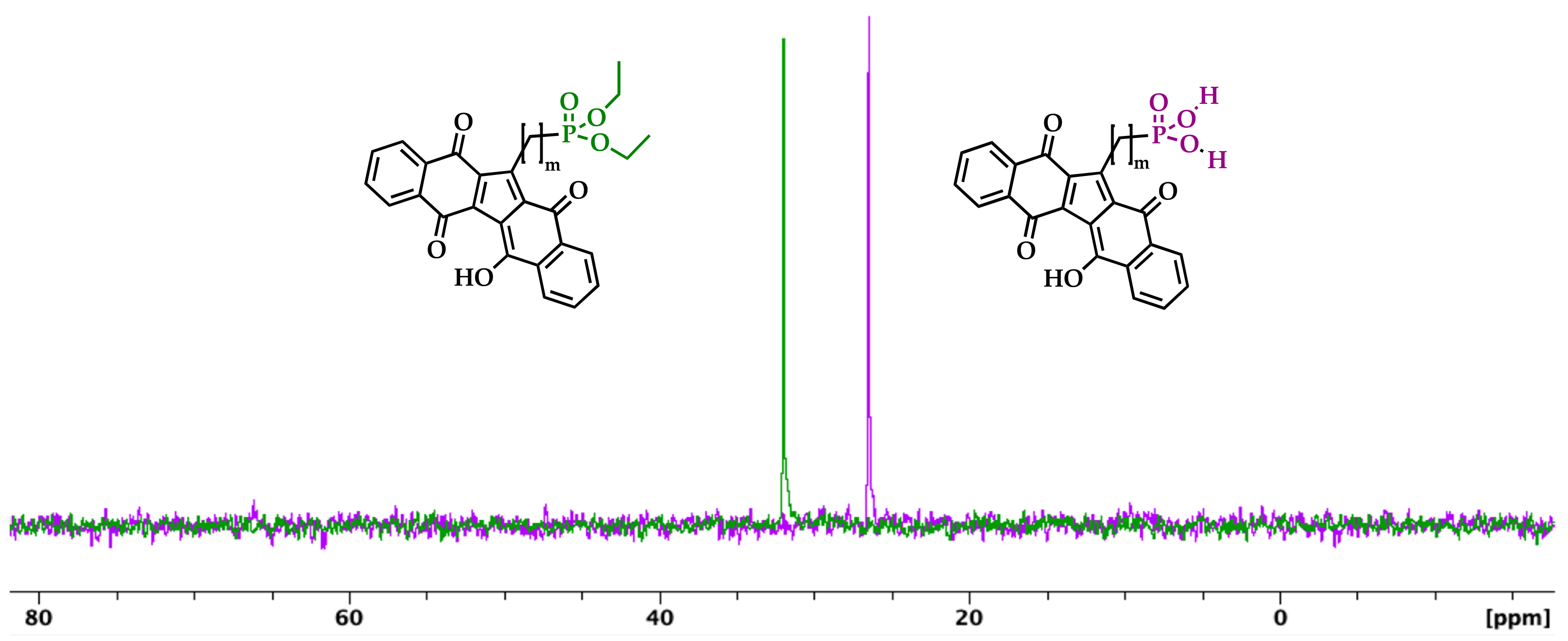

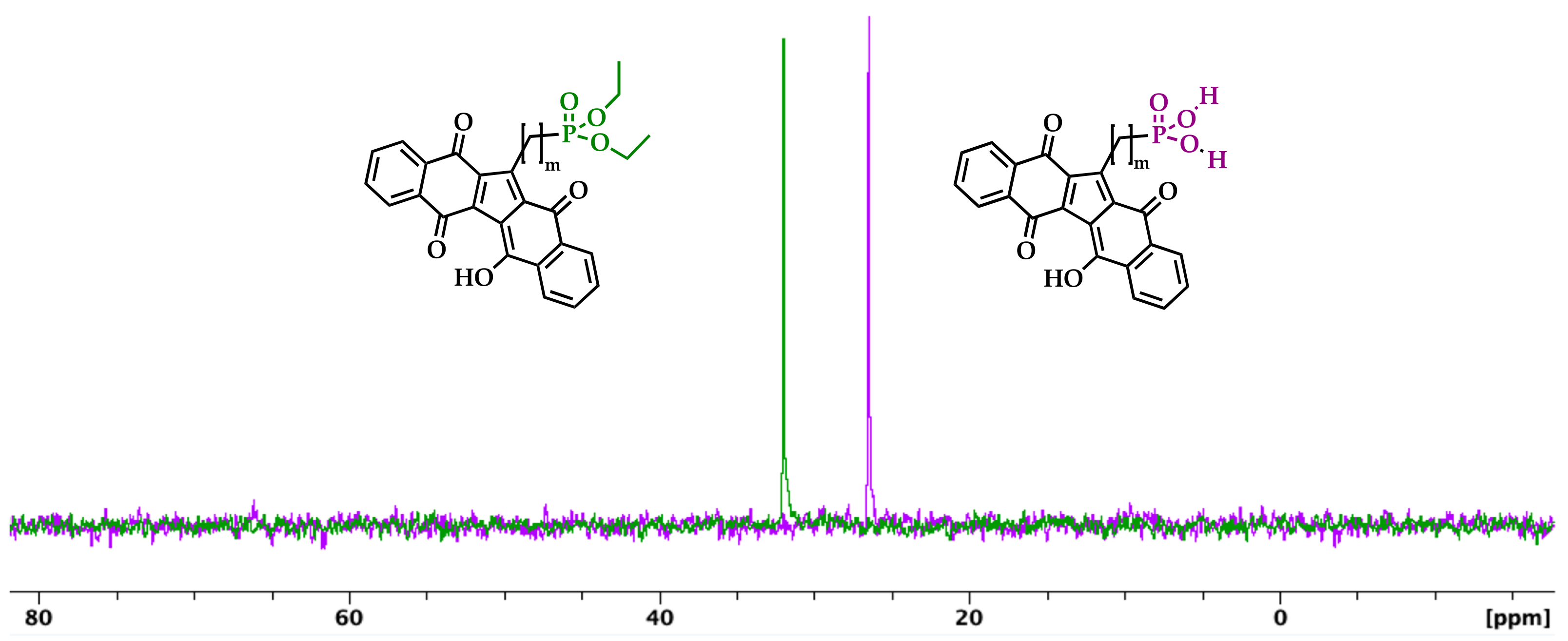{kind=link}
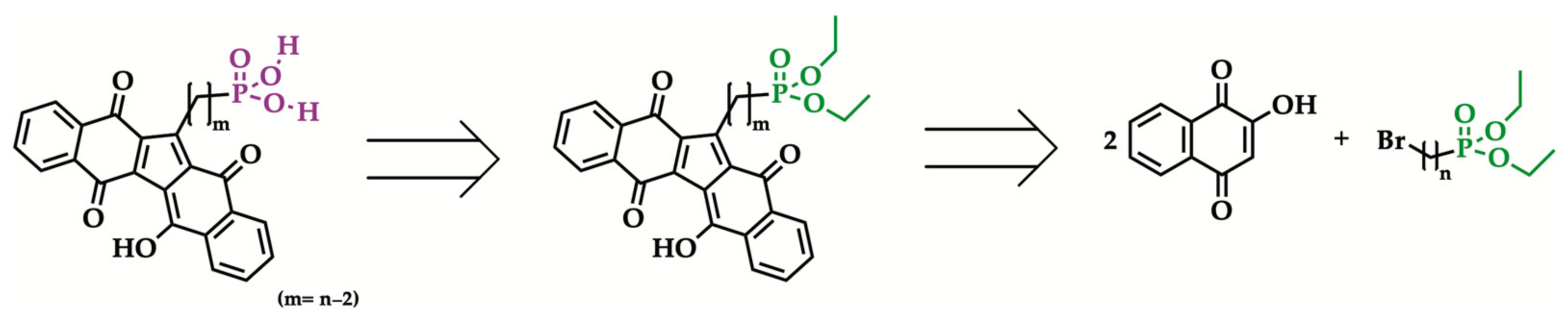{kind=link}
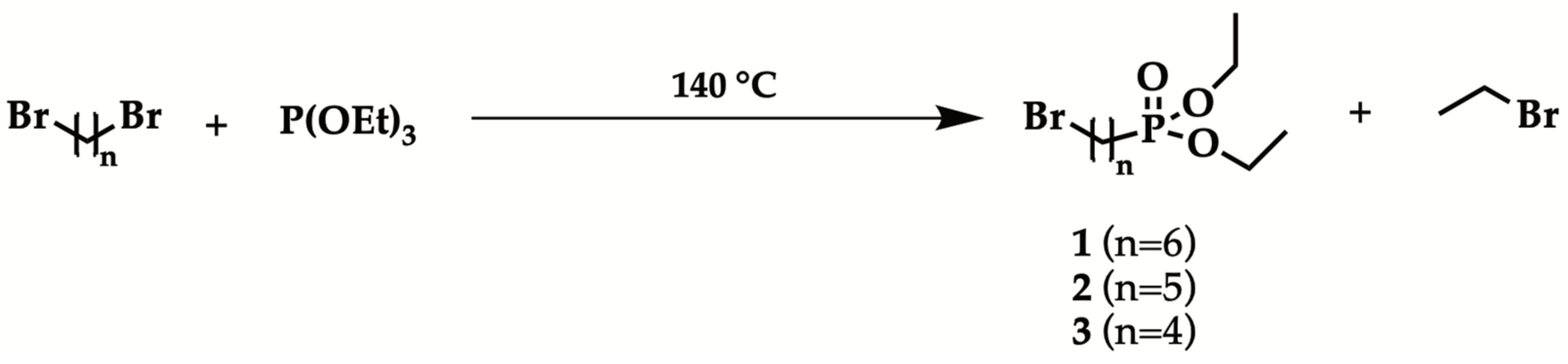{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Br(CH2)nBr | Reaction Time | Isolated Yield (%) |
|---|---|---|---|
| 1 | n = 4 | 12 h | - |
| 2 a | n = 4 | 12 h | - |
| 3 b | n = 4 | 5 min | - |
| 4 a,c | n = 6 | 90 min | 5 |
| 5 a,d | n = 6 | 3 h | 40 |
| 6 a,d | n = 5 | 3 h | 40 |
| 7 a,d | n = 4 | 3 h | 20 e |
| Reagent | Product | Yield (%) |
|---|---|---|
| 1 | 4 | 11 |
| 2 | 5 | 10 |
| 3 | 6 | 7 |
| Reagent | Product | Yield (%) |
|---|---|---|
| 4 | 7 | 100 |
| 5 | 8 | 95 |
| 6 | 9 | 97 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Forchetta, M.; Conte, V.; Fiorani, G.; Galloni, P.; Sabuzi, F. A Sustainable Improvement of ω-Bromoalkylphosphonates Synthesis to Access Novel KuQuinones. Organics 2021, 2, 107-117. https://doi.org/10.3390/org2020010
Forchetta M, Conte V, Fiorani G, Galloni P, Sabuzi F. A Sustainable Improvement of ω-Bromoalkylphosphonates Synthesis to Access Novel KuQuinones. Organics. 2021; 2(2):107-117. https://doi.org/10.3390/org2020010
Chicago/Turabian StyleForchetta, Mattia, Valeria Conte, Giulia Fiorani, Pierluca Galloni, and Federica Sabuzi. 2021. "A Sustainable Improvement of ω-Bromoalkylphosphonates Synthesis to Access Novel KuQuinones" Organics 2, no. 2: 107-117. https://doi.org/10.3390/org2020010
APA StyleForchetta, M., Conte, V., Fiorani, G., Galloni, P., & Sabuzi, F. (2021). A Sustainable Improvement of ω-Bromoalkylphosphonates Synthesis to Access Novel KuQuinones. Organics, 2(2), 107-117. https://doi.org/10.3390/org2020010