1. Introduction
Methyl esters are components of biodiesel used as a fuel in Compression Ignition (CI) engines, both in mixtures with fossil diesel and as a stand-alone fuel [
1]. Just like the fossil counterpart, biodiesel is composed of a wide range of hydrocarbon components with different carbon chain lengths, degree of saturation, and branching. Carbon chain length vary in the range 12 to 22, with the dominating components mostly having 16 or 18 carbon atoms and ranging from saturated to unsaturated with up to three double bonds [
2]. Efficient and environmentally friendly use of methyl ester biodiesel as a replacement for fossil diesel require a thorough understanding of the combustion characteristics and pollutant formation. Fundamental studies in laboratory setups [
3,
4,
5,
6,
7,
8,
9,
10] are used to increase the detailed understanding of chemical and physical properties, often in combination with modeling. Comprehensive chemical kinetics mechanisms for biodiesel combustion have been produced by a research group at Lawrence Livermore National Lab (LLNL) and the CRECK group at Politecnico Milan, the mechanisms include a number of common biodiesel constituents that are representative of the true, much larger, range of substances. The LLNL mechanism [
11] include five real biodiesel compounds and consist of about 4800 species and 20,000 reactions. The mechanism from the CRECK group include real biodiesel methyl esters in the size range C
13–C
23 and is simplified by lumping of isomers and intermediates and has a more modest size of about 420 species and 13,000 reactions [
2]. The large sizes of the biodiesel mechanisms are a result of the lack of symmetry in the methyl ester molecules; the number of possible unique radicals and products are very large in each step of the oxidation [
12,
13,
14]. An additional factor mainly affecting the size of the low-temperature combustion subset of reactions is isomerization of RO
2 components; at low temperatures, there are a large number of permitted H-shifts.
While fundamental studies provide detailed information about the combustion process, experiments and simulations in full engine systems are also needed, to optimize engine performance and quantify pollutant formation. Computational Fluid Dynamics (CFD) simulations are important tools in gaining an understanding that is not possible to achieve from experimental studies. CFD simulations are, however, computationally demanding, in particular the Large Eddy Simulations (LES) [
15,
16] where finite rate chemistry describing the fuel reactions is included. Due to the computational expense the chemical kinetics mechanisms implemented in LES have to be of a highly reduced size, preferentially with less than 30 species and number of reactions in the range 50 to 150. While it is a significant challenge to achieve such small mechanism for any fuel, it is close to incomprehensible for biodiesel for which a detailed mechanism of a simple surrogate mixture consists of thousands of species and tens of thousands of reactions.
Compression Ignition engine combustion rely on the auto-ignition properties of the fossil diesel or biodiesel fuel [
17], and accurate modeling of the chemistry of the ignition process is fundamental in engine CFD. Low-temperature combustion chemistry and ignition in the NTC (Negative Temperature Coefficient) region has been a challenge to understand and to model, for alkane fuels as well as for oxygenates as the methyl esters. Experiments in laboratory configurations and detailed kinetic modeling indicate that the presence of double bonds decrease the reactivity at low and intermediate temperatures and thus increase the ignition delay time at these conditions [
18].
Research and development concerning the combustion properties of biodiesel fuels has been intense in the last decade and the frontline of understanding of the chemistry and physics of methyl ester combustion has moved significantly forward [
11,
17]. Due to experimental challenges a large portion of the research has been performed using ester compounds that are smaller than the actual biodiesel components. This is also convenient from a modeling point of view since smaller fuel molecules require fewer species and reactions in the chemical kinetics mechanisms. For research purposes smaller methyl esters like methyl butanoate have been extensively studied [
19], which reveal important features of the ester moiety. However, the short hydrocarbon chain is not representative for that of a real biodiesel compound, which behave similar to a corresponding alkane [
17]. Among methyl esters, the saturated, straight chain compound methyl decanoate, C
11H
22O
2, (MD) is considered a suitable model fuel because of the relatively long alkane chain [
2,
13,
19,
20,
21,
22]. Experimental studies of ignition delay times over a wide range of conditions have been published [
5,
23,
24]. An important characteristic of MD is that it has the Negative Temperature Coefficient (NTC) behavior of a large methyl ester for ignition at low temperatures [
25], which is not the case for esters with a short hydrocarbon chain. The low-temperature chemistry is largely located in the hydrocarbon chain of large fuels, which means that large methyl esters can be expected to have a behavior similar to the alkanes [
17]. Several detailed mechanisms have been developed for MD [
6,
12,
13,
23,
24], the most well-known published already in 2008 by Herbinet et al. [
12], who improved it and extended it to two unsaturated methyl decenoate isomers in 2010 [
13]. A few years later, the CRECK group at Politecnico Milan published a versatile mechanism for methyl ester combustion, including both high- and low-temperature chemistry of MD [
2,
19,
20].
The mentioned comprehensive mechanisms are far too large to be used in CFD simulations, and efforts have been made to reduce the size of the mechanisms. For an outline of different methods, we refer to the book by Turanyi and Tomlin [
25]. Skeletal mechanism reduction methods are used to remove species and reactions of less importance and thus result in smaller mechanisms while still maintaining the “carbon backbone” of the reaction sequence [
25]. Popular methods for skeletal reduction are Direct Relation Graph (DRG) [
26] and various extensions of it. Most reduction methodologies do not, however, have the capacity to produce mechanisms that are of a size that make them suitable for CFD. An example of a typical mechanism reduction for large fuels was performed by Dievart et al. [
23] who presented a detailed mechanism for MD combustion, with 2276 species and 7086 reactions, and used Path Flux Analysis to achieve a reduced version with the same predictive capacity using 530 species and 2396 reactions. An efficient way to further reduce mechanism size is decoupling methodologies where the fuel breakdown chemistry is significantly reduced while the “base chemistry” of small H/O species and the smallest hydrocarbons are treated in more detail. This far it has mainly been applied to hydrocarbon fuels, for example the HyChem [
27,
28] mechanisms where the base chemistry consist of species up to C
4, and by Zettervall et al. [
29] who presented a kerosene mechanism with semi-global fuel breakdown and base chemistry consisting only of C
1 species. A decoupling methodology was applied to ester fuels by Chang et al. [
30] who made the fuel reaction subset as small as about 135 reactions, with a detailed base mechanism up to C
4, which makes it still too large for implementation in CFD. The complex low temperature chemistry is particularly difficult to represent with a highly reduced subset of reactions.
Mechanism reduction methods rely on the fundamental understanding ingrained in the detailed descriptions, by directly using the detailed mechanisms to produce a small version. This mean that the reduced mechanism strongly relies on the quality of the detailed versions, and potential deficiencies are likely to be inherited by the reduced mechanisms. When several comprehensive mechanisms exist, the choice of which one to use as basis for a reduction needs to be carefully considered in aspects like validation range and mathematical stiffness.
In the present work, we examine the potential to produce reduced mechanisms for ignition from comprehensive mechanisms of different complexity. The Ant Colony Reduction (ACR) methodology is used to select the important subset of reactions from two detailed mechanisms. The target fuels span important functionalities by including both a saturated methyl ester, methyl decanoate, and two isomers of the unsaturated methyl decenoate, methyl 9-decenoate (MDe9) and methyl 5-decenoate (MDe5). MDe9 has the double bond on the terminal position, furthest away from the ester group, while MDe5 has it halfway, on the fifth carbon from the ester group. The esters are also compared to the analogue alkane, n-decane (ND). As parent mechanisms, the highly detailed mechanisms of Herbinet et al. [
13] were used to produced reduced mechanisms for the three methyl esters MD, MDe5, and MDe9; and the lumped but still complex mechanism by the CRECK group [
2] was used to create mechanisms for MD and the ND. Mechanisms were created for a range of temperatures separated into high- and low-temperature parts, to identify the separate chemical subsets governing the phenomena. The aim is to establish reduced sets of chemistry that accurately predict ignition delay at high and low temperature. By testing two different parent mechanisms, the potential of these as foundation for mechanism reduction are investigated.
2. Model Development and Validation
Reduced mechanisms were produced for ignition delay time of high- and low-temperature cases. The two investigated temperature ranges were 600–1100 K and 1100–1500 K, and all mechanisms were constructed to include conditions covering equivalence ratios of 0.5, 1.0, and 1.5, and pressures of 5, 10, and 50 atm. Mechanisms are summarized in
Table 1 and are available as
Supplementary Material.
The parent mechanisms by Herbinet et al. [
13] and Saggese et al. [
2] (in the following called CRECK) have been shown to, despite significant differences in mechanism composition, produce very similar ignition delay times for high temperature ignition (900–1350 K) at elevated pressures (10, 20 atm), in good agreement with experimental data [
4]. The original MD methyl decanoate mechanism by Herbinet et al. [
12] was validated up to a pressure of 50 atm. There is no direct comparison made with experimental data in the present work; instead, we rely on the validation of the mechanisms performed by previous authors. Apart from the already-mentioned studies, experimental data at a pressure of 16 atm, for the three equivalence ratios used as targets here, were published by Wang and Oehlschlaeger [
5]. That study includes simulations and it is shown that the mechanisms of Herbinet et al. [
13] and Saggese et al. [
2] perform well at the investigated conditions.
The mechanisms by Herbinet et al. [
12,
13] are likely the most comprehensive mechanism available for the methyl esters considered here; all potential fuel radicals and intermediates are included, and no discrimination has been done with considerations on how common the reactions are. The mechanism for methyl decanoate consists of 9742 reversible reactions among 2878 species, and the mechanisms for the methyl decenoates are about the same size. The mechanism by Saggese et al. [
2] (CRECK) contain about 13,000 reversible reactions among 420 species but is made for a range of fuels and can therefore not be directly compared in size to the mechanisms by Herbinet et al. [
13], made for single fuels. CRECK has been constructed in a hierarchically way, moving from syngas up to large mechanisms like the ones used here. CRECK mechanisms are self-consistent, i.e., they include the previous sub-mechanisms. Detail on the development of the extensive CRECK mechanisms and the lumping procedures to decrease the total number of reactions can be found in the publications by Grana et al. [
19,
20] and Sagesse et al. [
2].
Mechanism reduction was done using the Ant-Colony Reduction (ACR) methodology. ACR has previously been applied to create a small reduced mechanism for methanol combustion at SI-engine conditions [
31], mechanisms for various aspects of n-heptane combustion [
32], and several skeletal mechanisms for atmospheric chemistry simulations [
33]. ACR use a semi-stochastic algorithm that selects reactions and species from a parent mechanism (commonly highly detailed) to form a skeletal mechanism. The skeletal mechanism is tested towards results of the parent mechanisms and A-factors of the Arrhenius expressions in the skeletal mechanism are optimized to reproduce the result of the parent mechanism. The optimization is done using a metaheuristic approach called differential evolution. In this approach, the new rate of reaction,
k’, is related to the original rate of reaction,
k, by having a value in the range [
k/λ, λ
k]. The value of λ was chosen as 1.2 for reactions only containing C
0-species and 3.0 for reactions containing C. The choice of λ is a crude estimate of uncertainty for reaction classes, based on considerations of common uncertainty ranges. The ACR is applied on simulations using the chemistry solver CANTERA 2.4 [
34]. A zero-dimensional homogenous reactor assuming an ideal gas was used. Common definitions of ignition delay time are related to OH radical concentrations and in the present work, the global maximum gradient of OH radicals was identified and defined as the ignition delay time. The simulation had a length of >0.05 s, at which an equilibrium concentration of OH had been found for all the simulated cases.
The reduced mechanisms produced here are summarized in
Table 1, where mechanism names include LT and HT to indicate that they were constructed for the temperature ranges 600–1100 K and 1100–1500 K, respectively. The mechanisms were developed based on the criteria to be the smallest possible mechanism with less than 80% average deviation from the parent mechanism. For these mechanisms, the ACR reduction procedure typically reduce species down to about 10% of that in the parent mechanism, and the reduction of number of reactions is down to about 1%. The high-temperature mechanisms are always smaller than the low-temperature mechanisms, both with respect to number of species and reactants and independent on parent mechanism. MD for which reduced mechanisms were constructed from both parent mechanisms is accurately represented by smaller mechanisms when CRECK [
2] is the parent mechanism compared to when the mechanism by Herbinet et al. [
13] was used. Comparison of the different mechanisms for methyl esters constructed from the mechanism by Herbinet et al. [
13] reveal that MD is the methyl ester for which the largest reduced mechanism is required to accurately reproduce the results from the parent mechanism, while the reduced mechanisms for MDe9 are the smallest. These trends are further investigated in the results and discussion section.
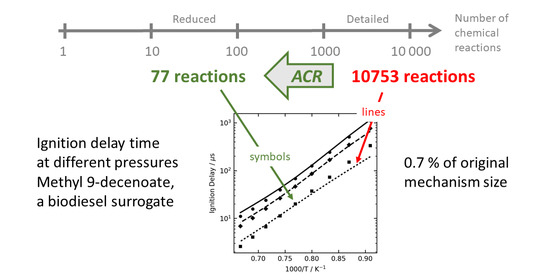
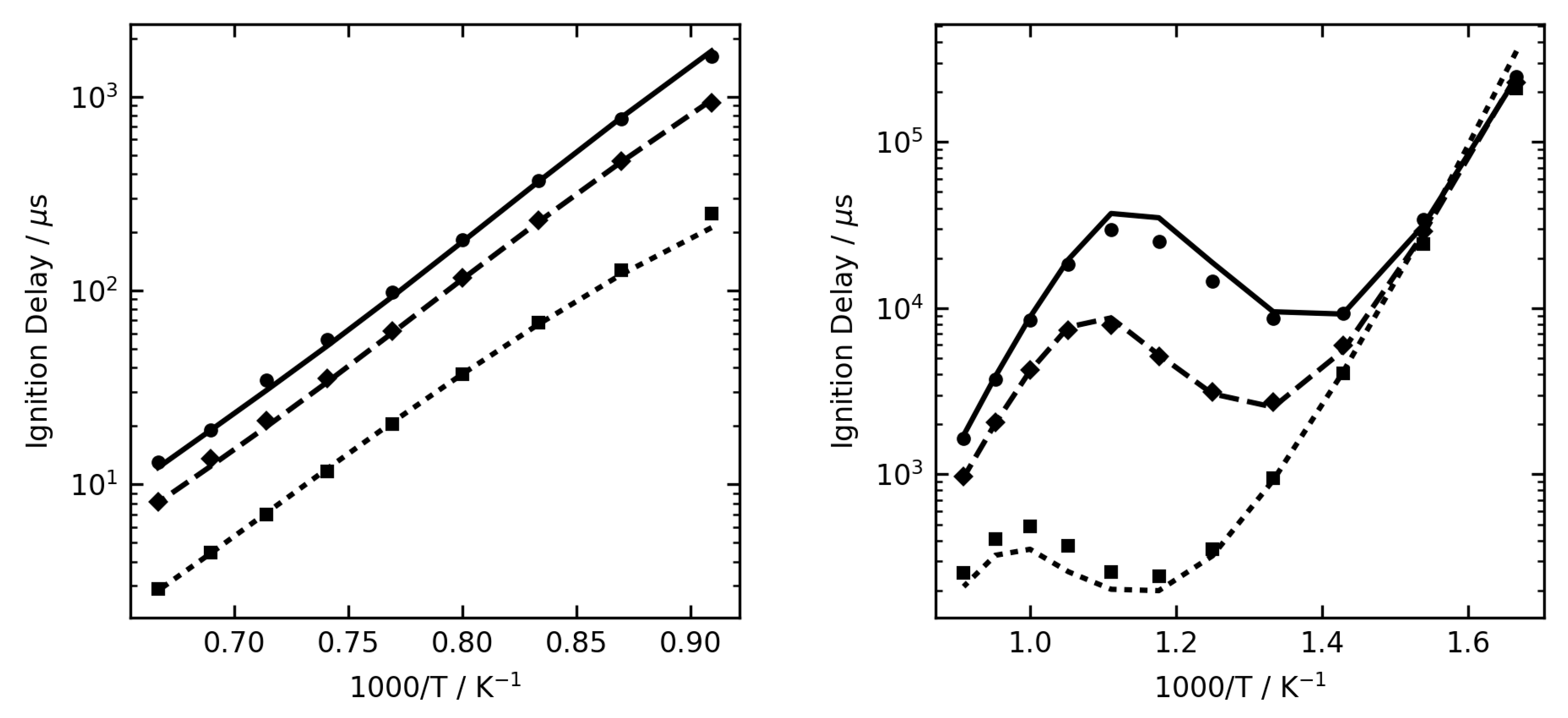
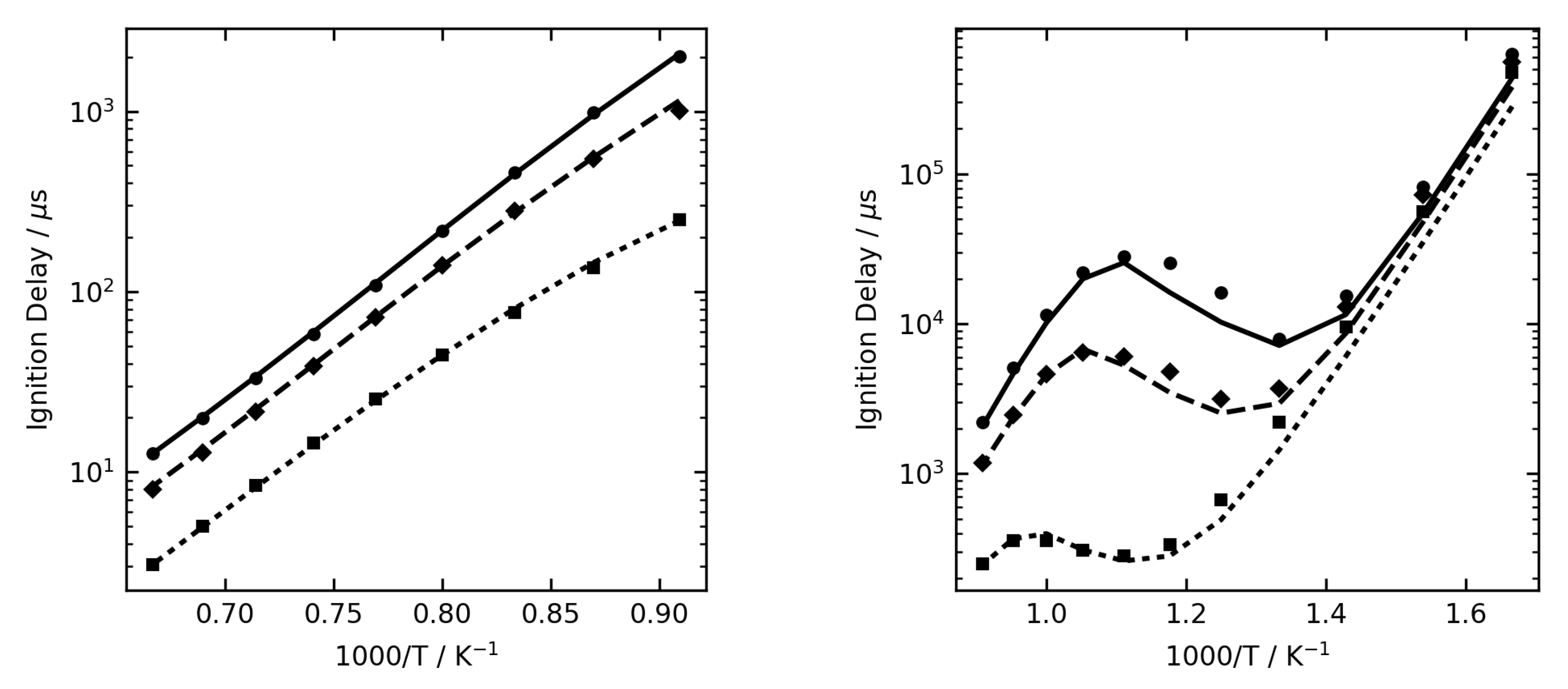
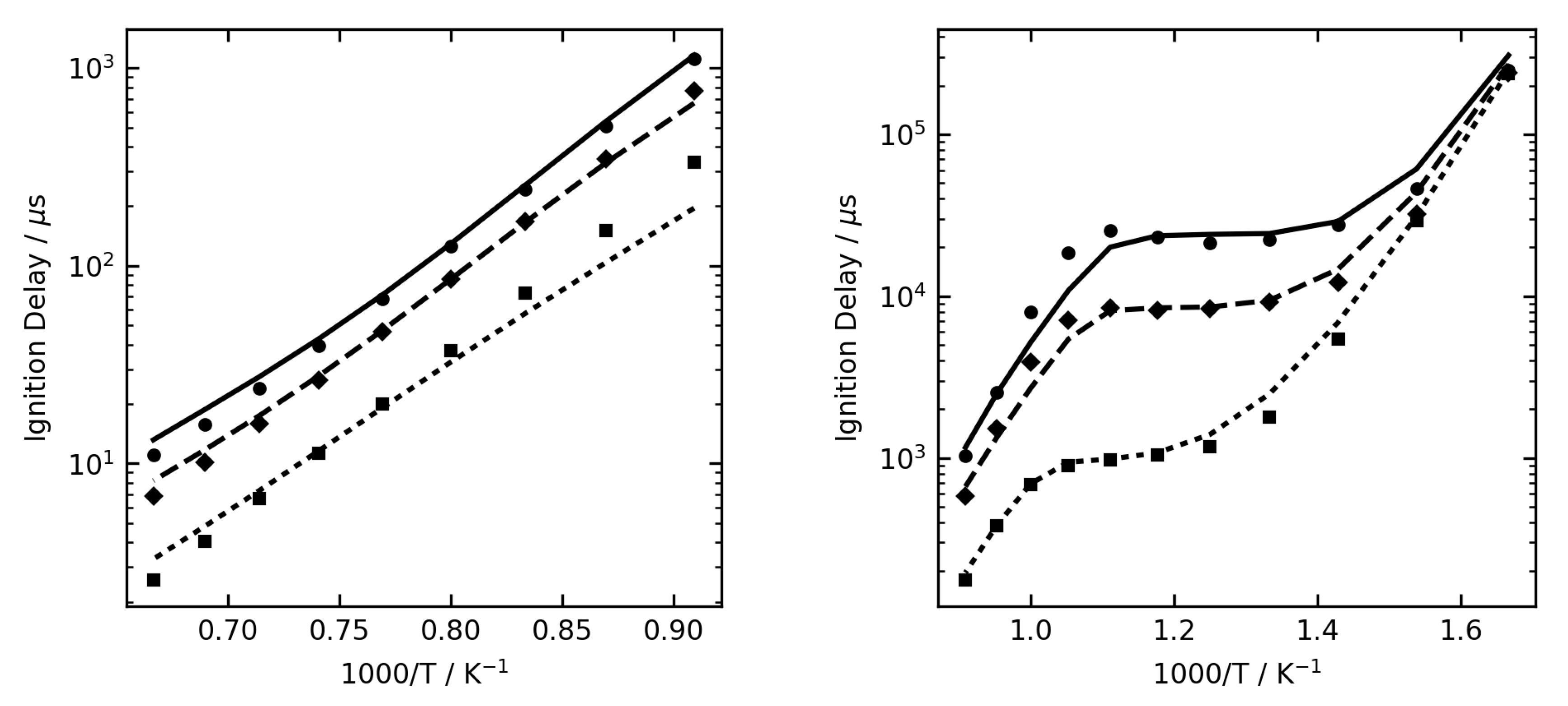
Figure 1,
Figure 2 and
Figure 3 show some examples of ignition delay time, allowing comparison between the complex (lines) and reduced (symbol) mechanisms. The overall agreement is very good, and the reduced mechanisms are seen to capture the NTC region very well.
3. Results and Discussion
In this section, the reduced mechanisms are analyzed to elucidate their composition, to provide information on the chemical subsets required to accurately reproduce ignition delay times using minimal reduced mechanisms. The composition of the reduced mechanisms listed in
Table 1 is, as a first step, analyzed with respect to the relative occurrence of reactions involving hydrocarbon compounds of different size, from the fuel C
11 down to C
0. This does, however, not give any information on the functionality of the compounds, and therefore also reaction path diagrams showing the most important hydrocarbon species are used when appropriate.
In the following, the parent mechanisms are referred to with the names CRECK and Herbinet, respectively, for the mechanisms developed by Saggesse et al. [
2] and Herbinet et al. [
13]. The abbreviations HT for high temperature ignition and LT for low temperature ignition are used in the text in results and discussion section.
3.1. Reduced High- and Low Temperature Mechanisms
As mentioned, the detailed mechanism by Herbinet et al. [
13] is constructed to contain all possible reaction pathways and has a total of 9742 reversible reactions among 2878 species for methyl decanoate, and about the same size for the two unsaturated esters. In the ACR reduction, only the most important reactions are kept, in the case of MD 258 irreversible reactions for the HT mechanism and 810 for the LT mechanism. In percent this means the size of these reduced mechanisms are 1.3% and 4.2% of that of the parent mechanism, for the HT and LT mechanisms, respectively.
In
Figure 4, it can be seen how the relative distribution of species of different size change upon reduction, for high- and low-temperature ignition of MD, Herbinet-MD-HT, and Herbinet-MD-LT.
Figure 4 reveals that in the parent mechanism. the reactions are distributed in all reaction classes, with the largest group being C-11, indicating an extensive fuel radical chemistry including around 3000 reversible reactions. The same trend with a large fraction of reactions in the C-11 class is seen for the reduced mechanisms, in particular for the high-temperature mechanism. In absolute numbers, the LT mechanism include more than 350 C-11 reactions, while the HT mechanism has less than 100 C-11 reactions. The reactions in the HT C-11 subset are mainly H-abstraction by OH, reactions also present in the LT subset. The many C-11 reactions in the LT mechanism reflect the complicated chemistry at these conditions, where H-abstraction is followed by O
2 addition and isomerization without breaking the carbon-carbon bonds.
The reduced mechanisms do not include any reactions with C-10 and few C-9. For the HT reduced mechanisms, the fraction of reactions in the C-0 and C-1 subsets are significant; in absolute numbers, 38 irreversible reactions. An important general difference between HT and LT mechanism is the absence of compounds with O-O group in the HT mechanism. The non-carbon species included are the same for both HT and LT mechanisms: OH, O, H, O2, H2O, HO2, H2O2, and H2.
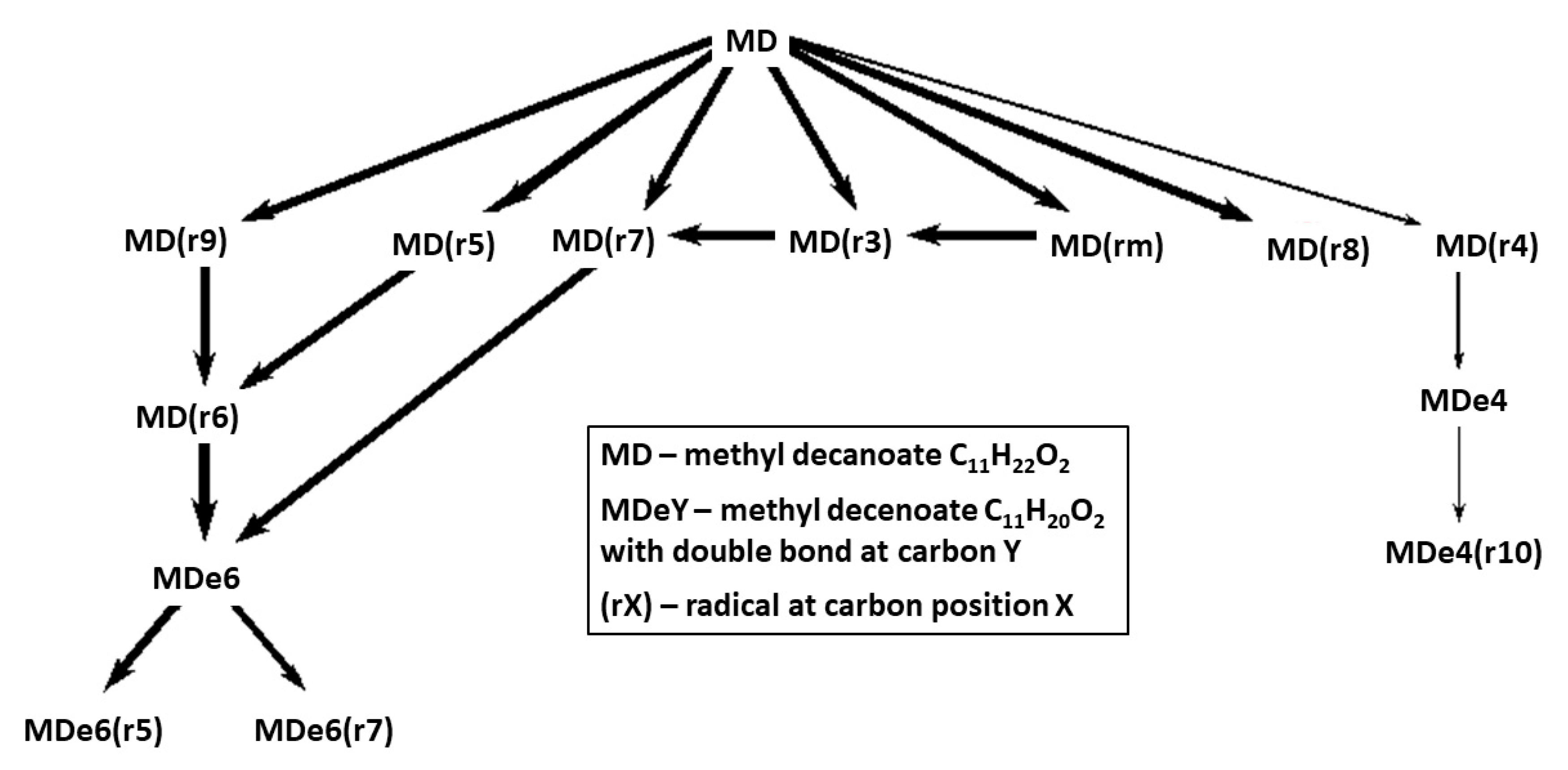
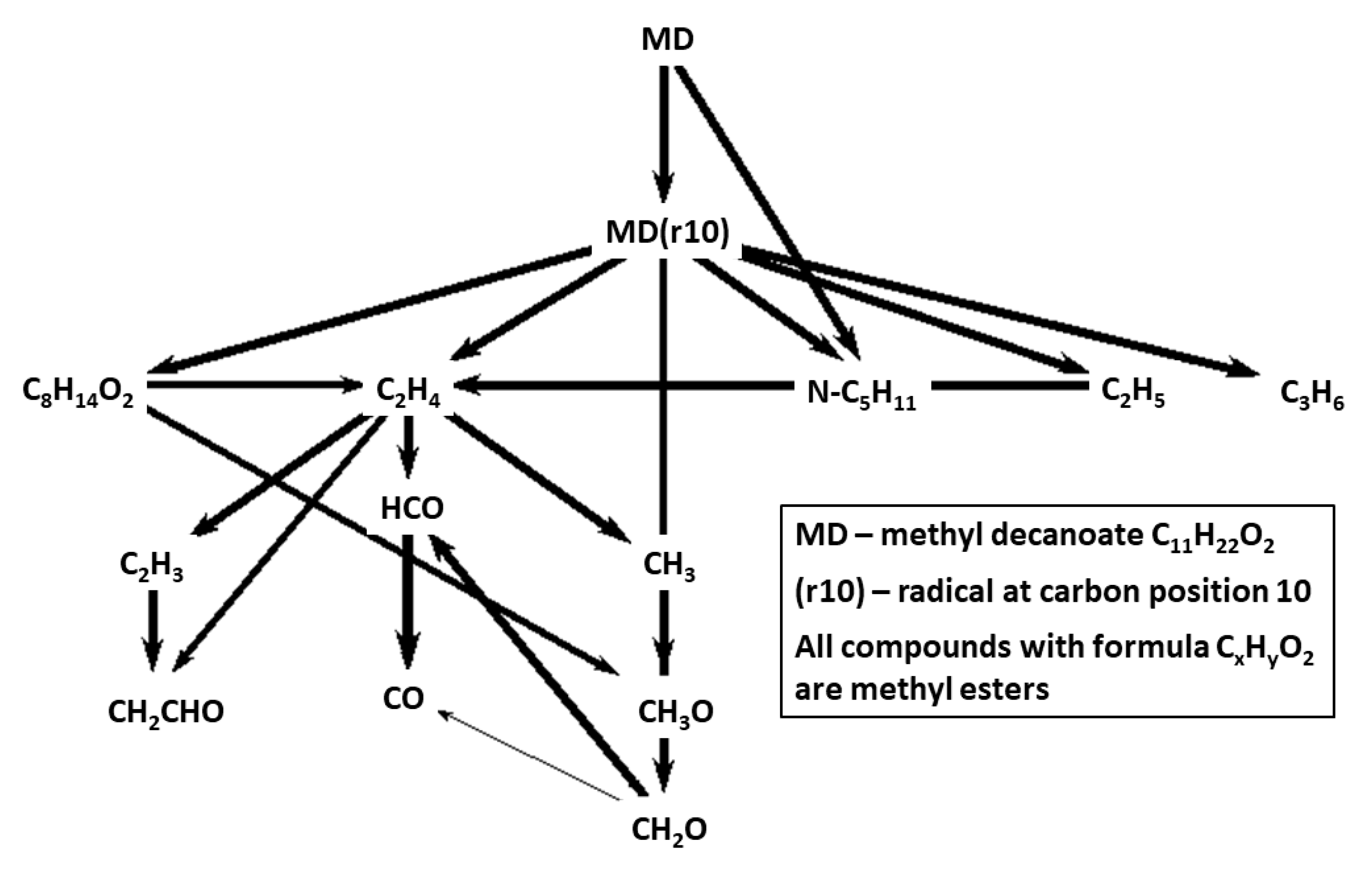
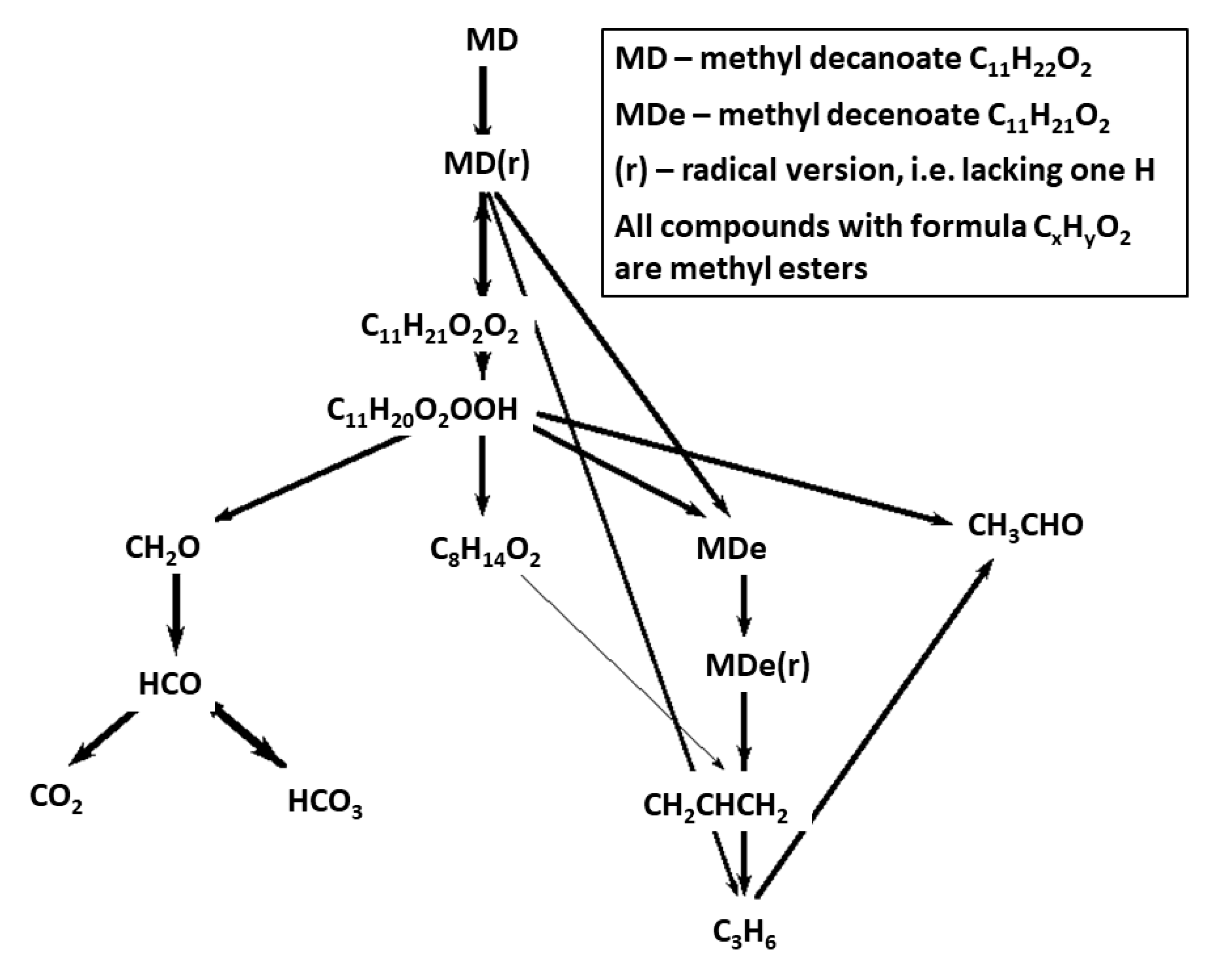
Reaction pathways for fuel breakdown in the mechanisms Herbinet-MD-HT and Herbinet-MD-LT are presented in
Figure 5 and
Figure 6. The diagrams include the 14 hydrocarbon species with highest concentration at 50% of fuel consumption, to reveal important fuel chemistry at a stage where a radical pool is established. An important difference between the HT and LT cases is that in the HT case, fuel radicals are transformed into unsaturated species, via several H-abstraction steps, while for the LT case, early addition of oxygen molecule to the fuel radicals is seen.
3.2. Comparison of Mechanisms from the Two Parent Mechanisms
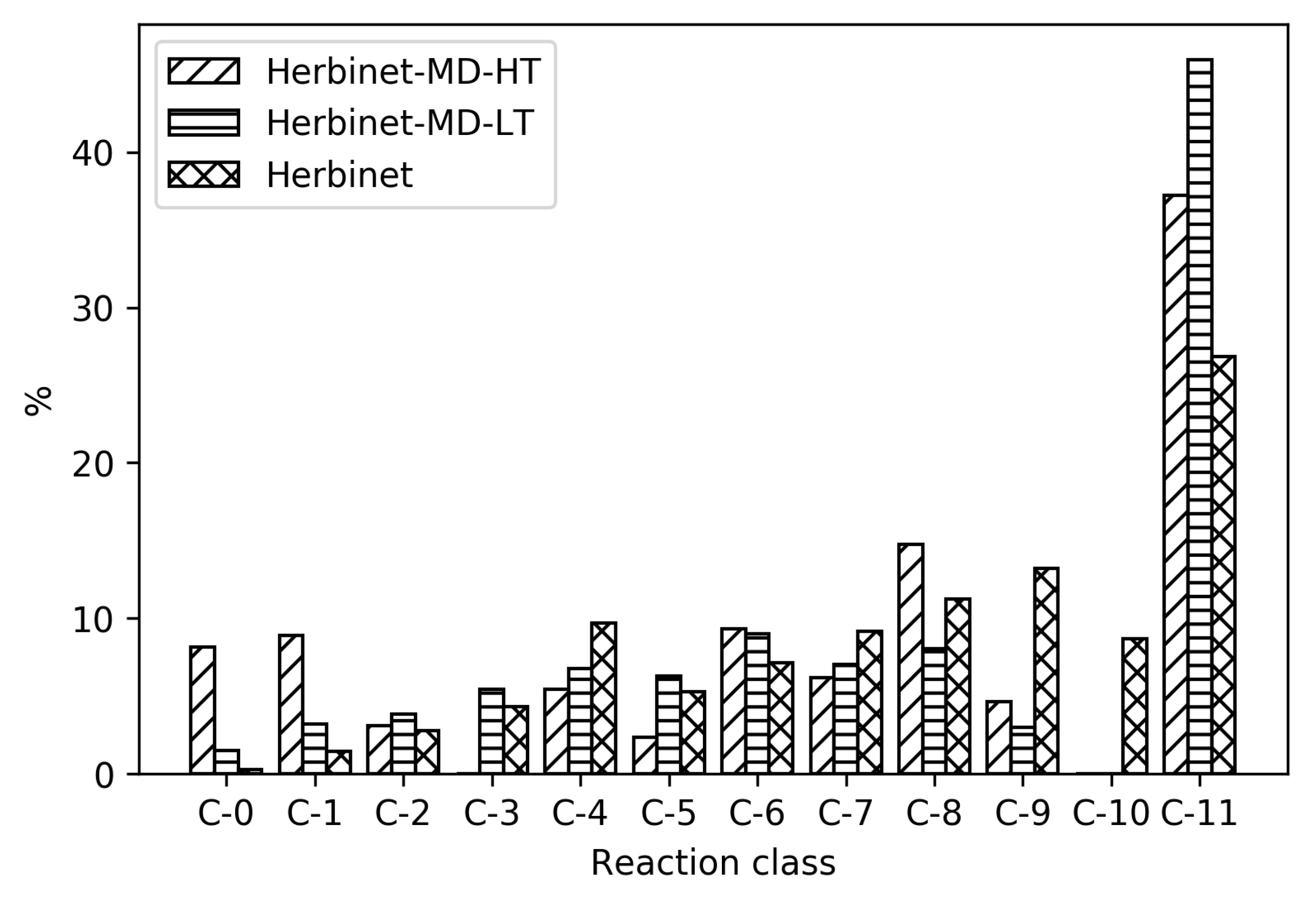
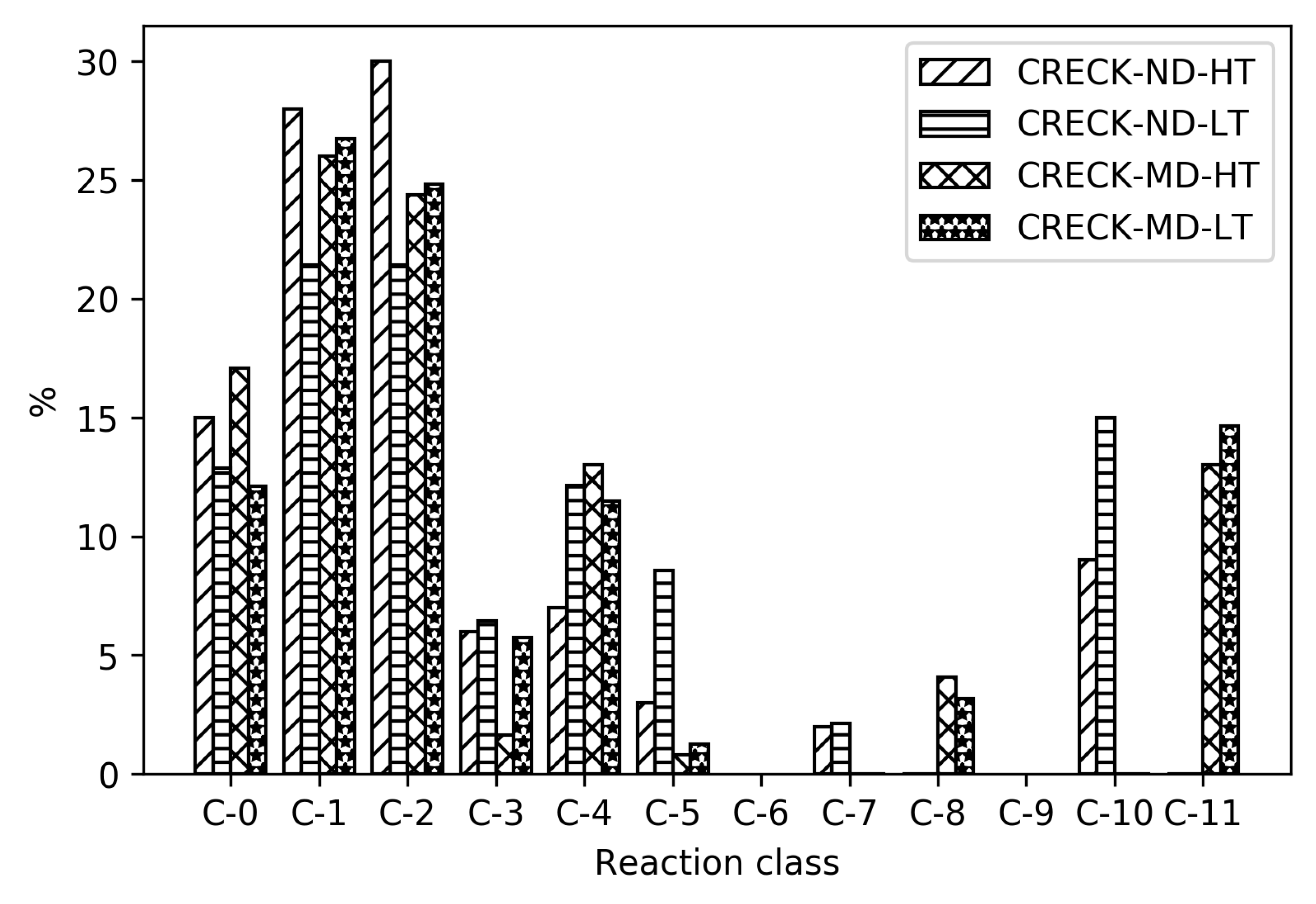
To reveal the differences between the reduced mechanisms produced from different parent mechanisms, reaction distribution for all the reduced mechanisms for MD are shown in
Figure 7. As seen in
Table 1, the absolute sizes of these mechanisms are different, with CRECK producing significantly smaller mechanisms. For the high temperatures, CRECK produce mechanisms about half the size of the mechanisms based on Herbinet, and for low temperatures, about a fifth of the size. This indicates that the lumping performed in the construction of the CRECK mechanism simplified a significant fraction of the low temperature chemistry. Recall that Herbinet include a large number of fuel radicals and their analysis [
13] of decomposition of fuel into fuel radicals show that in the case of MD, the different reaction paths have very similar importance. In the CRECK mechanism, the fuel radicals are lumped together to simplify the mechanism. This is clearly seen in
Figure 7 where the reduced mechanisms for MD with Herbinet mechanism as parent have a larger fraction of C-11 reactions and more also in the C-6 to C-9 range. Among the 810 reactions in the Herbinet-MD-LT mechanism more than 40% belong to C-11, while the corresponding mechanism created from CRECK has only about 15% out of 157 reactions belonging to the C-11 subset. If the absolute number of reactions in the C-1 and C-0 subsets are compared for the same two mechanisms, it is seen that they both contain about 40 reactions in these subsets.
The reduced mechanisms from both parent mechanisms give similarly good accuracy for ignition delay times. A conclusion to draw from the mechanism performance and the size, is that using a lumped mechanism like CRECK for a reduction is advantageous to create minimal mechanisms.
Figure 8 and
Figure 9 show reaction path diagrams including 14 most important carbon containing species for CRECK-MD-HT and CRECK-MD-LT mechanisms. When comparing the figures with the corresponding fuel-breakdown figures for Herbinet mechanism,
Figure 5 and
Figure 6, the lumping of fuel radicals become very evident. In the mechanisms based on CRECK, the fuel is transformed to a lumped radical, that in the HT case rapidly breaks down into smaller fragments and in the LT case becomes oxygenated.
3.3. Methyl Ester Compared to the Corresponding Alkane
The CRECK mechanism was used to create reduced mechanisms for methyl decanoate and n-decane, compounds with a straight chain saturated hydrocarbon part consisting of 10 carbon atoms. Low-temperature ignition chemistry of a saturated, straight-chain methyl ester is very similar to the corresponding alkane since the important chemistry is mainly involving the hydrocarbon chain. The dominating first reaction step at low temperature is H abstraction from the hydrocarbon chain, followed by addition of an oxygen molecule. The result of this is a reaction chain similar to the one described for n-decane, with the ester moiety acting only as a spectator. The high temperature chemistry does, however, follow a partly different path. At high temperatures, the fuel decomposes rapidly into smaller fragments, which in
Figure 10 can be seen for ND as a larger fraction of the smaller species in the HT case and more weight towards intermediate sizes in the LT case.
3.4. Mechanisms for Unsaturated Methyl Esters
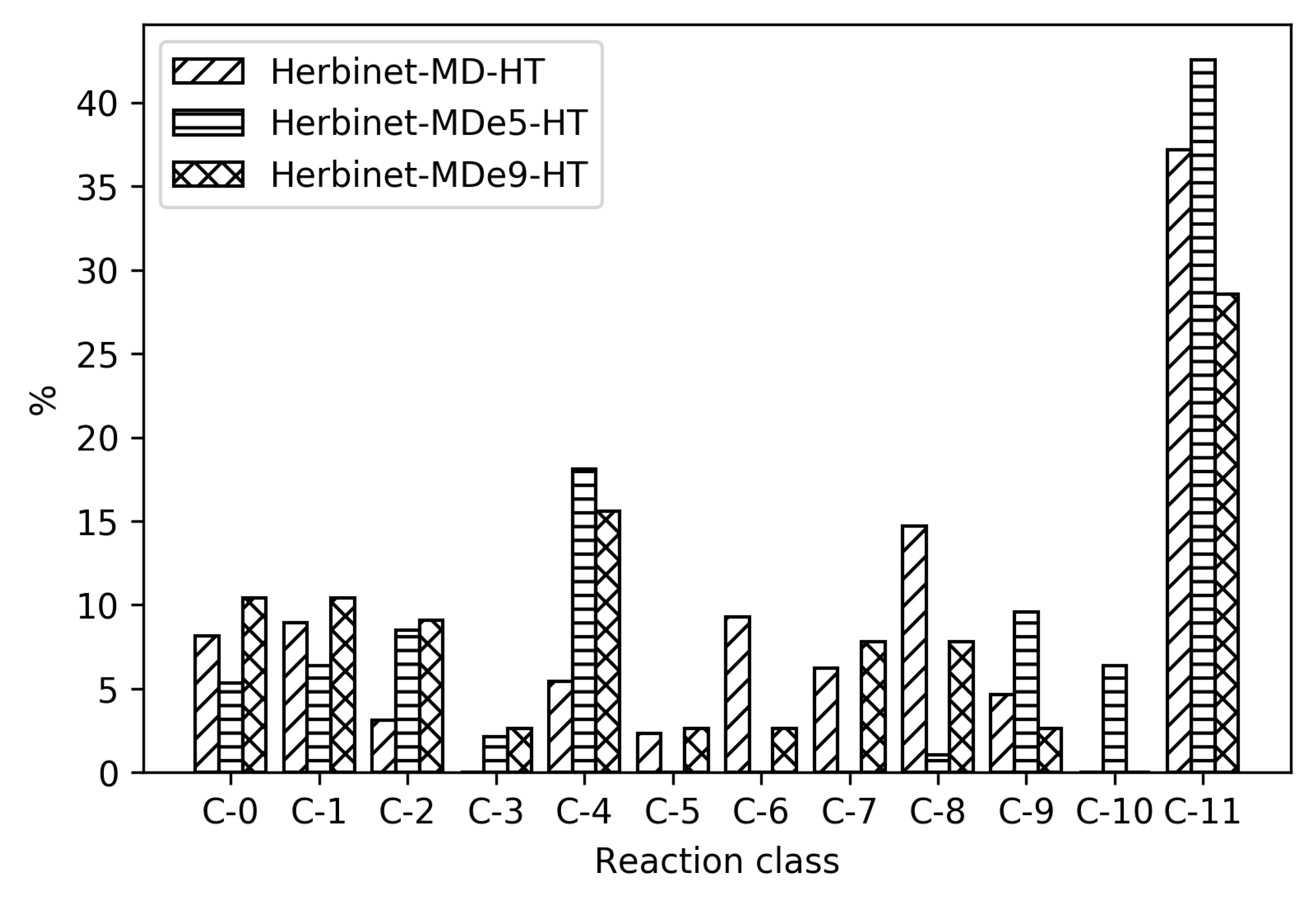
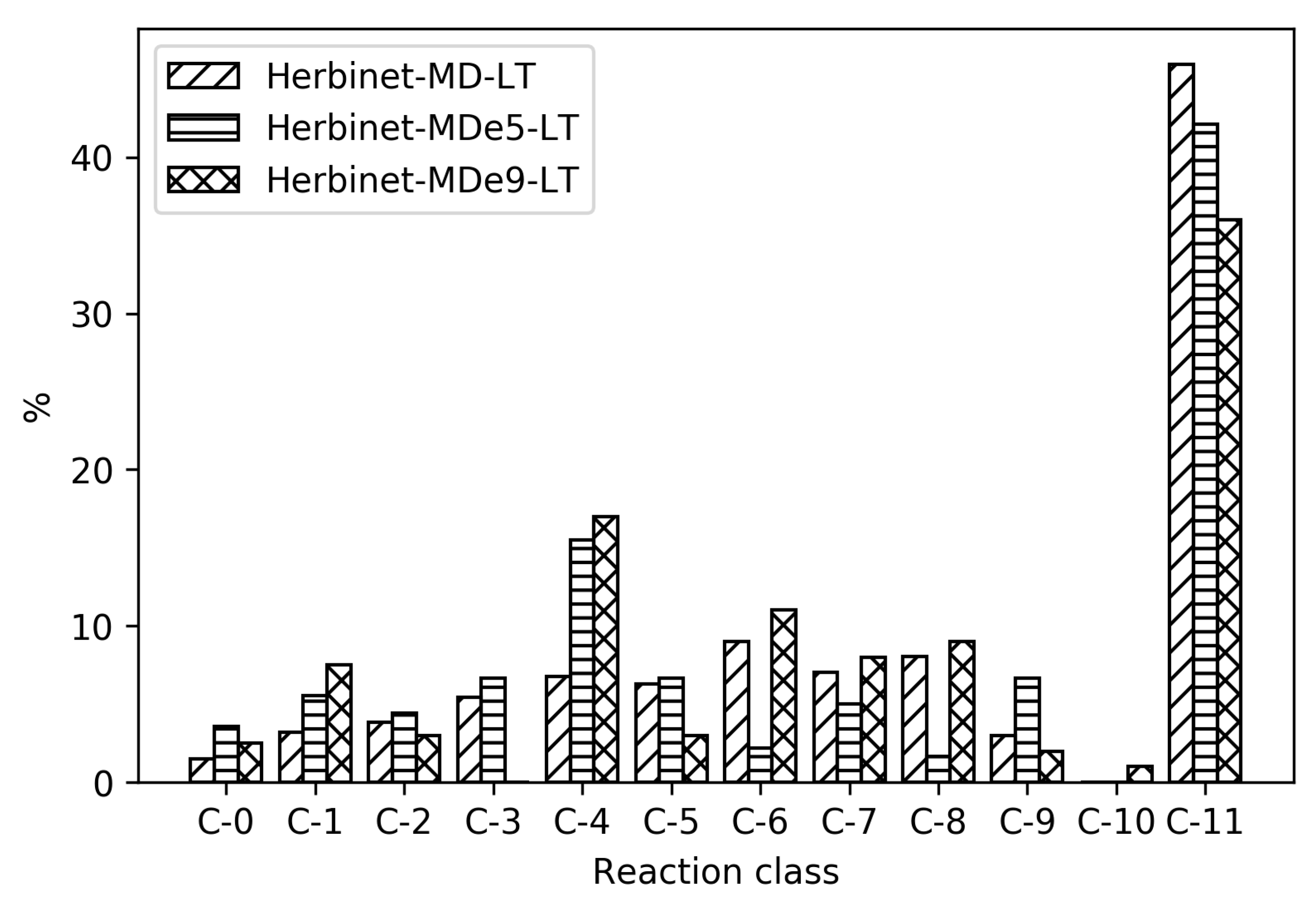
The decomposition of methyl decenoates, MDe5 and MDe9, is highly dependent on the position of the double bond. The double bond position is more sensitive to H-abstraction reaction in an initial part of the ignition event and this determine the relative abundance of different intermediates. In
Figure 11 and
Figure 12, the relative distribution of different intermediate size bins can be seen, for high- and low-temperature ignition, respectively. It is seen that the composition with respect to C-5 to C-10 species are very different between the three fuels, which is a result of different decomposition routes dependent on the double bod positions. The methyl decenoates have fewer abundant intermediates than MD, which explains why the MDs are accurately described by a smaller number of reactions. In the case of MD, there are many different reaction pathways of about equal importance when going to fuel radical.
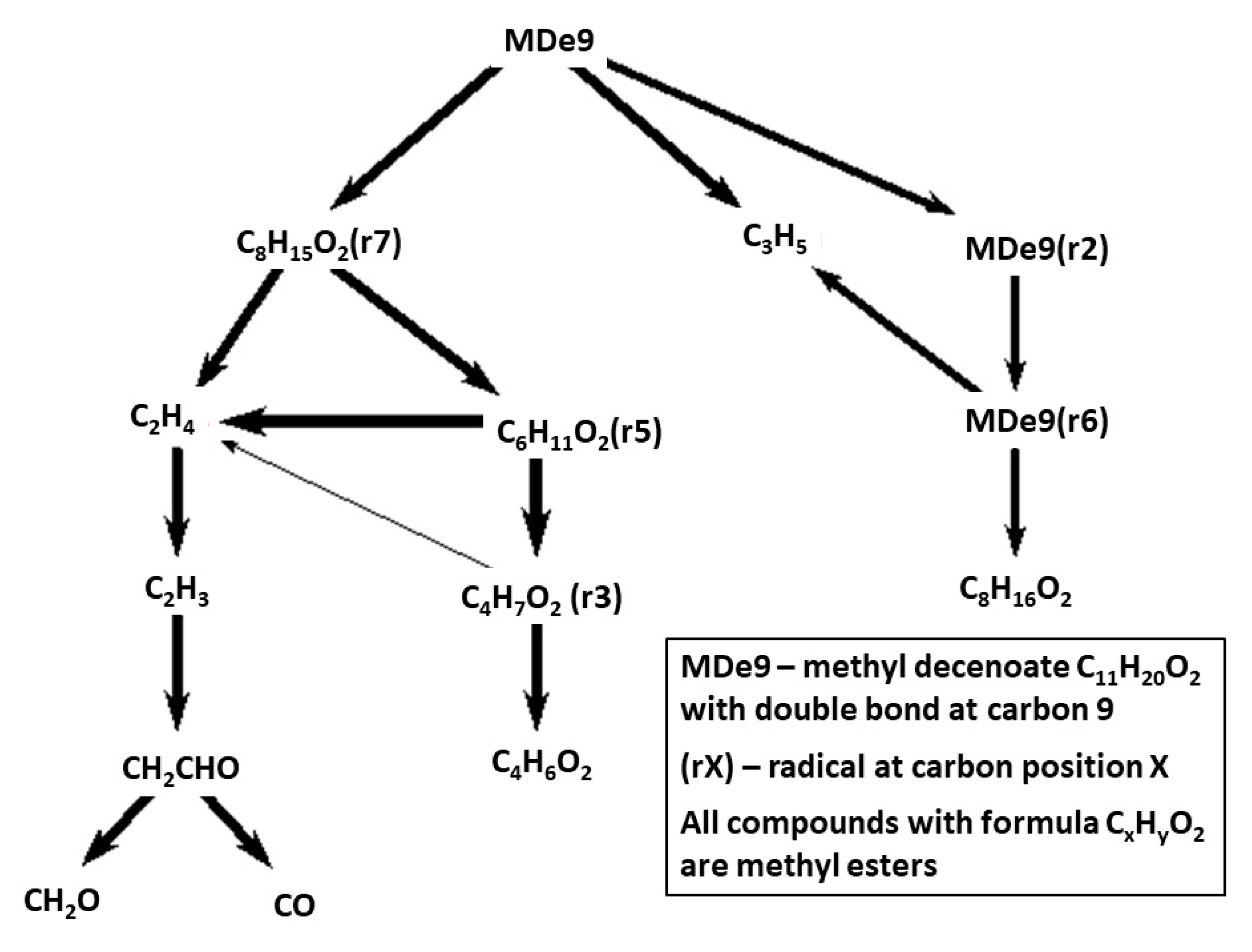
The reaction path diagram in
Figure 13 is for methyl 9-decenoate that due to the double bond has a quite different fuel breakdown pattern compared to the saturated methyl decanoate, for which HT fuel breakdown is presented in
Figure 5. Even though they originate from the similar Herbinet parent mechanisms, the MDe9 mechanism has been reduced to a much larger extent than the MD mechanism. While MD has a large number of important fuel radicals, MDe9 decompose via fewer routes with only one significant C-11 route and more rapid conversion into smaller fragments.
4. Conclusions
This work reveals some important characteristics of relevance for the development of reduced mechanisms. An important novelty of relevance for the chemical kinetics community is that two different parent mechanisms were used for mechanism reduction, and the implications for the final reduced mechanisms compared. Smallest reduced mechanisms are obtained when the CRECK mechanism is used as parent mechanism; it is possible to produce reduced mechanisms smaller than 150 reactions, small enough for implementation in CFD, while maintaining the predictive capacity for ignition delay time over the whole temperature and pressure range. The CRECK mechanism has already been reduced in size of the fuel radical and intermediate species, by a lumping approach, while the subsets of smaller hydrocarbon species are similar to the more detailed Herbinet mechanism. Indeed, the reduced mechanisms constructed from the two parent mechanisms are similar in the small hydrocarbon and H/O subsets of reactions, but quite different in the fuel breakdown subsets. The reduced mechanisms produced from the Herbinet mechanism are for some fuels still quite extensive, but it is important to notice that still the reduction of size is significant. These reduced mechanisms constructed from Herbinet mechanisms can be suitable for further reduction using methods that simplify the chemical paths.
The mechanisms developed within this work are available as
Supplementary Material, in CANTERA format. It is important to remember that the mechanisms are intended for use for combustion cases within the specified range of conditions with respect to temperature, pressure, and equivalence ratio. Highly reduced mechanisms like these shall not be used for conditions outside the range used in the reduction procedure.
The mechanisms presented in this work are only made to reproduce ignition delay and for versatile mechanisms for use in CFD also flame propagation and extinction must be accurately reproduced. It has been shown in numerous kinetic studies that the flame chemistry to some extent is different from the ignition chemistry and that some additional reactions will be necessary for a comprehensive mechanism. However, as shown in our recent publication on reduced n-heptane chemistry, the low-temperature ignition subset is the most extensive chemical subset needed in small reduced mechanisms. Reaction subsets for flame propagation and extinction are significantly smaller, as demonstrated in our previous work on n-heptane, and based on this we can assume that reduced mechanisms extended to the full scope of combustion will likely not be significantly larger than the mechanisms for ignition presented here.
The main importance of the present work is that the fundamental chemistry necessary to reproduce ignition delay of methyl esters at different conditions are revealed. Based on this understanding and using the ACR method, mechanisms for relevant mixtures of methyl esters representing real biodiesel can be constructed, which the authors intend to present in a future work.






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}