

Regulation of Phosphate Transporters and Novel Regulator of Phosphate Metabolism


Abstract
:1. Introduction
2. Phosphate Transporter Classification in the Body
3. Pi Transporters and Disease
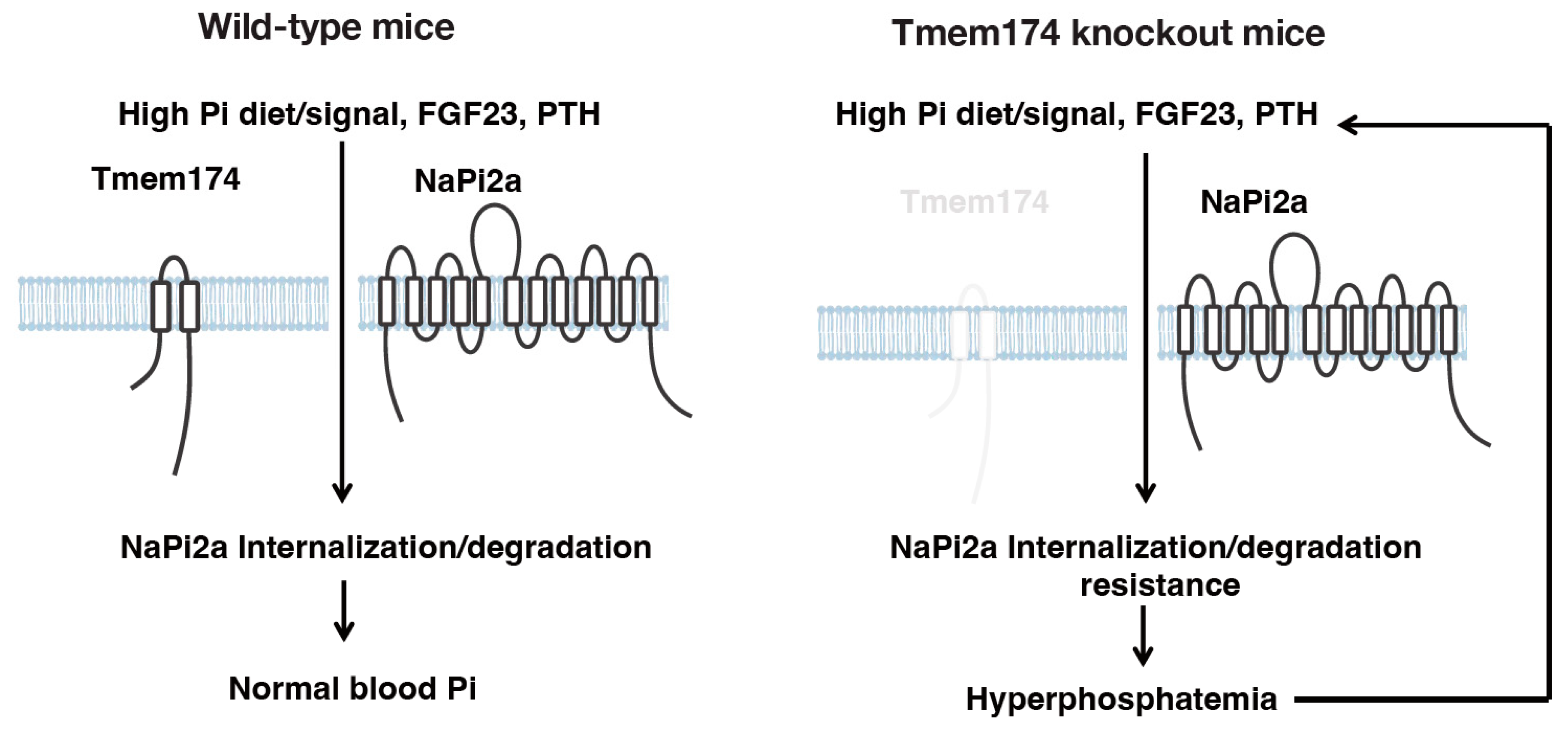
4. A Novel Regulator of Phosphate Metabolism
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Erem, S.; Razzaque, M.S. Dietary Phosphate Toxicity: An Emerging Global Health Concern. Histochem. Cell Biol. 2018, 150, 711–719. [Google Scholar] [CrossRef] [PubMed]
- Kalantar-Zadeh, K.; Gutekunst, L.; Mehrotra, R.; Kovesdy, C.P.; Bross, R.; Shinaberger, C.S.; Noori, N.; Hirschberg, R.; Benner, D.; Nissenson, A.R.; et al. Understanding Sources of Dietary Phosphorus in the Treatment of Patients with Chronic Kidney Disease. Clin. J. Am. Soc. Nephrol. 2010, 5, 519–530. [Google Scholar] [CrossRef] [PubMed]
- Uribarri, J. Phosphorus Homeostasis in Normal Health and in Chronic Kidney Disease Patients with Special Emphasis on Dietary Phosphorus Intake. Semin. Dial. 2007, 20, 295–301. [Google Scholar] [CrossRef] [PubMed]
- Qadeer, H.A.; Bashir, K. Physiology, Phosphate. In StatPearls; StatPearls Publishing LLC.: Treasure Island, FL, USA, 2023. [Google Scholar]
- Hernando, N.; Gagnon, K.; Lederer, E. Phosphate Transport in Epithelial and Nonepithelial Tissue. Physiol. Rev. 2021, 101, 1–35. [Google Scholar] [CrossRef]
- Klein, G.L. The Role of the Musculoskeletal System in Post-Burn Hypermetabolism. Metabolism 2019, 97, 81–86. [Google Scholar] [CrossRef]
- Goretti Penido, M.; Alon, U.S. Phosphate Homeostasis and its Role in Bone Health. Pediatr. Nephrol. 2012, 27, 2039–2048. [Google Scholar] [CrossRef]
- Miyagawa, A.; Tatsumi, S.; Takahama, W.; Fujii, O.; Nagamoto, K.; Kinoshita, E.; Nomura, K.; Ikuta, K.; Fujii, T.; Hanazaki, A.; et al. The Sodium Phosphate Cotransporter Family and Nicotinamide Phosphoribosyltransferase Contribute to the Daily Oscillation of Plasma Inorganic Phosphate Concentration. Kidney Int. 2018, 93, 1073–1085. [Google Scholar] [CrossRef]
- Nomura, K.; Tatsumi, S.; Miyagawa, A.; Shiozaki, Y.; Sasaki, S.; Kaneko, I.; Ito, M.; Kido, S.; Segawa, H.; Sano, M.; et al. Hepatectomy-Related Hypophosphatemia: A Novel Phosphaturic Factor in the Liver-Kidney Axis. J. Am. Soc. Nephrol. 2014, 25, 761–772. [Google Scholar] [CrossRef]
- Tatsumi, S.; Katai, K.; Kaneko, I.; Segawa, H.; Miyamoto, K.I. Nad Metabolism and the Slc34 Family: Evidence for a Liver-Kidney Axis Regulating Inorganic Phosphate. Pflug. Arch. 2019, 471, 109–122. [Google Scholar] [CrossRef]
- Pizzagalli, M.D.; Bensimon, A.; Superti-Furga, G. A Guide to Plasma Membrane Solute Carrier Proteins. FEBS J. 2021, 288, 2784–2835. [Google Scholar] [CrossRef]
- Bartoloni, L.; Antonarakis, S.E. The Human Sugar-Phosphate/Phosphate Exchanger Family Slc37. Pflug. Arch. 2004, 447, 780–783. [Google Scholar] [CrossRef]
- Cappello, A.R.; Curcio, R.; Lappano, R.; Maggiolini, M.; Dolce, V. The Physiopathological Role of the Exchangers Belonging to the Slc37 Family. Front. Chem. 2018, 6, 122. [Google Scholar] [CrossRef] [PubMed]
- Chou, J.Y.; Mansfield, B.C. The Slc37 Family of Sugar-Phosphate/Phosphate Exchangers. Curr. Top. Membr. 2014, 73, 357–382. [Google Scholar] [CrossRef]
- Chou, J.Y.; Sik Jun, H.; Mansfield, B.C. The Slc37 Family of Phosphate-Linked Sugar Phosphate Antiporters. Mol. Aspects Med. 2013, 34, 601–611. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Sun, H.; Luo, J.; Cheng, X.; Lv, W.; Luo, W.; Chen, W.J.; Xiong, Z.Q.; Liu, J.Y. The Pathology of Primary Familial Brain Calcification: Implications for Treatment. Neurosci. Bull. 2023, 39, 659–674. [Google Scholar] [CrossRef] [PubMed]
- Balck, A.; Schaake, S.; Kuhnke, N.S.; Domingo, A.; Madoev, H.; Margolesky, J.; Dobricic, V.; Alvarez-Fischer, D.; Laabs, B.H.; Kasten, M.; et al. Genotype-Phenotype Relations in Primary Familial Brain Calcification: Systematic Mdsgene Review. Mov. Disord. 2021, 36, 2468–2480. [Google Scholar] [CrossRef]
- Chande, S.; Bergwitz, C. Role of Phosphate Sensing in Bone and Mineral Metabolism. Nat. Rev. Endocrinol. 2018, 14, 637–655. [Google Scholar] [CrossRef]
- Jiang, Y.; Li, X.; Feng, J.; Li, M.; Wang, O.; Xing, X.P.; Xia, W.B. The Genetic Polymorphisms of Xpr1 and Scl34a3 Are Associated with Fanconi Syndrome in Chinese Patients of Tumor-Induced Osteomalacia. J. Endocrinol. Investig. 2021, 44, 773–780. [Google Scholar] [CrossRef]
- Xu, X.; Li, X.; Sun, H.; Cao, Z.; Gao, R.; Niu, T.; Wang, Y.; Ma, T.; Chen, R.; Wang, C.; et al. Murine Placental-Fetal Phosphate Dyshomeostasis Caused by an Xpr1 Deficiency Accelerates Placental Calcification and Restricts Fetal Growth in Late Gestation. J. Bone Miner. Res. 2020, 35, 116–129. [Google Scholar] [CrossRef]
- Ansermet, C.; Moor, M.B.; Centeno, G.; Auberson, M.; Hu, D.Z.; Baron, R.; Nikolaeva, S.; Haenzi, B.; Katanaeva, N.; Gautschi, I.; et al. Renal Fanconi Syndrome and Hypophosphatemic Rickets in the Absence of Xenotropic and Polytropic Retroviral Receptor in the Nephron. J. Am. Soc. Nephrol. 2017, 28, 1073–1078. [Google Scholar] [CrossRef]
- Magen, D.; Berger, L.; Coady, M.J.; Ilivitzki, A.; Militianu, D.; Tieder, M.; Selig, S.; Lapointe, J.Y.; Zelikovic, I.; Skorecki, K. A Loss-of-Function Mutation in Napi-IIa and Renal Fanconi’s Syndrome. N. Engl. J. Med. 2010, 362, 1102–1109. [Google Scholar] [CrossRef] [PubMed]
- Al-Sardar, H.; Al-Habbo, D.J.; Al-Hayali, R.M. Pulmonary Alveolar Microlithiasis: Report of Two Brothers with the Same Illness and Review of Literature. BMJ Case Rep. 2014, 2014. [Google Scholar] [CrossRef]
- Dandan, S.; Yuqin, C.; Wei, L.; Ziheng, P.; Dapeng, Z.; Jianzhu, Y.; Xin, X.; Yonghong, L.; Fengjun, T. Novel Deletion of Slc34a2 in Chinese Patients of Pam Shares Mutation Hot Spot with Fusion Gene Slc34a2-Ros1 in Lung Cancer. J. Genet. 2018, 97, 939–944. [Google Scholar] [CrossRef] [PubMed]
- Erel, F.; Güngör, C.; Sarıoğlu, N.; Aksu, G.D.; Turan, G.; Demirpolat, G. Spontaneous Pneumomediastinum and Subcutaneous Emphysema Secondary to Pulmonary Alveolar Microlithiasis. Tuberk. Toraks 2021, 69, 416–420. [Google Scholar] [CrossRef]
- Ma, T.; Ren, J.; Yin, J.; Ma, Z. A Pedigree with Pulmonary Alveolar Microlithiasis: A Clinical Case Report and Literature Review. Cell Biochem. Biophys. 2014, 70, 565–572. [Google Scholar] [CrossRef] [PubMed]
- Saito, A.; Nikolaidis, N.M.; Amlal, H.; Uehara, Y.; Gardner, J.C.; LaSance, K.; Pitstick, L.B.; Bridges, J.P.; Wikenheiser-Brokamp, K.A.; McGraw, D.W.; et al. Modeling Pulmonary Alveolar Microlithiasis by Epithelial Deletion of the Npt2b Sodium Phosphate Cotransporter Reveals Putative Biomarkers and Strategies for Treatment. Sci. Transl. Med. 2015, 7, 313ra181. [Google Scholar] [CrossRef]
- Bergwitz, C.; Miyamoto, K.I. Hereditary Hypophosphatemic Rickets with Hypercalciuria: Pathophysiology, Clinical Presentation, Diagnosis and Therapy. Pflug. Arch. 2019, 471, 149–163. [Google Scholar] [CrossRef] [PubMed]
- Bergwitz, C.; Roslin, N.M.; Tieder, M.; Loredo-Osti, J.C.; Bastepe, M.; Abu-Zahra, H.; Frappier, D.; Burkett, K.; Carpenter, T.O.; Anderson, D.; et al. Slc34a3 Mutations in Patients with Hereditary Hypophosphatemic Rickets with Hypercalciuria Predict a Key Role for the Sodium-Phosphate Cotransporter Napi-Iic in Maintaining Phosphate Homeostasis. Am. J. Hum. Genet. 2006, 78, 179–192. [Google Scholar] [CrossRef]
- Ichikawa, S.; Sorenson, A.H.; Imel, E.A.; Friedman, N.E.; Gertner, J.M.; Econs, M.J. Intronic deletions in the Slc34a3 Gene Cause Hereditary Hypophosphatemic Rickets with Hypercalciuria. J. Clin. Endocrinol. Metab. 2006, 91, 4022–4027. [Google Scholar] [CrossRef]
- Lorenz-Depiereux, B.; Benet-Pages, A.; Eckstein, G.; Tenenbaum-Rakover, Y.; Wagenstaller, J.; Tiosano, D.; Gershoni-Baruch, R.; Albers, N.; Lichtner, P.; Schnabel, D.; et al. Hereditary Hypophosphatemic Rickets with Hypercalciuria is Caused by Mutations in the Sodium-Phosphate Cotransporter Gene Slc34a3. Am. J. Hum. Genet. 2006, 78, 193–201. [Google Scholar] [CrossRef]
- Inden, M.; Iriyama, M.; Zennami, M.; Sekine, S.I.; Hara, A.; Yamada, M.; Hozumi, I. The Type Iii Transporters (Pit-1 and Pit-2) Are the Major Sodium-Dependent Phosphate Transporters in the Mice and Human Brains. Brain Res. 2016, 1637, 128–136. [Google Scholar] [CrossRef] [PubMed]
- Wagner, C.A. Pharmacology of Mammalian Na(+)-Dependent Transporters of Inorganic Phosphate. In Handbook of Experimental Pharmacology; Springer: Berlin/Heidelberg, Germany, 2023. [Google Scholar]
- Carecchio, M.; Mainardi, M.; Bonato, G. The Clinical and Genetic Spectrum of Primary Familial Brain Calcification. J. Neurol. 2023, 270, 3270–3277. [Google Scholar] [CrossRef] [PubMed]
- Bergwitz, C.; Jüppner, H. Fgf23 and Syndromes of Abnormal Renal Phosphate Handling. Adv. Exp. Med. Biol. 2012, 728, 41–64. [Google Scholar] [CrossRef] [PubMed]
- Christov, M.; Jüppner, H. Phosphate Homeostasis Disorders. Best. Pract. Res. Clin. Endocrinol. Metab. 2018, 32, 685–706. [Google Scholar] [CrossRef] [PubMed]
- Vanessa, H. Calcium, Phosphate and Magnesium Disorders. In Fluid and Electrolyte Disorders; Usman, M., Ed.; IntechOpen: Rijeka, Croatia, 2018. [Google Scholar]
- Peacock, M. Phosphate Metabolism in Health and Disease. Calcif. Tissue Int. 2021, 108, 3–15. [Google Scholar] [CrossRef]
- Tomoe, Y.; Segawa, H.; Shiozawa, K.; Kaneko, I.; Tominaga, R.; Hanabusa, E.; Aranami, F.; Furutani, J.; Kuwahara, S.; Tatsumi, S.; et al. Phosphaturic Action of Fibroblast Growth Factor 23 in Npt2 Null Mice. Am. J. Physiol. Renal Physiol. 2010, 298, F1341–F1350. [Google Scholar] [CrossRef] [PubMed]
- Segawa, H.; Kawakami, E.; Kaneko, I.; Kuwahata, M.; Ito, M.; Kusano, K.; Saito, H.; Fukushima, N.; Miyamoto, K. Effect of Hydrolysis-Resistant Fgf23-R179q on Dietary Phosphate Regulation of the Renal Type-Ii Na/Pi Transporter. Pflug. Arch. 2003, 446, 585–592. [Google Scholar] [CrossRef]
- Segawa, H.; Yamanaka, S.; Ohno, Y.; Onitsuka, A.; Shiozawa, K.; Aranami, F.; Furutani, J.; Tomoe, Y.; Ito, M.; Kuwahata, M.; et al. Correlation between Hyperphosphatemia and Type Ii Na-Pi Cotransporter Activity in Klotho Mice. Am. J. Physiol. Renal Physiol. 2007, 292, F769–F779. [Google Scholar] [CrossRef]
- Hanazaki, A.; Ikuta, K.; Sasaki, S.; Sasaki, S.; Koike, M.; Tanifuji, K.; Arima, Y.; Kaneko, I.; Shiozaki, Y.; Tatsumi, S.; et al. Role of Sodium-Dependent Pi Transporter/Npt2c on Pi Homeostasis in Klotho Knockout Mice Different Properties between Juvenile and Adult Stages. Physiol. Rep. 2020, 8, e14324. [Google Scholar] [CrossRef]
- Nakatani, T.; Sarraj, B.; Ohnishi, M.; Densmore, M.J.; Taguchi, T.; Goetz, R.; Mohammadi, M.; Lanske, B.; Razzaque, M.S. In Vivo Genetic Evidence for Klotho-Dependent, Fibroblast Growth Factor 23 (Fgf23)-Mediated Regulation of Systemic Phosphate Homeostasis. FASEB J. 2009, 23, 433–441. [Google Scholar] [CrossRef]
- Nakatani, T.; Ohnishi, M.; Razzaque, M.S. Inactivation of Klotho Function Induces Hyperphosphatemia Even in Presence of High Serum Fibroblast Growth Factor 23 Levels in a Genetically Engineered Hypophosphatemic (Hyp) Mouse Model. FASEB J. 2009, 23, 3702–3711. [Google Scholar] [CrossRef]
- Dërmaku-Sopjani, M.; Sopjani, M.; Saxena, A.; Shojaiefard, M.; Bogatikov, E.; Alesutan, I.; Eichenmüller, M.; Lang, F. Downregulation of Napi-Iia and Napi-Iib Na-Coupled Phosphate Transporters by Coexpression of Klotho. Cell Physiol. Biochem. 2011, 28, 251–258. [Google Scholar] [CrossRef] [PubMed]
- Prud’homme, G.J.; Kurt, M.; Wang, Q. Pathobiology of the Klotho Antiaging Protein and Therapeutic Considerations. Front. Aging 2022, 3, 931331. [Google Scholar] [CrossRef] [PubMed]
- Hu, M.C.; Shi, M.; Zhang, J.; Pastor, J.; Nakatani, T.; Lanske, B.; Razzaque, M.S.; Rosenblatt, K.P.; Baum, M.G.; Kuro-o, M.; et al. Klotho: A Novel Phosphaturic Substance Acting as an Autocrine Enzyme in the Renal Proximal Tubule. FASEB J. 2010, 24, 3438–3450. [Google Scholar] [CrossRef] [PubMed]
- Turner, M.E.; Rowsell, T.S.; Lansing, A.P.; Jeronimo, P.S.; Lee, L.H.; Svajger, B.A.; Zelt, J.G.; Forster, C.M.; Petkovich, M.P.; Holden, R.M. Vascular Calcification Maladaptively Participates in Acute Phosphate Homeostasis. Cardiovasc. Res. 2023, 119, 1077–1091. [Google Scholar] [CrossRef]
- Nguyen, N.T.; Nguyen, T.T.; Ly, D.D.; Xia, J.-B.; Qi, X.-F.; Lee, I.-K.; Cha, S.-K.; Park, K.-S. Oxidative Stress by Ca2+ Overload Is Critical for Phosphate-Induced Vascular Calcification. Am. J. Physiol. Heart Circ. Physiol. 2020, 319, H1302–H1312. [Google Scholar] [CrossRef]
- Seifert, E.L.; Ligeti, E.; Mayr, J.A.; Sondheimer, N.; Hajnóczky, G. The Mitochondrial Phosphate Carrier: Role in Oxidative Metabolism, Calcium Handling and Mitochondrial Disease. Biochem. Biophys. Res. Commun. 2015, 464, 369–375. [Google Scholar] [CrossRef]
- Hill Gallant, K.M.; Stremke, E.R.; Trevino, L.L.; Moorthi, R.N.; Doshi, S.; Wastney, M.E.; Hisada, N.; Sato, J.; Ogita, Y.; Fujii, N.; et al. EOS789, a Broad-Spectrum Inhibitor of Phosphate Transport, is Safe with an Indication of Efficacy in a Phase 1b Randomized Crossover Trial in Hemodialysis Patients. Kidney Int. 2021, 99, 1225–1233. [Google Scholar] [CrossRef]
- Tsuboi, Y.; Ichida, Y.; Murai, A.; Maeda, A.; Iida, M.; Kato, A.; Ohtomo, S.; Horiba, N. Eos789, Pan-Phosphate Transporter Inhibitor, Ameliorates the Progression of Kidney Injury in anti-GBM-Induced Glomerulonephritis Rats. Pharmacol. Res. Perspect. 2022, 10, e00973. [Google Scholar] [CrossRef]
- Tsuboi, Y.; Ohtomo, S.; Ichida, Y.; Hagita, H.; Ozawa, K.; Iida, M.; Nagao, S.; Ikegami, H.; Takahashi, T.; Horiba, N. Eos789, a Novel Pan-Phosphate Transporter Inhibitor, is Effective for the Treatment of Chronic Kidney Disease-Mineral Bone Disorder. Kidney Int. 2020, 98, 343–354. [Google Scholar] [CrossRef]
- Filipski, K.J.; Sammons, M.F.; Bhattacharya, S.K.; Panteleev, J.; Brown, J.A.; Loria, P.M.; Boehm, M.; Smith, A.C.; Shavnya, A.; Conn, E.L.; et al. Discovery of Orally Bioavailable Selective Inhibitors of the Sodium-Phosphate Cotransporter Napi2a (Slc34a1). ACS Med. Chem. Lett. 2018, 9, 440–445. [Google Scholar] [CrossRef]
- Clerin, V.; Saito, H.; Filipski, K.J.; Nguyen, A.H.; Garren, J.; Kisucka, J.; Reyes, M.; Jüppner, H. Selective Pharmacological Inhibition of the Sodium-Dependent Phosphate Cotransporter NPT2a Promotes Phosphate Excretion. J. Clin. Investig. 2020, 130, 6510–6522. [Google Scholar] [CrossRef]
- Tenenhouse, H.S.; Martel, J.; Gauthier, C.; Segawa, H.; Miyamoto, K. Differential Effects of Npt2a Gene Ablation and X-Linked Hyp Mutation on Renal Expression of Npt2c. Am. J. Physiol. Renal Physiol. 2003, 285, F1271–F1278. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, I.; Segawa, H.; Ikuta, K.; Hanazaki, A.; Fujii, T.; Tatsumi, S.; Kido, S.; Hasegawa, T.; Amizuka, N.; Saito, H.; et al. Eldecalcitol Causes Fgf23 Resistance for Pi Reabsorption and Improves Rachitic Bone Phenotypes in the Male Hyp Mouse. Endocrinology 2018, 159, 2741–2758. [Google Scholar] [CrossRef] [PubMed]
- Martins, J.S.; Liu, E.S.; Sneddon, W.B.; Friedman, P.A.; Demay, M.B. 1,25-Dihydroxyvitamin D Maintains Brush Border Membrane Napi2a and Attenuates Phosphaturia in Hyp Mice. Endocrinology 2019, 160, 2204–2214. [Google Scholar] [CrossRef]
- Friedman, P.A.; Sneddon, W.B.; Mamonova, T.; Montanez-Miranda, C.; Ramineni, S.; Harbin, N.H.; Squires, K.E.; Gefter, J.V.; Magyar, C.E.; Emlet, D.R.; et al. Rgs14 Regulates Pth-and Fgf23-Sensitive Npt2a-Mediated Renal Phosphate Uptake Via Binding to the Nherf1 Scaffolding Protein. J. Biol. Chem. 2022, 298, 101836. [Google Scholar] [CrossRef] [PubMed]
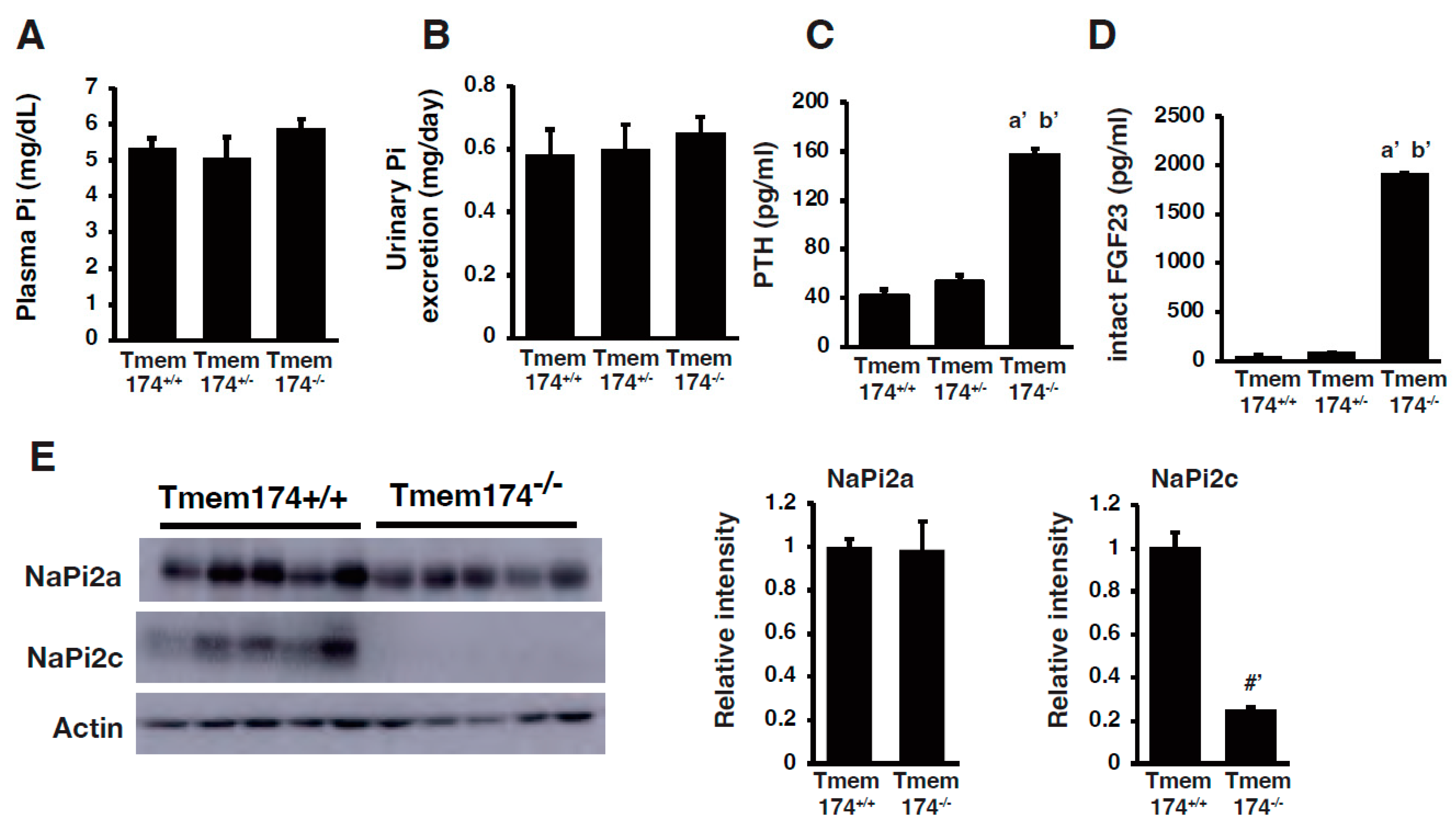
- Sasaki, S.; Shiozaki, Y.; Hanazaki, A.; Koike, M.; Tanifuji, K.; Uga, M.; Kawahara, K.; Kaneko, I.; Kawamoto, Y.; Wiriyasermkul, P.; et al. Tmem174, a Regulator of Phosphate Transporter Prevents Hyperphosphatemia. Sci. Rep. 2022, 12, 6353. [Google Scholar] [CrossRef]
- Miyazaki-Anzai, S.; Keenan, A.L.; Blaine, J.; Miyazaki, M. Targeted Disruption of a Proximal Tubule-Specific Tmem174 Gene in Mice Causes Hyperphosphatemia and Vascular Calcification. J. Am. Soc. Nephrol. 2022, 33, 1477–1486. [Google Scholar] [CrossRef]
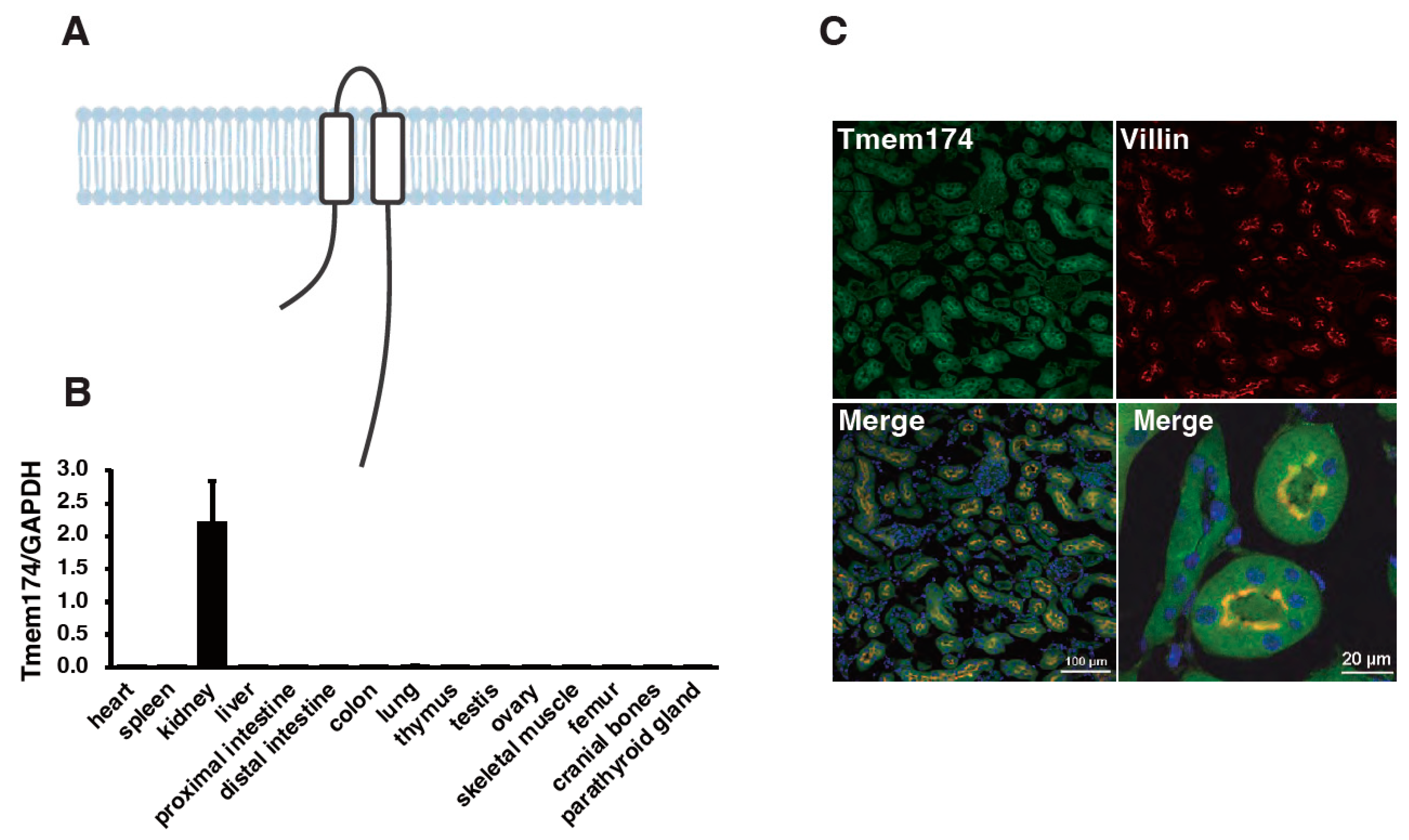


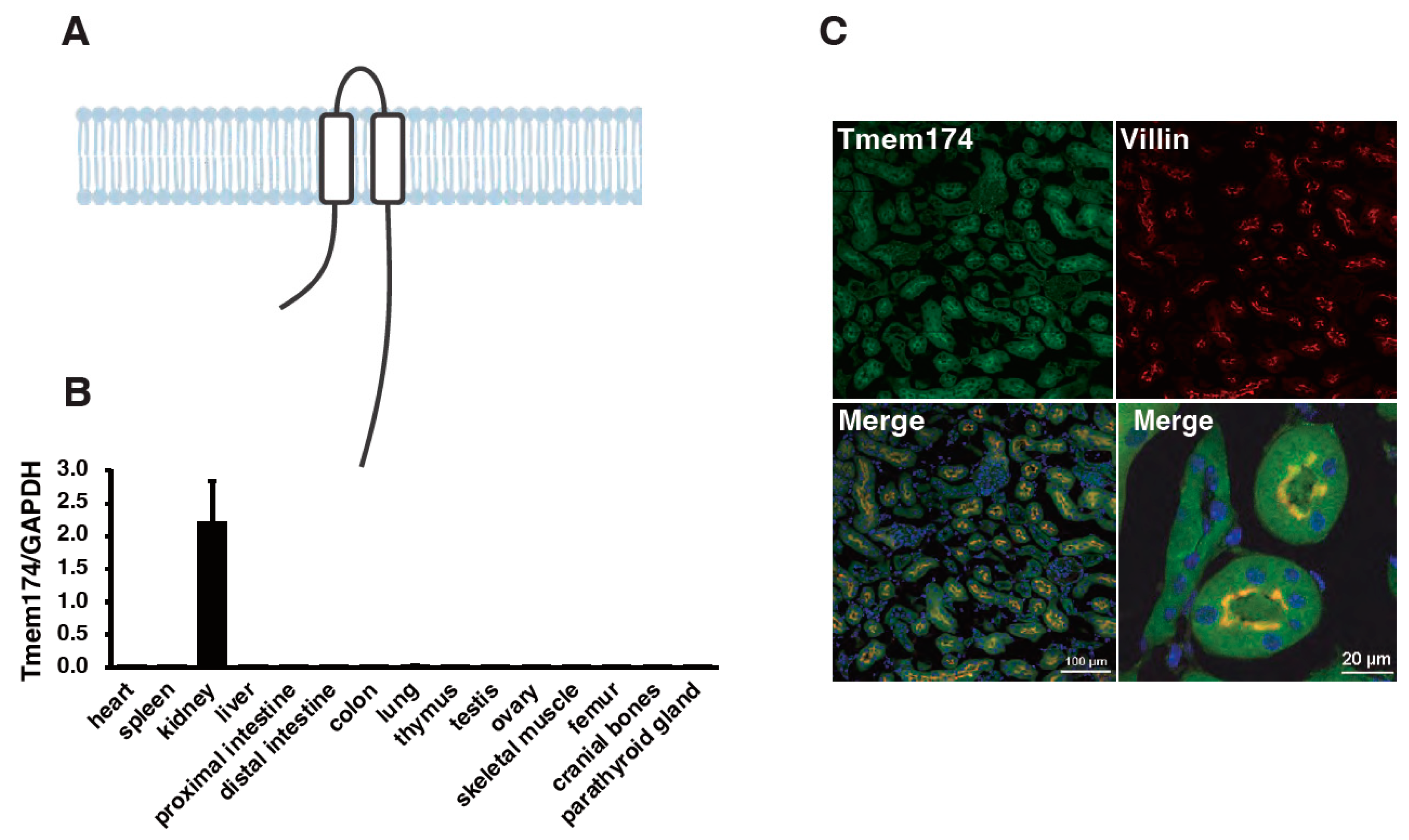{kind=link}
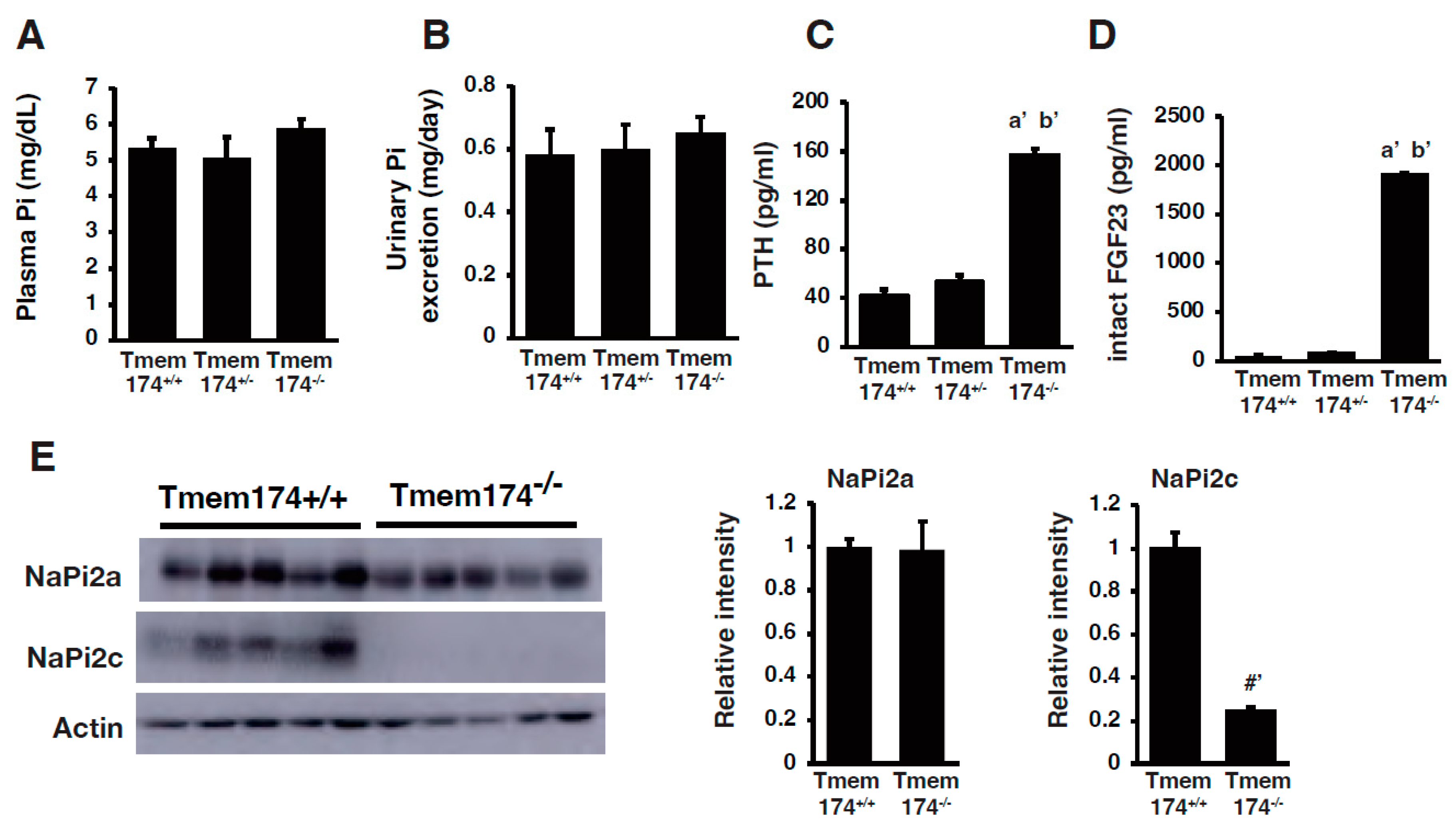{kind=link}
{kind=link}
| Name | Function | Substrates |
|---|---|---|
| SLC20 | Na+-phosphate cotransporter | Phosphate |
| SLC34 | Na+-phosphate cotransporter | Phosphate |
| SLC37 | Sugar-phosphate/phosphate exchanger | Glucose-6-phosphate/phosphate |
| SLC53 | Phosphate carriers | Phosphate |
| SLC62 | Pyrophosphate transporter | Pyrophosphate |
| SLC63 | Sphingosine-phosphate transporter | Sphingosine-phosphate |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Koike, M.; Uga, M.; Shiozaki, Y.; Miyamoto, K.-i.; Segawa, H. Regulation of Phosphate Transporters and Novel Regulator of Phosphate Metabolism. Endocrines 2023, 4, 607-615. https://doi.org/10.3390/endocrines4030043
Koike M, Uga M, Shiozaki Y, Miyamoto K-i, Segawa H. Regulation of Phosphate Transporters and Novel Regulator of Phosphate Metabolism. Endocrines. 2023; 4(3):607-615. https://doi.org/10.3390/endocrines4030043
Chicago/Turabian StyleKoike, Megumi, Minori Uga, Yuji Shiozaki, Ken-ichi Miyamoto, and Hiroko Segawa. 2023. "Regulation of Phosphate Transporters and Novel Regulator of Phosphate Metabolism" Endocrines 4, no. 3: 607-615. https://doi.org/10.3390/endocrines4030043
APA StyleKoike, M., Uga, M., Shiozaki, Y., Miyamoto, K.-i., & Segawa, H. (2023). Regulation of Phosphate Transporters and Novel Regulator of Phosphate Metabolism. Endocrines, 4(3), 607-615. https://doi.org/10.3390/endocrines4030043