Abstract
Thyroid diseases in children and adolescents include acquired or congenital conditions, including genetic disorders either isolated or part of a syndrome. Briefly, we will review the physiology and pathophysiology of the thyroid gland and its disorders. The aim of this chapter is to describe genetic abnormalities of the thyroid gland.
1. Thyroid Anatomy, Embryology, and Physiology
1.1. Thyroid Anatomy and Physiology
The thyroid gland is located in the anterior neck and consists of two lobes connected by the isthmus. Each lobe consists of follicles made from follicular and parafollicular cells with a central lumen filled with colloid containing thyroglobulin (TG) and enzymes such as thyroid peroxidase (TPO) that are responsible for organification, oxidation, and coupling reactions [1,2]. There are two main active thyroid hormones triiodothyronine (T3) and thyroxine (T4) that regulate:
- Basal metabolic rate by stimulating Na+/K+ ATPase activity resulting in increasing body temperature, respiratory rate (RR), and oxygen consumption
- lipolysis, gluconeogenesis, glycogenolysis
- neuronal differentiation, synapse development, myelination in the prenatal and newborn periods, regulating neurodevelopment
- brain maturation: coordination, gait
- psychiatric function: intellectual development
- growth and pubertal development
- beta 1-adrenoreceptor stimulation in the heart for control of heart rate (HR), cardiac output (CO), contractility, and stroke volume [1]
The hypothalamic peptide thyrotropin-releasing hormone (TRH) regulates pituitary thyroid-stimulating hormone (TSH) which then regulates thyroid hormone synthesis by G-protein coupled TSH receptors on the follicular cells [3]. Thyroid hormone synthesis is also regulated by negative feedback, as T3 levels rise, sensitivity to TRH is diminished and TSH release is lower.
After uptake of circulating iodide at the basolateral membrane, T4 (majority) and T3 are produced and released.
T4 is peripherally converted to T3 via enzymatic monodeiodination. There are three types of monodeiodinase enzymes responsible for the synthesis of T3, reverse T3, and diiodothyronine (T2) (Figure 1) [4].
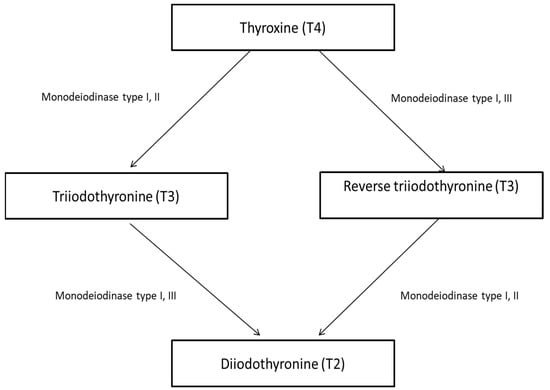
Figure 1.
Thyroid deiodinases.
Monodeiodinase type I predominantly expressed in the liver, kidney, and thyroid is responsible for phenolic or outer ring deiodination resulting in the production of active T3. Monodeiodinase type II located in the brain, pituitary, placenta, skeletal muscle, heart, thyroid, and brown adipose tissue also stimulates active T3 production. Monodeiodinase type III is responsible for the production of inactive reverse T3 by deiodination of the inner tyrosyl ring.
During fetal embryogenesis, there is a predominance of type 3 monodeiodinase activity, low type 1 monodeiodinase activity, and local activation of type 2 monodeiodinase activity in the brain and some other tissues to provide adequate local T3. This prevents brain developmental abnormalities and maintains an anabolic state in the fetus. During fetal embryogenesis, levels of rT3 are high due to the predominance of monodeiodinase type 3 activity [5,6,7].
Normally T3 and T4 are bound to thyroid-binding globulin (TBG) in the blood. TBG is decreased in liver failure due to loss of synthetic function. Increased estrogen levels during pregnancy and oral contraceptive pills (OCPs) use stimulate TBG synthesis, thereby increased T3 and T4 levels are not consistent with a thyrotoxic state as levels of free T3 and T4, the bioavailable hormones, are in the normal range [1,5,8].
Thyroid hormone enters neuronal cells via iodothyronine membrane transporters, a clinically significant one is the monocarboxylate transporter 8 (MCT8) expressed in the brain [9,10].
Intracellularly, thyroid hormone binds to nuclear thyroid receptors, TRα and TRβ. These receptors regulate DNA transcription downstream [5,11].
1.2. Thyroid Embryology
Thyroid embryogenesis starts with the expression of thyroid transcription factor-1 (TTF1) encoded by NK2 homeobox-1 (NKX2-1), thyroid transcription factor-2 (TTF2) encoded by Forkhead Box Protein-E1 (FOXE1), and Paired Box Gene 8 (PAX8) on embryonic thyroid stem cells [5].
The human embryonic forebrain and hypothalamus begin to differentiate by 3 weeks of gestation under the influence of homeodomain proteins or transcription factors. At 8–9 weeks of gestation, the hypothalamus, fetal gut, and pancreas begin to secrete TRH, which stimulates thyroid gland development.
The thyroid diverticulum consisting of spherical follicles made from follicular and parafollicular cells, arises from the ectodermal floor of the primitive pharynx (Rathke’s pouch) or the primitive forebrain. Follicular cells are derived from a median endodermal mass in the foramen cecum at the base of the tongue while parafollicular cells that produce calcitonin are derived from the 4th pharyngeal pouch [1,2,5]. By 50 days of gestation, both structures fuse and the thyroid gland descends from the foramen cecum (base of the tongue) into the anterior neck.
The thyroid will remain connected by the thyroglossal duct to the base of the tongue until full thyroid maturation. The thyroglossal duct usually disappears by the end of thyroid gland formation.
2. Laboratory
Thyroid function can be assessed by measuring T4 and T3 levels. These measurements can be affected by abnormal levels of thyroxine-binding protein, prealbumin, albumin, transthyretin, and all carrier proteins. The ratio of total to the free hormone is in the range of 1000 to 1. TSH levels are useful for the complete evaluation of the axis and to determine thyroid function. A high TSH level is detected when thyroid gland function is impaired and does not produce enough FT4, while normal or low TSH is usually associated with central hypothyroidism or hyperfunctioning thyroid. All thyroid assays should be evaluated by age-appropriate reference values [5,12,13].
Newborn screening (Figure 2) is recommended for every newborn regardless of symptoms at 48–72 h of life and includes detection of TSH and/or T4 levels in dried blood spots. Lab results in newborn screen could be affected by prematurity, low birth weight, illness, and multiple births, e.g., after twin-twin transfusion (which can obscure hypothyroidism in the recipient fetus). Many newborn screening tests are done soon after 24 h, requiring adjustment of the expected range and possibly introducing errors due to the dynamic of TSH recruitment after delivery [14,15]. Although very rare, the clinical presentation of congenital hypothyroidism may include prolonged sleep, protruding abdomen and umbilicus, puffy face and pale skin, macroglossia with feeding difficulties, hypotonia and delayed reflexes, delayed meconium elimination, prolonged jaundice due to conjugated hyperbilirubinemia, absence of femoral epiphysis on knee X-ray due to delayed skeletal maturation. Late manifestations of untreated congenital hypothyroidism include soft-tissue myxedema, delayed epiphyseal maturation, and developmental retardation.
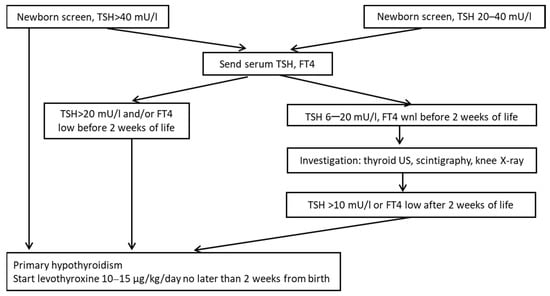
Figure 2.
Algorithm for congenital hypothyroidism screening and workup (adapted from European Society for. Pediatric Endocrinology. Consensus Guidelines on Screening, Diagnosis, and Management of Congenital Hypothyroidism 2014).
3. Imaging
Scintigraphy and ultrasound are the main diagnostic imaging tools for the evaluation of the thyroid gland. Ultrasonography is generally the first choice for diagnosing the presence or absence of a thyroid gland, size, echogenic texture, and structure, and for intrathyroid nodules as small as 2–3 mm in size. The addition of color and spectral Doppler imaging helps to determine the blood flow in the thyroid and screen for malignancy [16].
Radionuclide scintigraphy using 99 mTechnetium pertechnetate or 123-Iodine is used in the evaluation of athyreosis (absence of any uptake), hypoplasia of a gland in situ (with or without hemithyroid), a normal or large gland in situ, focal thyroid nodule on the basis of relative uptake of radioactive isotope by the nodule. 123-Iodine gives a clearer scan than 99 m Tc [17,18].
4. Congenital Disorders
4.1. Congenital Hypothyroidism
The most common cause of congenital hypothyroidism (CH) is thyroid dysgenesis which accounts for 85% of primary CH [13]. Ectopic thyroid accounts for 1:5000 cases and thyroid agenesis for 1:15,000 cases [5]. The most common location for ectopic thyroid is sublingual due to migration deficit. The thyroid gland is normally differentiated but is small in size. A rare passage of TSH receptor blocking antibodies from a mother who has an uncommon form of autoimmune hypothyroidism due to receptor blockade rather than cytotoxic destruction of thyroid cells results in neonatal hypothyroidism that resolves after the maternal antibodies are metabolized [19,20,21,22]. On occasion, treatment is needed until the blocking antibodies disappear.
Thyroid dysgenesis (abnormal thyroid gland development) is usually sporadic but may present as a familial disorder. Genetic evaluation of a child with CH is not routinely recommended but should be discussed case by case. Families with a history of CH, in a pattern consistent with autosomal recessive inheritance, should be considered for a genetic consultation for possible recurrence and other associated disorders [5,23,24,25].
Thyroid agenesis (complete absence of thyroid tissue) due to GLIS family zinc finger protein 3 (GLIS3) mutation includes also congenital glaucoma, deafness, liver, kidney, pancreas abnormalities [5,26] (Table 1). Thyroid ectopy (interrupted thyroid descent from the thyroid anlage to its final location in front of the trachea) has no known genetic predisposition. Inactivation of homeobox NKX2-1 results in thyroid aplasia [5,27]. PAX8 gene inactivation results in thyroid hypoplasia with a lack of thyroid follicular cells and the presence of C-cells [28,29,30].

Table 1.
Genetic mutations causing congenital hypothyroidism [23,31,32].
Thyroid dyshormonogenesis (a disorder of thyroid hormone synthesis) is diagnosed in 1:30,000 cases and is associated with defects of thyroperoxidase (TPO), which are the most common genetic mutations [5,23]. The first step of hormonal synthesis is iodine transport into the cells, which could be impaired due to the mutation of sodium/iodide symporter (SLC5A5) [33]. After diffusion of iodine to the apex of the cells, iodine leaves the thyroid cell through the chloride/iodide pump (Pendrin) which is responsible for transporting iodine out of the cell and into the follicular colloid. Mutations in the Pendrin gene lead to defects in the transport of iodine to colloid [34]. The same chloride/iodide transport occurs in the cochlea and the above mutation can lead to sensorineural hearing loss (Pendred syndrome). Iodine in the follicular colloid is oxidized by hydrogen peroxide and binds to tyrosine residues in TG to form iodotyrosine. Multiple mutations of the TPO gene located on chromosome 2 have been discovered including homozygous and heterozygous missense mutations, frame-shift mutations, single-nucleotide substitutions, base pair duplications, all of which lead to defects in oxidation [5,23,35].
H2O2 generation deficiency leads to a decrease in the substrate for iodine oxidation and further defect in iodotyrosine formation [36,37,38]. Thyroid dyshormonogenesis disorders may present as early as the neonatal period or later, up to 15 years of age. Treatment in general for all subtypes requires thyroxine supplementation.
Thyroid hormone resistance occurs in 1:40,000 cases and is usually due to autosomal dominant mutation of thyroid receptors with 15–20% developed sporadically [5]. THRA and THRB gene mutations include single-amino-acid deletions, substitutions, in-frame deletions, and frame-shift insertions [39,40,41]. Treatment of thyroid hormone resistance includes high dose thyroxine or thyroid hormone analog TRIAC, and triiodothyroacetic acid (tiratricol). TRIAC has effects similar to thyroid hormone, but acts more on bone turnover, resulting in higher sex hormone-binding globulin and lower serum cholesterol levels [39,40].
Another mechanism of thyroid resistance could be associated with MCT8 deficiency [42,43,44]. Thyroid hormone analog diiodothyropropionic acid (DITPA) is used for the treatment as it does not require MCT8 to enter cells. DITPA is a cardio-selective thyroid hormone analog, which acts to suppress TSH, and reduce T3 levels with further reverse of the hypermetabolism and weight loss [42].
Consumptive hypothyroidism is considered one of the thyroid resistance disorders due to the need of high dose thyroxine treatment. The condition develops early after birth once congenital hemangiomas, which expresses high levels of monodeiodinase type 3, begins to develop [45]. The condition usually resolves either after spontaneous involution or surgical treatment of the hemangioma [5,45].
Central hypothyroidism is rare and occurs in 1:21,000 infants due to a lack of TRH or TSH [14,31,35]. There are several case reports of hypothalamic hypothyroidism [46,47] caused by Isolated TRH deficiency due to TRH gene mutation located on 3q22 [48]. Mutations in the TRH receptor gene (TRHR) located on chromosome 8 can cause central hypothyroidism due to complete resistance to the thyrotropin-releasing hormone [49]. Children usually present with short stature and delayed bone maturation [50].
Isolated TSH deficiency was described in several families with undetectable TSH and no response to TRH stimulation. There are two genes known for being responsible for pituitary cell development and differentiation. PROP paired-like homeobox 1(PROP1) and POU class 1 homeobox 1 (POU1F1) mutations, which are terminal factors in the differentiation cascade of pituitary cells, can cause TSH deficiency and age-dependent pituitary hypoplasia [5,51]. PROP1 mutation leads to panhypopituitarism with growth hormone, TSH, luteinizing hormone, follicular stimulating hormone, prolactin, and adrenocorticotropin deficiencies. POU1F1 mutation leads to growth hormone, TSH, and prolactin deficiencies [52].
Mutations causing TSH receptor inactivation lead to the absence of follicular architecture, while normal differentiated thyroid cells exist [53].
Thyroid-binding globulin deficiency is an X-linked state with an incidence of 1 in 4000 males and usually presents with normal TSH and FT4 and low T4 levels [11]. This euthyroid state is confirmed by a low TBG level or normal Free T4. Treatment is not indicated.
Treatment
Weight-based dosing of levothyroxine 10–15 μg/kg/day is recommended for infants [51,54]. Assuming the infant weighs 3–4 kg, 50 mcg is a good starting dose. This dose normalizes the TSH and FT4 levels and also has the best neurodevelopmental outcome at age of 5. The level of FT4 should be slightly elevated in the mid to upper half of the normal range optimally between 0.5 and 2.0 ng/dL [54].
Levothyroxine should be given by crushing tablets and mixing with water or breast milk at the same time of the day. Repeat thyroid function should be obtained every 2 weeks during the first month after treatment initiation, followed by every 1–2 months during the first 6 months of life, every 3–4 months between 6 months and 3 years, every 6–12 months until growth completed [14]. Levothyroxine is recommended for continuous use for 3 years during the critical neurodevelopment period of the child’s life.
A trial off of levothyroxine can be considered after age 3 years if the patient had been euthyroid on the same dose of medication.
4.2. Congenital Hyperthyroidism
Neonatal Graves’ disease is a consequence of maternal Graves’ disease. It is caused by maternal TSH activating antibodies also known as TSI (thyroid-stimulating immunoglobulin) crossing into fetal circulation late in pregnancy and fetal hyperthyroidism manifests in the second part of gestation up until 6 months of age when serum IgG levels clear from neonatal circulation [55,56]. Treating the pregnant mother with propylthiouracil (PTU) should be started while monitoring for normalization of the fetal HR. PTU is recommended during the first trimester of pregnancy due to the possible teratogenic effect of methimazole (MMI).
Neonatal Graves’ is treated with a β-blocker (propranolol 1–2 mg/kg/day divided into four doses) for symptomatic relief and MMI (0.5–1 mg/kg/day divided into three doses) is the antithyroid medication of choice over PTU given PTU’s higher risk for hepatotoxicity [55,57,58].
Activating mutations of the TSH receptor, solitary toxic adenoma, or multinodular goiter are causes of autonomous hyperfunctioning thyroid. These TSH receptor mutations are usually not seen until childhood, adolescence, or adult life. Usually, the treatment of nonautoimmune hyperthyroidism is radioactive ablation or surgical thyroidectomy [12,59].
5. Acquired Disorders
5.1. Primary Hypothyroidism
Hashimoto’s disease/hypothyroidism is the most common type of acquired hypothyroidism [60]. It is an autoimmune disease associated with HLA-DR5. The course of the disease can start with transient thyrotoxicosis (Hashitoxicosis) as a result of follicular rupture and the release of premade hormones into the bloodstream. The thyroid could be enlarged, but nontender. Later during the hypothyroid state, children may present with learning problems, cold intolerance, bradycardia, facial puffiness, constipation, short stature with delayed bone age. Children may also present with precocious puberty due to elevated TSH levels which act as FSH receptor stimulator. The presence of TPO antibodies and TG antibodies as markers of autoimmunity supports the diagnosis of Hashimoto thyroiditis.
Children with Down syndrome and Turner syndrome should also be screened annually for Hashimoto thyroiditis more frequently than in the general population [61,62]. Treatment should be started once thyroid function shows low FT4 levels and TSH levels of more than 10 μU/mL. Current data do not support treatment when TSH < 10 μU/mL since there are no clinical or chemical benefits observed [54]. Levothyroxine is usually started with 1–2 μg/kg/day and titrated to achieve a euthyroid state in adults. In children the age-based dosing guideline is as follows: 1–3 years 4–6 μg/kg/d, for patients 3–10 years 3–5 μg/kg/d, and 10–16 years 2–4 μg/kg/d. Levothyroxine may also be dosed based on body surface area calculated at 100 μg/m2/d. Children with severe TSH elevation or FT4 suppression should be started on a third to half of the usual dose and monitored closely to prevent pseudotumor cerebri [63]. Patients should also be educated that excessive soy intake, iron supplements, and excessive fiber can interfere with the absorption of levothyroxine [14,51].
5.2. Hyperthyroidism
Graves’ Disease
Graves’ disease is an autoimmune condition leading to hyperthyroidism and is the most common cause of hyperthyroidism among children. The disease occurs approximately in 1 per 5000 children, more frequently in females than in males. It is a result of TSH receptor stimulation by autoantibodies (TSI) which leads to the overproduction of thyroid hormones by follicular cells. T lymphocytes release cytokines which lead to local inflammation and production of autoantibodies via dysregulated B-cells. The level of the TSI antibodies correlates with the level of FT4.
TSI uses Fischer rat thyroid follicular cell line FRTL 5 immortalized cell release of cAMP while thyroid-stimulating hormone receptor antibodies (TRab) are measured by competitive binding assay. TPO and TG antibodies may be useful for the confirmation of an autoimmune disease but are not diagnostic for Graves’.
Patients present with nonspecific signs early such as irritability, changes in concentration, learning problems, insomnia, excessive perspiration, weight loss, and diarrhea. Once the disease progresses children have lower performance in school, excessive nervousness, tremor, tachypnea, and neck enlargement. Children sometimes can complain of vision changes, palpitations, and swallowing discomfort [15,64].
The thyroid gland is symmetrically enlarged and rubbery to palpation, usually nontender, while bilateral eye proptosis is less common in children than in adults. Signs are positive for tachycardia, hypertension with widened pulse pressure, possible palpable thrill, and systolic ejection murmur due to functional mitral valve insufficiency, weight loss, and a growth spurt. Other findings include excessive nervousness on the exam and a positive straight arm test, which confirms postural tremor.
High TSI, presence of TG and TPO antibodies, low or normal TSH, 2–10 times elevated total T3 and free T4 levels are indicative of Graves’. Occasionally T3 only thyrotoxicosis without free T4 elevations is observed. Low potassium levels can be seen due to thyrotoxic hypokalemic paralysis. Thyroid ultrasound findings consist of enlarged size, hypervascularization, and normal or low echogenicity. Sinus tachycardia is seen, but in severe presentation, signs of hypokalemia, prolonged QT, and atrial fibrillation could be observed.
In case of accelerated growth, bone age is done to assess growth and height prediction. Advanced bone age and osteopenia in severe cases could be appreciated on X-ray [12,15,64].
Rarely do patients present with thyroid storm, which consists of fever, agitation, delirium, psychosis stupor or coma, tachycardia, cardiac arrhythmias, congestive cardiac failure, vomiting, diarrhea, and hepatic failure. It is characterized by multisystem decompensation with a mortality rate of 8–25%. Burch–Wartofsky Point Scale (BWPS) is usually used for diagnosis of thyroid storm with ≥45 points consistent with diagnosis [8]. In thyrotoxic hypokalemic paralysis, patients suddenly have lower extremity weakness and difficulty walking due to excessive Na+/K+-ATPase activity from high thyroxine levels [65,66].
First, correction of potentially life-threatening conditions such as cardiac arrhythmias, symptomatic hypertension, and hypokalemia are needed. Cardioselective β-blockers are the first medications for controlling the rhythm, elevated blood pressure, and widened pulse pressure. Propranolol or atenolol is started with age-appropriate dose (2 mg/kg/day) and kept until stabilized on antithyroid medications. Second and third line antihypertensives include ACE-inhibitors, or Ca-channel blockers when β-blockers are contraindicated (asthma).
Antithyroid drugs are started at the time of the diagnosis and should be continued for at least 2–3 years with longer treatment suggested in children than adults. Of historical significance, PTU is the most active drug which inhibits both TPO iodination of the tyrosine residues of TG and blocks the conversion of thyroxine to triiodothyronine by inhibition of 5′deiodininase. MMI inhibits TPO only, and has a more favorable side effect profile since it is not associated with drug-associated hepatitis or the production of cytoplasmic antineutrophil antibodies [67]. Both PTU and MMI can cause agranulocytosis. Patients should have their baseline absolute neutrophil counts (ANC) and regular ANC counts checked with they have signs and symptoms such as fever, and acute infection. PTU is currently not recommended for pediatric use due to severe liver toxicity. Side effects include rash, jaundice, arthralgia, gastrointestinal problems and are dose-dependent.
The dose of MMI starts at 0.5–1 mg/kg/day up to 40 mg daily in divided doses and varies with the clinical and chemical response. Usually, after 2–3 weeks of initial treatment dose, the biochemical response (total T3, freeT4) should be repeated with the dose decreased by 30–50% when euthyroid function is achieved. Clinical response (normalization of vital signs, weight gain, growth velocity, and musculoskeletal activity) is usually achieved after 3 months of continuous daily treatment. Further regular reevaluation of thyroid function is recommended every 3–6 weeks to avoid hyper- and hypothyroidism [55,68].
Definitive therapy with total thyroidectomy or radioiodine ablation is recommended if antithyroid medication is unable to achieve a euthyroid state (either due to poor compliance or relapse after completing full course of antithyroid treatment) or there is an absolute contraindication for medical therapy due to toxicity. Post-ablative or post-surgical hypothyroidism is treated with levothyroxine [15,57].
Surgery is usually recommended for children younger than 5 years old and for those with poor response to radioactive iodine therapy when the thyroid gland is more than 80 g. Total or near-total thyroidectomy has better outcomes compared with subtotal thyroidectomy which is associated with higher risks of relapse. Patients should be monitored for acute hypocalcemia after the surgery which could be a complication of either intentional or inadvertent parathyroid gland(s) removal or interruption of the blood flow to the glands. Hypocalcemia is the most frequent complication and occurs in 40% of thyroidectomies in children, but this risk can be reduced by consulting a high-volume thyroid surgeon [57,69].
Radioactive iodine therapy is considered in children who do not respond to medication or surgery. Use of 131I is recommended only in children > 5 years old due to higher risks of thyroid cancer. The maximum irradiation dose is <10 mCi in children < 10 years old. The dose of 131I is calculated per gland size in grams, which is estimated with imaging. A dose of 150 uCi per gram is considered when the thyroid gland is less than 30 g and 200 uCi with a higher weight up to 80 g. Higher irradiation doses lead to hypothyroidism in 95% after 3 months of the procedure with 5% retreatment needed if hyperthyroidism persists after 6 months. When gland size is more than 80 g, radioactive iodine is usually less effective and surgery should be considered as a preferred option. Low dose radiation in children younger than 20 years is associated with increased rates of thyroid neoplasms compared with high dose radiation >150 uCi of 131I per gram of thyroid tissue [70,71].
6. Goiter
Toxic multinodular goiter is the second most common cause of hyperthyroidism and is associated with a mutation in the TSH receptor which leads to hyperfunctioning of follicular cells. TSH receptor acts independent of TSH stimulation. Multinodular goiter could also be a part of McCune-Albright syndrome due to activation of the G protein receptor stimulatory alpha subunit that activates adenylyl cyclase. Hyperplasia of the thyroid precedes the formation of nodules [72].
7. Thyroid Nodules
The prevalence of thyroid nodules among children (11–13%) is much less than in adults. Every thyroid nodule should be assessed for thyroid cancer especially in children and adolescents [59].
There are high-risk groups for thyroid nodules and/or thyroid cancer development (Table 2) [59,73]. It is recommended that they be screened annually with a thyroid exam (nodules and cervical lymphadenopathy) and sonogram if there are concerns on the exam.
Table 2.
Disorders associated with thyroid nodule formation.
Nodule evaluation (Figure 3) includes thyroid function assessment with TSH, FT4, total T3 levels, and calcitonin levels for possible medullary thyroid cancer. Ultrasonography features that are suspicious include: the presence of microcalcifications, irregular margins, increased intranodular blood flow and hypoechogenicity, and presence of lymph nodes. In children, the size of the nodule is less likely to be indicative of malignancy as in adults who use a cutoff value of 1 cm, but the ultrasound characteristics are just as important. The most important and necessary diagnostic tool is ultrasound-guided fine needle aspiration (FNA) of the nodule which is recommended to be done independently of thyroid nodule size among children [59].
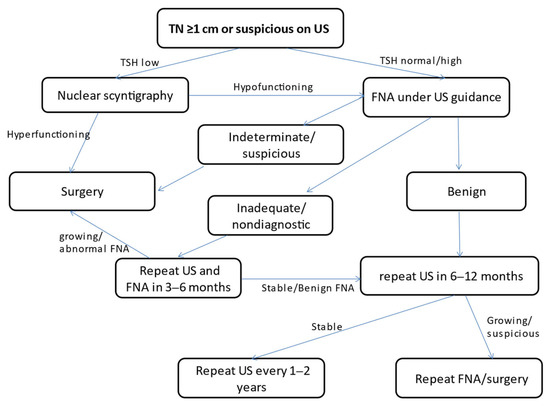
Figure 3.
Pathogenesis, diagnosis, and management of thyroid nodules in children (adapted from Management Guidelines for Children with Thyroid Nodules and Differentiated Thyroid Cancer [74]).
FNA cytopathology findings are interpreted according to The Bethesda six-tier system for reporting thyroid cytopathology, which is used for both children and adults according to American Thyroid Association (ATA):
- Nondiagnostic or unsatisfactory (specimen with limited cellularity: fewer than six follicular cell groups each containing 10–15 cells per group from at least two separate aspirates; absence of follicular cells or poor fixation and preservation
- Benign
- atypia of undetermined significance (AUS) or follicular lesion of undetermined significance (FLUS)
- follicular/Hurthle neoplasm or suspicious for a follicular/Hurthle neoplasm
- suspicious for malignancy
- malignant [59]
According to the multicentral study by Canberk et al. [75], the distribution of cytopathology findings among 405 FNA specimens were: 44 (11%) for nondiagnostic, 204 (50%) for benign category, 40 (10%) for AUS/FLUS, 36 (9%) for follicular neoplasm/suspicious for a follicular neoplasm, 24 (6%) for suspicious for malignancy and 57 (14%) for malignancy categories. The actual risk of malignancy in nodules surgically excised among adults is 20% for nondiagnostic, 2.5% for benign category, 14% for AUS/FLUS, 25% for follicular neoplasm/suspicious for a follicular neoplasm, 70% for suspicious for malignancy and 99% for malignancy categories [76].
Thyroid Cancer
Differentiated thyroid cancer (DTC) usually presents at late stage with complications in children. Follicular thyroid cancers (FTC) and papillary thyroid cancers (PTC) are rare, and occur in 0.54 cases per 100,000 persons [77,78]. At diagnosis, 40–60% with PTC already have lymph node metastases in the neck at diagnosis, however, the prognosis in children is usually excellent due to its slow-growing nature and its response to radiation treatment.
There are several genetic alterations (Table 3) leading to activating of the mitogen-activated protein kinase (MAPK) pathway, which is responsible for cell division. MAPK pathway includes activating ret proto-oncogene/neurotrophic receptor tyrosine kinase 1 (RET/NTRK1 tyrosine kinases), activating mutations of RAS type family, BRAF, and MET proto-oncogenes.
Table 3.
Gene mutations and associated thyroid cancer variants (adapted from Management Guidelines for Children with Thyroid Nodules and Differentiated Thyroid Cancer).
PTCs are responsible for 90% of childhood thyroid cancers and present as a solitary nodule. Children with newly diagnosed PTC should undergo a sonogram of the thyroid and cervical lymph nodes prior to total thyroidectomy (with or without neck dissection depending on staging and preoperative imaging). Further imaging should also be considered to assess for distant metastasis, usually to the lungs or bones. A high volume-neck surgeon decreases the risk for post-operative complications [59].
FTCs are responsible for 5–10% of childhood cancers. FTC also can spread locally to lymph nodes in the neck but are more likely to spread to distant organs compared to PTC.
Medullary thyroid carcinoma (MTC) arises from calcitonin-secreting cells, C cells, or parafollicular cells. About 30% of MTC are part of the genetic syndrome multiple endocrine neoplasia (MEN) type 2A (MEN2A) and 2B (MEN2B), which is a consequence of a missense gain-of function autosomal-dominant mutation of RET oncogene on chromosome 10 (Table 4 and Table 5) [59,79,80].
Table 4.
MEN2A and MEN2B clinical features and common mutations (adapted from Update on Multiple Endocrine Neoplasia Type 2: Focus on Medullary Thyroid Carcinoma; Revised American Thyroid Association Guidelines for the Management of Medullary Thyroid Carcinoma).
Table 5.
RET oncogene mutation and risk categorization.
Familial MTC is diagnosed when two or more family members have MTC and usually occurs in adults without MEN2A or 2B features, and is inherited in an autosomal dominant fashion. Genetic testing for RET mutation is recommended.
Diagnosis of MTC is confirmed by elevated calcitonin level, which is at least doubled from the upper limit of normal, and further genetic testing should be done. Patients diagnosed with hereditary MTC are screened for pheochromocytoma, hyperparathyroidism, and for other manifestations. Patients are then stratified by the risk of thyroid malignancy [79].
Postoperative monitoring of calcitonin for recurrent or persistent disease is recommended every 3 months during the first year after the surgery with longer intervals 6–12 months later if undetectable [79]. The management of thyroid carcinomas is beyond the scope of this review.
8. Conclusions
In this review, we discussed the approach to the diagnosis and management of thyroid diseases in children. We emphasized the importance of genetics in understanding the causes of most congenital and acquired disorders, such as congenital hypothyroidism, thyroid nodules, and thyroid cancers. Further investigation is needed for possible prevention and early goal-directed therapy.
Author Contributions
Conceptualization and original draft preparation and editing, I.G.; review and editing, I.G., V.L.C. and L.F.; supervision, V.L.C. and L.F.; All authors have read and agreed to the published version of the manuscript.
Funding
This review received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Armstrong, M.; Asuka, E.; Fingeret, A. Physiology, Thyroid Function. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2021. [Google Scholar]
- Pirahanchi, Y.; Tariq, M.A.; Jialal, I. Physiology, Thyroid. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2021. [Google Scholar]
- Mariotti, S.; Beck-Peccoz, P. Physiology of the Hypothalamic-Pituitary-Thyroid Axis. In Endotext; Feingold, K.R., Anawalt, B., Boyce, A., Chrousos, G., de Herder, W.W., Dhatariya, K., Dungan, K., Grossman, A., Hershman, J.M., Hofland, J., et al., Eds.; MDText.com, Inc.: South Dartmouth, MA, USA, 2000. [Google Scholar]
- Hernandez, A.; St Germain, D.L. Thyroid hormone deiodinases: Physiology and clinical disorders. Curr. Opin. Pediatr. 2003, 15, 416–420. [Google Scholar] [CrossRef] [PubMed]
- Sperling, M.A.V.G.V.; Deladoey, J. Pediatric Endocrinology. Disorders of the Thyroid in the Newborn and Infant; Elsevier: Philadelphia, PA, USA, 2014. [Google Scholar]
- Eng, L.; Lam, L. Thyroid Function During the Fetal and Neonatal Periods. Neoreviews 2020, 21, e30–e36. [Google Scholar] [CrossRef] [PubMed]
- Sterrett, M. Maternal and Fetal Thyroid Physiology. Clin. Obstet. Gynecol. 2019, 62, 302–307. [Google Scholar] [CrossRef] [PubMed]
- Burrow, G.N.; Fisher, D.A.; Larsen, P.R. Maternal and fetal thyroid function. N. Engl. J. Med. 1994, 331, 1072–1078. [Google Scholar] [CrossRef]
- Bernal, J. Action of thyroid hormone in brain. J. Endocrinol. Investig. 2002, 25, 268–288. [Google Scholar] [CrossRef]
- Zoeller, R.T.; Rovet, J. Timing of thyroid hormone action in the developing brain: Clinical observations and experimental findings. J. Neuroendocrinol. 2004, 16, 809–818. [Google Scholar] [CrossRef]
- Braun, D.; Schweizer, U. Thyroid Hormone Transport and Transporters. Vitam. Horm. 2018, 106, 19–44. [Google Scholar] [CrossRef]
- Ross, D.S.; Burch, H.B.; Cooper, D.S.; Greenlee, M.C.; Laurberg, P.; Maia, A.L.; Rivkees, S.A.; Samuels, M.; Sosa, J.A.; Stan, M.N.; et al. 2016 American Thyroid Association Guidelines for Diagnosis and Management of Hyperthyroidism and Other Causes of Thyrotoxicosis. Thyroid 2016, 26, 1343–1421. [Google Scholar] [CrossRef]
- Weiner, A.; Oberfield, S.; Vuguin, P. The Laboratory Features of Congenital Hypothyroidism and Approach to Therapy. Neoreviews 2020, 21, e37–e44. [Google Scholar] [CrossRef]
- American Academy of Pediatrics; Susan, R.; Rose, M.D.; The Section on Endocrinology and Committee on Genetics; American Thyroid Association; Rosalind, S.; Brown, M.D.; The Public Health Committee; Lawson Wilkins Pediatric Endocrine Society. Update of newborn screening and therapy for congenital hypothyroidism. Pediatrics 2006, 117, 2290–2303. [Google Scholar] [CrossRef]
- Kahaly, G.J.; Bartalena, L.; Hegedus, L.; Leenhardt, L.; Poppe, K.; Pearce, S.H. 2018 European Thyroid Association Guideline for the Management of Graves’ Hyperthyroidism. Eur. Thyroid J. 2018, 7, 167–186. [Google Scholar] [CrossRef] [PubMed]
- Clerc, J. Imaging the thyroid in children. Best Pract. Res. Clin. Endocrinol. Metab. 2014, 28, 203–220. [Google Scholar] [CrossRef] [PubMed]
- Chun, S.; Lee, Y.S.; Yu, J. Thyroid imaging study in children with suspected thyroid dysgenesis. Ann. Pediatr. Endocrinol. Metab. 2021, 26, 53–59. [Google Scholar] [CrossRef] [PubMed]
- Gandhi, A.; Wong, K.K.; Gross, M.D.; Avram, A.M. Lingual Thyroid Ectopia: Diagnostic SPECT/CT Imaging and Radioactive Iodine Treatment. Thyroid 2016, 26, 573–579. [Google Scholar] [CrossRef]
- Ozon, A.; Tekin, N.; Siklar, Z.; Gulcan, H.; Kara, C.; Tastekin, A.; Demir, K.; Koc, E.; Evliyaoglu, O.; Kurtoglu, S. Neonatal effects of thyroid diseases in pregnancy and approach to the infant with increased TSH: Turkish Neonatal and Pediatric Endocrinology and Diabetes Societies consensus report. Turk. Arch. Pediatrics/Türk Pediatri Arşivi 2018, 53, S209–S223. [Google Scholar] [CrossRef] [PubMed]
- Van Trotsenburg, A.S.P. Management of neonates born to mothers with thyroid dysfunction, and points for attention during pregnancy. Best Pract. Res. Clin. Endocrinol. Metab. 2020, 34, 101437. [Google Scholar] [CrossRef] [PubMed]
- Matsuura, N.; Yamada, Y.; Nohara, Y.; Konishi, J.; Kasagi, K.; Endo, K.; Kojima, H.; Wataya, K. Familial neonatal transient hypothyroidism due to maternal TSH-binding inhibitor immunoglobulins. N. Engl. J. Med. 1980, 303, 738–741. [Google Scholar] [CrossRef]
- Brown, R.S.; Bellisario, R.L.; Mitchell, E.; Keating, P.; Botero, D. Detection of thyrotropin binding inhibitory activity in neonatal blood spots. J. Clin. Endocrinol. Metab. 1993, 77, 1005–1008. [Google Scholar] [CrossRef][Green Version]
- Grasberger, H.; Refetoff, S. Genetic causes of congenital hypothyroidism due to dyshormonogenesis. Curr. Opin. Pediatr. 2011, 23, 421–428. [Google Scholar] [CrossRef]
- Zdraveska, N.; Kocova, M.; Nicholas, A.K.; Anastasovska, V.; Schoenmakers, N. Genetics of Gland-in-situ or Hypoplastic Congenital Hypothyroidism in Macedonia. Front. Endocrinol. 2020, 11, 413. [Google Scholar] [CrossRef]
- Castanet, M.; Polak, M.; Bonaiti-Pellie, C.; Lyonnet, S.; Czernichow, P.; Leger, J.; Afdphe. Nineteen years of national screening for congenital hypothyroidism: Familial cases with thyroid dysgenesis suggest the involvement of genetic factors. J. Clin. Endocrinol. Metab. 2001, 86, 2009–2014. [Google Scholar] [CrossRef]
- Senee, V.; Chelala, C.; Duchatelet, S.; Feng, D.; Blanc, H.; Cossec, J.C.; Charon, C.; Nicolino, M.; Boileau, P.; Cavener, D.R.; et al. Mutations in GLIS3 are responsible for a rare syndrome with neonatal diabetes mellitus and congenital hypothyroidism. Nat. Genet. 2006, 38, 682–687. [Google Scholar] [CrossRef] [PubMed]
- Dentice, M.; Cordeddu, V.; Rosica, A.; Ferrara, A.M.; Santarpia, L.; Salvatore, D.; Chiovato, L.; Perri, A.; Moschini, L.; Fazzini, C.; et al. Missense mutation in the transcription factor NKX2-5: A novel molecular event in the pathogenesis of thyroid dysgenesis. J. Clin. Endocrinol. Metab. 2006, 91, 1428–1433. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, A.; Hermanns, P.; Rodrigues, A.L.; Sousa, I.; Anselmo, J.; Bikker, H.; Cabral, R.; Pereira-Duarte, C.; Mota-Vieira, L.; Pohlenz, J. A new PAX8 mutation causing congenital hypothyroidism in three generations of a family is associated with abnormalities in the urogenital tract. Thyroid 2013, 23, 1074–1078. [Google Scholar] [CrossRef] [PubMed]
- Iwahashi-Odano, M.; Fujisawa, Y.; Ogata, T.; Nakashima, S.; Muramatsu, M.; Narumi, S. Identification and functional characterization of a novel PAX8 mutation (p.His39Pro) causing familial thyroid hypoplasia. Clin. Pediatr. Endocrinol. 2020, 29, 173–178. [Google Scholar] [CrossRef] [PubMed]
- Vilain, C.; Rydlewski, C.; Duprez, L.; Heinrichs, C.; Abramowicz, M.; Malvaux, P.; Renneboog, B.; Parma, J.; Costagliola, S.; Vassart, G. Autosomal dominant transmission of congenital thyroid hypoplasia due to loss-of-function mutation of PAX8. J. Clin. Endocrinol. Metab. 2001, 86, 234–238. [Google Scholar] [CrossRef]
- Cherella, C.E.; Wassner, A.J. Update on congenital hypothyroidism. Curr. Opin. Endocrinol. Diabetes Obes. 2020, 27, 63–69. [Google Scholar] [CrossRef]
- Vono-Toniolo, J.; Rivolta, C.M.; Targovnik, H.M.; Medeiros-Neto, G.; Kopp, P. Naturally occurring mutations in the thyroglobulin gene. Thyroid 2005, 15, 1021–1033. [Google Scholar] [CrossRef]
- Pohlenz, J.; Rosenthal, I.M.; Weiss, R.E.; Jhiang, S.M.; Burant, C.; Refetoff, S. Congenital hypothyroidism due to mutations in the sodium/iodide symporter. Identification of a nonsense mutation producing a downstream cryptic 3′ splice site. J. Clin. Investig. 1998, 101, 1028–1035. [Google Scholar] [CrossRef]
- Ladsous, M.; Vlaeminck-Guillem, V.; Dumur, V.; Vincent, C.; Dubrulle, F.; Dhaenens, C.M.; Wémeau, J.L. Analysis of the thyroid phenotype in 42 patients with Pendred syndrome and nonsyndromic enlargement of the vestibular aqueduct. Thyroid 2014, 24, 639–648. [Google Scholar] [CrossRef]
- Rastogi, M.V.; LaFranchi, S.H. Congenital hypothyroidism. Orphanet J. Rare Dis. 2010, 5, 17. [Google Scholar] [CrossRef] [PubMed]
- Muzza, M.; Rabbiosi, S.; Vigone, M.C.; Zamproni, I.; Cirello, V.; Maffini, M.A.; Maruca, K.; Schoenmakers, N.; Beccaria, L.; Gallo, F.; et al. The clinical and molecular characterization of patients with dyshormonogenic congenital hypothyroidism reveals specific diagnostic clues for DUOX2 defects. J. Clin. Endocrinol. Metab. 2014, 99, E544–E553. [Google Scholar] [CrossRef] [PubMed]
- Vigone, M.C.; Fugazzola, L.; Zamproni, I.; Passoni, A.; Di Candia, S.; Chiumello, G.; Persani, L.; Weber, G. Persistent mild hypothyroidism associated with novel sequence variants of the DUOX2 gene in two siblings. Hum. Mutat. 2005, 26, 395. [Google Scholar] [CrossRef] [PubMed]
- Moreno, J.C.; Bikker, H.; Kempers, M.J.; van Trotsenburg, A.S.; Baas, F.; de Vijlder, J.J.; Vulsma, T.; Ris-Stalpers, C. Inactivating mutations in the gene for thyroid oxidase 2 (THOX2) and congenital hypothyroidism. N. Engl. J. Med. 2002, 347, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Bottcher, Y.; Paufler, T.; Stehr, T.; Bertschat, F.L.; Paschke, R.; Koch, C.A. Thyroid hormone resistance without mutations in thyroid hormone receptor beta. Med. Sci. Monit. 2007, 13, CS67–CS70. [Google Scholar] [CrossRef]
- Moran, C.; Agostini, M.; Visser, W.E.; Schoenmakers, E.; Schoenmakers, N.; Offiah, A.C.; Poole, K.; Rajanayagam, O.; Lyons, G.; Halsall, D.; et al. Resistance to thyroid hormone caused by a mutation in thyroid hormone receptor (TR)alpha1 and TRalpha2: Clinical, biochemical, and genetic analyses of three related patients. Lancet Diabetes Endocrinol. 2014, 2, 619–626. [Google Scholar] [CrossRef]
- Van Gucht, A.L.; Meima, M.E.; Zwaveling-Soonawala, N.; Visser, W.E.; Fliers, E.; Wennink, J.M.; Henny, C.; Visser, T.J.; Peeters, R.P.; van Trotsenburg, A.S. Resistance to Thyroid Hormone Alpha in an 18-Month-Old Girl: Clinical, Therapeutic, and Molecular Characteristics. Thyroid 2016, 26, 338–346. [Google Scholar] [CrossRef]
- Grijota-Martinez, C.; Barez-Lopez, S.; Gomez-Andres, D.; Guadano-Ferraz, A. MCT8 Deficiency: The Road to Therapies for a Rare Disease. Front. Neurosci. 2020, 14, 380. [Google Scholar] [CrossRef]
- Van Geest, F.S.; Gunhanlar, N.; Groeneweg, S.; Visser, W.E. Monocarboxylate Transporter 8 Deficiency: From Pathophysiological Understanding to Therapy Development. Front. Endocrinol. 2021, 12, 723750. [Google Scholar] [CrossRef]
- Friesema, E.C.; Grueters, A.; Biebermann, H.; Krude, H.; von Moers, A.; Reeser, M.; Barrett, T.G.; Mancilla, E.E.; Svensson, J.; Kester, M.H.; et al. Association between mutations in a thyroid hormone transporter and severe X-linked psychomotor retardation. Lancet 2004, 364, 1435–1437. [Google Scholar] [CrossRef]
- Huang, S.A.; Tu, H.M.; Harney, J.W.; Venihaki, M.; Butte, A.J.; Kozakewich, H.P.; Fishman, S.J.; Larsen, P.R. Severe hypothyroidism caused by type 3 iodothyronine deiodinase in infantile hemangiomas. N. Engl. J. Med. 2000, 343, 185–189. [Google Scholar] [CrossRef] [PubMed]
- Katakami, H.; Kato, Y.; Inada, M.; Imura, H. Hypothalamic hypothyroidism due to isolated thyrotropin-releasing hormone (TRH) deficiency. J. Endocrinol. Investig. 1984, 7, 231–233. [Google Scholar] [CrossRef] [PubMed]
- Niimi, H.; Inomata, H.; Sasaki, N.; Nakajima, H. Congenital isolated thyrotrophin releasing hormone deficiency. Arch. Dis. Child. 1982, 57, 877–878. [Google Scholar] [CrossRef]
- Yamada, M.; Radovick, S.; Wondisford, F.E.; Nakayama, Y.; Weintraub, B.D.; Wilber, J.F. Cloning and structure of human genomic DNA and hypothalamic cDNA encoding human prepro thyrotropin-releasing hormone. Mol. Endocrinol. 1990, 4, 551–556. [Google Scholar] [CrossRef] [PubMed]
- Bonomi, M.; Busnelli, M.; Beck-Peccoz, P.; Costanzo, D.; Antonica, F.; Dolci, C.; Pilotta, A.; Buzi, F.; Persani, L. A family with complete resistance to thyrotropin-releasing hormone. N. Engl. J. Med. 2009, 360, 731–734. [Google Scholar] [CrossRef] [PubMed]
- Persani, L.; Cangiano, B.; Bonomi, M. The diagnosis and management of central hypothyroidism in 2018. Endocr. Connect. 2019, 8, R44–R54. [Google Scholar] [CrossRef]
- Leger, J.; Olivieri, A.; Donaldson, M.; Torresani, T.; Krude, H.; van Vliet, G.; Polak, M.; Butler, G.; ESPE-PES-SLEP-JSPE-APEG-APPES-ISPAE; The Congenital Hypothyroidism Consensus Conference Group. European Society for Paediatric Endocrinology consensus guidelines on screening, diagnosis, and management of congenital hypothyroidism. J. Clin. Endocrinol. Metab. 2014, 99, 363–384. [Google Scholar] [CrossRef]
- Pine-Twaddell, E.; Romero, C.J.; Radovick, S. Vertical transmission of hypopituitarism: Critical importance of appropriate interpretation of thyroid function tests and levothyroxine therapy during pregnancy. Thyroid 2013, 23, 892–897. [Google Scholar] [CrossRef]
- Sunthornthepvarakul, T.; Gottschalk, M.E.; Hayashi, Y.; Refetoff, S. Brief report: Resistance to thyrotropin caused by mutations in the thyrotropin-receptor gene. N. Engl. J. Med. 1995, 332, 155–160. [Google Scholar] [CrossRef]
- Jonklaas, J.; Bianco, A.C.; Bauer, A.J.; Burman, K.D.; Cappola, A.R.; Celi, F.S.; Cooper, D.S.; Kim, B.W.; Peeters, R.P.; Rosenthal, M.S.; et al. Guidelines for the treatment of hypothyroidism: Prepared by the american thyroid association task force on thyroid hormone replacement. Thyroid 2014, 24, 1670–1751. [Google Scholar] [CrossRef]
- Kaplowitz, P.B.; Vaidyanathan, P. Update on pediatric hyperthyroidism. Curr. Opin. Endocrinol. Diabetes Obes. 2020, 27, 70–76. [Google Scholar] [CrossRef] [PubMed]
- Van der Kaay, D.C.; Wasserman, J.D.; Palmert, M.R. Management of Neonates Born to Mothers With Graves’ Disease. Pediatrics 2016, 137, e20151878. [Google Scholar] [CrossRef] [PubMed]
- Barczynski, M. Current approach to surgical management of hyperthyroidism. Q. J. Nucl. Med. Mol. Imaging 2021, 65, 124–131. [Google Scholar] [CrossRef] [PubMed]
- De Luca, F.; Salzano, G.; Zirilli, G.; Calafiore, M.; Corica, D.; Sferlazzas, C. Management of hyperthyroidism in children. Expert Rev. Endocrinol. Metab. 2016, 11, 301–309. [Google Scholar] [CrossRef] [PubMed]
- Niedziela, M. Pathogenesis, diagnosis and management of thyroid nodules in children. Endocr. Relat. Cancer 2006, 13, 427–453. [Google Scholar] [CrossRef] [PubMed]
- Foley, T.P., Jr.; Abbassi, V.; Copeland, K.C.; Draznin, M.B. Brief report: Hypothyroidism caused by chronic autoimmune thyroiditis in very young infants. N. Engl. J. Med. 1994, 330, 466–468. [Google Scholar] [CrossRef]
- Van Trotsenburg, A.S.; Kempers, M.J.; Endert, E.; Tijssen, J.G.; de Vijlder, J.J.; Vulsma, T. Trisomy 21 causes persistent congenital hypothyroidism presumably of thyroidal origin. Thyroid 2006, 16, 671–680. [Google Scholar] [CrossRef]
- Bull, M.J. Health supervision for children with Down syndrome. Pediatrics 2011, 128, 393–406. [Google Scholar] [CrossRef]
- Van Dop, C.; Conte, F.A.; Koch, T.K.; Clark, S.J.; Wilson-Davis, S.L.; Grumbach, M.M. Pseudotumor cerebri associated with initiation of levothyroxine therapy for juvenile hypothyroidism. N. Engl. J. Med. 1983, 308, 1076–1080. [Google Scholar] [CrossRef]
- Leung, A.K.C.; Leung, A.A.C. Evaluation and Management of Children with Thyrotoxicosis. Recent Pat. Endocr. Metab. Immune Drug Discov. 2017, 11, 22–31. [Google Scholar] [CrossRef]
- Burch, H.B.; Wartofsky, L. Life-threatening thyrotoxicosis. Thyroid storm. Endocrinol. Metab. Clin. N. Am. 1993, 22, 263–277. [Google Scholar] [CrossRef]
- Reddy, V.; Taha, W.; Kundumadam, S.; Khan, M. Atrial fibrillation and hyperthyroidism: A literature review. Indian Heart J. 2017, 69, 545–550. [Google Scholar] [CrossRef] [PubMed]
- Abdi, H.; Amouzegar, A.; Azizi, F. Antithyroid Drugs. Iran J. Pharm. Res. 2019, 18, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Leger, J.; Kaguelidou, F.; Alberti, C.; Carel, J.C. Graves’ disease in children. Best Pract. Res. Clin. Endocrinol. Metab. 2014, 28, 233–243. [Google Scholar] [CrossRef]
- Sosa, J.A.; Tuggle, C.T.; Wang, T.S.; Thomas, D.C.; Boudourakis, L.; Rivkees, S.; Roman, S.A. Clinical and economic outcomes of thyroid and parathyroid surgery in children. J. Clin. Endocrinol. Metab. 2008, 93, 3058–3065. [Google Scholar] [CrossRef]
- Dolphin, G.W. The risk of thyroid cancers following irradiation. Health Phys. 1968, 15, 219–228. [Google Scholar] [CrossRef]
- Boice, J.D., Jr. Thyroid disease 60 years after Hiroshima and 20 years after Chernobyl. JAMA 2006, 295, 1060–1062. [Google Scholar] [CrossRef]
- Alaguvelsamy, S.; Pal Singh, S.; Ramalingam, R.; Kombupalayam Komarappa Gounder, R. Giant toxic multinodular goiter with dyspnea: A case report. Int. J. Surg. Case Rep. 2020, 73, 190–195. [Google Scholar] [CrossRef]
- Chernock, R.D.; Rivera, B.; Borrelli, N.; Hill, D.A.; Fahiminiya, S.; Shah, T.; Chong, A.S.; Aqil, B.; Mehrad, M.; Giordano, T.J.; et al. Poorly differentiated thyroid carcinoma of childhood and adolescence: A distinct entity characterized by DICER1 mutations. Mod. Pathol. 2020, 33, 1264–1274. [Google Scholar] [CrossRef]
- Francis, G.L.; Waguespack, S.G.; Bauer, A.J.; Angelos, P.; Benvenga, S.; Cerutti, J.M.; Dinauer, C.A.; Hamilton, J.; Hay, I.D.; Luster, M.; et al. Management Guidelines for Children with Thyroid Nodules and Differentiated Thyroid Cancer. Thyroid 2015, 25, 716–759. [Google Scholar] [CrossRef]
- Canberk, S.; Barroca, H.; Girao, I.; Aydin, O.; Uguz, A.; Erdogan, K.; Tastekin, E.; Bongiovanni, M.; Soares, P.; Maximo, V.; et al. Performance of the Bethesda System for Reporting Thyroid Cytology in Multi-Institutional Large Cohort of Pediatric Thyroid Nodules: A Detailed Analysis. Diagnostics 2022, 12, 179. [Google Scholar] [CrossRef] [PubMed]
- Haugen, B.R.; Alexander, E.K.; Bible, K.C.; Doherty, G.M.; Mandel, S.J.; Nikiforov, Y.E.; Pacini, F.; Randolph, G.W.; Sawka, A.M.; Schlumberger, M.; et al. 2015 American Thyroid Association Management Guidelines for Adult Patients with Thyroid Nodules and Differentiated Thyroid Cancer: The American Thyroid Association Guidelines Task Force on Thyroid Nodules and Differentiated Thyroid Cancer. Thyroid 2016, 26, 1–133. [Google Scholar] [CrossRef] [PubMed]
- Sperling, M.A.R.S.A. Pediatric Endocrinology. In Thyroid Disorders in Children and Adolescents; Elsivier: Philadelphia, PA, USA, 2014. [Google Scholar]
- Vaccarella, S.; Lortet-Tieulent, J.; Colombet, M.; Davies, L.; Stiller, C.A.; Schüz, J.; Togawa, K.; Bray, F.; Franceschi, S.; Dal Maso, L.; et al. Global patterns and trends in incidence and mortality of thyroid cancer in children and adolescents: A population-based study. Lancet Diabetes Endocrinol. 2021, 9, 144–152. [Google Scholar] [CrossRef]
- Wells, S.A., Jr.; Asa, S.L.; Dralle, H.; Elisei, R.; Evans, D.B.; Gagel, R.F.; Lee, N.; Machens, A.; Moley, J.F.; Pacini, F.; et al. Revised American Thyroid Association guidelines for the management of medullary thyroid carcinoma. Thyroid 2015, 25, 567–610. [Google Scholar] [CrossRef] [PubMed]
- Bauer, A.J. Pediatric Thyroid Cancer: Genetics, Therapeutics and Outcome. Endocrinol. Metab. Clin. N. Am. 2020, 49, 589–611. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).