Pathological Maintenance and Evolution of Breast Cancer: The Convergence of Irreversible Biological Actions of ER Alpha


{kind=link}
Abstract
:1. Introduction
2. Biological Irreversibility: A Concept Applied to Regulatory Functions
3. Relationship between Cell Cycle Progression and ERα Expression in Breast Cancer Cells
4. Factors Implicated in the Irreversible Character of ERα-Mediated Transcriptions
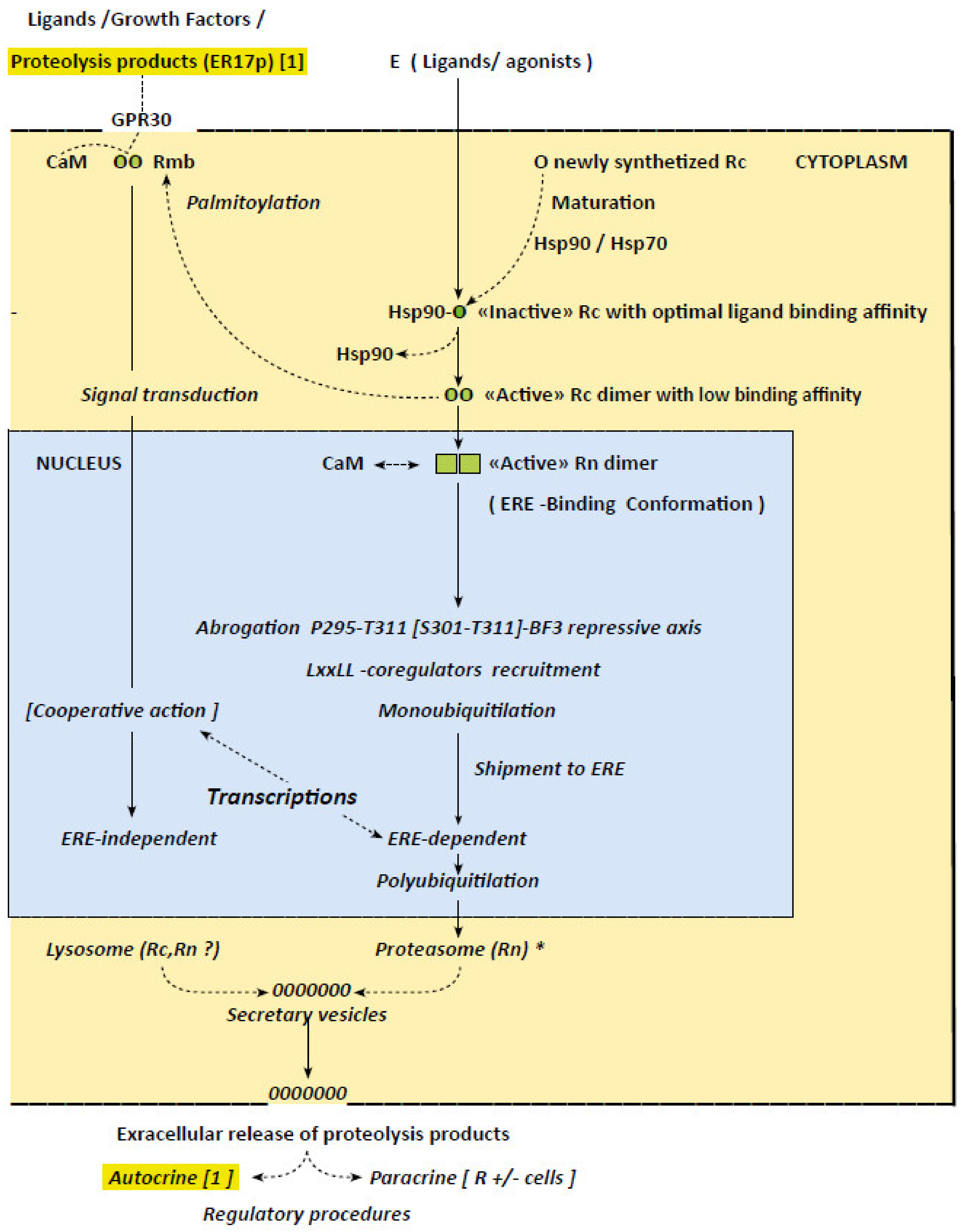
5. ERα “Activation”: Loss of Ligand Binding Capacity and Related Receptor Dimerization
6. Allosteric ERα Activation Implying Successive Coregulator Recruitments
7. Implication of Proteolytic ERα Fragments in Autocrine and Paracrine Regulations
8. Concluding Remarks and Conclusions
Supplementary Materials
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AF | Activation function |
| AP 1 | Activation Protein 1 |
| BF3 | Binding Function 3 |
| CDK | Cyclin-Dependent Kinase |
| ER | Estrogen Receptor |
| ERE | Estrogen Response Element |
| ERK | Extra Celluar Regulated Kinase |
| ERα 17p | Peptide corresponding to the P295-T311 amino acid sequence of ERα |
| GPR | G-Protein Coupled Receptor |
| HER 2 | Human Epidermal Growth Factor 2 |
| Hsp | Heat Shock Protein |
| Ki-67 | Protein identified with an antibody raised against a Hodgkin’ lymphoma protein |
| LxxLL | binding motif of coregulators L refers to Leucine, x to any other amino acid |
| PRNC | Proline-rich Nuclear Receptor Regulatory Protein |
| PR | Progesterone Receptor |
| pRb | Retinoblastoma Protein |
| SERM | Selective Estrogen Receptor Modulator |
References
- Arnal, J.-F.; Lenfant, F.; Metivier, R.; Flouriot, G.; Henrion, D.; Adlanmerini, M.; Gourdy, P.; Chambon, P.; Katzenellenbogen, B.; Katzenellenbogen, J. Membrane and nuclear estrogen receptor alpha actions: From tissue specificity to medical implications. Physiol. Rev. 2017, 17, 1045–1087. [Google Scholar] [CrossRef] [PubMed]
- Weikum, E.R.; Liu, X.; Ortlund, E.A. The nuclear receptor superfamily: A structural perspective. Protein Sci. 2018, 27, 1876–1892. [Google Scholar] [CrossRef] [PubMed]
- Hager, G.L.; Lim, C.M.; Bauman, C.T. Trafficking of nuclear receptors in living cells. J. Steroid Biochem. Mol. Biol. 2000, 74, 249–254. [Google Scholar] [CrossRef]
- Levin, E.R. Extranuclear steroid receptors are essential for steroid hormone actions. Annu. Rev. Med. 2015, 66, 271–280. [Google Scholar] [CrossRef] [PubMed]
- Clark, J.H.; Peck, E.J., Jr. Female Sex Steroids. Receptors and Functions. In Monographs in Endocrinology; Springer: Berlin/Heidelberg, Germany, 1979; Volume 14. [Google Scholar]
- Leclercq, G. Calcium-induced activation of estrogen captor alpha—New insight. Steroids 2012, 77, 924–927. [Google Scholar] [CrossRef]
- Lippman, M.E.; Dickson, R.B.; Kassid, A.; Gelmann, R.E.; Davidson, N.; McManaway, M.; Huff, K.; Bonzert, D.; Bates, S.; Swain, S.; et al. Autocrine and paracrine growth regulation of human breast cancer. J. Steroid Biochem. 1986, 24, 147–154. [Google Scholar] [CrossRef]
- Leclercq, G.; Lacroix, M.; Laïos, I.; Laurent, G. Estrogen receptor alpha: Impact of Ligands on intracellular shuttling and turnover rate in breast cancer cells. Curr. Cancer Drug Targets 2006, 6, 39–64. [Google Scholar] [CrossRef] [Green Version]
- Kampa, M.; Pelekanou, V.; Notas, G.; Stathopoulos, E.N.; Castanas, E. The estrogen receptor: Two or more molecules, multiple variants, diverse localizations, signaling and functions. Are we undergoing a paradigm-shift as regards their significance in breast cancer? Hormones 2013, 12, 69–85. [Google Scholar] [CrossRef]
- Seo, H.-S.; Leclercq, G. Evaluation of potential implication of membrane estrogen binding sites on ERE-dependent transcriptional activity and intracellular estrogen receptor-alpha regulation in MCF-7 breast cancer cells. J. Steroid Biochem. Mol. Biol. 2002, 80, 109–123. [Google Scholar] [CrossRef]
- La Rosa, P.; Perisi, V.; Leclercq, G.; Marino, M.; Acconcia, F. Palmitoylation regulates 17β-estradiol-induced estrogen receptor-a degradation and transcriptional activity. Mol. Endocrinol. 2012, 26, 762–764. [Google Scholar] [CrossRef] [Green Version]
- Winkeldfeld, S.R.; Lin, F.D.E. Communication between genomic and non-genomic signaling events coordinate steroid hormone actions. Steroids 2018, 133, 2–7. [Google Scholar]
- Watson, G.H.; Muldoon, T.G. Specific binding of estrogen and estrogen receptor complex by microsomes from estrogen responsive tissues of the rat. Endocrinology 1985, 117, 1341–1349. [Google Scholar] [CrossRef] [PubMed]
- Klinge, C.M. Estrogens regulate life and death in mitochondria. J. Bioenerg. 2017, 49, 307–324. [Google Scholar] [CrossRef] [PubMed]
- Kushner, P.J.; Agard, D.A.; Greene, G.L.; Scanlan, T.S.; Shiau, A.K.; Uth, R.M.; Webb, P. Estrogen receptor pathways to AP-1. J. Steroid Biochem. Mol. Biol. 2000, 74, 311–317. [Google Scholar] [CrossRef]
- Safe, S.; Kim, K. Non classical ER/Sp and AR/AP-1 signaling pathways. J. Mol. Endocrinol. 2008, 41, 263–275. [Google Scholar] [CrossRef]
- Holding, A.N.; Cullen, A.E.; Markowitz, M. Genome-Wide estrogen receptor alpha activation is sustained not cyclical. Elife. 2018, 7, e40854. [Google Scholar] [CrossRef]
- Vic, P.; Garcia, M.; Humeau, C.; Rochefort, H. Early effect of estrogen on chromatin ultrastructure in endometrial nuclei. Mol. Cell. Endocrinol. 1980, 19, 79–92. [Google Scholar] [CrossRef]
- Verrijdt, A.; Leclercq, G.; Devleeschouwer, N.; Danguy, A. Tritiated actinomycin-D staining method: A valuable tool to study estrogen receptor-induced modifications of transcriptional activity in normal and neoplastic cells. Arch. Int. Physiol. Biochim. 1985, 93, 65–73. [Google Scholar] [CrossRef]
- Leclercq, G.; Hulin, N.; Heuson, J.-C. Interaction of activated of estradiol receptor complex and chromatin in isolated uterine nuclei. Eur. J. Cancer. 1973, 9, 681–685. [Google Scholar] [CrossRef]
- El Khissin, A.; Leclercq, G. Exchange of bound estrogens and antiestrogens in MCF-7 cells: Evidence for ligand–induced stable configurations of the estrogen receptor. Steroids 1998, 63, 65–574. [Google Scholar] [CrossRef]
- Laïos, I.; Journé, F.; Laurent, G.; Nonclercq, D.; Toillon, R.-A.; Seo, H.-S.; Leclercq, G. Mechanism governing the accumulation of estrogen receptor alpha in MCF-7 breast cancer cells treated with hydroxytamoxifen and related antiestrogens. J. Steroid Biochem. Mol. Biol. 2003, 87, 207–221. [Google Scholar] [CrossRef] [PubMed]
- Madeddu, L.; Legros, N.; De Vleeschouwer, N.; Bosman, C.; Piccart, M.J.; Leclercq, G. Estrogen receptor status and estradiol sensitivity of MCF-7 cells in exponential growth phase. Eur. J. Cancer Clin. Oncol. 1988, 23, 185–190. [Google Scholar] [CrossRef]
- Metivier, R.; Penot, G.; Hubner, M.R.; Reid, G.; Rand, H.; Kos, M.; Gannon, F. Estrogen receptor-alpha directs ordered, cyclic and combinatorial recruitment of cofactors on a natural target promotor. Cell 2003, 115, 751–763. [Google Scholar] [CrossRef] [Green Version]
- Inwald, E.C.; Klinkhammer-Schalke, M.; Hofstädter, F.; Zeman, F.; Koller, M.; Gersternhauer, M.; Ortmann, O. Ki-67 is a prognostic parameter in breast cancer patients: Results of a large population-based cohort of cancer registry. Brest Cancer Res Treat. 2013, 139, 539–552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, F.; Wu, S.; Zhou, J.; Sun, J.; Lin, Q.; Lin, H.; Guan, X.; He, Z. Prognostic value of Ki-67 in breast cancer patients with positive lymph nodes: A retrospective cohort study. PLoS ONE 2014, 9, e87264. [Google Scholar] [CrossRef]
- Sun, X.; Kaufman, P.D. Ki-67: More than a proliferation marker. Chromosoma 2018, 127, 175–186. [Google Scholar] [CrossRef] [PubMed]
- Doisneau-Sixou, S.F.; Sergio, S.M.; Caroll, J.S.; Hui, R.; Mushgrove, E.A.; Sutherland, R.L. Estrogens and antiestrogens regulation of cell cycle progression in breast cancer cells. Endocr. Relat. Cancer 2003, 10, 179–186. [Google Scholar] [CrossRef]
- Zheng, Y.; Murphy, L.C. Regulation of steroid hormones receptors and co regulators during cycle highlights potential novel function in addition to roles as transcription factors. Nuclear Recept. Signal. 2016, 14, e001. [Google Scholar] [CrossRef] [Green Version]
- Maynadier, M.; Ramirez, J.-M.; Cathiard, A.-M.; Platet, N.; Gras, D.; Geizes, M.M.; Saeed, S.; Nirde, P.; Garcia, M. Unliganded estrogen receptor alpha inhibits breast cancer cell growth through interaction with a cyclin-dependent kinase inhibitor (p21(WAF1)). FASEB J. 2007, 22, 671–681. [Google Scholar] [CrossRef]
- Wasierska-Gadek, J.; Mauritz, M. Why (multi) targeting of cyclin-dependent kinases is a promising therapeutic option for hormone-positive breast cancer and beyond. Future Med. Chem. 2016, 8, 55–72. [Google Scholar] [CrossRef]
- Rostagno, P.; Moll, J.I.; Birtwisle-Peyrottes, I.; Ettore, F.; Caldani, C. Cell cycle expression of estrogen receptors determined by image analysis on human breast cancer cells in vitro and in vivo. Breast Cancer Res. Treat. 1996, 39, 147–154. [Google Scholar] [CrossRef] [PubMed]
- Danguy, A.; Kiss, R.; Leclercq, G.; Heuson, J.C.; Pasteels, J.L. Morphology of MXT mammary tumors. Correlation with growth characteristics and hormone sensitivity. Eur. J. Cancer Clin. Oncol. 1986, 22, 69–75. [Google Scholar] [PubMed]
- Piccart, M.J.; Tivedi, S.; Maaroufi, Y.; Debbaudt, A.; Leclercq, G. Evolution towards hormone independence of the MXT mouse mammary tumor is associated with a gradual change in its estrogen receptor molecular polymorphism. Cancer Biochem. Biophys. 1998, 16, 169–182. [Google Scholar] [PubMed]
- El Khissiin, A.; Journé, F.; Laïos, I.; Seo, H.-S.; Leclercq, G. Evidence of an estrogen receptor form devoid of estrogen binding ability in MCF-7 cell. Steroids 2000, 65, 903–913. [Google Scholar] [CrossRef]
- JavanModoghadam, S.; Weihua, Z.; Hunt, K.K.; Keyomarsi, K. Estrogen receptor alpha is cell cycle–regulated and regulates cell cycle in a ligand-dependent fashion. Cell Cycle 2016, 15, 1579–1590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saceda, M.; Lindsey, R.K.; Solomon, H.; Angeloni, S.; Martin, M.B. Estradiol regulates estrogen receptor mRNA stability. J. Steroid Biochem. Mol. Biol. 1998, 66, 113–120. [Google Scholar] [CrossRef]
- Laïos, I.; Journe, F.; Nonclercq, D.; Salazar Vidal, D.; Toillon, R.-A.; Leclercq, G. Role of the proteasome in the regulation of estrogen receptor α turnover and function in MCF-7 breast cancer cells. J. Steroids Biochem. Molec. Biol. 2005, 94, 347–359. [Google Scholar] [CrossRef] [PubMed]
- El Khissiin, A.; Leclercq, G. Implication of proteasome in estrogen receptor degradation. FEBS Lett. 1999, 448, 160–166. [Google Scholar] [CrossRef] [Green Version]
- Nonclercq, D.; Journé, F.; Laïos, I.; Chaboteaux, C.; ToillonR, A.; Leclercq, G.; Laurent, G. Effect of nuclear receptor inhibition on estrogen rececptor regulation in breast cancer cells. J. Mol. Endocrocrinol. 2007, 39, 105–118. [Google Scholar] [CrossRef] [Green Version]
- Leclercq, G.; Legros, N.; Piccart, M.J. Accumulation of a non-binding form of estrogen receptor in MCF-7 cells under hydroxytamoxifen treatment. J. Steroid Biochem. Mol. Biol. 1992, 41, 545–552. [Google Scholar] [CrossRef]
- Tecalco-Cruz, A.C.; Pérez-Alvarado, I.A.; Ramirez-Jarquin, J.O. Nucleo-cytoplasmic transport of estrogen receptor alpha in breast cancer cells. Cell. Signal. 2017, 34, 121–132. [Google Scholar] [CrossRef] [PubMed]
- Kondavoka, I.V.; Shashova, E.E.; Sidenko, E.A.; Astahova, T.M.; Zakarova, L.A.; Sharova, N.P. Estrogen receptors and ubiquitin proteasome system. Mutual regulation. Biomolecules 2020, 10, 500. [Google Scholar]
- Powers, G.L.; Rajbhandari, P.; Solodin, N.M.; Brickford, B.; Alarid, E.T. The proteasome inhibitor Bortesomib induces an inhibitory chromatin environment at a distal enhancer of the estrogen receptor-α gene. PLoS ONE 2013, 8, e81110. [Google Scholar] [CrossRef] [PubMed]
- Ayaz, G.; Vasar, P.; Karakya, B.; Kars, G.; Razizaden, N.; Yavuz, K.; Turan, G.; Muyan, M. Dynamic transcriptional events mediated by estrogen receptor alpha. Front. Biosci. 2019, 24, 245–276. [Google Scholar]
- Le Romancer, M.; Poulard, C.; Cohen, P.; Sentis, S.; Renoir, J.-M.; Corbo, L. Cracking the estrogen receptors posttranslational code in breast tumors. Endocr. Rev. 2011, 32, 597–622. [Google Scholar] [CrossRef] [Green Version]
- Le Dilly, F.; Beato, M. Signaling by steroid hormones in the 3D nuclear space. Int. J. Mol. Sci. 2018, 19, 306. [Google Scholar] [CrossRef]
- Pesiri, V.; Di Muzio, E.; Polticelli, F.; Acconcia, F. Selective binding of estrogen receptor α. IUBMB Life 2016, 68, 497–501. [Google Scholar] [CrossRef] [Green Version]
- Tecalco-Cruz, A.C.; Ramirez-Jarquin, J.O. Polyubiquitination inhibition of esytogen receptor alpha and its implication in breast cancer. World J. Clin. Oncol. 2018, 9, 60–70. [Google Scholar] [CrossRef]
- Tecalco-Cruz, A.C.; Ramirez-Jarquin, J.O.; Cruz-Ramos, E. Estrogen receptor alpha and its ubiquitination in breast cancer. Curr. Drug Targets 2019, 20, 690–704. [Google Scholar] [CrossRef]
- Gallo, D.; Haddad, I.; Laurent, G.; Vinh, J.; Jacquemotte, F.; Jacquot, Y.; Leclercq, G. Regulatory function of the P295-T311 motif of the estrogen receptor α—Does proteosomal degradation of the receptor induce peptides implicated in estrogenic responses? Nuclear Recept. Signal. 2018, e007. [Google Scholar] [CrossRef] [Green Version]
- Totta, P.; Perisi, V.; Marino, M.; Acconcia, F. Lysosomial function is involved in 17 bêta estradiol-induced estrogen receptor alpha degradation and cell proliferation. PLoS ONE 2014, 9, e94880. [Google Scholar] [CrossRef] [PubMed]
- Totta, P.; Busonero, C.; Leone, S.; Marino, M.; Acconcia, F. Dynamin II is required for 17β-estradiol signaling and autophagy-based ERα degradation. Sci. Rep. 2018, 6, 23727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sampayo, R.G.; Toscani, A.M.; Rubashkin, M.G.; Thi, K.; Masullo, L.A.; Violi, I.L.; Lakins, J.N.; Caceres, A.A.; Hines, W.C.; Leskow, F.C.; et al. Fibronectine rescues estrogen receptor α from lysosomal degradation in breast cancer cells. J. Cell. Biol. 2018, 217, 2777–2798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eick, G.; Thornton, J.W. Evolution of steroid receptors from an estrogen-sensitive ancestral receptor. Mol. Cell. Endocrinol. 2011, 334, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Albini, A.; Rosano, C.; Angelini, A.; Amaro, A.; Esposito, A.I.; Maramotti, S.; Noonan, D.M.; Pfeffer, U. Exogenous hormonal regulation in breast cancer cells by phytoestrogens and endocrine disruptors. Curr. Med. Chem. 2014, 21, 1129–1149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fiss, A.F.; Benzeno, S.; Rao, J.; Caplan, A.J. Control of estrogen ligand binding by Hsp90. J. Steroid Biochem. Mol. Biol. 2000, 72, 223–230. [Google Scholar]
- Lee, M.O.; Kim, E.O.; Kwon, H.J.; Kim, Y.M.; Kang, H.J.; Kang, H.; Lee, J.L. Radicicol represses transcriptional activity of estrogen receptor the by suppressing the stabilization of the receptor by heat shock protein 90. Mol. Cell. Endocrinol. 2002, 188, 47–54. [Google Scholar] [CrossRef]
- Nonclercq, D.; Journé, F.; Body, J.-C.; Leclercq, G.; Laurent, G. Ligand-independent and agonist-mediated degradation of estrogen receptor -α in breast carcinoma cells; evidence for distinct degradative pathways. Mol. Cell. Endocrinol. 2004, 227, 53–65. [Google Scholar] [CrossRef]
- Leclercq, G.; Jensen, E.V. Dimeric structure of unoccupied; activated estrogen receptor. In Proceedings of the Fifth International CBT Symposium on Intracellular Hormone Receptors and Ligand-Dependent Transcriptional Regulation, Huddinnge, Sweden, 6–8 September 1993; p. 91. [Google Scholar]
- Klinge, C.M. Estrogen binding to estrogen response elements slows ligand dissociation and synergically activates reporter gene expression. Mol. Cell Endocrinol. 1999, 150, 99–111. [Google Scholar] [CrossRef]
- Sonoda, M.T.; Martinez, L.; Webb, P.; Skaf, M.S.; Polikarpov, I. Ligand dissociation from estrogen receptor is mediated by receptor dimerization: Evidence from molecular dynamics simulations. Molec. Endorinol. 2008, 22, 1565–1578. [Google Scholar] [CrossRef] [Green Version]
- Leclercq, G.; Laïos, I.; Elie-Caille, C.; Leiber, D.; Laurent, G.; Lesnivska, E.; Tanfin, Z.; Jacquot, Y. ERα dimerisation: A key factor for the weak estrogenic activity of an ERα modulator unable to compete with estradiol in binding assays. J. Recept. Signal Transdcuct. Res. 2017, 37, 149–166. [Google Scholar] [CrossRef] [PubMed]
- Razandi, M.; Pedram, A.; Merchenthaler, I.; Greene, G.L.; Levin, E.R. Plasma membrane estrogens receptors exist and function as dimers. Mol. Endocrinol. 2004, 18, 2854–2865. [Google Scholar] [CrossRef] [PubMed]
- Jacquot, Y.; Spaggiari, D.; Schappler, J.; Lesniewska, E.; Rudaz, S.; Leclercq, G. ERE-dependent transcription and cell proliferation: Independence of these two processes mediated by the introduction of a sulfone function into the weak estrogen estrothazine. Eur. J. Pharm. Sci. 2017, 109, 169–181. [Google Scholar] [CrossRef] [PubMed]
- Jacquot, Y.; Laïos, I.; Cleeren, A.; Nonclercq, D.; Bermont, L.; Refoulevet, B.; Boubekeur, K.; Xicluna, A.; Leclercq, G.; Laurent, G. Synthesis, structure, and estrogenic activity of 4-amino-3-(2 methylbenzyl) coumarins on human breast cancer cells. Bioorg. Med. Chem. 2007, 15, 2269–2282. [Google Scholar] [CrossRef] [PubMed]
- Laïos, I.; Cleeren, A.; Leclercq, G.; Nonclercq, D.; Laurent, G.; Schlenk, M.; Wellner, A.; Gust, R. Effects of (R/S)/(S,R)-4,5-bis(2-chloro_4-Hydroxyphenyl)piperazines on estrogen receptor alpha level and transcriptional activity in MCF-7 cells. Biochem. Pharmacol. 2000, 74, 1029–1038. [Google Scholar]
- Jacquot, Y.; Gallo, D.; Leclercq, G. Estrogen receptor alpha—Identification by a modeling approach of a potential polyproline II recognizing domain within the AF-2 region of the receptor that would play a role of prime importance in its mechanism of action. J. Steroid Biochem. Mol. Biol. 2007, 104, 1–10. [Google Scholar] [CrossRef]
- Gallo, D.; Leclercq, G.; Jacquot, Y. The N-Terminal part of the ligand binding domain of the the human estrogen receptor α: A new target for estrogen disruptors. In Medical Chemistry Research Progress; Colombo, G.P., Ricci, S., Eds.; Nova Science Publishers: Hauppauge, NY, USA, 2008. [Google Scholar]
- Buzon, V.; Carbo, L.R.; Estruch, S.B.; FLetterick, R.J.; Estebanez-Perpina, E. A conserved surface on the ligand binding domain of nuclear receptors for allosteric controls. Mol. Cell. Endocrinol. 2012, 348, 394–402. [Google Scholar] [CrossRef]
- Mackinmon, J.A.; Gallastegui, N.; Osguthorpe, D.J.; Hagler, A.T.; Estebanez-Perpina, E. Allosteric mechanisms of nuclear receptors: Insights from computional simulations. Mol. Cell. Endocrinol. 2014, 393, 75–78. [Google Scholar] [CrossRef]
- Li, I.; Sacks, D.B. Functional interactions between calmodulin and estrogen receptor-alpha. Cell Signal. 2007, 19, 439–443. [Google Scholar] [CrossRef]
- Gallo, D.; Jacquot, Y.; Laurent, G.; Leclercq, G. Calmodulin, a regulatory partner of the estrogen receptor alpha in breast cancer cells. Mol. Cell. Endocrinol. 2008, 268, 20–26. [Google Scholar] [CrossRef] [Green Version]
- Carlier, L.; Byrne, C.; Miclet, E.; Bourgouin-Voillard, S.; Nicaise, M.; Tabet, J.C.; Desmadrol, M.; Leclercq, G.; Lequin, O.; Jacquot, Y. Studies of the interaction between calmodulin and the R287-T311 region of human estrogen receptor α reveal an atypical binding process. Biochem. Biophys. Res. Commun. 2012, 419, 356–361. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Zhang, Y.; Hedman, A.C.; Ames, J.B.; Sacks, D.B. Calmodulin lobes facilitate dimerization and activation of estrogen receptor-α. J. Biol. Chem. 2017, 292, 4614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bourgouin-Voillard, S.; Fournier, F.; Alfonso, C.; Jacquot, Y.; Leclercq, G.; Tabet, J.C. Calmodulin association with the synthetic ERα 17p peptide investigated by Mass Spectrometry. Int. J. Mass Spectrom. 2011, 305, 87–94. [Google Scholar] [CrossRef]
- Gallo, D.; Haddad, I.; Duvillier, H.; Jacquemotte, F.; Laïos, I.; Laurent, G.; Jacquot, Y.; Vinh, H.; Leclercq, G. Trophic effect in MCF-7 cells of ERalpha17p, a peptide corresponding to a platform regulatory motif of the estrogen receptor alpha–Underlying mechanism. J. Steroid Mol. Biol. 2008, 109, 138–149. [Google Scholar] [CrossRef]
- Byrne, C.; Miclet, E.; Broutin, I.; Gallo, D.; Pelekanou, V.; Kampa, M.; Leclercq, G.; Jacquot, Y. Identification of polyproline II regions derived from the proline-rich nuclear coactivators PNRC and PNRC2: New insights for ERα coactivators interactions. Cherality 2013, 25, 628–642. [Google Scholar] [CrossRef]
- Leiber, D.; Burlina, F.; Byrne, C.; Robin, P.; Piesse, C.; Gonzalez, L.; Leclercq, G.; Tafin, Z.; Jacquot, Y. The sequence Pro298-Thr311 of the hinge of oestrogen receptor a involved in ERK1/2 activation via GPR30 in leiomyoma cells. Biochem. J. 2015, 472, 97–104. [Google Scholar] [CrossRef]
- Norman, A.W.; Mizicki, M.T.; Norman, D.P. Steroid-hormone rapid actions. Membrane receptors and conformational ensemble model. Nat. Rev. Drug Discov. 2004, 3, 27–41. [Google Scholar] [CrossRef]
- Jordan, V.C.; Schafer, J.M.; Levenson, A.S.; Liu, H.; Pease, K.M.; Simons, L.A.; Zapf, J.W. Molecular classification of estrogens. Cancer Res. 2001, 61, 6619–6623. [Google Scholar]
- Bai, Z.; Gust, R. Breast cancer, estrogen receptor and ligands. Arch. Pharm. 2009, 342, 133–149. [Google Scholar] [CrossRef]
- Bourgoin-Voillard, S.; Gallo, D.; Laïos, I.; Cleeren, A.; El Bali, L.; Jacquot, Y.; Nonclercq, D.; Laurent, G.; Tabet, J.-C.; Leclercq, G. Capacity of type I and II to confer to estrogen receptor alpha an appropriate conformation for the recruitment of coactivators containing a LxxLL motif—Relationship with the regulation of receptor level and ERE-dependent transcription in MCF-7 cells. Bichem. Pharmacol. 2010, 79, 746–757. [Google Scholar] [CrossRef] [Green Version]
- Cheng, S.-B.; Graber, C.T.; Quinn, J.A.; Filardo, E.J. Retrograde transport of the membrane estrogen receptor, G-protein-coupled-receptor-30 (GPR30/GPR) from the plasma membrane towards the nucleus. Steroids 2011, 76, 892–896. [Google Scholar] [PubMed]
- Mairesse, N.; Devleeschouwer, N.; Leclercq, G.; Galand, P. Estogen-induced synthesis and secretion of proteins in the human breast cancer cell line MCF-7. J. Steroid Biochem. 1981, 15, 375–381. [Google Scholar] [CrossRef]
- Olea-Serrano, N.; Leclercq, G.; Mairesse, N.; Heuson, J.-C. Bulk protein secretion induced by estradiol in MCF-7 cells. Eur. J. Cancer Clin. Oncol. 1985, 21, 1267–1271. [Google Scholar] [CrossRef]
- Linares, P.M.; Algaba, A.; Urzainqui, A.; Guijarro-Rojas, M.; Fernandez-Contreras, M.E.; Garrido, J.; Chaparro, M.; Gisbert, J.P.; Bermejo, F.; Guerra, I.; et al. Ratio of circulating estrogen receptors bêta and alpha (ERβ/ERα) indicates endoscopic activity in patients with Crohn’s disease. Dig. Dis. Sci. 2017, 62, 2744–2754. [Google Scholar] [CrossRef] [PubMed]
- Notas, G.; Kampa, M.; Pelekanou, V.; Toullinaki, M.; Jacquot, Y.; Leclercq, G.; Castanas, E. Whole transcriptome analysis of the ERα synthetic fragment P295-T311 (ERa17p) identifies specific ERα-isoform (ERα, ERα36)-dependent and independent actions in breast cancer cells. Mol. Oncol. 2013, 7, 595–610. [Google Scholar] [CrossRef]
- Maselli, A.; Cappoccia, S.; Pugliese, P.; Raggi, C.; Cirulli, F.; Fabi, A.; Maloni, W.; Perdomicini, M.; Ortona, E. Antibodies specific to estrogen receptor alpha act as estrogen agonists and their levels correlate with breast cancer cell proliferation. Oncoimmunology 2015, 12, e107375. [Google Scholar]
- Leclercq, G. Natural anti-estrogen receptor alpha antibodies able to induce estrogenic responses in breast cancer cells: Hypotheses concerning their mechanisms of action and emergence. Int. J. Mol Sci. 2018, 30, 411. [Google Scholar] [CrossRef] [Green Version]
- Maselli, A.; Perlato, S.; Pugglisi, R.; Riggi, C.; Spada, M.; Macchia, D.; Pontecorvi, G.; Iessi, E.; Pagano, M.T.; Cirulli, F.; et al. Antibodies specific to ERα are involved in tamoxifen resistance in hormone positive breast cancer. Cells 2019, 8, 750. [Google Scholar] [CrossRef] [Green Version]
- Herynck, M.H.; Fuqua, S.A.W. Estrogen receptor mutations in human disease. Endocr. Rev. 2004, 25, 869–898. [Google Scholar] [CrossRef] [Green Version]
- Barone, I.; Brusco, L.; Fuqua, S.A.W. Estrogen receptor mutations and changes in downstream gene expression signaling. Clin. Cancer Res. 2010, 16, 2702–2708. [Google Scholar] [CrossRef] [Green Version]
- Reinert, T.; Saad, E.A.; Barrios, C.H.; Bines, J. Cinical implications of ESR1 mutations in hormone receptor-positive advanced breast cancers. Front. Oncol. 2017, 7, e00026. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jordan, V.C.; Corpan, R.; Maximov, P.Y. Estrogen receptor mutations found in breast cancer metastases integrated with molecular pharmacology of selective ER modulators. J. Natl. Cancer Inst. 2015, 107, 075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.-Y.; Yin, L. Estrogen receptor alpha-36 (ER-α36): Anew player in human breast cancer. Mol. Cell. Endocrinol. 2015, 418, 193–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pelekanou, V.; Notas, G.; Kampa, M.; Tsentelierou, E.; Radojiric, J.; Leclercq, G.; Castanas, E.; Stathopoulos, E. ERα, a new variant of the ERα is expressed in triple negative breast carcinomas and has a specific transcriptomic signature in breast cancer cell lines. Steroids 2012, 77, 928–934. [Google Scholar] [CrossRef] [PubMed]
- Chaudhri, R.A.; Haddadi, A.; Lobachev, K.S.; Schwartz, Z.; Boyan, B.D. Estrogen receptor-alpha 36 mediates the ant-apoptotic effect of estradiol in triple negative cancer cells via a membrane-Aassociated mechanism. Biochim. Biophys. Acta Mol. Res. 2014, 1843, 2796–2806. [Google Scholar] [CrossRef] [Green Version]
- Pagano, M.T.; Ortona, E.; Dupuis, M.L. A role for estrogen receptor alpha36 in cancer progression. Font. Endocrinol. 2020, 11, e00506. [Google Scholar] [CrossRef]
- Merenbakh-Lamin, K.; Ben-Baruch, N.; Yesheskel, A.; Dvir, A.; Soussan-Gutman, L.; Jeselsohn, R.; Yelensky, R.; Brown, M.; Miller, V.A.; Sarid, D.; et al. D538G mutation in estrogen receptor-α: A novel mechanism for acquired endocrine resistance in breast cancer. Cancer Res. 2013, 73, 6856–6864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fanning, S.W.; Mayne, C.G.; Dharmarajan, V.; Carlson, K.E.; Martin, T.A.; Novick, S.J.; Toy, W.; Green, B.; Panchamukhi, S.; Katzenellenbogen, B.S.; et al. Estrogen receptor alpha somatic Mutations Y537S and D538G confer breast cancer endocrine resistance by stabilising the activating function2 binding conformation. Elife 2016, 5, e122792. [Google Scholar] [CrossRef] [PubMed]
- Pavlin, M.; Gelsomino, L.; Barone, I.; Spinello, A.; Catalano, S.; Ando, S.; Magisrato, A. Structural Thermodynamic and kinetic traits of antiestogen-componds selectivity targeting the Y537S mutant estrogen receptor α transcritional activity in breast cancer cell lines. Front. Chem. 2019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giordano, G.; Cui, Y.; Barone, I.; Ando, S.; Mancini, A.M.; Berno, V.; Fuqua, A.W. Growth factor-indued to tamoxifen is associated with a mutation of estrogen receptor alpha and its phosphorylation at serine 305. Breast Cancer Res. Treat. 2010, 119, 71–85. [Google Scholar] [CrossRef] [Green Version]
- Barone, I.; Iacopetta, D.; Covington, K.R.; Cui, Y.; Tsimelzon, A.; Beyer, A.; Ando, S.; Fuqua, A.W. Phosphorylation of the mutant K303R estrogen receptor alpha at serune 305 affects aromatase inhibitor activity. Oncogene 2010, 29, 404–414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leclercq, G.; Gallo, D.; Cossy, J.; Laïos, I.; Larsimont, D.; Laurent, G.; Jacquot, Y. Peptides targeting estrogen receptor alpha–potential applications for breast cancer treatment. Curr. Pharm. Des. 2011, 17, 2632–2653. [Google Scholar] [CrossRef] [PubMed]
- Busoner, C.; Leone, S.; Bartolini, S.; Acconcia, F. Strategies to degrade estrogen receptor α in primary and ESR1 mutant-expressing metastatic breast cancer. Mol. Cell. Endocrinol. 2019, 480, 107–121. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Leclercq, G. Pathological Maintenance and Evolution of Breast Cancer: The Convergence of Irreversible Biological Actions of ER Alpha. Endocrines 2021, 2, 1-14. https://doi.org/10.3390/endocrines2010001
Leclercq G. Pathological Maintenance and Evolution of Breast Cancer: The Convergence of Irreversible Biological Actions of ER Alpha. Endocrines. 2021; 2(1):1-14. https://doi.org/10.3390/endocrines2010001
Chicago/Turabian StyleLeclercq, Guy. 2021. "Pathological Maintenance and Evolution of Breast Cancer: The Convergence of Irreversible Biological Actions of ER Alpha" Endocrines 2, no. 1: 1-14. https://doi.org/10.3390/endocrines2010001
APA StyleLeclercq, G. (2021). Pathological Maintenance and Evolution of Breast Cancer: The Convergence of Irreversible Biological Actions of ER Alpha. Endocrines, 2(1), 1-14. https://doi.org/10.3390/endocrines2010001