Abstract
Copy number variations (CNVs) have been implicated in various conditions of differences of sexual development (DSD). Generally, larger genomic aberrations are more often considered disease-causing or clinically relevant, but over time, smaller CNVs have been associated with various forms of DSD. The main objective of this study is to identify small CNVs and the smallest regions of overlap (SROs) in patients with atypical female genitalia (AFG) and build a CNV map of AFG. We queried the DECIPHER database for recurrent duplications and/or deletions detected across the genome of AFG individuals. From these data, we constructed a chromosome map consisting of SROs and investigated such regions for genes that may be associated with the development of atypical female genitalia. Our study identified 180 unique SROs (7.95 kb to 45.34 Mb) distributed among 22 chromosomes. The most SROs were found in chromosomes X, 17, 11, and 22. None were found in chromosome 3. From these SROs, we identified 22 genes as potential candidates. Although none of these genes are currently associated with AFG, a literature review indicated that almost half were potentially involved in the development and/or function of the reproductive system, and only one gene was associated with a disorder that reported an individual patient with ambiguous genitalia. Our data regarding novel SROs requires further functional investigation to determine the role of the identified candidate genes in the development of atypical female genitalia, and this paper should serve as a catalyst for downstream molecular studies that may eventually affect the genetic counseling, diagnosis, and management of these DSD patients.
1. Introduction
The development of female genital structures is controlled by a set of intricate biological processes. Disruptions at the genetic, endocrine, structural, and/or environmental level can lead to atypical or differences of sexual development (DSD), conditions associated with malformation and/or dysfunction of the reproductive system.
At the genetic level, development of the internal female reproductive tract depends on three finely-regulated processes: development of the Müllerian ducts, regression of the Wolffian ducts, and differentiation of the Müllerian ducts [1,2,3]. The reproductive tract is initially undifferentiated in the form of the Wolffian duct, which is derived from the mesonephros of the urinary tract and present in all embryos [4]. Expression of genes such as WNT4 induce formation of the Müllerian duct, which uses the preexisting Wolffian duct as a scaffold [2]. The presence of a Y chromosome, specifically the SRY gene, determines whether the primitive gonads differentiate into ovaries or testes. Without SRY, no anti-Müllerian hormone (AMH) or testosterone is produced, resulting in the maintenance of the Müllerian ducts and regression of the Wolffian ducts [2,3,4,5]. Maturation of the Müllerian ducts further depends on additional genetic pathways which result in development of the uterus, oviducts, cervix, and upper third of the vagina. Improper fusion of the Müllerian ducts or a failure of primitive tissue regression can lead to atypical structures such as bicornuate uterus or uterine or vaginal septum [1,4]. Development of the external female genitalia is less understood; however, TP63 has been identified as a major genetic player in the differentiation of urogenital mucosa and septation of the cloaca [6].
Despite tight genetic regulation, environmental factors can throw female reproductive tract development into disarray. Endocrine disrupting chemicals, which mimic or antagonize estrogens and androgens, are found throughout the environment, from nature and wildlife to commerce and laboratories. These xenoestrogens can affect fetal development in utero and even during pre-conception. For example, in the mid-1900s, physicians prescribed diethylstilbestrol (DES) to prevent miscarriage—it was later discovered that females exposed to DES in utero developed “T-shaped” uteruses which in turn predisposed them to worse pregnancy outcomes [2,7]. Furthermore, changes at the epigenetic level (e.g., DNA methylation, histone acetylation, etc.) can affect gene expression pre- and postnatally, leading to atypical reproductive structures and function [8].
Typically, DSD is classified into three major groups: 46, XY DSD, 46, XX DSD, and sex chromosome DSD [9,10]. Additionally, DSD can result from a standalone genetic change (i.e., isolated) or as a clinical feature of a greater disorder with congenital abnormalities unrelated to the reproductive system (i.e., syndromic) [3,11]. Some specific conditions include Mayer-Rokitansky-Küster-Hauser (MRKH) syndrome, congenital adrenal hyperplasia (CAH), ovotesticular DSD, and nonsyndromic testicular DSD, among others [1,10]. Overall, it has been estimated that DSD occurs in approximately 1 in 4500–5500 newborns [12,13,14].
While the rate of molecular diagnosis is higher in 46, XX DSD patients compared to 46, XY DSD patients (64% vs. 12% respectively), the majority, if not all, of those are diagnosed with CAH [11,15,16]. The remaining 46, XX DSD patients often do not know the genetic etiology of their conditions, and historically, such patients have been diagnosed using phenotypic presentation. Indeed, geneticists have begun focusing on patient comorbidities and malformative syndromes with documented DSD for clues into the genetic origin of atypical genital development [17,18].
Copy Number Variations. Chromosomal microarray analysis (CMA) has been utilized to uncover copy number variants (CNVs) that are potentially significant in the manifestation of DSD. For example, previous studies have shown that duplications of SOX9 [19,20] and NR0B1 [21,22] are relevant in the development of 46, XX and 46, XY DSD, respectively. Throughout the years, an abundance of CNV data has been published and stored in various public databases such as DECIPHER [23]. Utilization of CNV data from these resources allowed for the characterization and delineation of the smallest regions of overlap (SROs) that often result in the identification of candidate genes that may have clinical relevance.
In this retrospective study, we queried the DECIPHER database for genomic regions containing recurrent duplications and/or deletions in patients with atypical development of female external and internal genital structures. From these data, we constructed a whole genome map of SROs and further investigated such regions for candidate genes that may be associated with various forms of atypical female genitalia. We also expanded our phenotype analysis to include other non-genital anomalies that may be comorbid with atypical female sex development.
To the best of our knowledge, this is the first study to create a whole genome CNV map of atypical female genitalia and to identify corresponding candidate genes. Data from this study can be utilized for downstream research, and findings from all above-mentioned efforts may provide a better understanding of the genetic etiology of atypical female genitalia, facilitate diagnosis and genetic counseling, and improve patient management. Lastly, our data will further contribute to the development of a whole genome map for DSD overall.
2. Materials and Methods
2.1. Data Source and Collection
We searched DECIPHER’s open-access database (accessed 16 June 2020) to identify patients with documented “abnormality of the female genitalia” (thereafter referred to as atypical female genitalia when possible). Additionally, we categorized the phenotypes by whether they affected internal genitalia, external genitalia, or both. Some patients were categorized as “male” because they exhibited phenotypes such as hypospadias, cryptorchidism, and male “pseudohermaphroditism” (Supplementary Table S1).
2.2. Delineation of SROs
Using the chromosome map function “Browser” in DECIPHER, recurrent and overlapping copy number variable regions across the entire genome were identified. Within each CNV cluster (≥2 CNVs), an SRO map was created. Singular CNVs were disregarded. SROs were further classified as unabridged or extrapolated (Supplementary Figure S1). Unabridged SROs are defined by one patient’s full CNV and are completely contained within the confines of CNVs from all other patients in a given interval. Extrapolated SROs are not defined by one complete CNV—instead, CNVs from two individual patients establish the boundaries of the SRO (i.e., one CNV establishes the beginning of the SRO while another establishes the end).
Locus and size of each SRO were further determined using the UCSC Genome Browser (https://genome.ucsc.edu/; accessed on 16 June 2020) [24]. Chromosomal locations for each SRO were manually entered into Chromosome Analysis Suite 4.1 (ChAS; Affymetrix, Inc., Santa Clara, CA, USA) to generate the SRO CNV map. Gene content was determined by accessing each patient profile and their respective gene list. For extrapolated SROs, we obtained gene content by organizing genes by location and manually counting those, including non-protein coding, within the boundaries of the SRO. Based on DECIPHER data, genes were categorized as candidates for atypical female genitalia if they displayed a high likelihood of haploinsufficiency (%HI; 0.00–10.00) and/or the probability of loss-of-function (LoF) intolerance (pLI; 0.90–1.00).
2.3. Definition and Identification of Candidate Genes
In this study, we defined a candidate gene as one that has yet to undergo functional studies that prove causality between it and the development of AFG. The initial list of candidates contained hundreds of genes. To further narrow our list of candidate genes, we focused on small SROs (≤500 kb) that contained five or fewer haploinsufficient and/or LoF intolerant genes. We investigated whether these genes had previously been associated with DSD or the development/function of the reproductive system. PubMed was searched to identify studies that had associated the candidate genes with atypical development of the female genitalia. We searched using the following terms: (gene name) and (disorders of sexual development OR DSD OR intersex). A similar approach was used with Google. To confirm each gene’s novelty status, we accessed their individual OMIM phenotypes through their respective DECIPHER profiles. Candidate regions are SROs that contain no transcribed genes and may hold regulatory sequences, such as promoters, enhancers, silencers, etc.
2.4. Comorbidities
Within SROs, similarities in patients’ DSD phenotypes were assessed. Phenotypes were classified based on the structure/organ affected. Certain phenotypes, such as uterine neoplasm, primary amenorrhea, etc., received individual categories. The proportion of patients that exhibited the same phenotypes of abnormal female genitalia as the reference patient, whose CNV determined the SRO, was noted. For extrapolated SROs, of the two CNVs defining its boundaries, the patient who had the fewest number of overall phenotypes was selected as the reference patient. DSD phenotypes not exhibited by the reference patient but exhibited by non-reference patients were also recorded. An analogous process was used for non-DSD comorbidities which were listed in order of prevalence. Atypical female genitalia phenotypes were then grouped by affected region and assessed for recurring comorbidities.
3. Results
Genotypic and Phenotypic Characterization. We identified 300 patients (access date: 16 June 2020) with a spectrum of phenotypic findings including, but not limited to, aplasia/hypoplasia of reproductive structures, endocrine dysfunction, neoplasms, and cysts (Supplementary Table S1). There were 191 patients that only exhibited 1 DSD phenotype, while 108 exhibited two or more. One patient did not have any apparent DSD phenotypes. Additionally, 137 patients had DSD phenotypes that solely affected the internal genitalia while 94 patients had DSD phenotypes that only affected the external genitalia (Figure 1). Both internal and external genitalia were atypical in 25 patients. Very few patients (n = 5) exhibited atypical male phenotypes. 39 patients were classified as unspecified due to ambiguity in their reported phenotypes. Most of the patients (n = 264) that exhibited atypical female genitalia had a 46, XX sex chromosome complement, while 23 individuals were 46, XY (Figure 1). The remaining 13 did not have a known sex (n = 9) or were classified as “other” (n = 4). Patients exhibited a variety of phenotypes, with abnormality of the labia, abnormality of the uterus, and abnormality of the female genitalia being the most common (Figure 2).
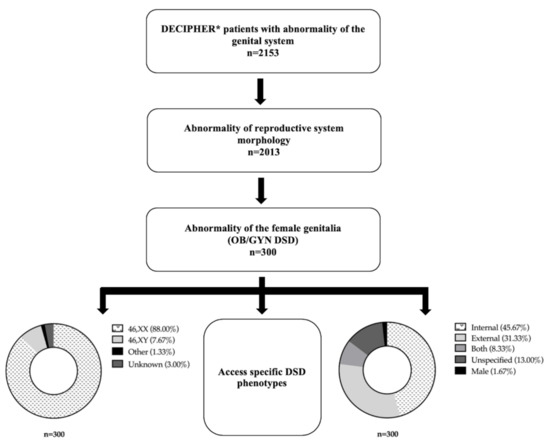
Figure 1.
Methodology for identifying and categorizing patients with atypical female genitalia. * Access date: 16 June 2020.
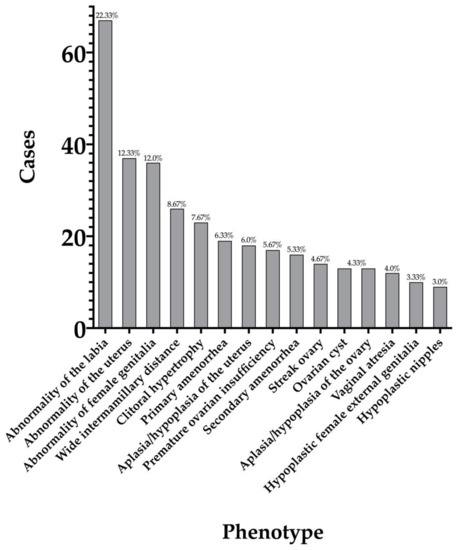
Figure 2.
Most frequent DSD phenotypes seen in patients with atypical female genitalia.
3.1. Delineation of SROs
We were able to identify 56 CNV clusters across the genome. Within those clusters, we identified 180 SROs throughout 22 pairs of chromosomes (Figure 3). The X chromosome contained the highest number of SROs (n = 35) while chromosomes 14 and Y had the least number of SROs with one each (Figure 4). Chromosome 3 was the only chromosome that did not contain any SROs. The majority (64%) of SROs were extrapolated while 65 were unabridged (Supplementary Figure S1). SROs tended to consist of a combination of deletions and duplications, with 64% containing both. Of the remaining SROs, 33% only contained deletions while a mere 3% solely contained duplications (Figure 5). SROs ranged from 7.95 kb to 45 Mb in size. Most SROs were larger than 1 Mb (70.00%), with only 10.56% (n = 19) being less than 250 kb (Figure 5).
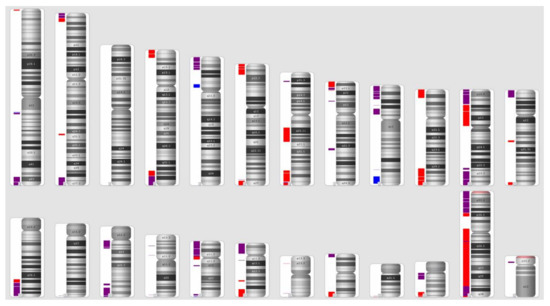
Figure 3.
SRO Distribution across the Genome. Red bars indicate SROs composed entirely of deletions while blue bars indicate SROs composed solely of duplications. Purple bars are SROs that feature both deletions and duplications.
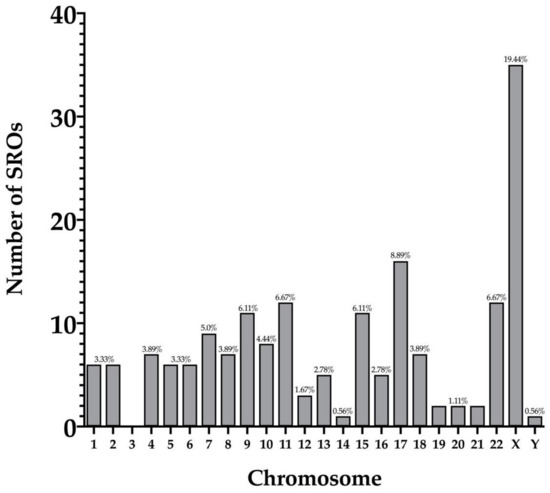
Figure 4.
Number of SROs per chromosome.
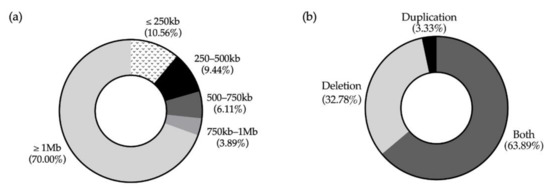
Figure 5.
Characteristics of SROs. (a) Breakdown of SROs by size. (b) Breakdown of SROs by CNV content.
3.2. Previously Described Genes and Regions
There were 25 SROs that overlapped with genes or regions that are known to cause or are associated with atypical female genitalia (Table 1). Only one region (CNV 22q11.21), containing six adjacent SROs, overlapped with candidate genes identified in our analysis.

Table 1.
Genes Known to be Associated with DSD 1.
3.3. Candidate Genes and Regions
Within the 180 SROs, 335 genes displayed a high probability of haploinsufficiency while 660 genes were considered extremely loss-of-function (LoF) intolerant (Table 2). Applying the SRO size and gene quantity limitations, we were left with 22 candidates to further investigate. Although none of the genes were widely associated with DSD, a literature review indicated that almost half were potentially involved in the development and/or function of the reproductive system (Table 3). Most of these genes did not exist in regions that were linked to DSD. In exception, GGNBP2, MAZ, SCARF2, MED15, UBE2L3, and MAPK1 overlapped with CNVs that are associated with MRKH syndrome types I and II [25,26]; however, none of these genes correlated to phenotypes/syndromes in Online Mendelian Inheritance in Man (OMIM). Additionally, six candidate genes had associated OMIM phenotypes, while the remaining 16 had no associated phenotypes. Of the six genes with OMIM phenotypes, only one was associated with a disorder that reported an individual patient with ambiguous genitalia [27].
Table 2.
List of SROs.
Table 3.
Candidate Genes.
3.4. Gene-Desert SROs
Four SROs did not contain any genes, including one that was considered for candidate genes (SRO087). They ranged from 29.28 kb to 684.32 kb. 75% of these SROs were extrapolated and consisted of both deletions and duplications. The most common DSD phenotype seen in these patients were abnormalities of the external genitalia (50%).
3.5. Comorbidities
The majority of our DSD patient population exhibited additional phenotypes unrelated to the reproductive system. Intellectual disability (47%), short stature (35%), micrognathia (27%), microcephaly (24%), and low-set ears (23%) were frequently encountered (Supplementary Table S2); however, certain female reproductive organs presented with unique comorbidities. Not infrequently, abnormality of the pinna, hypertelorism, atrial septal defect, short neck, and premature birth were seen with abnormalities of the uterus, while anal atresia and hypertelorism were common in patients with abnormalities of the vagina. Patients with abnormalities of the ovaries tended to be small for gestational age and have hypertelorism, while patients with abnormalities of the breasts and nipples tended to have muscular hypotonia. Comorbidities in patients with labial abnormalities and clitoral abnormalities aligned with those of the overall patient population, and there was only one patient with an abnormality of the fallopian tubes, so we could not determine the significance of the present comorbidities.
4. Discussion
Human sexual development requires a complicated synchronization of many biological elements that affect female reproductive structures and endocrine function. Dysregulation of a single gene or gene networks can lead to the atypical development and function of the reproductive system. Historically, clinicians relied on visible phenotypes to diagnose patients with 46, XX DSD. As advanced genomic technologies evolved, diagnoses of various forms of DSD have accelerated, resulting in a wealth of CNV data. Such data are used for further investigation of candidate genes and regions as well as downstream functional studies to elucidate clinical relevance.
4.1. DECIPHER
The database proved to be instrumental in the creation of our SRO map and our search for novel candidate genes. DECIPHER is unique in that persons and institutions can freely access its data and submit genetic information that is available for proceeding use. This quality makes it invaluable not only for research of DSD but also for research of all genetic variations. Additionally, DECIPHER’s inclusion of sex chromosome information that is more expansive than “male” and “female” takes away one of the obstacles faced by DSD researchers while also being affirmative to patient sex identity [28].
4.2. Delineation of SROs
It has already been shown that small CNVs are potentially involved in the development of DSD [29,30,31]. Therefore, we focused on SROs less than 500 kb in size because they are often overlooked when assessing the genome for candidate genes. Although many SROs were large in size (>500 kb) and therefore outside our range of focus, these data can be used in future investigations. With further analysis, these larger CNVs can be partitioned into smaller regions that eventually pinpoint the etiology of various AFG.
4.3. Candidate Genes and Regions
Clinical medicine has often excluded small CNVs in the genomic analysis of DSD. Over the past decade, the identification of small deletions and duplications has improved diagnostics given that approximately 30% of all DSD patients cannot be classified as sex chromosome, 46, XY, or 46, XX DSD [32]. Furthermore, 19–21% of 46, XX individuals receive a molecular diagnosis [33,34]. Previous studies have shown that small CNV analysis can reveal genes/regions that are relevant in sex development [29,30]. For instance, a 5.2 kb region upstream of SOX9 was found to contain regulatory elements for sex development in both 46, XX and 46, XY individuals [31]. Our study further demonstrates the promise of small (<500 kb) CNVs when performing genetic testing for patients with atypical female genitalia. We would argue that size does not matter, but gene content does.
We identified 36 small SROs (<500 kb) that contained 22 candidate genes with %HI < 10% and pLI scores > 0.90 (Table 3). The majority of these genes did not overlap with genes or regions that were previously documented in cases of 46, XX DSD. Identification of these regions further endorses the importance of deletions and duplications in sexual development, and given the use of HI and pLI as predictive tools, our data can steer scientists towards relevant genes for functional studies. Some SROs (SRO102, SRO115, SROs135-141) overlapped with CNVs that have been identified as potentially pathogenic [25,26,35]; however, studies don’t often address these novel genes, and the candidates have yet to be associated with DSD phenotypes in OMIM. For example, GGNBP2 is located within the region 17q12, which is commonly associated with MRKH types I and II [36,37,38], but remains to be further explored as a candidate gene despite its role in testes morphology and spermatogenesis [39]. Other dosage-sensitive genes housed within common CNVs have also yet to be thoroughly investigated. MAZ, located in region 16p11.2, is a dosage-sensitive transcription factor that is responsible, in part, for genitourinary development [40]. While one CNV study briefly mentions MAZ in its methodology [37], it exists in the periphery of the study’s focus, and the literature is otherwise devoid of information on MAZ’s role in GU development.
Within regions 22q11.21 and 16p11.2 exist two genes, MAPK1 and MAPK3, respectively, that have the potential to become more explicitly associated with DSD. MAPK1 and MAPK3 function as kinases downstream of MAP3K1, which has an established role in DSD. Increased phosphorylation of MAPK1 and MAPK3 due to gain-of-function mutations in MAP3K1 leads to upregulation of FOXL2 and FST [41,42], additional genes with roles in ovary formation [43,44,45]. It is also known that MAPK1/3 plays an important role in luteinizing hormone signal transduction during ovulation [46]. The dosage-sensitivity of MAPK1 and MAPK3 further supports evidence that CNVs in these regions lead to the development of ambiguous genitalia.
SCARF2 presents an interesting case: Despite its presence on an individual academic center’s DSD panel [47], there is no evidence explicitly stating the gene’s role in development of AFG. However, like GGNBP2, it is located in a region (22q11.21) that commonly exhibits CNVs that are associated with ambiguous genitalia and MRKH types I and II [26,35]. Currently, it is known that mutations in SCARF2 cause the rare, autosomal recessive disorder Van Den Ende-Gupta syndrome (VDEGS; OMIM: 600920) which is mainly characterized by skeletal and craniofacial phenotypes [48]; however, a single case of VDEGS reportedly exhibited ambiguous genitalia [27]. Interestingly, VDEGS maps to distal 22q11.2, which contains the critical region responsible for DiGeorge syndrome (OMIM: 188400). Similar to VDEGS, patients with DiGeorge syndrome have distinct abnormal facies but with added cardiac defects, thymic hypoplasia, and hypocalcemia [49]. Less frequently, 46, XX patients may exhibit genitourinary anomalies such as absent uterus or uterine didelphys [50,51]. While DiGeorge syndrome’s distinguishing phenotypes can be attributed to a haploinsufficiency of TBX1 [49], the genetic etiology of the genitourinary phenotypes is not well understood. Further investigation into the function of SCARF2 may resolve the association between CNV 22q11.2 and abnormalities of female genitalia while also shedding light as to why 46, XX patients with VDEGS and DiGeorge syndrome exhibit genital anomalies.
Outside of identified but unexplored CNVs, there still exist genes that are prime for inquiry. PAPPA is an insulin-like growth factor binding protein (IGFBP) protease that increases bioavailable insulin growth factor which, in turn, promotes the development of a dominant ovarian follicle [52,53]. Predictably, when PAPPA is knocked out, follicular development and ovarian function in female murine models is disrupted, resulting in decreased fertility [54] and suggesting that the gene plays a significant role in the ovary’s functional integrity. Nonetheless, to our knowledge, PAPPA is not yet under consideration as a potential candidate gene for 46, XX DSD.
SENP3 and EIF4A1 are jointly transcribed and eventually spliced into their respective, individual transcripts. Despite their different functions at the molecular level, they both seem to play a unique role in fertility. During meiosis, SENP3, a deSUMOylation protease, is required for the G2-M transition. Downregulation of the gene disrupts spindle assembly and germinal vesicle breakdown, which prevents the first polar body extrusion and, therefore, proper oocyte maturation [55]. Unlike SENP3, it is suggested that EIF4A1 plays a critical role post-fertilization. As a translation initiation factor, EIF4A1 produces proteins that are necessary for early embryonic cell division. Sans EIF4A1, there is inadequate growth of the blastocyst, leading to implantation failure and decreased fertility [56]. Additionally, it should be noted that point mutations in the EIF4B family transcription factor family can result in gonadal dysgenesis and amenorrhea in 46, XX individuals [57]. SENP3 and EIF4A1 flock the point of fertilization and seem to play significant roles in oocyte integrity. The potential of these genes leads us to suggest further investigation into their effects on female reproductive system development.
4.4. Gene-Desert SROs
SROs lacking expressed genes should also not be ignored, for they may contain DNA sequences that are necessary for the regulation of genes involved in the development of the female reproductive tract. In 2011, Benko et al. identified a 78 kb non-coding regulatory region upstream of SOX9. A few years later, this sex-determining region was whittled down to approximately 5.2 kb [31,58]. Similarly, rearrangements in the regulatory region of SOX3 have been found to be associated with sex reversal in 46, XX males [59]. Despite the landscape being devoid of genes, non-coding regions of the genome hold unfound potential in understanding human sex development.
4.5. Comorbidities
Although anomalies affecting every organ system were revealed, the most common comorbidities were intellectual disability, short stature, and micrognathia. Interestingly, we found that intellectual disability and other developmental delays occurred notably less frequently in other studies, and when separated by sex, 46, XX patients were found to have no incidences of short stature [11,17]. The prevalence of these comorbidities in our patient population is important to note, given that DSD diagnosis has historically relied on clinical presentation. Turner syndrome is often the first diagnosis considered in female-presenting DSD patients when short stature, intellectual disability, and cardiac anomalies are present; karyotypes are typically used to diagnose this condition [60,61]. Turner patients also tend to exhibit smaller mandible size, along with retrognathia and mandibular posterior rotation [62,63,64]. Additionally, most individuals with alpha-thalassemia X-linked intellectual disability (ATRX) syndrome have comorbid undifferentiated streak gonads [18]. Furthermore, MRKH phenotypes can occur in Silver-Russel syndrome, which is characterized by abnormalities of the skeletal system such as short stature and micrognathia with narrow chin [65,66,67]. Given the concurrence of these phenotypes, clinicians should perhaps approach DSD with a wider scope and consider other whole genome technologies, such as CMA, when assessing patients and performing targeted analysis of DSD genes and regions.
Although our study focused on patients with atypical female genitalia who were presumably 46, XX, a small proportion (7.67%) of patients were 46, XY. These patients displayed a wide range of DSD phenotypes, but most common were abnormalities of the ovary, uterus, and labia. Five of these patients had what would be considered male phenotypes such as cryptorchidism, hypospadias, and male DSD (“pseudohermaphroditism”). The remaining 18 patients only exhibited female DSD phenotypes. Interestingly, despite the fair number of patients with 46, XY phenotype, we found only one SRO on chromosome Y, and it contained no genes with high likelihood of haploinsufficiency or loss-of-function intolerance. Regardless of the presence of candidate genes, it is nonetheless necessary to follow-up with these patients. 46, XY females with gonadal dysgenesis are at higher risk of developing germ cell tumors (e.g., gonadoblastoma, dysgerminoma, Sertoli cell tumor, etc.) [68,69]. Endocrine, gastrointestinal, and congenital comorbidities are also frequent, and these patients should be assessed and counseled accordingly [70].
4.6. Limitations and Recommendations
The generation of our SRO map depended on the properties of the CNVs reported in the DECIPHER database. Despite our expectations, many of the deletions and/or duplications found in patients were large, which in turn led to massive SROs outside the scope of this study. There are a few likely reasons for the skewed presentation of size; the current standard for CMA tends to disregard smaller CNVs potentially leading to underreporting. Additionally, DECIPHER collects data from institutions internationally which utilize different array technologies and techniques when collecting patients’ genetic information. It is for these reasons that we present our SRO map not as a complete and conclusive coordinate system but as a guide towards regions of the genome that are promising for future study. Furthermore, we would argue that these larger regions often contain an overabundance of data that is difficult to sieve through. Larger CNVs must be resolved at finer detail, and we suggest that subsequent studies focus on smaller regions in order to pinpoint significant candidate genes.
Not infrequently, DECIPHER was lacking in data regarding the haploinsufficiency and loss of function effects of genes. Therefore, it is possible that potential candidate genes were missed given our method of selection. Regardless, our study provides adequate fodder for inquiry, allowing scientists to utilize our data to predict gene HI and pLI in future functional studies.
When combing the literature for previously established associations with atypical female genitalia, it was impossible to perform a complete and total search on each candidate gene. There is a slight possibility that we overlooked a study that tied a candidate gene to DSD, especially if the study indicated a CNV region but didn’t address the genes within. However, given our search protocol, we believe we adequately assessed the literature for existing evidence.
It is still necessary to discuss an apparent disconnect between studies on specific genes and those on CNVs. We found quite a few studies that investigated large CNVs that are associated with the manifestation of MRKH [36,37,38], yet there existed no follow-up studies probing potential candidate genes found in those regions. These CNV regions proved valuable in better understanding the etiology of the disorder; nonetheless, it is important to resolve the genes in said regions as to understand why certain CNVs lead to pathological states.
Our final recommendation regards neither genes nor CNVs. Instead, it addresses DECIPHER’s classification system. While analyzing the database, we came across DSD phenotypes that included the term “hermaphroditism”. Only a few patients were cataloged using this terminology; nonetheless, using “hermaphroditism” to describe patients is both unhelpful and pejorative. Many intersex patients consider the term outdated and stigmatizing [14], and clinically, “hermaphroditism” is problematic given that it focuses on gonadal anatomy and does not consider other aspects of atypical genital development nor gender identity [9,71,72]. Classifications centered on “hermaphroditism” should be removed from DECIPHER, and instead, clinicians should record phenotypes that are specific to organ morphology and dysfunction [28]. This follows for other classifications such as general “abnormality of the female genitalia” and “ambiguous genitalia, female”. Since these classifications provide no insight into the patient’s biology or lived experience, we recommend a more affirmative term such as “atypical” instead of “abnormal”. Understandably, it may be impossible to dispose of these classifications altogether, depending on the extent of data submitted; nevertheless, physicians should be encouraged to enter more detailed phenotypes when adding patient information into DECIPHER.
5. Conclusions
This study successfully identified small regions of overlap in patients with atypical development of female genitalia. Our findings suggest that CMA can be used to identify smaller, clinically significant CNVs which have historically been disregarded. The genes within these regions have an untapped potential which, upon further investigation, may provide a better understanding of the genetic etiology of the development of atypical female genitalia. We are hopeful that this study will inspire exploration of these candidate genes and lead to better diagnosis and management of individuals with AFG.
Supplementary Materials
The following are available online at https://www.mdpi.com/article/10.3390/reprodmed3020014/s1, Figure S1: Examples of Clusters, Table S1: DSD Characteristics of Patients with AFG, Table S2: Common Comorbidities in Patients with AFG.
Author Contributions
I.E.A. conceptualized and designed the study and reviewed manuscript; A.U.A. collected and analyzed data and wrote the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
A.U.A. received a $5275 stipend from Washington University School of Medicine’s Office of Medical Student Research through the Dean’s Fellowship.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Informed consent was obtained through the DECIPHER Project.
Data Availability Statement
The data presented in this study are available on request from the corresponding author. The data are not publicly available due to data usage agreement with DECIPHER Project.
Acknowledgments
This study makes use of data generated by the DECIPHER community. A full list of centres who contributed to the generation of the data is available from https://deciphergenomics.org/about/stats (accessed on 16 June 2020) and via email from contact@deciphergenomics.org. Funding for the DECIPHER project was provided by Wellcome. Those who carried out the original data collection and analysis bear no responsibility for later data interpretation or analysis. The first author received funding through the Dean’s Fellowship via the Washington University School of Medicine’s Office of Medical Student Research.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Connell, M.T.; Owen, C.M.; Segars, J.H. Genetic Syndromes and Genes Involved in the Development of the Female Reproductive Tract: A Possible Role for Gene Therapy. J. Genet. Syndr. Gene Ther. 2013, 4, 127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Massé, J.; Watrin, T.; Laurent, A.; Deschamps, S.; Guerrier, D.; Pellerin, I. The Developing Female Genital Tract: From Genetics to Epigenetics. Int. J. Dev. Biol. 2009, 53, 411–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ono, M.; Harley, V.R. Disorders of Sex Development: New Genes, New Concepts. Nat. Rev. Endocrinol. 2013, 9, 79–91. [Google Scholar] [CrossRef] [PubMed]
- Horii, M.; Boyd, T.K.; Quade, B.J.; Crum, C.P.; Parast, M.M. Chapter 1—Female Genital Tract Development and Disorders of Childhood. In Diagnostic Gynecologic and Obstetric Pathology; Elsevier: Philadelphia, PA, USA, 2018; pp. 1–21. ISBN 9780323447324. [Google Scholar]
- MacLaughlin, D.T.; Teixeira, J.; Donahoe, P.K. Perspective: Reproductive Tract Development—New Discoveries and Future Directions. Endocrinology 2001, 142, 2167–2172. [Google Scholar] [CrossRef]
- Ince, T.A.; Cviko, A.P.; Quade, B.J.; Yang, A.; McKeon, F.D.; Mutter, G.L.; Crum, C.P. P63 Coordinates Anogenital Modeling and Epithelial Cell Differentiation in the Developing Female Urogenital Tract. Am. J. Pathol. 2002, 161, 1111–1117. [Google Scholar] [CrossRef] [Green Version]
- Kaufman, R.H.; Adam, E.; Binder, G.L.; Gerthoffer, E. Upper Genital Tract Changes and Pregnancy Outcome in Offspring Exposed in Utero to Diethylstilbestrol. Am. J. Obstet. Gynecol. 1980, 137, 299–308. [Google Scholar] [CrossRef]
- Ho, S.M.; Cheong, A.; Adgent, M.A.; Veevers, J.; Suen, A.A.; Tam, N.N.C.; Leung, Y.K.; Jefferson, W.N.; Williams, C.J. Environmental Factors, Epigenetics, and Developmental Origin of Reproductive Disorders. Reprod. Toxicol. 2017, 68, 85–104. [Google Scholar] [CrossRef] [Green Version]
- Dreger, A.D.; Chase, C.; Sousa, A.; Gruppuso, P.A.; Frader, J. Changing the Nomenclature/Taxonomy for Intersex: A Scientific and Clinical Rationale. J. Pediatric Endocrinol. Metab. 2005, 18, 729–733. [Google Scholar] [CrossRef]
- Lee, P.A.; Houk, C.P.; Ahmed, S.F.; Hughes, I.A.; Achermann, J.; Ahmed, F.; Baskin, L.; Berenbaum, S.; Bertelloni, S.; Brock, J.; et al. Consensus Statement on Management of Intersex Disorders. Pediatrics 2006, 118, e488–e500. [Google Scholar] [CrossRef] [Green Version]
- Heeley, J.M.; Hollander, A.S.; Austin, P.F.; Merritt, D.F.; Wesevich, V.G.; Amarillo, I.E. Risk Association of Congenital Anomalies in Patients with Ambiguous Genitalia: A 22-Year Single-Center Experience. J. Pediatric Urol. 2018, 14, 153.e1–153.e7. [Google Scholar] [CrossRef]
- Sax, L. How Common Is Intersex? A Response to Anne Fausto-Sterling. J. Sex Res. 2002, 39, 174–178. [Google Scholar] [CrossRef]
- Thyen, U.; Lanz, K.; Holterhus, P.M.; Hiort, O. Epidemiology and Initial Management of Ambiguous Genitalia at Birth in Germany. Horm. Res. 2006, 66, 195–203. [Google Scholar] [CrossRef]
- Lee, P.A.; Nordenström, A.; Houk, C.P.; Ahmed, S.F.; Auchus, R.; Baratz, A.; Baratz Dalke, K.; Liao, L.M.; Lin-Su, K.; Looijenga, L.H.J.; et al. Global Disorders of Sex Development Update since 2006: Perceptions, Approach and Care. Horm. Res. Paediatr. 2016, 85, 158–180. [Google Scholar] [CrossRef]
- Öcal, G.; Berberoğlu, M.; Siklar, Z.; Aycan, Z.; Hacihamdioglu, B.; Erdeve, Ş.S.; Çamtosun, E.; Kocaay, P.; Ruhi, H.I.; Kiliç, B.G.; et al. Clinical Review of 95 Patients with 46,Xx Disorders of Sex Development Based on the New Chicago Classification. J. Pediatric Adolesc. Gynecol. 2015, 28, 6–11. [Google Scholar] [CrossRef]
- De Paula, G.B.; Barros, B.A.; Carpini, S.; Tincani, B.J.; Mazzola, T.N.; Sanches Guaragna, M.; Piveta, C.S.D.C.; De Oliveira, L.C.; Andrade, J.G.R.; Guaragna-Filho, G.; et al. 408 Cases of Genital Ambiguity Followed by Single Multidisciplinary Team during 23 Years: Etiologic Diagnosis and Sex of Rearing. Int. J. Endocrinol. 2016, 2016, 4963574. [Google Scholar] [CrossRef] [Green Version]
- Cox, K.; Bryce, J.; Jiang, J.; Rodie, M.; Sinnott, R.; Alkhawari, M.; Arlt, W.; Audi, L.; Balsamo, A.; Bertelloni, S.; et al. Novel Associations in Disorders of Sex Development: Findings from the I-DSD Registry. J. Clin. Endocrinol. Metab. 2014, 99, E348–E355. [Google Scholar] [CrossRef] [Green Version]
- Hutson, J.M.; Grover, S.R.; O’Connell, M.; Pennell, S.D. Malformation Syndromes Associated with Disorders of Sex Development. Nat. Publ. Group 2014, 10, 476–487. [Google Scholar] [CrossRef]
- Huang, B.; Wang, S.; Ning, Y.; Lamb, A.N.; Bartley, J. Autosomal XX Sex Reversal Caused by Duplication of SOX9. Am. J. Med. Genet. 1999, 87, 349–353. [Google Scholar] [CrossRef]
- Refai, O.; Friedman, A.; Terry, L.; Jewett, T.; Pearlman, A.; Perle, M.A.; Ostrer, H. De Novo 12;17 Translocation Upstream of Sox9 Resulting in 46,Xx Testicular Disorder of Sex Development. Am. J. Med. Genet. Part A 2010, 152, 422–426. [Google Scholar] [CrossRef]
- Bardoni, B.; Zanaria, E.; Guioli, S.; Floridia, G.; Worley, K.C.; Tonini, G.; Ferrante, E.; Chiumello, G.; McCabe, E.R.B.; Fraccaro, M.; et al. A Dosage Sensitive Locus at Chromosome Xp21 Is Involved in Male to Female Sex Reversal. Nat. Genet. 1994, 7, 497–501. [Google Scholar] [CrossRef]
- Smyk, M.; Berg, J.S.; Pursley, A.; Curtis, F.K.; Fernandez, B.A.; Bien-Willner, G.A.; Lupski, J.R.; Cheung, S.W.; Stankiewicz, P. Male-to-Female Sex Reversal Associated with an ∼250 Kb Deletion Upstream of NR0B1 (DAX1). Hum. Genet. 2007, 122, 63–70. [Google Scholar] [CrossRef]
- Firth, H.V.; Richards, S.M.; Bevan, A.P.; Clayton, S.; Corpas, M.; Rajan, D.; van Vooren, S.; Moreau, Y.; Pettett, R.M.; Carter, N.P. DECIPHER: Database of Chromosomal Imbalance and Phenotype in Humans Using Ensembl Resources. Am. J. Hum. Genet. 2009, 84, 524–533. [Google Scholar] [CrossRef] [Green Version]
- Kent, W.; Sugnet, C.; Furey, T.; Roskin, K.; Pringle, T.; Zahler, A.; Haussler, D. UCSC Genome Browser. Available online: https://genome.ucsc.edu/cite.html (accessed on 10 January 2021).
- Demir Eksi, D.; Shen, Y.; Erman, M.; Chorich, L.P.; Sullivan, M.E.; Bilekdemir, M.; Yllmaz, E.; Luleci, G.; Kim, H.G.; Alper, O.M.; et al. Copy Number Variation and Regions of Homozygosity Analysis in Patients with Müllerian Aplasia. Mol. Cytogenet. 2018, 11, 13. [Google Scholar] [CrossRef]
- Audí, L.; Ahmed, S.F.; Krone, N.; Cools, M.; McElreavey, K.; Holterhus, P.M.; Greenfield, A.; Bashamboo, A.; Hiort, O.; Wudy, S.A.; et al. Genetics in Endocrinology: Approaches to Molecular Genetic Diagnosis in the Management of Differences/Disorders of Sex Development (DSD): Position Paper of EU COST Action BM 1303 “DSDnet”. Eur. J. Endocrinol. 2018, 179, R197–R206. [Google Scholar] [CrossRef] [Green Version]
- Bistritzer, T.; Fried, K.; Lahat, E.; Dvir, M.; Goldberg, M. Congenital Contractural Arachnodactyly in Two Double Second Cousins: Possible Homozygosity. Clin. Genet. 1993, 44, 15–19. [Google Scholar] [CrossRef]
- Theisen, J.G.; Amarillo, I.E. Creating Affirmative and Inclusive Practices When Providing Genetic and Genomic Diagnostic and Research Services to Gender-Expansive and Transgender Patients. J. Appl. Lab. Med. 2021, 6, 142–154. [Google Scholar] [CrossRef]
- Hagan, A.; Amarillo, I.E. Small Copy-Number Variations Involving Genes of the FGF Pathway in Differences in Sex Development. Hum. Genome Var. 2017, 4, 17011. [Google Scholar] [CrossRef] [Green Version]
- Amarillo, I.E.; Nievera, I.; Hagan, A.; Huchthagowder, V.; Heeley, J.; Hollander, A.; Koenig, J.; Austin, P.; Wang, T. Integrated Small Copy Number Variations and Epigenome Maps of Disorders of Sex Development. Hum. Genome Var. 2016, 3, 16012. [Google Scholar] [CrossRef]
- Croft, B.; Ohnesorg, T.; Hewitt, J.; Bowles, J.; Quinn, A.; Tan, J.; Corbin, V.; Pelosi, E.; van den Bergen, J.; Sreenivasan, R.; et al. Human Sex Reversal Is Caused by Duplication or Deletion of Core Enhancers Upstream of SOX9. Nat. Commun. 2018, 9, 5319. [Google Scholar] [CrossRef] [Green Version]
- García-Acero, M.; Moreno-Niño, O.; Suárez-Obando, F.; Molina, M.; Manotas, M.C.; Prieto, J.C.; Forero, C.; Céspedes, C.; Pérez, J.; Fernandez, N.; et al. Disorders of Sex Development: Genetic Characterization of a Patient Cohort. Mol. Med. Rep. 2020, 21, 97–106. [Google Scholar] [CrossRef] [Green Version]
- Kohva, E.; Miettinen, P.J.; Taskinen, S.; Hero, M.; Tarkkanen, A.; Raivio, T. Disorders of Sex Development: Timing of Diagnosis and Management in a Single Large Tertiary Center. Endocr. Connect. 2018, 7, 595–603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eggers, S.; Sadedin, S.; van den Bergen, J.A.; Robevska, G.; Ohnesorg, T.; Hewitt, J.; Lambeth, L.; Bouty, A.; Knarston, I.M.; Tan, T.Y.; et al. Disorders of Sex Development: Insights from Targeted Gene Sequencing of a Large International Patient Cohort. Genome Biol. 2016, 17, 243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tannour-Louet, M.; Han, S.; Corbett, S.T.; Louet, J.F.; Yatsenko, S.; Meyers, L.; Shaw, C.A.; Kang, S.H.L.; Cheung, S.W.; Lamb, D.J. Identification of De Novo Copy Number Variants Associated with Human Disorders of Sexual Development. PLoS ONE 2010, 5, e15392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheroki, C.; Krepischi-Santos, A.C.V.; Szuhai, K.; Brenner, V.; Kim, C.A.E.; Otto, P.A.; Rosenberg, C. Genomic Imbalances Associated with Müllerian Aplasia. J. Med. Genet. 2008, 45, 228–232. [Google Scholar] [CrossRef]
- Nik-Zainal, S.; Strick, R.; Storer, M.; Huang, N.; Rad, R.; Willatt, L.; Fitzgerald, T.; Martin, V.; Sandford, R.; Carter, N.P.; et al. High Incidence of Recurrent Copy Number Variants in Patients with Isolated and Syndromic Müllerian Aplasia. J. Med. Genet. 2011, 48, 197–204. [Google Scholar] [CrossRef] [Green Version]
- Williams, L.S.; Demir Eksi, D.; Shen, Y.; Lossie, A.C.; Chorich, L.P.; Sullivan, M.E.; Phillips, J.A.; Erman, M.; Kim, H.G.; Alper, O.M.; et al. Genetic Analysis of Mayer-Rokitansky-Kuster-Hauser Syndrome in a Large Cohort of Families. Fertil. Steril. 2017, 108, 145–151.e2. [Google Scholar] [CrossRef] [Green Version]
- Chen, A.; Li, J.; Song, L.; Ji, C.; Böing, M.; Chen, J.; Brand-Saberi, B. GGNBP2 Is Necessary for Testis Morphology and Sperm Development. Sci. Rep. 2017, 7, 2998. [Google Scholar] [CrossRef]
- Haller, M.; Au, J.; O’Neill, M.; Lamb, D.J. 16p11.2 Transcription Factor MAZ Is a Dosage-Sensitive Regulator of Genitourinary Development. Proc. Natl. Acad. Sci. USA 2018, 115, E1849–E1858. [Google Scholar] [CrossRef] [Green Version]
- Chamberlin, A.; Huether, R.; Machado, A.Z.; Groden, M.; Liu, H.M.; Upadhyay, K.; Vivian, O.; Gomes, N.L.; Lerario, A.M.; Nishi, M.Y.; et al. Mutations in MAP3K1 That Cause 46,XY Disorders of Sex Development Disrupt Distinct Structural Domains in the Protein. Hum. Mol. Genet. 2019, 28, 1620–1628. [Google Scholar] [CrossRef]
- Loke, J.; Pearlman, A.; Radi, O.; Zuffardi, O.; Giussani, U.; Pallotta, R.; Camerino, G.; Ostrer, H. Mutations in MAP3K1 Tilt the Balance from SOX9/FGF9 to WNT/β-Catenin Signaling. Hum. Mol. Genet. 2014, 23, 1073–1083. [Google Scholar] [CrossRef] [Green Version]
- Uda, M.; Ottolenghi, C.; Crisponi, L.; Garcia, J.E.; Deiana, M.; Kimber, W.; Forabosco, A.; Cao, A.; Schlessinger, D.; Pilia, G. Foxl2 Disruption Causes Mouse Ovarian Failure by Pervasive Blockage of Follicle Development. Hum. Mol. Genet. 2004, 13, 1171–1181. [Google Scholar] [CrossRef] [Green Version]
- Ottolenghi, C.; Omari, S.; Garcia-Ortiz, J.E.; Uda, M.; Crisponi, L.; Forabosco, A.; Pilia, G.; Schlessinger, D. Foxl2 Is Required for Commitment to Ovary Differentiation. Hum. Mol. Genet. 2005, 14, 2053–2062. [Google Scholar] [CrossRef]
- Yao, H.H.C.; Matzuk, M.M.; Jorgez, C.J.; Menke, D.B.; Page, D.C.; Swain, A.; Capel, B. Follistatin Operates Downstream of Wnt4 in Mammalian Ovary Organogenesis. Dev. Dyn. 2004, 230, 210–215. [Google Scholar] [CrossRef] [Green Version]
- Fan, H.Y.; Liu, Z.; Shimada, M.; Sterneck, E.; Johnson, P.F.; Hedrick, S.M.; Richards, J.S. MAPK3/1 (ERK1/2) in Ovarian Granulosa Cells Are Essential for Female Fertility. Science 2009, 324, 938–941. [Google Scholar] [CrossRef] [Green Version]
- Abnormal/Ambiguous Genitalia Panel|The University of Chicago Genetic Services. Available online: https://dnatesting.uchicago.edu/tests/abnormalambiguous-genitalia-panel (accessed on 13 September 2020).
- Anastasio, N.; Ben-Omran, T.; Teebi, A.; Ha, K.C.H.; Lalonde, E.; Ali, R.; Almureikhi, M.; der Kaloustian, V.M.; Liu, J.; Rosenblatt, D.S.; et al. Mutations in SCARF2 Are Responsible for van Den Ende-Gupta Syndrome. Am. J. Hum. Genet. 2010, 87, 553–559. [Google Scholar] [CrossRef] [Green Version]
- Jerome, L.A.; Papaioannou, V.E. DiGeorge Syndrome Phenotype in Mice Mutant for the T-Box Gene, Tbx1. Nat. Genet. 2001, 27, 286–291. [Google Scholar] [CrossRef]
- Sundaram, U.T.; McDonald-McGinn, D.M.; Huff, D.; Emanuel, B.S.; Zackai, E.H.; Driscoll, D.A.; Bodurtha, J. Primary Amenorrhea and Absent Uterus in the 22q11.2 Deletion Syndrome. Am. J. Med. Genet. Part A 2007, 143, 2016–2018. [Google Scholar] [CrossRef] [Green Version]
- Scheuerle, A. Teenager with Uterine Didelphys, Absent Kidney and 22q11.2 Deletion. Am. J. Med. Genet. Part A 2008, 146, 800–801. [Google Scholar] [CrossRef]
- Conover, C.A.; Faessen, G.F.; Ilg, K.E.; Chandrasekher, Y.A.; Christiansen, M.; Overgaard, M.T.; Oxvig, C.; Giudice, L.C. Pregnancy-Associated Plasma Protein-A Is the Insulin-like Growth Factor Binding Protein-4 Protease Secreted by Human Ovarian Granulosa Cells and Is a Marker of Dominant Follicle Selection and the Corpus Luteum. Endocrinology 2001, 142, 2155. [Google Scholar] [CrossRef]
- Hourvitz, A.; Kuwahara, A.; Hennebold, J.D.; Tavares, A.B.; Negishi, H.; Lee, T.H.; Erickson, G.F.; Adashi, E.Y. The Regulated Expression of the Pregnancy-Associated Plasma Protein-A in the Rodent Ovary: A Proposed Role in the Development of Dominant Follicles and of Corpora Lutea. Endocrinology 2002, 143, 1833–1844. [Google Scholar] [CrossRef]
- Nyegaard, M.; Overgaard, M.T.; Su, Y.Q.; Hamilton, A.E.; Kwintkiewicz, J.; Hsieh, M.; Nayak, N.R.; Conti, M.; Conover, C.A.; Giudice, L.C. Lack of Functional Pregnancy-Associated Plasma Protein-A (PAPPA) Compromises Mouse Ovarian Steroidogenesis and Female Fertility. Biol. Reprod. 2010, 82, 1129–1138. [Google Scholar] [CrossRef] [Green Version]
- Huang, C.J.; Wu, D.; Khan, F.A.; Huo, L.J. The SUMO Protease SENP3 Orchestrates G2-M Transition and Spindle Assembly in Mouse Oocytes. Sci. Rep. 2015, 5, 15600. [Google Scholar] [CrossRef] [Green Version]
- Plaks, V.; Gershon, E.; Zeisel, A.; Jacob-Hirsch, J.; Neeman, M.; Winterhager, E.; Rechavi, G.; Domany, E.; Dekel, N. Blastocyst Implantation Failure Relates to Impaired Translational Machinery Gene Expression. Reproduction 2014, 148, 87–98. [Google Scholar] [CrossRef] [Green Version]
- Yatsenko, S.A.; Witchel, S.F. Genetic Approach to Ambiguous Genitalia and Disorders of Sex Development: What Clinicians Need to Know. Semin. Perinatol. 2017, 41, 232–243. [Google Scholar] [CrossRef]
- Benko, S.; Gordon, C.T.; Mallet, D.; Sreenivasan, R.; Thauvin-Robinet, C.; Brendehaug, A.; Thomas, S.; Bruland, O.; David, M.; Nicolino, M.; et al. Disruption of a Long Distance Regulatory Region Upstream of SOX9 in Isolated Disorders of Sex Development. J. Med. Genet. 2011, 48, 825–830. [Google Scholar] [CrossRef]
- Sutton, E.; Hughes, J.; White, S.; Sekido, R.; Tan, J.; Arboleda, V.; Rogers, N.; Knower, K.; Rowley, L.; Eyre, H.; et al. Identification of SOX3 as an XX Male Sex Reversal Gene in Mice and Humans. J. Clin. Investig. 2011, 121, 328–341. [Google Scholar] [CrossRef] [Green Version]
- Gravholt, C.H.; Andersen, N.H.; Conway, G.S.; Dekkers, O.M.; Geffner, M.E.; Klein, K.O.; Lin, A.E.; Mauras, N.; Quigley, C.A.; Rubin, K.; et al. Clinical Practice Guidelines for the Care of Girls and Women with Turner Syndrome: Proceedings from the 2016 Cincinnati International Turner Syndrome Meeting. Eur. J. Endocrinol. 2017, 177, G1–G70. [Google Scholar] [CrossRef]
- Mazzocco, M.M.M.; Baumgardner, T.; Freund, L.S.; Reiss, A.L. Social Functioning among Girls with Fragile X or Turner Syndrome and Their Sisters. J. Autism Dev. Disord. 1998, 28, 509–517. [Google Scholar] [CrossRef]
- Grön, M.; Pietilä, K.; Alvesalo, L. The Craniofacial Complex in 45,X/46,XX Females. Arch. Oral Biol. 1999, 44, 1077–1084. [Google Scholar] [CrossRef]
- Babić, M.; Šćepan, I.; Mićić, M. Comparative Cephalometric Analysis in Patients with X-Chromosome Aneuploidy. Arch. Oral Biol. 1993, 38, 179–183. [Google Scholar] [CrossRef]
- Dumancic, J.; Kaic, Z.; Varga, M.L.; Lauc, T.; Dumic, M.; Milosevic, S.A.; Brkic, H. Characteristics of the Craniofacial Complex in Turner Syndrome. Arch. Oral Biol. 2010, 55, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Howard M Saal; Madeleine D Harbison; Irene Netchine Silver-Russell Syndrome. Available online: https://www-ncbi-nlm-nih-gov.beckerproxy.wustl.edu/books/NBK1324/ (accessed on 25 January 2021).
- Bruce, S.; Hannula-Jouppi, K.; Peltonen, J.; Kere, J.; Lipsanen-Nyman, M. Clinically Distinct Epigenetic Subgroups in Silver-Russell Syndrome: The Degree of H19 Hypomethylation Associates with Phenotype Severity and Genital and Skeletal Anomalies. J. Clin. Endocrinol. Metab. 2009, 94, 579–587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abraham, M.B.; Carpenter, K.; Baynam, G.S.; MacKay, D.J.G.; Price, G.; Choong, C.S. Report and Review of Described Associations of Mayer-Rokitansky-Küster-Hauser Syndrome and Silver-Russell Syndrome. J. Paediatr. Child Health 2015, 51, 555–560. [Google Scholar] [CrossRef] [PubMed]
- Latyshev, O.Y.; Samsonova, L.N.; Kasatkina, E.P.; Okminyan, G.F.; Kiselyova, Y.v.; Timofeyeva, Y.S. The Risk for Developing Tumor in Patients with Gonadal Dysgenesis 46,XY. Bull. Sib. Med. 2015, 14, 41–46. [Google Scholar] [CrossRef]
- Huang, H.; Wang, C.; Tian, Q. Gonadal Tumour Risk in 292 Phenotypic Female Patients with Disorders of Sex Development Containing Y Chromosome or Y-Derived Sequence. Clin. Endocrinol. 2017, 86, 621–627. [Google Scholar] [CrossRef]
- Berglund, A.; Johannsen, T.H.; Stochholm, K.; Viuff, M.H.; Fedder, J.; Main, K.M.; Gravholt, C.H. Morbidity, Mortality, and Socioeconomics in Females with 46,XY Disorders of Sex Development: A Nationwide Study. J. Clin. Endocrinol. Metab. 2018, 103, 1418–1428. [Google Scholar] [CrossRef] [Green Version]
- Vilain, E.; Achermann, J.C.; Eugster, E.A.; Harley, V.R.; Morel, Y.; Wilson, J.D.; Hiort, O. We Used to Call Them Hermaphrodites. Genet. Med. 2007, 9, 65–66. [Google Scholar] [CrossRef]
- Hughes, I.A. Disorders of Sex Development: A New Definition and Classification. Best Pract. Res. Clin. Endocrinol. Metab. 2008, 22, 119–134. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).