

The Microvascular–Immune Interface in Cardiovascular Disease: A Stage-Based Framework of Microvascular Failure

Abstract
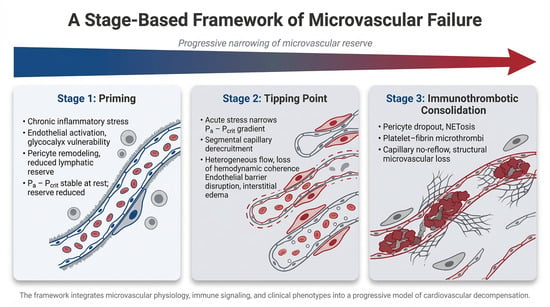
1. Introduction
2. Literature Review
2.1. Stage 1: The Primed Microvascular–Immune Unit
2.1.1. The Microvascular–Immune Unit in Cardiovascular Disease
2.1.2. Structural Components of Microvascular Vulnerability
Endothelial Surface: Baseline Vulnerability
Pericyte Remodeling: Chronic Sensitization
Lymphatic Reserve: Impaired Clearance Capacity
Circulating Elements: Lowered Activation Threshold
2.1.3. Hemodynamic and Clinical Conditioning
Hemodynamic Vulnerability
Clinical Drivers
Gut–Vascular Axis
Conditioning for Failure
2.2. Stage 2: The Acute Tipping Point—Loss of Microvascular Control
2.2.1. Established Physiology: The Arterial Waterfall and Critical Closing Pressure
2.2.2. Effects of Acute Stressors on Microvascular Parameters
2.2.3. Threshold Instability: A Framework Interpretation
2.2.4. Barrier Failure and Fluid Geometry
2.2.5. Lymphatic Overload and Congestive Amplification
2.2.6. Clinical Recognition of Microvascular Instability
2.3. Stage 3: Amplification and Consolidation—The Immunothrombotic Cascade
2.3.1. Pericyte Dysfunction as Amplifier
2.3.2. Pericytes in HFpEF and Coronary Microvascular Dysfunction
2.3.3. Pericytes in Post-Myocardial Infarction Remodeling
2.3.4. Cytokine Escalation and Inflammatory Cell Death
2.3.5. Septic Cardiomyopathy as Prototype
2.3.6. NETosis and Immunothrombosis
2.3.7. Distinguishing Stage 2 from Early Stage 3 in Practice
2.3.8. Potential for Bidirectional Movement and Reversibility
2.4. Clinical Cardiovascular Syndromes
2.4.1. Type 2 Myocardial Infarction
2.4.2. HFpEF Decompensation
2.4.3. Targeted Anti-Inflammatory Therapy (CANTOS)
2.5. Therapeutic and Research Implications
2.5.1. Preserving the Microvascular–Immune Unit
2.5.2. Glycocalyx Stabilization and Endothelial Preservation
2.5.3. Acute Barrier Support and Fluid Strategy
2.5.4. Mechanical Circulatory Support and Microvascular Coherence
2.5.5. Future Directions
3. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Ince, C. The microcirculation is the motor of sepsis. Crit. Care 2005, 9, S13–S19. [Google Scholar] [CrossRef] [PubMed]
- Ince, C. Hemodynamic coherence and the rationale for monitoring the microcirculation. Crit. Care 2015, 19, S8. [Google Scholar] [CrossRef]
- Młynarska, E.; Bojdo, K.; Frankenstein, H.; Krawiranda, K.; Kustosik, N.; Lisińska, W.; Rysz, J.; Franczyk, B. Endothelial Dysfunction as the Common Pathway Linking Obesity, Hypertension and Atherosclerosis. Int. J. Mol. Sci. 2025, 26, 10096. [Google Scholar] [CrossRef]
- Paulus, W.J.; Tschöpe, C. A Novel Paradigm for Heart Failure with Preserved Ejection Fraction. J. Am. Coll. Cardiol. 2013, 62, 263–271. [Google Scholar] [CrossRef]
- Libby, P.; Loscalzo, J.; Ridker, P.M.; Farkouh, M.E.; Hsue, P.Y.; Fuster, V.; Hasan, A.A.; Amar, S. Inflammation, Immunity, and Infection in Atherothrombosis. J. Am. Coll. Cardiol. 2018, 72, 2071–2081. [Google Scholar] [CrossRef]
- Rossitto, G.; Mary, S.; McAllister, C.; Neves, K.B.; Haddow, L.; Rocchiccioli, J.P.; Lang, N.N.; Murphy, C.L.; Touyz, R.M.; Petrie, M.C.; et al. Reduced Lymphatic Reserve in Heart Failure with Preserved Ejection Fraction. J. Am. Coll. Cardiol. 2020, 76, 2817–2829. [Google Scholar] [CrossRef]
- Han, J.-H. Immuno-metabolic diseases and therapeutics: Molecular mechanisms via inflammasome signaling. Cell Commun. Signal 2025, 23, 373. [Google Scholar] [CrossRef]
- Swirski, F.K.; Nahrendorf, M. Cardioimmunology: The immune system in cardiac homeostasis and disease. Nat. Rev. Immunol. 2018, 18, 733–744. [Google Scholar] [CrossRef] [PubMed]
- Isath, A.; Mehra, M.R. Cardio-systemic Stress and Trained Innate Immune Memory in Heart Failure. Transplantation 2025, 109, 565–568. [Google Scholar] [CrossRef] [PubMed]
- Isath, A.; Desai, A.S.; Mehra, M.R. Beyond the Pump. JACC Heart Fail. 2026, 14, 102711. [Google Scholar] [CrossRef]
- Woodcock, T.E.; Woodcock, T.M. Revised Starling equation and the glycocalyx model of transvascular fluid exchange: An improved paradigm for prescribing intravenous fluid therapy. Br. J. Anaesth. 2012, 108, 384–394. [Google Scholar] [CrossRef]
- Levick, J.R. Revision of the Starling principle: New views of tissue fluid balance. J. Physiol. 2004, 557, 704. [Google Scholar] [CrossRef] [PubMed]
- Uchimido, R.; Schmidt, E.P.; Shapiro, N.I. The glycocalyx: A novel diagnostic and therapeutic target in sepsis. Crit. Care 2019, 23, 16. [Google Scholar] [CrossRef]
- Lee, L.L.; Chintalgattu, V. Pericytes in the Heart. In Pericyte Biology in Different Organs; Birbrair, A., Ed.; Springer International Publishing: Cham, Switzerland, 2019; pp. 187–210. [Google Scholar] [CrossRef]
- Simmonds, S.J.; Grootaert, M.O.J.; Cuijpers, I.; Carai, P.; Geuens, N.; Herwig, M.; Baatsen, P.; Hamdani, N.; Luttun, A.; Heymans, S.; et al. Pericyte loss initiates microvascular dysfunction in the development of diastolic dysfunction. Eur. Heart J. Open 2023, 4, oead129. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Fu, J.; Huang, Y.; Duan, R.; Zhang, W.; Wang, C.; Wang, S.; Hu, X.; Zhao, H.; Wang, L.; et al. Biology and function of pericytes in the vascular microcirculation. Anim. Models Exp. Med. 2023, 6, 337–345. [Google Scholar] [CrossRef]
- Cooper, S.T.E.; Lokman, A.B.; Riley, P.R. Role of the Lymphatics in Cardiac Disease. Arter. Thromb. Vasc. Biol. 2024, 44, 1181–1190. [Google Scholar] [CrossRef]
- Fudim, M.; Salah, H.M.; Sathananthan, J.; Bernier, M.; Pabon-Ramos, W.; Schwartz, R.S.; Rodés-Cabau, J.; Côté, F.; Khalifa, A.; Virani, S.A.; et al. Lymphatic Dysregulation in Patients with Heart Failure. J. Am. Coll. Cardiol. 2021, 78, 66–76. [Google Scholar] [CrossRef]
- Koupenova, M.; Clancy, L.; Corkrey, H.A.; Freedman, J.E. Circulating Platelets as Mediators of Immunity, Inflammation, and Thrombosis. Circ. Res. 2018, 122, 337–351. [Google Scholar] [CrossRef]
- Engelmann, B.; Massberg, S. Thrombosis as an intravascular effector of innate immunity. Nat. Rev. Immunol. 2013, 13, 34–45. [Google Scholar] [CrossRef] [PubMed]
- Mann, D.L. Innate Immunity and the Failing Heart: The Cytokine Hypothesis Revisited. Circ. Res. 2015, 116, 1254–1268. [Google Scholar] [CrossRef]
- Hotamisligil, G.S. Inflammation and metabolic disorders. Nature 2006, 444, 860–867. [Google Scholar] [CrossRef] [PubMed]
- Hotamisligil, G.S. Foundations of Immunometabolism and Implications for Metabolic Health and Disease. Immunity 2017, 47, 406–420. [Google Scholar] [CrossRef]
- Libby, P. Mechanisms of Acute Coronary Syndromes and Their Implications for Therapy. N. Engl. J. Med. 2013, 368, 2004–2013. [Google Scholar] [CrossRef]
- Fajgenbaum, D.C.; June, C.H. Cytokine Storm. N. Engl. J. Med. 2020, 383, 2255–2273. [Google Scholar] [CrossRef]
- Jung, C.; Fuernau, G.; De Waha, S.; Eitel, I.; Desch, S.; Schuler, G.; Figulla, H.R.; Thiele, H. Intraaortic balloon counterpulsation and microcirculation in cardiogenic shock complicating myocardial infarction: An IABP-SHOCK II substudy. Clin. Res. Cardiol. 2015, 104, 679–687. [Google Scholar] [CrossRef]
- Dubin, A.; Pozo, M.O.; Casabella, C.A.; Pálizas, F.; Murias, G.; Moseinco, M.C.; Kanoore Edul, V.S.; Pálizas, F.; Estenssoro, E.; Ince, C. Increasing arterial blood pressure with norepinephrine does not improve microcirculatory blood flow: A prospective study. Crit. Care 2009, 13, R92. [Google Scholar] [CrossRef]
- Castro, R.; Retamal, J.; Hernández, G.; Kattan, E.; Pinsky, M.R. Critical closing pressure in the circulation: Understanding the vascular waterfall phenomenon. J. Crit. Care 2026, 93, 155485. [Google Scholar] [CrossRef]
- Michel, C.C.; Curry, F.E. Microvascular Permeability. Physiol. Rev. 1999, 79, 703–761. [Google Scholar] [CrossRef] [PubMed]
- Miller, A.; Rola, P.; Spiegel, R.; Haycock, K. Arteriolar Collapse and Haemodynamic Incoherence in Shock: Rethinking Critical Closing Pressure. J. Pers. Med. 2026, 16, 78. [Google Scholar] [CrossRef] [PubMed]
- Hørsdal, O.K. Can utilization of the venous-to-arterial carbon dioxide difference improve patient outcomes in cardiogenic shock? A narrative review. Am. Heart J. Plus 2025, 50, 100504. [Google Scholar] [CrossRef]
- Chommeloux, J.; Montero, S.; Franchineau, G.; Bréchot, N.; Hékimian, G.; Lebreton, G.; Le Guennec, L.; Bourcier, S.; Nieszkowska, A.; Leprince, P.; et al. Microcirculation Evolution in Patients on Venoarterial Extracorporeal Membrane Oxygenation for Refractory Cardiogenic Shock. Crit. Care Med. 2020, 48, e9–e17. [Google Scholar] [CrossRef]
- De Backer, D.; Orbegozo Cortes, D.; Donadello, K.; Vincent, J.-L. Pathophysiology of microcirculatory dysfunction and the pathogenesis of septic shock. Virulence 2014, 5, 73–79. [Google Scholar] [CrossRef] [PubMed]
- Garapati, H.N.; Dandamudi, S.; Kalagara, H. Venous excess ultrasound score (VExUS). Best Pract. Res. Clin. Anaesthesiol. 2025, 39, 351–359. [Google Scholar] [CrossRef]
- Schemmelmann, M.; Kelm, M.; Jung, C. The microcirculation in cardiogenic shock. Eur. Heart J. Acute Cardiovasc. Care 2024, 13, 802–809. [Google Scholar] [CrossRef] [PubMed]
- Chapman, A.R.; Taggart, C.; Boeddinghaus, J.; Mills, N.L.; Fox, K.A.A. Type 2 myocardial infarction: Challenges in diagnosis and treatment. Eur. Heart J. 2025, 46, 504–517. [Google Scholar] [CrossRef] [PubMed]
- Levy, J.H.; Alexander, P.M.A.; Wolberg, A.S.; McCarty, O.J.T.; Pusateri, A.E.; Bartz, R.R.; Bergmeier, W.; Cohen, M.J.; Connors, J.M.; Morrissey, J.H.; et al. ECMO-induced coagulopathy: Strategic initiatives for research and clinical practice (a workshop report of the NHLBI). Blood Vessel. Thromb. Hemost. 2025, 2, 100064. [Google Scholar] [CrossRef]
- Bourguignon, C.; Grouthier, V.; Chapouly, C.; Couffinhal, T.; Renault, M.-A. Cardiovascular risk factors have an impact on the biology of pericytes. Arch. Cardiovasc. Dis. 2025, 118, S180. [Google Scholar] [CrossRef]
- Hamilton, N.B. Pericyte-mediated regulation of capillary diameter: A component of neurovascular coupling in health and disease. Front. Neuroenerg. 2010, 2, 5. [Google Scholar] [CrossRef]
- Schrimpf, C.; Teebken, O.E.; Wilhelmi, M.; Duffield, J.S. The Role of Pericyte Detachment in Vascular Rarefaction. J. Vasc. Res. 2014, 51, 247–258. [Google Scholar] [CrossRef]
- Moro, M.; Balestrero, F.C.; Grolla, A.A. Pericytes: Jack-of-all-trades in cancer-related inflammation. Front. Pharmacol. 2024, 15, 1426033. [Google Scholar] [CrossRef]
- Van Splunder, H.; Villacampa, P.; Martínez-Romero, A.; Graupera, M. Pericytes in the disease spotlight. Trends Cell Biol. 2024, 34, 58–71. [Google Scholar] [CrossRef]
- Brandt, M.M.; Cheng, C.; Merkus, D.; Duncker, D.J.; Sorop, O. Mechanobiology of Microvascular Function and Structure in Health and Disease: Focus on the Coronary Circulation. Front. Physiol. 2021, 12, 771960. [Google Scholar] [CrossRef]
- Sato, R.; Nasu, M. A Review of Sepsis-Induced Cardiomyopathy. J. Intensive Care 2015, 3, 48. [Google Scholar] [CrossRef] [PubMed]
- Moore-Morris, T.; Evans, S.M. Cardiac Pericyte Diversity in Infarct Remodeling: Not Just Vascular Support Cells? Circulation 2023, 148, 899–902. [Google Scholar] [CrossRef] [PubMed]
- Boissier, F.; Aissaoui, N. Septic cardiomyopathy: Diagnosis and management. J. Intensive Med. 2022, 2, 8–16. [Google Scholar] [CrossRef]
- Karki, R.; Sharma, B.R.; Tuladhar, S.; Williams, E.P.; Zalduondo, L.; Samir, P.; Zheng, M.; Sundaram, B.; Banoth, B.; Malireddi, R.K.S.; et al. Synergism of TNF-α and IFN-γ Triggers Inflammatory Cell Death, Tissue Damage, and Mortality in SARS-CoV-2 Infection and Cytokine Shock Syndromes. Cell 2021, 184, 149–168.e17. [Google Scholar] [CrossRef]
- Bautista-Becerril, B.; Campi-Caballero, R.; Sevilla-Fuentes, S.; Hernández-Regino, L.M.; Hanono, A.; Flores-Bustamante, A.; González-Flores, J.; García-Ávila, C.A.; Aquino-Gálvez, A.; Castillejos-López, M.; et al. Immunothrombosis in COVID-19: Implications of Neutrophil Extracellular Traps. Biomolecules 2021, 11, 694. [Google Scholar] [CrossRef]
- Vazquez-Garza, E.; Jerjes-Sanchez, C.; Navarrete, A.; Joya-Harrison, J.; Rodriguez, D. Venous thromboembolism: Thrombosis, inflammation, and immunothrombosis for clinicians. J. Thromb. Thrombolysis 2017, 44, 377–385. [Google Scholar] [CrossRef] [PubMed]
- Smilowitz, N.R.; Toleva, O.; Chieffo, A.; Perera, D.; Berry, C. Coronary Microvascular Disease in Contemporary Clinical Practice. Circ. Cardiovasc. Interv. 2023, 16, e012568. [Google Scholar] [CrossRef]
- Nikolopoulou, L.; Dimitriadis, K.; Pyrpyris, N.; Tatakis, F.; Iliakis, P.; Thomopoulos, C.; Konstantinidis, D.; Rallidis, L.; Tousoulis, D.; Tsioufis, K. The Effect of Renal Denervation on Capillary Density in Patients with Uncontrolled Hypertension. Microcirculation 2025, 32, e70015. [Google Scholar] [CrossRef]
- Liu, L.; Wang, X.-X.; Wang, S.-X.; Yang, H.; Xiao, X.; Li, N.; Chai, H.-J.; Wang, H.-X. Attenuation of myocardial ischemia–reperfusion injury in mice through CD80/86 deficiency: Improved microvascular obstruction via reduced macrophage and T lymphocyte infiltration. Basic Res. Cardiol. 2025, 120, 873–888. [Google Scholar] [CrossRef]
- Itkin, M.; Rockson, S.G.; Burkhoff, D. Pathophysiology of the Lymphatic System in Patients with Heart Failure. J. Am. Coll. Cardiol. 2021, 78, 278–290. [Google Scholar] [CrossRef]
- Landi, I.; Guerritore, L.; Iannaccone, A.; Ricotti, A.; Rola, P.; Garrone, M. Assessment of venous congestion with venous excess ultrasound score in the prognosis of acute heart failure in the emergency department: A prospective study. Eur. Heart J. Open 2024, 4, oeae050. [Google Scholar] [CrossRef]
- Ridker, P.M.; Everett, B.M.; Thuren, T.; MacFadyen, J.G.; Chang, W.H.; Ballantyne, C.; Fonseca, F.; Nicolau, J.; Koenig, W.; Anker, S.D.; et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N. Engl. J. Med. 2017, 377, 1119–1131. [Google Scholar] [CrossRef]
- Lagrange, J.; Jahangiri, M.; Baudry, G.; Monzo, L.; Guerci, P.; Ter Maaten, J.M.; Heymans, S.; Mercier, N.; Girerd, N. Updates on the endothelial glycocalyx in heart failure with preserved ejection fraction. Am. J. Physiol.-Heart Circ. Physiol. 2025, 329, H1316–H1330. [Google Scholar] [CrossRef]
- Ponikowska, B.; Fudim, M.; Iwanek, G.; Zymliński, R.; Biegus, J. Harnessing the lymphatic system. Heart Fail. Rev. 2024, 30, 673–683. [Google Scholar] [CrossRef]
- McLean, P.; Bennett, J.; Trey Woods, E.; Chandrasekhar, S.; Newman, N.; Mohammad, Y.; Khawaja, M.; Rizwan, A.; Siddiqui, R.; Birnbaum, Y.; et al. SGLT2 inhibitors across various patient populations in the era of precision medicine: The multidisciplinary team approach. npj Metab. Health Dis. 2025, 3, 29. [Google Scholar] [CrossRef]
- Durante, W.; Behnammanesh, G.; Peyton, K.J. Effects of Sodium-Glucose Co-Transporter 2 Inhibitors on Vascular Cell Function and Arterial Remodeling. Int. J. Mol. Sci. 2021, 22, 8786. [Google Scholar] [CrossRef] [PubMed]
- Roman-Pepine, D.; Serban, A.M.; Capras, R.-D.; Cismaru, C.M.; Filip, A.G. A Comprehensive Review: Unraveling the Role of Inflammation in the Etiology of Heart Failure. Heart Fail. Rev. 2025, 30, 931–954. [Google Scholar] [CrossRef] [PubMed]
- Jacquet, C.; Gustafsson, R.; Patel, A.K.; Hansson, M.; Rankin, G.; Bano, F.; Byström, J.W.; Blomberg, A.; Rasmuson, J.; Satchell, S.; et al. Matrix Metalloproteinase-9 Mediates Endothelial Glycocalyx Degradation and Correlates with Severity of Hemorrhagic Fever with Renal Syndrome. iScience 2025, 28, 113262. [Google Scholar] [CrossRef] [PubMed]
- Meuwese, M.C.; Mooij, H.L.; Nieuwdorp, M.; Van Lith, B.; Marck, R.; Vink, H.; Kastelein, J.J.P.; Stroes, E.S.G. Partial recovery of the endothelial glycocalyx upon rosuvastatin therapy in patients with heterozygous familial hypercholesterolemia. J. Lipid Res. 2009, 50, 148–153. [Google Scholar] [CrossRef]
- Banerjee, S.; Mwangi, J.G.; Stanley, T.K.; Mitra, R.; Ebong, E.E. Regeneration and Assessment of the Endothelial Glycocalyx to Address Cardiovascular Disease. Ind. Eng. Chem. Res. 2021, 60, 17328–17347. [Google Scholar] [CrossRef]
- Sawashita, Y.; Kazuma, S.; Tokinaga, Y.; Kikuchi, K.; Hirata, N.; Masuda, Y.; Yamakage, M. Albumin protects the ultrastructure of the endothelial glycocalyx of coronary arteries in myocardial ischemia-reperfusion injury in vivo. Biochem. Biophys. Res. Commun. 2023, 666, 29–35. [Google Scholar] [CrossRef] [PubMed]
- Aldecoa, C.; Llau, J.V.; Nuvials, X.; Artigas, A. Role of albumin in the preservation of endothelial glycocalyx integrity and the microcirculation: A review. Ann. Intensive Care 2020, 10, 85. [Google Scholar] [CrossRef]
- Kuhn, M. Keeps Cardiac Pericytes in Good Shape: Regulator of G-Protein Signaling-5. Circ. Res. 2024, 134, 1256–1258. [Google Scholar] [CrossRef]
- Dimitriadis, K.; Adamopoulou, E.; Pyrpyris, N.; Sakalidis, A.; Leontsinis, I.; Manta, E.; Mantzouranis, E.; Beneki, E.; Soulaidopoulos, S.; Konstantinidis, D.; et al. The effect of SGLT2 inhibitors on the endothelium and the microcirculation: From bench to bedside and beyond. Eur. Heart J.-Cardiovasc. Pharmacother. 2023, 9, 741–757. [Google Scholar] [CrossRef] [PubMed]
- Barrera-Chimal, J.; Bonnard, B.; Jaisser, F. Roles of Mineralocorticoid Receptors in Cardiovascular and Cardiorenal Diseases. Annu. Rev. Physiol. 2022, 84, 585–610. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Domain | Stage 1: Priming | Stage 2: Functional Hemodynamic Incoherence | Early Stage 3: Immunothrombotic Consolidation |
|---|---|---|---|
| Dominant biology | Endothelial activation, glycocalyx vulnerability, pericyte sensitisation, reduced lymphatic reserve; compensated microvascular unit. | Narrowing of the Pa − Pcrit gradient, heterogeneous capillary flow, loss of hemodynamic coherence; perfusion defects are functional and potentially reversible. | Endothelial injury, sustained pericyte contraction/dropout, NET–fibrin deposition; perfusion defects become structural and less responsive to pressure restoration. |
| Clinical pattern | High-risk cardiometabolic, inflammatory, hypertensive, renal, or HFpEF phenotype; perfusion preserved at rest, reserve reduced. | Hypoperfusion signs (delayed capillary refill, mottling, oliguria) despite acceptable systemic blood pressure and cardiac output; symptoms may be subtle. | Persistent hypoperfusion despite normalisation of MAP/cardiac output; end-organ dysfunction (e.g., worsening oliguria, altered mentation); may overlap with overt shock. |
| Candidate markers/correlates | hs-CRP, IL-6; subclinical glycocalyx shedding (mild syndecan-1, heparan sulfate). (Natriuretic peptides may be elevated but reflect myocardial strain, not microvascular priming per se.) | ΔPCO2 widening (>6 mmHg) with normal/high SvO2 (>70%); impaired lactate clearance; dynamic mottling and VExUS that improve with hemodynamic optimisation. | Rising syndecan-1, heparan sulfate, D-dimer, CitH3 (citrullinated histone H3); emerging NET-associated markers (e.g., MPO-DNA, cell-free DNA) may also be elevated. |
| Imaging/bedside microcirculation | No validated bedside staging test; impaired vasodilatory reserve may be inferred from coronary flow reserve (CFR) or stress perfusion imaging in specialised settings. | Sublingual videomicroscopy: reduced perfused vessel density (PVD) or proportion of perfused vessels (PPV) that improves with correction of the triggering insult. VExUS often dynamic. | Persistent reduction in PVD/PPV despite macro-hemodynamic correction; fixed no-reflow pattern on microcirculatory imaging; VExUS may remain elevated despite decongestion. |
| Therapeutic posture | Preventive: preserve microvascular integrity, reduce endothelial inflammatory signalling, and optimise cardiometabolic status. | Restore hemodynamic coherence: optimise the Pa–Pcrit gradient, relieve congestion, and prevent immunothrombotic consolidation. | Rescue/supportive: maintain systemic perfusion, limit secondary microvascular injury; investigational immunomodulatory or NET-targeted strategies may be considered. |
| Representative therapeutic categories | SGLT2 inhibitors, MRAs, RAAS inhibitors, statins; cardiometabolic risk reduction; cautious decongestion when appropriate. | Individualised fluid/vasopressor/decongestion strategy guided by ΔPCO2, capillary refill, and VExUS; barrier-supportive approaches (albumin, sphingosine-1-phosphate) remain investigational. | MCS/ECMO when clinically indicated; meticulous anticoagulation; albumin/barrier-support hypotheses; investigational anti-NET or immunomodulatory agents (none yet proven). |
| Validation status | Hypothesis-generating; no prospective staging system exists. All candidate markers and thresholds are proposed and require validation. | No validated transition thresholds; proposed physiological signatures must be tested in prospective cohorts. | Supportive of structural immunothrombotic progression but not diagnostic as stand-alone criteria; reversibility likely limited once this stage is reached. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license.
Share and Cite
Panneflek, J.; Lauzea, B.; Barbarawi, M.; Greenaway, A. The Microvascular–Immune Interface in Cardiovascular Disease: A Stage-Based Framework of Microvascular Failure. Hearts 2026, 7, 17. https://doi.org/10.3390/hearts7020017
Panneflek J, Lauzea B, Barbarawi M, Greenaway A. The Microvascular–Immune Interface in Cardiovascular Disease: A Stage-Based Framework of Microvascular Failure. Hearts. 2026; 7(2):17. https://doi.org/10.3390/hearts7020017
Chicago/Turabian StylePanneflek, Jathniel, Béatrice Lauzea, Mahmoud Barbarawi, and Atari Greenaway. 2026. "The Microvascular–Immune Interface in Cardiovascular Disease: A Stage-Based Framework of Microvascular Failure" Hearts 7, no. 2: 17. https://doi.org/10.3390/hearts7020017
APA StylePanneflek, J., Lauzea, B., Barbarawi, M., & Greenaway, A. (2026). The Microvascular–Immune Interface in Cardiovascular Disease: A Stage-Based Framework of Microvascular Failure. Hearts, 7(2), 17. https://doi.org/10.3390/hearts7020017