


Dynamic Behavior of Catalysts Under Operating Conditions: Part 1—Origin and Development of the Ideas †


Abstract
| “The progress of knowledge is to be measured not by the questions that it has answered but by the questions that it provokes us to ask.” |
| Arthur Eddington, New Pathways in Science, 1934 |
1. Introduction
2. The Early Stage of Catalysis Research
2.1. Homogeneous Catalysis
2.2. Heterogeneous Catalysis
- The chemical theory of catalysis put forward by Paul Sabatier [71,72,73]. According to this theory, a catalytic reaction proceeds through the chemical interaction of a catalyst with one of the reactants to form a temporary unstable intermediate compound that participates in further transformations but is absent from the final products. For example, according to Sabatier’s theory, the dehydration of saturated alcohols to alkenes over aluminum oxide proceeds via unstable aluminum alcoholates [73] (p. 53):Al2O3 + 2CnH2n+1OH → Al2O2(OCnH2n+1)2 + H2O,Al2O2(OCnH2n+1)2 → 2CnH2n + Al2O2(OH)2,Al2O2(OH)2 → Al2O3 + H2O.The reaction rate reaches a maximum at an optimal strength of interaction between catalyst and reactant, which should be neither too weak nor too strong. This important statement is known as the Sabatier principle (the Sabatier–Balandin volcano plots [40]). The chemical theory made it possible to explain the selectivity of solid catalysts based on their chemical nature, regardless of their physical state. Obviously, Sabatier’s theory largely repeats the theory of intermediate compounds, which was adopted by organic chemists to elucidate homogeneous catalytic reactions. This is not surprising given that Sabatier, being a student of Berthelot, adhered to the above-mentioned ideas of his teacher [72] (p. 4). The invaluable contribution made by Sabatier, among other things, lies in extending the theory of intermediate compounds to a wide range of heterogeneous catalytic reactions. According to Sabatier, surface intermediate compounds have a definite stoichiometric composition, as in homogeneous catalysis. However, efforts to detect them were unsuccessful until the 1930s.
- The physical theory of catalysis developed primarily by Max Bodenstein and Walther Nernst [74,75] on the basis of earlier physical theories of heterogeneous processes and Ostwald’s concept (see also Ref. [76]). According to this theory, no chemical involvement of the catalyst is implied, and its role is limited merely to condensing (concentrating) the reacting substances around the surface. For this reason, the overall rate of catalytic process is governed by the diffusion of reactants and products through the thick layer of adsorbed molecules. In a later version of this theory, Bodenstein argued that the surface of a solid catalyst not only concentrates but also “deforms” the reactant molecules with its “force fields”, thereby causing a catalytic reaction [77]. Nikolai D. Zelinsky held the same view on the essence of heterogeneous catalysis, rejecting the concept of intermediate compounds [78].
- The adsorption theory worked out by Irving Langmuir [67,79,80,81]. An extremely important statement of this theory is that the primary act of heterogeneous catalysis is chemisorption which consists of the valence interaction of reactant molecules with the catalyst crystal. The “checkerboard” model with a uniform distribution of static adsorption sites in accordance with the crystal lattice structure was assumed to describe the catalyst surface. In essence, Langmuir’s theory first combined the physical and chemical aspects of heterogeneous catalysis, considering catalytic phenomena at the solid surface from a molecular-kinetic point of view.
3. The Concept of Active Sites and Its Evolution
3.1. The Emergence of the Concept
3.2. The Armstrong–Hilditch Hypothesis
3.3. Active Site as a Cluster of Atoms and Kobozev’s Theory
3.4. Active Sites from the Standpoint of Electronic Concepts and Beyond
4. Toward Modern Concepts on the Dynamic Nature of Heterogeneous Catalysts
4.1. Boreskov’s Concept of the Effect of Reaction Medium on the Composition, Structure, and Properties of Solid Catalysts
- Chemical changes leading to phase transformations of the active component of the catalyst. As a result, the catalyst performance usually changes dramatically, which makes it easy to detect such phenomena and to take them into account when studying catalysts and choosing reaction conditions.
- Changes in the catalyst composition not accompanied by phase transformations. These phenomena are quite common, but the influence of such changes on catalytic performance may be less pronounced and is often ignored. The reaction medium can alter the ratio of catalyst components, e.g., by partially or even completely removing one of them. These changes in composition can spread throughout the catalyst bulk.
- Changes in the composition of the catalyst surface layers, occurring only up to a certain depth.
4.2. Role of Surface Science in Elucidating the Dynamic Nature of Working Catalysts
5. The Concept of Cocktail-Type Catalysis
- A cocktail of catalysts. Such a system contains metal complexes, clusters, and NPs, which make comparable contributions to a catalytic reaction;
- A cocktail of species. Such a system also contains metal complexes, clusters, and NPs, but only one of them makes a major contribution to a catalytic reaction.
 |
 |
6. Conclusions and Outlook
- What are the limits of applicability of existing concepts on the catalyst dynamics?
- Can these concepts be formulated as a single unified concept or theory?
- Can a universal concept of the dynamic behavior of catalysts play a key unifying role for the modern science of catalysis?
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| acac | Acetylacetonate |
| AES | Auger electron spectroscopy |
| BIMe | 1,3-Dimethyl-1,3-dihydro-2H-benzimidazol-2-ylidene |
| COD | 1,5-Cyclooctadiene |
| dba | Dibenzylideneacetone |
| DFT | Density functional theory |
| DMF | N,N-Dimethylformamide |
| EDS | Energy-dispersive X-ray spectroscopy |
| ESI-HMRS | Electrospray ionization high-resolution mass spectrometry |
| Et | Ethyl |
| HREELS | High-resolution electron energy loss spectroscopy |
| ICP-AES | Inductively coupled plasma atomic emission spectroscopy |
| LEED | Low-energy electron diffraction |
| Me | Methyl |
| NHCs | N-heterocyclic carbenes |
| NMR | Nuclear magnetic resonance |
| NPs | Nanoparticles |
| Ph | Phenyl |
| Py | Pyridine |
| SEM | Scanning electron microscopy |
| t-Bu | tert-butyl |
| TEM | Transmission electron microscopy |
| TOF | Turnover frequency |
| UHV | Ultra-high vacuum |
| XAS | X-ray absorption spectroscopy |
| XPS | X-ray photoelectron spectroscopy |
References
- Kung, H.H.; Kung, M.C. Inspiration from nature for heterogeneous catalysis. Catal. Lett. 2014, 144, 1643–1652. [Google Scholar] [CrossRef]
- Coppens, M.-O.; Weissenberger, T.; Zhang, Q.; Ye, G. Nature-inspired, computer-assisted optimization of hierarchically structured zeolites. Adv. Mater. Interfaces 2021, 8, 2001409. [Google Scholar] [CrossRef]
- Wang, K.-Y.; Zhang, J.; Hsu, Y.-C.; Lin, H.; Han, Z.; Pang, J.; Yang, Z.; Liang, R.-R.; Shi, W.; Zhou, H.-C. Bioinspired framework catalysts: From enzyme immobilization to biomimetic catalysis. Chem. Rev. 2023, 123, 5347–5420. [Google Scholar] [CrossRef]
- Wang, Y.; Jia, G.; Yang, P.; Zhang, L.; Wong, K.-Y. Bioinspired design of heterogenous single-atomic-site catalysts for electrocatalysis and photocatalysis. Adv. Mater. 2025, 37, 2502131. [Google Scholar] [CrossRef] [PubMed]
- Chorkendorff, I.; Niemantsverdriet, J.W. Concepts of Modern Catalysis and Kinetics, 3rd ed.; Wiley-VCH: Weinheim, Germany, 2017. [Google Scholar]
- Poovan, F.; Chandrashekhar, V.G.; Natte, K.; Jagadeesh, R.V. Synergy between homogeneous and heterogeneous catalysis. Catal. Sci. Technol. 2022, 12, 6623–6649. [Google Scholar] [CrossRef]
- Reschetilowski, W. Einführung in die Heterogene Katalyse, 2nd ed.; Springer Spektrum: Berlin/Heidelberg, Germany, 2025. (In German) [Google Scholar]
- Taylor, H. Some aspects of catalytic science. Adv. Catal. Relat. Subj. 1957, 9, 1–7. [Google Scholar]
- van Santen, R.A. Mechanisms in Heterogeneous Catalysis; World Scientific Publishing Europe: London, UK, 2024; Chapter 7; pp. 646–691. [Google Scholar]
- Zamaraev, K.I. Catalysis by molecular design. Top. Catal. 1996, 3, 1–76. [Google Scholar] [CrossRef]
- Védrine, J.C. Revisiting active sites in heterogeneous catalysis: Their structure and their dynamic behavior. Appl. Catal. A Gen. 2014, 474, 40–50. [Google Scholar] [CrossRef]
- Schlögl, R. Heterogeneous catalysis. Angew. Chem. Int. Ed. 2015, 54, 3465–3520. [Google Scholar] [CrossRef]
- van Santen, R.A. Modern Heterogeneous Catalysis: An Introduction; Wiley-VCH: Weinheim, Germany, 2017. [Google Scholar]
- Tanaka, K. Dynamic Chemical Processes on Solid Surfaces: Chemical Reactions and Catalysis; Springer Nature: Singapore, 2017. [Google Scholar]
- Pan, Y.; Shen, X.; Yao, L.; Bentalib, A.; Peng, Z. Active sites in heterogeneous catalytic reaction on metal and metal oxide: Theory and practice. Catalysts 2018, 8, 478. [Google Scholar] [CrossRef]
- Vogt, C.; Weckhuysen, B.M. The concept of active site in heterogeneous catalysis. Nat. Rev. Chem. 2022, 6, 89–111. [Google Scholar] [CrossRef]
- Durán Pachón, L.; Rothenberg, G. Transition-metal nanoparticles: Synthesis, stability and the leaching issue. Appl. Organomet. Chem. 2008, 22, 288–299. [Google Scholar] [CrossRef]
- Moulijn, J.A.; van Diepen, A.E.; Kapteijn, F. Deactivation and regeneration. In Handbook of Heterogeneous Catalysis, 2nd ed.; Ertl, G., Knözinger, H., Schüth, F., Weitkamp, J., Eds.; Wiley-VCH: Weinheim, Germany, 2008; Volume 4, pp. 1829–1846. [Google Scholar]
- Argyle, M.D.; Bartholomew, C.H. Heterogeneous catalyst deactivation and regeneration: A review. Catalysts 2015, 5, 145–269. [Google Scholar] [CrossRef]
- Sádaba, I.; López Granados, M.; Riisager, A.; Taarning, E. Deactivation of solid catalysts in liquid media: The case of leaching of active sites in biomass conversion reactions. Green Chem. 2015, 17, 4133–4145. [Google Scholar] [CrossRef]
- Liu, S.; Zhu, M.; Iqbal, M. Research progress on stability of solid acid catalysts. Catal. Surv. Asia 2020, 24, 196–206. [Google Scholar] [CrossRef]
- Martín, A.J.; Mitchell, S.; Mondelli, C.; Jaydev, S.; Pérez-Ramírez, J. Unifying views on catalyst deactivation. Nat. Catal. 2022, 5, 854–866. [Google Scholar] [CrossRef]
- Redón, R.; Peña, N.G.G.; Crescencio, F.R. Leaching in metal nanoparticle catalysis. Recent Pat. Nanotechnol. 2014, 8, 31–51. [Google Scholar] [CrossRef] [PubMed]
- Bourouina, A.; Meille, V.; de Bellefon, C. About solid phase vs. liquid phase in Suzuki-Miyaura reaction. Catalysts 2019, 9, 60. [Google Scholar] [CrossRef]
- Galushko, A.S.; Ilyushenkova, V.V.; Burykina, J.V.; Shaydullin, R.R.; Pentsak, E.O.; Ananikov, V.P. The fast formation of a highly active homogeneous catalytic system upon the soft leaching of Pd species from a heterogeneous Pd/C precursor. Inorganics 2023, 11, 260. [Google Scholar] [CrossRef]
- Flytzani-Stephanopoulos, M.; Schmidt, L.D. Morphology and etching processes on macroscopic metal catalysts. Prog. Surf. Sci. 1979, 9, 83–111. [Google Scholar] [CrossRef]
- Wei, T.-C.; Phillips, J. Thermal and catalytic etching: Mechanisms of metal catalyst reconstruction. Adv. Catal. 1996, 41, 359–421. [Google Scholar]
- Vinnik, M.I. Mechanism of acid catalysis in solutions. Kinet. Catal. 1980, 21, 111–127. [Google Scholar]
- Kazanskii, V.B. Modern ideas about the mechanisms of homogeneous and heterogeneous acid catalysis: Similarities and differences. Russ. Chem. Rev. 1988, 57, 1109–1123. [Google Scholar] [CrossRef]
- Agarwal, P.K.; Bernard, D.N.; Bafna, K.; Doucet, N. Enzyme dynamics: Looking beyond a single structure. ChemCatChem 2020, 12, 4704–4720. [Google Scholar] [CrossRef]
- Corbella, M.; Pinto, G.P.; Kamerlin, S.C.L. Loop dynamics and the evolution of enzyme activity. Nat. Rev. Chem. 2023, 7, 536–547. [Google Scholar] [CrossRef]
- Aykac Fas, B.; Akbas Buz, Z.E.; Haliloglu, T. Global dynamics behind enzyme catalysis, evolution, and design. Curr. Opin. Struct. Biol. 2025, 94, 103131. [Google Scholar] [CrossRef]
- Ipatieff, V.N.; Blokh, M.A. The Catalytic Phenomena in Nature; Scientific Chemical and Technical Publishing House: Petrograd, RSFSR, 1922; pp. 12–76. (In Russian) [Google Scholar]
- Mittasch, A.; Theis, E. Von Davy und Döbereiner Bis Deacon, Ein Halbes Jahrhundert Grenzflächenkatalyse; Verlag Chemie: Berlin, Germany, 1932. (In German) [Google Scholar]
- Mittasch, A. Kurze Geschichte der Katalyse in Praxis und Theorie; Julius Springer: Berlin, Germany, 1939. (In German) [Google Scholar]
- Kuznetsov, V.I. The Development of the Science of Catalysis; Nauka: Moscow, Russia, 1964. (In Russian) [Google Scholar]
- Zecchina, A.; Califano, S. The Development of Catalysis: A History of Key Processes and Personas in Catalytic Science and Technology; John Wiley & Sons: Hoboken, NJ, USA, 2017. [Google Scholar]
- Kuznetsov, V.I. The development of basic ideas in the field of catalysis. Chymia 1966, 11, 179–204. [Google Scholar] [CrossRef]
- Laidler, K.J. The development of theories of catalysis. Arch. Hist. Exact Sci. 1986, 35, 345–374. [Google Scholar] [CrossRef]
- Davis, B.H. Development of the science of catalysis. In Handbook of Heterogeneous Catalysis, 2nd ed.; Ertl, G., Knözinger, H., Schüth, F., Weitkamp, J., Eds.; Wiley-VCH: Weinheim, Germany, 2008; Volume 1, pp. 16–37. [Google Scholar]
- Smith, J.K. History of catalysis. In Encyclopedia of Catalysis; Horváth, I.T., Ed.; John Wiley & Sons: Hoboken, NJ, USA, 2003; Volume 3, pp. 447–479. [Google Scholar]
- van Santen, R. Catalysis in perspective: Historic review. In Catalysis: From Principles to Applications; Beller, M., Renken, A., van Santen, R., Eds.; Wiley-VCH: Weinheim, Germany, 2012; Chapter 1; pp. 3–19. [Google Scholar]
- Augustine, R.L. Whither goest thou, catalysis. Catal. Lett. 2016, 146, 2393–2416. [Google Scholar] [CrossRef]
- Balandin, A.A. Present state of the catalysis problem and the theoretical basis of the search for catalysts. Bull. Acad. Sci. USSR Div. Chem. Sci. 1955, 4, 557–570. [Google Scholar] [CrossRef]
- Berzelius, J. Årsberättelse om Framstegen i Fysik Och Kemi; P. A. Norstedt & Söner: Stockholm, Sweden, 1835; pp. 239–247. (In Swedish) [Google Scholar]
- Berzelius. Some ideas on a new force acting in the combinations of organic compounds. Lond. Edinb. Philos. Mag. J. Sci. 1837, 10, 490–492. [Google Scholar]
- Armstrong, H.E. Opening address to the Chemical Section of the British Association [continued]. Nature 1885, 32, 467–472. [Google Scholar]
- Mitscherlich, E. Lehrbuch der Chemie, 2nd ed.; Ernst Siegfried Mittler: Berlin, Germany, 1833; Volume 1, pp. 97–107. (In German) [Google Scholar]
- Mitscherlich, E. Ueber die Aetherbildung. Ann. Phys. Chem. 1834, 31, 273–282. (In German) [Google Scholar] [CrossRef]
- Mitscherlich, E. Ueber die chemische Zersetzung und Verbindung mittelst Contactsubstanzen. Ann. Phys. Chem. 1842, 55, 209–229. (In German) [Google Scholar] [CrossRef]
- Désormes, C.B.; Clément, N. Théorie de la fabrication de l’acide sulfurique. Ann. Chim. 1806, 59, 329–339. (In French) [Google Scholar]
- Liebig, J. Ueber die Erscheinungen der Gährung, Fäulniß und Verwesung und ihre Ursachen. Ann. Pharm. 1839, 30, 250–287. (In German) [Google Scholar] [CrossRef]
- Liebig, J. Handbuch der Chemie mit Rücksicht auf Pharmacie: Als Neue Bearbeitung des Ersten Bandes von Geiger’s Handbuch der Pharmacie; C. F. Winter: Heidelberg, Germany, 1843; Volume 1, pp. 37–84. (In German) [Google Scholar]
- Hjelt, E. Geschichte der Organischen Chemie von ältester Zeit bis zur Gegenwart; Friedr. Vieweg & Sohn: Braunschweig, Germany, 1916; Chapter 17; pp. 362–401. (In German) [Google Scholar]
- Berthelot, M. La Synthèse Chimique; Germer Baillière: Paris, France, 1876; Book 1; pp. 64–98. (In French) [Google Scholar]
- Laidler, K.J. The World of Physical Chemistry; Oxford University Press: New York, NY, USA, 1995. [Google Scholar]
- Denisov, E.T.; Sarkisov, O.M.; Likhtenshtein, G.I. Chemical Kinetics: Fundamentals and New Developments; Elsevier Science: Amsterdam, The Netherlands, 2003; Chapter 1; pp. 1–15. [Google Scholar]
- Ostwald, W. Ueber Katalyse. In Verhandlungen der Gesellschaft Deutscher Naturforscher und Ärzte, 73. Versammlung zu Hamburg, 22–28 September 1901; F.C.W. Vogel: Leipzig, Germany, 1901; Part 1; pp. 184–202. (In German) [Google Scholar]
- Ostwald. Catalysis. Nature 1902, 65, 522–526. [Google Scholar] [CrossRef]
- Ostwald, W. Über Katalyse. Vortrag gehalten in Stockholm am 12. Dezember 1909 nach Empfang des Nobelpreises für Chemie. In Les Prix Nobel en 1909; Hasselberg, K.B., Pettersson, S.O., Mörner, K.A.H., af Wirsén, C.D., Santesson, M.C.G., Eds.; P. A. Norstedt & Söner: Stockholm, Sweden, 1910. (In German) [Google Scholar]
- Schwab, G.-M. Catalysis from the Standpoint of Chemical Kinetics; D. Van Nostrand Company: New York, NY, USA, 1937. [Google Scholar]
- Brönsted, J.N. Acid and basic catalysis. Chem. Rev. 1928, 5, 231–338. [Google Scholar] [CrossRef]
- Spitalsky, E. Über die kinetischen Gesetze der homogenen Katalyse. Z. Phys. Chem. Stöchiom. Verwandtschaftsl. 1926, 122, 257–286. (In German) [Google Scholar] [CrossRef]
- Spitalsky, E.; Koboseff, N. Über die kinetischen Gesetze der homogenen Katalyse. Experimentelle Untersuchung des inneren Mechanismus einer homogenen katalytischen Reaktion. Z. Phys. Chem. Stöchiom. Verwandtschaftsl. 1927, 127, 129–177. (In German) [Google Scholar] [CrossRef]
- Faraday, M. Experimental researches in electricity—Sixth series. Philos. Trans. R. Soc. Lond. 1834, 124, 55–76. [Google Scholar] [CrossRef]
- Playfair, L. On transformations produced by catalytic bodies. Mem. Proc. Chem. Soc. Lond. 1848, 3, 348–370. [Google Scholar]
- Langmuir, I. The mechanism of the catalytic action of platinum in the reactions 2CO + O2 = 2CO2 and 2H2 + O2 = 2H2O. Trans. Faraday Soc. 1922, 17, 621–654. [Google Scholar] [CrossRef]
- Lloyd, L. Handbook of Industrial Catalysts; Springer Science + Business Media: New York, NY, USA, 2011. [Google Scholar]
- Maxted, E.B. Catalysis and Its Industrial Applications; J. & A. Churchill: London, UK, 1933. [Google Scholar]
- Kuznetsov, V.I. The Development of Catalytic Organic Synthesis; Nauka: Moscow, Russia, 1964. (In Russian) [Google Scholar]
- Sabatier, P. La catalyse par les métaux communs. Rev. Gen. Sci. Pures Appl. 1905, 16, 842–850. (In French) [Google Scholar]
- Sabatier, P. La méthode d’hydrogénation directe par catalyse. Conférence faite à Stockholm le 11 décembre 1912 devant l’Académie des Sciences de Suède. In Les Prix Nobel en 1912; Hasselberg, K.B., Pettersson, S.O., Mörner, K.A.H., Santesson, M.C.G., Eds.; P. A. Norstedt & Söner: Stockholm, Sweden, 1913. (In French) [Google Scholar]
- Sabatier, P. Catalysis in Organic Chemistry; D. Van Nostrand Company: New York, NY, USA, 1922; Chapter 3; pp. 37–57. [Google Scholar]
- Nernst, W. Theorie der Reaktionsgeschwindigkeit in heterogenen Systemen. Z. Phys. Chem. Stöchiom. Verwandtschaftsl. 1904, 47, 52–55. (In German) [Google Scholar] [CrossRef]
- Bodenstein, M.; Fink, C.G. Heterogene katalytische Reaktionen. V. Allgemeine Bemerkungen. Z. Phys. Chem. Stöchiom. Verwandtschaftsl. 1907, 60, 46–69. (In German) [Google Scholar] [CrossRef]
- Bodenstein, M. Katalyse und Katalysatoren. Chem.-Ztg. 1902, 26, 1075–1079. (In German) [Google Scholar]
- Bodenstein, M. Ein Beitrag zur Theorie der katalytischen Hydrierung durch Platin. Justus Liebigs Ann. Chem. 1924, 440, 177–185. (In German) [Google Scholar] [CrossRef]
- Zelinsky, N.D. Katalyse und Formänderung der Molekeln. Ber. Dtsch. Chem. Ges. 1925, 58, 2755–2763. (In German) [Google Scholar] [CrossRef]
- Langmuir, I. The constitution and fundamental properties of solids and liquids. Part I. Solids. J. Am. Chem. Soc. 1916, 38, 2221–2295. [Google Scholar] [CrossRef]
- Langmuir, I. The adsorption of gases on plane surfaces of glass, mica and platinum. J. Am. Chem. Soc. 1918, 40, 1361–1403. [Google Scholar] [CrossRef]
- Langmuir, I. Chemical reactions on surfaces. Trans. Faraday Soc. 1922, 17, 607–620. [Google Scholar] [CrossRef]
- Taylor, H.S. A theory of the catalytic surface. Proc. R. Soc. Lond. Ser. A 1925, 108, 105–111. [Google Scholar] [CrossRef]
- Taylor, H.S. Fourth report of the Committee on contact catalysis. J. Phys. Chem. 1926, 30, 145–171. [Google Scholar] [CrossRef]
- Rideal, E.K. Concepts in catalysis. The contributions of Paul Sabatier and of Max Bodenstein. J. Chem. Soc. 1951, 1640–1647. [Google Scholar] [CrossRef]
- Lattes, A. De l’hydrogénation catalytique à la théorie chimique de la catalyse: Paul Sabatier, chimiste de génie, apôtre de la décentralisation. C. R. Chim. Acad. Sci. Ser. IIc Chim. 2000, 3, 705–709. (In French) [Google Scholar] [CrossRef]
- Ertl, G. Reactions at surfaces: Bodenstein’s impact and some current aspects. In Gas Phase Chemical Reaction Systems: Experiments and Models 100 Years After Max Bodenstein; Wolfrum, J., Volpp, H.-R., Rannacher, R., Warnatz, J., Eds.; Springer: Berlin/Heidelberg, Germany, 1996; pp. 245–252. [Google Scholar]
- Roberts, M.W. Development of the industrial relevance of catalysis and its physiochemical basis (1860–1940). Catal. Lett. 2000, 67, 5–13. [Google Scholar] [CrossRef]
- Swenson, H.; Stadie, N.P. Langmuir’s theory of adsorption: A centennial review. Langmuir 2019, 35, 5409–5426. [Google Scholar] [CrossRef]
- Rideal, E.K.; Taylor, H.S. Catalysis in Theory and Practice, 2nd ed.; Macmillan and Co.: London, UK, 1926. [Google Scholar]
- Mitchell, J.A. Heterogeneous catalysis. J. Chem. Educ. 1932, 9, 261–271. [Google Scholar] [CrossRef]
- Rideal, E.K. The hydrogenation of ethylene in contact with nickel. J. Chem. Soc. Trans. 1922, 121, 309–318. [Google Scholar] [CrossRef]
- Burwell, R.L., Jr. Manual of symbols and terminology for physicochemical quantities and units—Appendix II. Definitions, terminology and symbols in colloid and surface chemistry. Part II: Heterogeneous catalysis. Pure Appl. Chem. 1976, 46, 71–90. [Google Scholar]
- Taylor, H.S. Über aktive Stellen an Katalysatoren. Z. Elektrochem. Angew. Phys. Chem. 1929, 35, 542–549. (In German) [Google Scholar] [CrossRef]
- Armstrong, E.F.; Hilditch, T.P. A study of catalytic actions at solid surfaces. Part XII.—Some observations relative to those particles of a catalyst which participate in chemical change. Proc. R. Soc. Lond. Ser. A 1925, 108, 111–120. [Google Scholar] [CrossRef]
- Armstrong, E.F.; Hilditch, T.P. Catalysis at solid surfaces. Chem. Ind. 1925, 44, 701–709. [Google Scholar]
- Armstrong, E.F.; Hilditch, T.P. La catalyse par les surfaces solides. In Structure et Activité Chimiques: Deuxième Conseil de Chimie Tenu à Bruxelles du 16 au 24 Avril 1925; Gauthier-Villars: Paris, France, 1926; pp. 493–518. (In French) [Google Scholar]
- Hulett, G.A.; Berger, H.W. Volatilization of platinum. J. Am. Chem. Soc. 1904, 26, 1512–1515. [Google Scholar] [CrossRef]
- Salanov, A.N.; Serkova, A.N.; Zhirnova, A.S.; Isupova, L.A.; Parmon, V.N. Formation of etching structures on the surface of polycrystalline platinum in the course of catalytic oxidation of ammonia with air at 1133 K. Kinet. Catal. 2025, 66, 88–109. [Google Scholar] [CrossRef]
- Tyrański, M.; Bujalski, J.M.; Orciuch, W.; Makowski, Ł. Study of the ammonia oxidation process on platinum gauze and catalyst degradation phenomenon—CFD simulation with surface reaction kinetics and catalyst entrained particles motion and deposition tracking. Chem. Eng. Res. Des. 2025, 217, 416–439. [Google Scholar] [CrossRef]
- Simonne, D.; Coati, A.; Vlad, A.; Garreau, Y.; Voisin, B.; Richard, M.I.; Resta, A. Linking surface reconstructions to catalytic selectivity on Pt(100) during ammonia oxidation. ACS Catal. 2026, 16, 599–613. [Google Scholar] [CrossRef]
- Schwab, G.-M.; Pietsch, E. Zur Topochemie der Kontaktkatalyse. Z. Phys. Chem. Abt. B 1928, 1, 385–408. (In German) [Google Scholar] [CrossRef]
- Schwab, G.-M.; Pietsch, E. Die Lokalisierung der Katalysatorwirkung vom Standpunkt des Experiments. Zur Topochemie der Kontaktkatalyse. III. Z. Elektrochem. Angew. Phys. Chem. 1929, 35, 573–582. (In German) [Google Scholar] [CrossRef]
- Zelinsky, N.D.; Balandin, A.A. Kinetik der katalytischen Dehydrogenisation des Dekahydronaphthalins. Z. Phys. Chem. Stöchiom. Verwandtschaftsl. 1927, 126, 267–289. (In German) [Google Scholar] [CrossRef]
- Balandin, A.A. Zur Theorie der heterogenen katalytischen Reaktionen. Multipletthypothese. Modell der Dehydrierungskatalyse. Z. Phys. Chem. Abt. B 1929, 2, 289–316. (In German) [Google Scholar] [CrossRef]
- Balandin, A.A. Spaltungsreaktionen bei der Hydrierungskatalyse in Gegenwart von Nickel. Über die Rolle des Katalysators in der heterogenen Katalyse. Z. Phys. Chem. Abt. B 1929, 3, 167–194. (In German) [Google Scholar] [CrossRef]
- Balandin, A.A. Academician Nikolai Dmitrievich Zelinsky (on his 85th birthday). Vestn. Akad. Nauk SSSR 1946, 16, 79–90. (In Russian) [Google Scholar]
- Balandin, A.A. The nature of active centers and the kinetics of catalytic dehydrogenation. Adv. Catal. Relat. Subj. 1958, 10, 96–129. [Google Scholar]
- Balandin, A.A. Modern state of the multiplet theory of heterogeneous catalysis. Adv. Catal. Relat. Subj. 1969, 19, 1–210. [Google Scholar]
- Balandin, A.A. Development of a unified theory of catalysis. Regarding the structural and energy factors. Khim. Nauka Prom-St. 1959, 4, 655–661. (In Russian) [Google Scholar]
- Kobosev, N.I. A theory of the formation of catalytically active “ensembles” on surfaces. I. Acta Physicochim. URSS 1938, 9, 805–844. [Google Scholar]
- Kobosev, N.; Nikolaev, L.; Zubovich, I.; Goldfeld, J. The “ensemble” and “aggravation” principles in catalysis. I. Fluctuation analysis of active centres catalysis on the region of precrystalline structures. Acta Physicochim. URSS 1946, 21, 289–320. [Google Scholar]
- Kobozev, N.I. Physical and mathematical basis for the theory of active centers. Usp. Khim. 1956, 25, 545–631. (In Russian) [Google Scholar]
- Kobosew, N.I.; Lebedew, W.P.; Malzew, A.N. Die Bestimmung der Zusammensetzung, Zahl und absoluten Wirksamkeit katalytisch aktiver Zentren. Z. Phys. Chem. 1961, 217, 1–41. (In German) [Google Scholar] [CrossRef]
- Kobozev, N.I. Adsorption catalysts and the theory of active centers. An essay on the development of the theory of active ensembles and aggravation. In Modern Problems of Physical Chemistry; Gerasimov, Y.I., Akishin, P.A., Eds.; Moscow University Publishing: Moscow, Russia, 1968; Volume 3, pp. 3–60. (In Russian) [Google Scholar]
- Bussière, P.; Domanski, B.; Eyraud, C.; Prettre, M. Sur l’évaporation du platine au cours de la combustion du méthane (utilisation de 197Pt). C. R. Hebd. Seances Acad. Sci. 1956, 243, 1870–1872. (In French) [Google Scholar]
- Danchevskaya, M.N.; Kobozev, N.I. Catalysis by metal vapours. I. Catalytic properties of zinc and cadmium vapours. Russ. J. Phys. Chem. 1960, 34, 823–826. [Google Scholar]
- Danchevskaya, M.N.; Panasyuk, G.P.; Kobozev, N.I. Mass-spectrometric investigation of the mechanism of the dehydrogenation of methyl alcohol in zinc vapour. Russ. J. Phys. Chem. 1961, 35, 1044–1047. [Google Scholar]
- Danchevskaya, M.N.; Kobozev, N.I.; Moiseev, Y.V. Catalysis by metal vapours. II. Kinetics and mechanism of the decomposition of methanol in zinc and cadmium vapours. Russ. J. Phys. Chem. 1962, 36, 1171–1175. [Google Scholar]
- Danchevskaya, M.N.; Kobozev, N.I.; Pankrushev, Y.A. Catalysis by metal vapours. III. Catalytic cracking of hydrocarbons in zinc vapour. Russ. J. Phys. Chem. 1964, 38, 231–234. [Google Scholar]
- Peretyaka, A.R.; Danchevskaya, M.N.; Kobozev, N.I. Catalysis by metal vapours. IV. Homomolecular hydrogen isotope exchange reaction. Russ. J. Phys. Chem. 1971, 45, 216–218. [Google Scholar]
- Peretyaka, A.R.; Danchevskaya, M.N.; Kobozev, N.I. Catalysis by metal vapours. V. Para-ortho hydrogen conversion. Russ. J. Phys. Chem. 1971, 45, 333–335. [Google Scholar]
- Danchevskaya, M.N.; Kobozev, N.I.; Moiseev, Y.V.; Torbin, S.N. Catalysis by metal vapours. VI. Dehydrogenation of isopropyl alcohol. Russ. J. Phys. Chem. 1972, 46, 527–529. [Google Scholar]
- Danchevskaya, M.N.; Kobozev, N.I.; Galan-Sanches, N.I.; Torbin, S.N. Catalysis by metal vapours. VII. Catalytic decomposition of butyl and isobutyl alcohols in zinc vapour. Russ. J. Phys. Chem. 1974, 48, 435–436. [Google Scholar]
- Danchevskaya, M.N.; Torbin, S.N. The mechanism of the dehydrogenation of ethanol catalysed by atomic zinc vapour. Russ. J. Phys. Chem. 1978, 52, 254–255. [Google Scholar]
- Dolgoplosk, B.A.; Oreshkin, I.A.; Soboleva, T.V. The reactions of hydrocarbons under the influence of transition metal atoms and ions in the gas phase at room temperature. Russ. Chem. Rev. 1987, 56, 28–35. [Google Scholar] [CrossRef]
- Fialko, E.F.; Kikhtenko, A.V.; Goncharov, V.B.; Zamaraev, K.I. Ion cyclotron resonance study of CO oxidation in the gas phase in the presence of rhenium cations with carbonyl and oxygen ligands. Comparison with heterogeneous catalysis. Catal. Lett. 1996, 41, 7–11. [Google Scholar] [CrossRef]
- Böhme, D.K.; Schwarz, H. Gas-phase catalysis by atomic and cluster metal ions: The ultimate single-site catalysts. Angew. Chem. Int. Ed. 2005, 44, 2336–2354. [Google Scholar] [CrossRef] [PubMed]
- Lang, S.M.; Bernhardt, T.M. Gas phase metal cluster model systems for heterogeneous catalysis. Phys. Chem. Chem. Phys. 2012, 14, 9255–9269. [Google Scholar] [CrossRef]
- Li, X.-N.; Zou, X.-P.; He, S.-G. Metal-mediated catalysis in the gas phase: A review. Chin. J. Catal. 2017, 38, 1515–1527. [Google Scholar] [CrossRef]
- Panchenkov, G.M.; Lebedev, V.P. Chemical Kinetics and Catalysis; Mir Publishers: Moscow, Russia, 1976; Chapter 11; pp. 458–534. [Google Scholar]
- Nikolaev, L. Théorie des ensembles actifs de Kobozev. J. Chim. Phys. Phys.-Chim. Biol. 1954, 51, 777–780. (In French) [Google Scholar]
- Gil’debrand, E.I. Low concentration catalysts on carriers. Int. Chem. Eng. 1966, 6, 449–480. [Google Scholar]
- Mal’tsev, A.N. Theory of active ensembles for catalysis by N. I. Kobozev (on the 50th anniversary of the theory). Vestn. Mosk. Univ. Ser. 2 Khim. 1989, 30, 323–329. (In Russian) [Google Scholar]
- Carrà, S.; Ragaini, V.; Guella, F. Active centers in cyclohexene disproportionation on supported palladium. J. Catal. 1967, 8, 261–271. [Google Scholar] [CrossRef]
- Carrà, S.; Ragaini, V. On the mechanism of cyclohexene disproportionation catalyzed by supported palladium. Tetrahedron Lett. 1967, 8, 1079–1082. [Google Scholar] [CrossRef]
- Ragaini, V.; Forni, L.; Mao, L.V. Kinetic study and catalyst structural analysis for the dehydrogenation-hydrogenolysis of n-pentane over Ru-Al2O3 catalysts. J. Catal. 1975, 37, 339–347. [Google Scholar] [CrossRef]
- Krishnasamy, V.; Mathur, P.; Balasubramanian, K. Ensembling parameters for the dehydrogenation of Δ3-carene, α-pinene, β-pinene and camphene over platinum-alumina catalyst. Chem. Eng. Commun. 1983, 23, 115–123. [Google Scholar] [CrossRef]
- Krishnasamy, V.; Balasubramanian, K. Ensembling and monolayer parameters for the dehydrogenation of cyclohexane over Pt/Al2O3 catalyst. J. Catal. 1984, 90, 351–357. [Google Scholar] [CrossRef]
- Pilic, B.; Stoiljkovic, D.; Bakocevic, I.; Jovanovic, S.; Panic, D.; Korugic-Kara, L. Charge percolation mechanism of Ziegler—Natta polymerization: Part II: Importance of support nanoparticles. In New Polymeric Materials; Korugic-Karasz, L.C., MacKnight, W.J., Martuscelli, E., Eds.; American Chemical Society: Washington, DC, USA, 2005; Chapter 16; pp. 215–228. [Google Scholar]
- Boudart, M.; Aldag, A.W.; Ptak, L.D.; Benson, J.E. On the selectivity of platinum catalysts. J. Catal. 1968, 11, 35–45. [Google Scholar] [CrossRef]
- Boudart, M. Catalysis by supported metals. Adv. Catal. Relat. Subj. 1969, 20, 153–166. [Google Scholar]
- Dowden, D.A. Electronic structure and ensembles in chemisorption and catalysis by binary alloys. In Catalysis: Proceedings of the Fifth International Congress on Catalysis, Miami Beach, FL, USA, 20–26 August 1972; Hightower, J.W., Ed.; North-Holland Publishing Company: Amsterdam, The Netherlands, 1973; Volume 1, pp. 621–631. [Google Scholar]
- Ponec, V.; Sachtler, W.M.H. The reactions between cyclopentane and deuterium on nickel and nickel-copper alloys. J. Catal. 1972, 24, 250–261. [Google Scholar] [CrossRef]
- Sachtler, W.M.H.; van Santen, R.A. Surface composition and selectivity of alloy catalysts. Adv. Catal. 1977, 26, 69–119. [Google Scholar]
- Slinkin, A.A. Problems in the study of supported metal catalysts. Russ. Chem. Rev. 1980, 49, 404–413. [Google Scholar] [CrossRef]
- Slinkin, A.A. Structure and catalytic properties of deposited metals. Itogi Nauk. Tekh. Ser. Kinet. Katal. 1982, 10, 5–114. (In Russian) [Google Scholar]
- Martin, G.A. A quantitative approach to the ensemble model of catalysis by metals. Catal. Rev.-Sci. Eng. 1988, 30, 519–562. [Google Scholar] [CrossRef]
- Martin, G.A. Variation of the number of metal atoms involved in active sites and of the true activation energy of hydrocarbon conversion and CO hydrogenation over metals. Bull. Soc. Chim. Belg. 1996, 105, 131–137. [Google Scholar] [CrossRef]
- Sachtler, W.M.H. Ensemble and ligand effects in metal catalysis. In Handbook of Heterogeneous Catalysis, 2nd ed.; Ertl, G., Knözinger, H., Schüth, F., Weitkamp, J., Eds.; Wiley-VCH: Weinheim, Germany, 2008; Volume 3, pp. 1585–1593. [Google Scholar]
- Guo, Y.; Wang, M.; Zhu, Q.; Xiao, D.; Ma, D. Ensemble effect for single-atom, small cluster and nanoparticle catalysts. Nat. Catal. 2022, 5, 766–776. [Google Scholar] [CrossRef]
- Nakaya, Y.; Furukawa, S. Catalysis of alloys: Classification, principles, and design for a variety of materials and reactions. Chem. Rev. 2023, 123, 5859–5947. [Google Scholar] [CrossRef]
- Pisarzhevskii, L.V. On the mechanism of catalysis by metals and metal oxides. Zh. Russ. Fiz.-Khim. O-Va Chast. Khim. 1929, 61, 1609–1634. (In Russian) [Google Scholar]
- Lennard-Jones, J.E. Processes of adsorption and diffusion on solid surfaces. Trans. Faraday Soc. 1932, 28, 333–359. [Google Scholar] [CrossRef]
- Taylor, H.S. The activation energy of adsorption processes. J. Am. Chem. Soc. 1931, 53, 578–597. [Google Scholar] [CrossRef]
- Taylor, H.S. Chemical reactions at surfaces. Chem. Rev. 1931, 9, 1–46. [Google Scholar] [CrossRef]
- Schwab, G.-M. Metal electrons and catalysis. Trans. Faraday Soc. 1946, 42, 689–697. [Google Scholar] [CrossRef]
- Dowden, D.A. Heterogeneous catalysis. Part I. Theoretical basis. J. Chem. Soc. 1950, 242–265. [Google Scholar] [CrossRef]
- Dowden, D.A.; Reynolds, P.W. Some reactions over alloy catalysts. Discuss. Faraday Soc. 1950, 8, 184–190. [Google Scholar] [CrossRef]
- Burch, R. Importance of electronic ligand effects in metal alloy catalysts. Acc. Chem. Res. 1982, 15, 24–31. [Google Scholar] [CrossRef]
- Ponec, V. Alloy catalysts: The concepts. Appl. Catal. A Gen. 2001, 222, 31–45. [Google Scholar] [CrossRef]
- Chen, W.; Qian, G.; Wang, H.; Chen, D.; Zhou, X.; Yuan, W.; Duan, X. Rationalizing the d-band model from theory to practice in catalyst design. J. Am. Chem. Soc. 2025, 147, 46729–46744. [Google Scholar] [CrossRef]
- Vol’kenshtein, F.F. Effect of small quantities of impurities on the catalytic activity of ionic catalysts. Zh. Fiz. Khim. 1950, 24, 1068–1082. (In Russian) [Google Scholar]
- Boudart, M. Electronic chemical potential in chemisorption and catalysis. J. Am. Chem. Soc. 1952, 74, 1531–1535. [Google Scholar] [CrossRef]
- Hauffe, K. Über den Mechanismus von Gasreaktionen an Oberflächen halbleitender Katalysatoren. Adv. Catal. Relat. Subj. 1957, 9, 187–203. (In German) [Google Scholar]
- Wolkenstein, T. The electron theory of catalysis on semiconductors. Adv. Catal. Relat. Subj. 1960, 12, 189–264. [Google Scholar]
- Vol’kenshtein, F.F. Experiment and the electronic theory of catalysis. Russ. Chem. Rev. 1966, 35, 537–546. [Google Scholar] [CrossRef]
- Vol’kenshtein, F.F. The Electronic Theory of Catalysis on Semiconductors; The Macmillan Company: New York, NY, USA, 1963. [Google Scholar]
- Thon, N.; Taylor, H.A. Kinetics of active centers in surface-catalyzed reactions. J. Am. Chem. Soc. 1953, 75, 2747–2750. [Google Scholar] [CrossRef]
- Védrine, J.C. The role of redox, acid-base and collective properties and of cristalline state of heterogeneous catalysts in the selective oxidation of hydrocarbons. Top. Catal. 2002, 21, 97–106. [Google Scholar] [CrossRef]
- Taylor, H.S. Active centers. In Twelfth Report of the Committee on Catalysis, National Research Council; John Wiley & Sons: New York, NY, USA, 1940; Chapter 4; pp. 41–52. [Google Scholar]
- Taylor, H.S. Contact catalysis between two world wars. Am. Sci. 1946, 34, 553–572. [Google Scholar]
- Taylor, H.S. The heterogeneity of catalyst surfaces for chemisorption. Adv. Catal. Relat. Subj. 1948, 1, 1–26. [Google Scholar]
- Taylor, H.S. 5th Spiers Memorial Lecture. Catalysis: Retrospect and prospect. Discuss. Faraday Soc. 1950, 8, 9–18. [Google Scholar] [CrossRef]
- Taylor, H. Surface catalysis. Annu. Rev. Phys. Chem. 1961, 12, 127–150. [Google Scholar] [CrossRef]
- Boudart, M. Four decades of active centers. Am. Sci. 1969, 57, 97–111. [Google Scholar]
- Boudart, M. Concepts in heterogeneous catalysis. In Interactions on Metal Surfaces; Gomer, R., Ed.; Springer: Berlin/Heidelberg, Germany, 1975; Chapter 7; pp. 275–298. [Google Scholar]
- Boudart, M. Heterogeneous molecular catalysis: Oxymoron or reality? J. Mol. Catal. A Chem. 1997, 120, 271–280. [Google Scholar] [CrossRef]
- Rideal, E.K. Concepts in Catalysis; Academic Press: London, UK, 1968; Chapter 2; pp. 4–42. [Google Scholar]
- Kemball, C. Hugh Stott Taylor. 6 February 1890–17 April 1974. Biogr. Mem. Fellows R. Soc. 1975, 21, 517–547. [Google Scholar] [CrossRef][Green Version]
- Likholobov, V.A. On the 100th anniversary of the introduction of the concept of “active sites in catalysis”. Katal. Byull. 2025, 5–13. Available online: https://elibrary.ru/item.asp?id=82384837 (accessed on 31 May 2026). (In Russian)
- Likholobov, V.A. Development of the concept of “active sites in catalysis”: Michel Boudart. Katal. Byull. 2025, 9–28. Available online: https://elibrary.ru/item.asp?id=82943353 (accessed on 31 May 2026). (In Russian)
- Balandin, A.A.; Kotelkov, N.Z. Dehydrogenation and decomposition of cyclohexane at high temperatures over metal catalysts. Zh. Prikl. Khim. 1942, 15, 139–150. (In Russian) [Google Scholar]
- Tovbin, M.V.; Baram, O.M. On the role of polymorphic transformations in the mechanism of heterogeneous catalytic reactions. Ukr. Khim. Zh. 1957, 23, 567–572. (In Russian) [Google Scholar]
- Leidheiser, H., Jr.; Gwathmey, A.T. The catalytic reaction of hydrogen and oxygen on plane faces of a single crystal of copper. J. Am. Chem. Soc. 1948, 70, 1200–1206. [Google Scholar] [CrossRef] [PubMed]
- Gwathmey, A.T.; Cunningham, R.E. The influence of crystal face in catalysis. Adv. Catal. Relat. Subj. 1958, 10, 57–95. [Google Scholar]
- Tamaru, K. Adsorption measurements during surface catalysis. Adv. Catal. Relat. Subj. 1964, 15, 65–90. [Google Scholar]
- Boreskov, G.K. Mechanism of action of solid catalysts. In Heterogeneous Catalysis in the Chemical Industry: Proceedings of the All-Union Conference of 1953; Boreskov, G.K., Naberezhnykh, M.E., Pshezhetskii, S.Y., Slin’ko, M.G., Temkin, M.I., Cherednichenko, V.M., Eds.; Goskhimizdat: Moscow, Russia, 1955; pp. 5–28. (In Russian) [Google Scholar]
- Boreskov, G.K. The development of the concepts of the nature of heterogeneous catalysis. Kinet. Catal. 1977, 18, 910–918. [Google Scholar]
- Boreskov, G.K. Heterogeneous Catalysis; Zamaraev, K.I., Khasin, A.V., Eds.; Nova Science Publishers: New York, NY, USA, 2003. [Google Scholar]
- Boreskov, G.K. Interaction between the catalyst and the reacting system. Zh. Fiz. Khim. 1958, 32, 2739–2747. (In Russian) [Google Scholar]
- Boreskov, G.K. The effects of the interaction of the reactants with the catalyst on the kinetics of catalysed reactions. Russ. J. Phys. Chem. 1959, 33, 243–246. [Google Scholar]
- Boreskov, G.K.; Ven’yaminov, S.A.; Sazonova, N.N. Investigation of the effect of the composition of the reaction mixture on the composition and properties of the catalyst in the oxidative dehydrogenation of butene-1 over an iron/antimony catalyst. Dokl. Akad. Nauk SSSR 1978, 240, 619–622. (In Russian) [Google Scholar]
- Sazonova, N.N.; Ven’yaminov, S.A.; Boreskov, G.K. Investigation of the mechanism of mild oxidation of hydrocarbons on solid oxide catalysts. I. Influence of the reaction medium on the catalytic properties of an iron oxide catalyst in the oxidative dehydrogenation of butylenes. Kinet. Katal. 1973, 14, 1169–1174. (In Russian) [Google Scholar]
- Andrushkevich, T.V.; Popova, G.Y. Mechanism of heterogeneous oxidation of acrolein to acrylic acid. Russ. Chem. Rev. 1991, 60, 1023–1034. [Google Scholar] [CrossRef]
- Andrushkevich, T.V. Heterogeneous catalytic oxidation of acrolein to acrylic acid: Mechanism and catalysts. Catal. Rev.-Sci. Eng. 1993, 35, 213–259. [Google Scholar] [CrossRef]
- Boreskov, G.K. Influence of changes in the composition of the catalyst on the kinetics of reactions of heterogeneous catalysis. Kinet. Catal. 1972, 13, 493–502. [Google Scholar]
- Boreskov, G.K. Influence of the reaction medium on the properties of solid catalysts. Kinet. Catal. 1980, 21, 1–10. [Google Scholar]
- Boreskov, G.K. Kinetics of heterogeneous catalytic reactions taking into account the influence of reaction mixtures on catalysts. In Proceedings of the 8th International Congress on Catalysis, Berlin (West), Germany, 2–6 July 1984; Verlag Chemie: Weinheim, Germany, 1984; Volume 3, pp. 231–242. [Google Scholar]
- Boreskov, G.K. Changes in the properties of solid catalysts in a reaction medium and their effect on the kinetics of heterogeneous catalytic reactions. Izv. Khim. 1984, 17, 60–70. (In Russian) [Google Scholar]
- Boudart, M.; Djéga-Mariadassou, G. Kinetics of Heterogeneous Catalytic Reactions; Princeton University Press: Princeton, NJ, USA, 1984. [Google Scholar]
- Boudart, M. Turnover rates in heterogeneous catalysis. Chem. Rev. 1995, 95, 661–666. [Google Scholar] [CrossRef]
- Boreskov, G.K.; Matros, Y.S. Unsteady-state performance of heterogeneous catalytic reactions. Catal. Rev.-Sci. Eng. 1983, 25, 551–590. [Google Scholar] [CrossRef]
- Matros, Y.S. Catalytic Processes Under Unsteady-State Conditions; Elsevier Science Publishers: Amsterdam, The Netherlands, 1989. [Google Scholar]
- Zagoruiko, A.N. Unsteady catalytic processes and sorption-catalytic technologies. Russ. Chem. Rev. 2007, 76, 639–654. [Google Scholar] [CrossRef]
- Zagoruiko, A.N.; Bobrova, L.; Vernikovskaya, N.; Zazhigalov, S. Unsteady-state operation of reactors with fixed catalyst beds. Rev. Chem. Eng. 2021, 37, 193–225. [Google Scholar] [CrossRef]
- Zazhigalov, S.; Elyshev, A.; Zagoruiko, A. Modeling of unsteady-state catalytic and adsorption–catalytic processes: Novel reactor designs. In Mathematical Modeling of Complex Reaction Systems in the Oil and Gas Industry; Ancheyta, J., Zagoruiko, A., Elyshev, A., Eds.; John Wiley & Sons: Hoboken, NJ, USA, 2024; Chapter 4; pp. 138–167. [Google Scholar]
- Shchukin, E.D. Correlation of a break and rearrangement of interatomic bonds in a solid phase and molecules of a medium during catalysis. In Mechanism of Catalysis; Boreskov, G.K., Andrushkevich, T.V., Eds.; Nauka: Novosibirsk, Russia, 1984; Part 2; pp. 142–154. (In Russian) [Google Scholar]
- Shchukin, E.D.; Bessonov, A.I.; Kontorovich, S.I.; Polukarova, Z.M.; Savenko, V.I.; Sokolova, L.N.; Margolis, L.Y.; Amelina, E.A.; Burenkova, L.N.; Romanovskii, B.V. Physicochemical mechanics of fine-pore catalysts in active media. Mater. Sci. 2003, 39, 334–350. [Google Scholar] [CrossRef]
- Rozovskii, A.Y. The influence of the reaction medium on a catalyst under catalysis conditions. Dokl. Phys. Chem. 1963, 151, 749–751. [Google Scholar]
- Rozovskii, A.Y. Action of the reaction mixture on a catalyst, and the self-regulation effect in catalysis. Kinet. Catal. 1967, 8, 974–987. [Google Scholar]
- Rozovskii, A.Y. Catalyst and Reaction Medium; Kolbanovskii, Y.A., Ed.; Nauka: Moscow, Russia, 1988. (In Russian) [Google Scholar]
- Parmon, V.N. A life devoted to catalysis. On the centenary of the birth of Academician G. K. Boreskov. Her. Russ. Acad. Sci. 2007, 77, 275–282. [Google Scholar] [CrossRef]
- Andrushkevich, T.V.; Bukhtiyarov, V.I. Scientific heritage of Georgii Konstantinovich Boreskov. Kinet. Catal. 2019, 60, 123–136. [Google Scholar] [CrossRef]
- Roginskii, S.Z.; Tret’yakov, I.I.; Shekhter, A.B. Electron microscopic study of the surface of working catalysts. I. Application of the method of shadow replicas to studying changes of catalysts under the influence of the catalyzed reaction. Zh. Fiz. Khim. 1949, 23, 50–56. (In Russian) [Google Scholar]
- Roginskii, S.Z.; Tret’yakov, I.I.; Shekhter, A.B. Electron microscopic study of the surface of working catalysts. II. Change of the surface of bulk palladium during catalytic oxidation of hydrogen. Zh. Fiz. Khim. 1949, 23, 1152–1160. (In Russian) [Google Scholar]
- Shekhter, A.B.; Tret’yakov, I.I. Electron microscopic investigation of changes in the surface of solid catalysts during operation. Bull. Acad. Sci. USSR Div. Chem. Sci. 1953, 2, 397–402. [Google Scholar] [CrossRef]
- Germer, L.H. A new electron diffraction technique, potentially applicable to research in catalysis. Adv. Catal. Relat. Subj. 1962, 13, 191–201. [Google Scholar]
- Farnsworth, H.E. The clean single-crystal-surface approach to surface reactions. Adv. Catal. Relat. Subj. 1964, 15, 31–63. [Google Scholar]
- Roberts, M.W. The impact of surface physics and the emergence of new experimental methods (1945–1965). Catal. Lett. 2000, 67, 23–29. [Google Scholar] [CrossRef]
- Roberts, M.W. Surface sensitive spectroscopies and their impact on catalysis (1970–1999). Catal. Lett. 2000, 67, 41–53. [Google Scholar] [CrossRef]
- Sinfelt, J.H. Role of surface science in catalysis. Surf. Sci. 2002, 500, 923–946. [Google Scholar] [CrossRef]
- Altman, E.I.; Freund, H.-J.; Bahnemann, D.W.; Chaudret, B.; Fout, A.R.; Gellman, A.J.; Ward, T.R.; Zaera, F.; Cremer, P.; Yang, P.; et al. In memory of Gabor Somorjai (1935–2025). Catal. Lett. 2025, 155, 397. [Google Scholar] [CrossRef]
- Hagstrom, S.; Lyon, H.B.; Somorjai, G.A. Surface structures on the clean platinum (100) surface. Phys. Rev. Lett. 1965, 15, 491–493. [Google Scholar] [CrossRef]
- Somorjai, G.A. Surface science and heterogeneous catalysis. Chimia 1981, 35, 1–9. [Google Scholar] [CrossRef]
- Somorjai, G.A. The surface science of heterogeneous catalysis. Surf. Sci. 1994, 299–300, 849–866. [Google Scholar] [CrossRef]
- Somorjai, G.A. Modern surface science and surface technologies: An introduction. Chem. Rev. 1996, 96, 1223–1235. [Google Scholar] [CrossRef]
- Somorjai, G.A. The development of molecular surface science and the surface science of catalysis: The Berkeley contribution. J. Phys. Chem. B 2000, 104, 2969–2979. [Google Scholar] [CrossRef]
- Somorjai, G.A.; Li, Y. Impact of surface chemistry. Proc. Natl. Acad. Sci. USA 2011, 108, 917–924. [Google Scholar] [CrossRef] [PubMed]
- Somorjai, G.A. Introduction to Surface Chemistry and Catalysis; John Wiley & Sons: New York, NY, USA, 1994. [Google Scholar]
- Kesmodel, L.L.; Stair, P.C.; Baetzold, R.C.; Somorjai, G.A. Surface structure and bonding of acetylene to the platinum (111) surface. Phys. Rev. Lett. 1976, 36, 1316–1319. [Google Scholar] [CrossRef]
- Kesmodel, L.L.; Dubois, L.H.; Somorjai, G.A. LEED analysis of acetylene and ethylene chemisorption on the Pt(111) surface: Evidence for ethylidyne formation. J. Chem. Phys. 1979, 70, 2180–2188. [Google Scholar] [CrossRef]
- Somorjai, G.A.; Van Hove, M.A. Surface restructuring as a mechanism for bond breaking and catalytic reactions at metal surfaces. Catal. Lett. 1988, 1, 433–437. [Google Scholar] [CrossRef]
- Somorjai, G.A.; Van Hove, M.A. Adsorbate-induced restructuring of surfaces. Prog. Surf. Sci. 1989, 30, 201–231. [Google Scholar] [CrossRef]
- Somorjai, G.A. Surface reconstruction and catalysis. Annu. Rev. Phys. Chem. 1994, 45, 721–751. [Google Scholar] [CrossRef]
- Van Hove, M.A.; Somorjai, G.A. Adsorption and adsorbate-induced restructuring: A LEED perspective. Surf. Sci. 1994, 299–300, 487–501. [Google Scholar] [CrossRef]
- Somorjai, G.A. The flexible surface. Correlation between reactivity and restructuring ability. Langmuir 1991, 7, 3176–3182. [Google Scholar] [CrossRef]
- Somorjai, G.A.; Rupprechter, G. The flexible surface: Molecular studies explain the extraordinary diversity of surface chemical properties. J. Chem. Educ. 1998, 75, 161–176. [Google Scholar] [CrossRef]
- Ertl, G. Oscillatory catalytic reactions at single-crystal surfaces. Adv. Catal. 1990, 37, 213–277. [Google Scholar]
- Ertl, G. Restructuring and self-organization in reactions on metal surfaces. J. Mol. Catal. 1992, 74, 1–9. [Google Scholar] [CrossRef]
- Ertl, G. Non-linear dynamics: Oscillatory kinetics and spatio-temporal pattern formation. In Handbook of Heterogeneous Catalysis, 2nd ed.; Ertl, G., Knözinger, H., Schüth, F., Weitkamp, J., Eds.; Wiley-VCH: Weinheim, Germany, 2008; Volume 3, pp. 1492–1516. [Google Scholar]
- Ertl, G. Reactions at surfaces: From atoms to complexity (Nobel Lecture). Angew. Chem. Int. Ed. 2008, 47, 3524–3535. [Google Scholar] [CrossRef]
- Ertl, G. Reactions at Solid Surfaces; John Wiley & Sons: Hoboken, NJ, USA, 2009. [Google Scholar]
- Newton, M.A. Dynamic adsorbate/reaction induced structural change of supported metal nanoparticles: Heterogeneous catalysis and beyond. Chem. Soc. Rev. 2008, 37, 2644–2657. [Google Scholar] [CrossRef]
- Sievers, C.; Noda, Y.; Qi, L.; Albuquerque, E.M.; Rioux, R.M.; Scott, S.L. Phenomena affecting catalytic reactions at solid-liquid interfaces. ACS Catal. 2016, 6, 8286–8307. [Google Scholar] [CrossRef]
- Wang, H.; Wu, Y.; Luo, Q.; Wu, H.; Xiao, F.-S.; Wang, L. Managing dynamic catalyst changes to upgrade reactors and reaction processes. Nat. Chem. Eng. 2025, 2, 169–180. [Google Scholar] [CrossRef]
- Du, X.; Fan, Y.; Fan, J.; Wang, X.; Li, R.; Bao, W.; Fu, Q. Dynamic evolution of catalyst structure for tuned catalytic performance in CO2 hydrogenation. JACS Au 2026, 6, 2100–2114. [Google Scholar] [CrossRef]
- Mizoroki, T.; Mori, K.; Ozaki, A. Arylation of olefin with aryl iodide catalyzed by palladium. Bull. Chem. Soc. Jpn. 1971, 44, 581. [Google Scholar] [CrossRef]
- Mori, K.; Mizoroki, T.; Ozaki, A. Arylation of olefin with iodobenzene catalyzed by palladium. Bull. Chem. Soc. Jpn. 1973, 46, 1505–1508. [Google Scholar] [CrossRef]
- Sheldon, R.A.; Wallau, M.; Arends, I.W.C.E.; Schuchardt, U. Heterogeneous catalysts for liquid-phase oxidations: Philosophers’ stones or Trojan horses? Acc. Chem. Res. 1998, 31, 485–493. [Google Scholar] [CrossRef]
- Biffis, A.; Zecca, M.; Basato, M. Palladium metal catalysts in Heck C–C coupling reactions. J. Mol. Catal. A Chem. 2001, 173, 249–274. [Google Scholar] [CrossRef]
- Gladysz, J.A. The experimental assay of catalyst recovery: General concepts. In Recoverable and Recyclable Catalysts; Benaglia, M., Ed.; John Wiley & Sons: Chichester, UK, 2009; Chapter 1; pp. 1–14. [Google Scholar]
- Parshall, G.W. Industrial applications of homogeneous catalysis. A review. J. Mol. Catal. 1978, 4, 243–270. [Google Scholar] [CrossRef]
- Cornils, B.; Herrmann, W.A.; Rasch, M. Otto Roelen, pioneer in industrial homogeneous catalysis. Angew. Chem. Int. Ed. Engl. 1994, 33, 2144–2163. [Google Scholar] [CrossRef]
- Hummel, H.G. Walter Reppe, 1892–1969. Chem. Ber. 1984, 117, I–XXI. (In German) [Google Scholar]
- Talsi, E.P.; Klimov, O.V.; Zamaraev, K.I. Characterization with 95Mo, 17O, 1H NMR and EPR of alkylperoxo, alkoxo, peroxo and diolo molybdenum (VI) complexes formed in the course of catalytic epoxidation of cyclohexene with organic hydroperoxides. J. Mol. Catal. 1993, 83, 329–346. [Google Scholar] [CrossRef]
- Zamaraev, K. New possibilities of NMR in mechanistic studies of homogeneous and heterogeneous catalysis. J. Mol. Catal. 1993, 82, 275–324. [Google Scholar] [CrossRef]
- Tsuji, J.; Fairlamb, I.J.S. Tris(dibenzylideneacetone)dipalladium–chloroform. In Handbook of Reagents for Organic Synthesis. Catalyst Components for Coupling Reactions; Molander, G.A., Ed.; John Wiley & Sons: Chichester, UK, 2008; pp. 602–607. [Google Scholar]
- Zalesskiy, S.S.; Ananikov, V.P. Pd2(dba)3 as a precursor of soluble metal complexes and nanoparticles: Determination of palladium active species for catalysis and synthesis. Organometallics 2012, 31, 2302–2309. [Google Scholar] [CrossRef]
- Ananikov, V.P.; Beletskaya, I.P. Toward the ideal catalyst: From atomic centers to a “cocktail” of catalysts. Organometallics 2012, 31, 1595–1604. [Google Scholar] [CrossRef]
- Kashin, A.S.; Ananikov, V.P. Catalytic C–C and C–heteroatom bond formation reactions: In situ generated or preformed catalysts? Complicated mechanistic picture behind well-known experimental procedures. J. Org. Chem. 2013, 78, 11117–11125. [Google Scholar] [CrossRef]
- Eremin, D.B.; Ananikov, V.P. Understanding active species in catalytic transformations: From molecular catalysis to nanoparticles, leaching, “Cocktails” of catalysts and dynamic systems. Coord. Chem. Rev. 2017, 346, 2–19. [Google Scholar] [CrossRef]
- Chernyshev, V.M.; Denisova, E.A.; Eremin, D.B.; Ananikov, V.P. The key role of R–NHC coupling (R = C, H, heteroatom) and M–NHC bond cleavage in the evolution of M/NHC complexes and formation of catalytically active species. Chem. Sci. 2020, 11, 6957–6977. [Google Scholar] [CrossRef]
- Prima, D.O.; Kulikovskaya, N.S.; Galushko, A.S.; Mironenko, R.M.; Ananikov, V.P. Transition metal ‘cocktail’-type catalysis. Curr. Opin. Green Sustain. Chem. 2021, 31, 100502. [Google Scholar] [CrossRef]
- Galushko, A.S.; Kashin, A.S.; Eremin, D.B.; Polynski, M.V.; Pentsak, E.O.; Chernyshev, V.M.; Ananikov, V.P. Introduction to dynamic catalysis and the interface between molecular and heterogeneous catalysts. In Nanoparticles in Catalysis: Advances in Synthesis and Applications; Philippot, K., Roucoux, A., Eds.; Wiley-VCH: Weinheim, Germany, 2021; Chapter 2; pp. 13–42. [Google Scholar]
- Chernyshev, V.M.; Ananikov, V.P. Nickel and palladium catalysis: Stronger demand than ever. ACS Catal. 2022, 12, 1180–1200. [Google Scholar] [CrossRef]
- Kostyukovich, A.Y.; Sahharova, L.T. Dynamic ‘cocktail’-type catalytic systems in C–N bond formation reactions. Russ. Chem. Rev. 2025, 94, RCR5181. [Google Scholar] [CrossRef]
- Prima, D.O.; Vatsadze, S.Z. Cocktail-type catalysis: An emerging concept in metal-mediated transformations. Organometallics 2025, 44, 1337–1357. [Google Scholar] [CrossRef]
- Galushko, A.S.; Cherepanova, V.A.; Ananikov, V.P. Totally defined catalysis as a new paradigm for describing catalytic systems by combining advanced analytics and machine learning techniques. Adv. Organomet. Chem. 2025, 84, 255–285. [Google Scholar]
- Egorov, M.P.; Lee, V.Y.; Alabugin, I.V. Cocktail of catalysts: A dynamic advance in modern catalysis. Chemistry 2025, 7, 109. [Google Scholar] [CrossRef]
- Maximov, A.L.; Egorov, M.P. Discovery and development of cocktail-type catalysis. Chin. J. Catal. 2025, 78, 7–24. [Google Scholar] [CrossRef]
- Pagliaro, M.; Muscolo, A.; Russo, M.; Mauriello, F.; Avellone, G.; Calabrò, P.S.; Ciriminna, R. How are discoveries in chemistry made? Insight from three discoveries and their impact. Chemistry 2025, 7, 200. [Google Scholar] [CrossRef]
- Galushko, A.S.; Gordeev, E.G.; Kashin, A.S.; Zubavichus, Y.V.; Ananikov, V.P. Visualization of catalyst dynamics and development of a practical procedure to study complex “cocktail”-type catalytic systems. Faraday Discuss. 2021, 229, 458–474. [Google Scholar] [CrossRef]
- Polynski, M.V.; Ananikov, V.P. Modeling key pathways proposed for the formation and evolution of “cocktail”-type systems in Pd-catalyzed reactions involving ArX reagents. ACS Catal. 2019, 9, 3991–4005. [Google Scholar] [CrossRef]
- Patil, E.D.; Burykina, J.V.; Eremin, D.B.; Boiko, D.A.; Shepelenko, K.E.; Ilyushenkova, V.V.; Chernyshev, V.M.; Ananikov, V.P. Quantitative determination of active species transforming the R-NHC coupling process under catalytic conditions. Inorg. Chem. 2024, 63, 2967–2976. [Google Scholar] [CrossRef]
- Saybulina, E.R.; Mironenko, R.M.; Galushko, A.S.; Ilyushenkova, V.V.; Izmailov, R.R.; Ananikov, V.P. Mechanistic analysis of transformative Pd/NHC catalyst evolution in the 1,2-diphenylacetylene semihydrogenation using molecular hydrogen. J. Catal. 2024, 430, 115293. [Google Scholar] [CrossRef]
- Peris, E. Smart N-heterocyclic carbene ligands in catalysis. Chem. Rev. 2017, 118, 9988–10031. [Google Scholar] [CrossRef]
- Nahra, F.; Cazin, C.S.J. Sustainability in Ru- and Pd-based catalytic systems using N-heterocyclic carbenes as ligands. Chem. Soc. Rev. 2021, 50, 3094–3142. [Google Scholar] [CrossRef]
- Panova, Y.S.; Kashin, A.S.; Vorobev, M.G.; Degtyareva, E.S.; Ananikov, V.P. Nature of the copper-oxide-mediated C–S cross-coupling reaction: Leaching of catalytically active species from the metal oxide surface. ACS Catal. 2016, 6, 3637–3643. [Google Scholar] [CrossRef]
- Gjermestad, C.S.; Seki, R.; Kusumoto, S.; Occhipinti, G.; Le Roux, E.; Nozaki, K.; Jensen, V.R. Stirred, not shaken: Pre-made catalyst cocktail enables arene-selective hydrodeoxygenation of phenols. ACS Catal. 2026, 16, 6228–6233. [Google Scholar] [CrossRef]
- Gates, B.C.; Katzer, J.R.; Schuit, G.C.A. Chemistry of Catalytic Processes; McGraw-Hill: New York, NY, USA, 1979; Chapter 2; pp. 112–183. [Google Scholar]
- van Leeuwen, P.W.N.M. Homogeneous Catalysis: Understanding the Art; Kluwer Academic Publishers: Dordrecht, The Netherlands, 2004; Chapter 2; pp. 29–61. [Google Scholar]
- Jangid, P.; Chaudhary, S. Stochastic dynamics of nanoparticle catalysis: A discrete-state perspective. Mater. Horiz. 2026, 13, 2152–2172. [Google Scholar] [CrossRef]
- Shooshtari Gugtapeh, H.; Aghaii, A.H.; Simchi, A. A critical review on the role of structural defects in electrochemical nitrate reduction catalysts: From mechanisms to formation strategies. J. Environ. Chem. Eng. 2025, 13, 120221. [Google Scholar] [CrossRef]
- Anekwe, I.M.S.; Isa, Y.M. Unlocking catalytic longevity: A critical review of catalyst deactivation pathways and regeneration technologies. Energy Adv. 2025, 4, 1075–1113. [Google Scholar] [CrossRef]
- Kityk, A.; Sadeghi, B.; Pavlik, V.; Hnatko, M.; Balog, M. Unveiling structure-performance relationships in HER catalysts for sustainable and scalable hydrogen production. Mater. Rep. Energy 2026, 6, 100407. [Google Scholar] [CrossRef]
- Zhan, Y.; Yang, T.; Liu, S.; Yang, L.; Wang, E.; Yu, X.; Wang, H.; Chou, K.-C.; Hou, X. In situ characterization method to reveal the surface reconstruction process of an electrocatalyst. Nanomaterials 2025, 15, 917. [Google Scholar] [CrossRef]
- Araújo, T.P.; Mitchell, S.; Pérez-Ramírez, J. Design principles of catalytic materials for CO2 hydrogenation to methanol. Adv. Mater. 2024, 36, 2409322. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Farzinpour, F.; Kornienko, N. Dynamic active sites behind Cu-based electrocatalysts: Original or restructuring-induced catalytic activity. Chem 2025, 11, 102575. [Google Scholar] [CrossRef]
- Feng, Y.; Chen, Y.; Zheng, L.; Chen, X.; Li, T.; Zhao, W. Descriptors construction and application in catalytic site design. iScience 2025, 28, 113080. [Google Scholar] [CrossRef]
- Wang, B.; Zheng, S.; Wu, J.; Li, J.; Pan, F. Inverse design of catalytic active sites via interpretable topology-based deep generative models. npj Comput. Mater. 2025, 11, 147. [Google Scholar] [CrossRef]
- Li, N.; Xu, J.; Gao, W.; Wan, S.; Xu, X.; Yan, B.; Wang, J.; Chen, G. Machine learning-enabled advanced oxidation processes: Catalytic active site engineering, oxidation pathway regulation, and reaction condition optimization. Chem. Eng. J. 2026, 528, 172413. [Google Scholar] [CrossRef]
- Hu, Y.; Tian, S.; Li, Y. Machine learning empowered single particle collision electrochemistry: A bridge from reaction mechanisms to energy and environmental applications. J. Environ. Chem. Eng. 2026, 14, 123032. [Google Scholar] [CrossRef]
- Li, A.; Oh, R.; Ren, Y.; Zhao, L.; Huang, X.J. Machine learning for advanced oxidation catalysis: From descriptors to inverse design. Mol. Catal. 2026, 596, 115889. [Google Scholar] [CrossRef]
- Chen, L.; Ding, X.; Wang, Z.; Xu, S.; Jiang, Q.; Dun, C.; Urban, J.J. Advances in in situ/operando techniques for catalysis research: Enhancing insights and discoveries. Surf. Sci. Technol. 2024, 2, 9. [Google Scholar] [CrossRef]
- Zhang, H.; Chen, L.; Dong, F.; Lu, Z.; Lv, E.; Dong, X.; Li, H.; Yuan, Z.; Peng, X.; Yang, S.; et al. Dynamic transformation of active sites in energy and environmental catalysis. Energy Environ. Sci. 2024, 17, 6435–6481. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
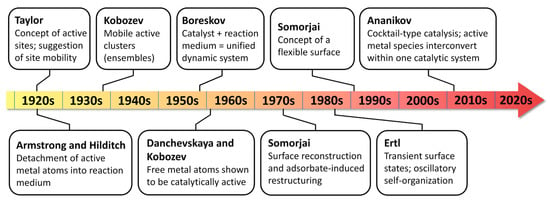
| Time Period | Main Concept | Main Conceptual Contribution | Relevance to Catalyst Dynamics |
|---|---|---|---|
| 1830s | Berzelius’ catalysis and Mitscherlich’s contact action | Catalysis was recognized as a distinct class of chemical phenomena | The catalyst was identified as a special participant of chemical transformation, although its internal changes were not yet considered |
| Late 19th–early 20th century | Ostwald’s kinetic definition and the theory of intermediate compounds | Catalysis was placed within chemical kinetics and thermodynamics | The catalyst was mainly treated as a rate-modifying component regenerated in the catalytic cycle |
| 1900s–1920s | Sabatier’s chemical theory and Langmuir’s adsorption theory | Surface interaction and chemisorption became central to heterogeneous catalysis | The surface became the key object for explaining catalytic action |
| 1925 | Taylor’s active-site concept | Catalytic activity was attributed to specific unsaturated surface atoms | Active sites were proposed as non-equivalent and potentially mobile entities |
| 1925–1930s | Armstrong–Hilditch hypothesis and Balandin’s multiplet theory | Detached atoms and geometrically defined atomic groups were considered as catalytic entities | The catalyst surface was no longer viewed as a rigid uniform plane |
| 1938–1970s | Kobozev’s theory of active ensembles | Catalytic activity was assigned to mobile atomic ensembles on the support surface | Active sites were treated as dynamic clusters capable, in some cases, of leaving the surface |
| 1950s–1960s | Electronic theory of catalysis | Active sites were assigned to unbound electrons and electron holes in solids | Active sites—unbound electrons and electron holes—were treated as dynamic entities that are generated under the action of reaction medium |
| 1960s–1980s | Boreskov’s concept of the catalyst–reaction medium system | The catalyst and reaction medium were considered as a unified interacting system | Catalyst composition and surface state were recognized as dependent on operating conditions |
| 1970s–2000s | Surface science, flexible surfaces, oscillatory catalysis | Adsorbate-induced restructuring and self-organization of surfaces were directly studied | Dynamic reconstruction became experimentally observable |
| 2012s–present | Ananikov’s cocktail-type catalysis | Molecular, cluster, nanoparticle, and surface-bound forms were treated as interconverting catalytic components | Homogeneous and heterogeneous catalysis became linked through dynamic catalyst evolution |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license.
Share and Cite
Mironenko, R.M.; Likholobov, V.A.; Lavrenov, A.V. Dynamic Behavior of Catalysts Under Operating Conditions: Part 1—Origin and Development of the Ideas. Chemistry 2026, 8, 79. https://doi.org/10.3390/chemistry8060079
Mironenko RM, Likholobov VA, Lavrenov AV. Dynamic Behavior of Catalysts Under Operating Conditions: Part 1—Origin and Development of the Ideas. Chemistry. 2026; 8(6):79. https://doi.org/10.3390/chemistry8060079
Chicago/Turabian StyleMironenko, Roman M., Vladimir A. Likholobov, and Aleksandr V. Lavrenov. 2026. "Dynamic Behavior of Catalysts Under Operating Conditions: Part 1—Origin and Development of the Ideas" Chemistry 8, no. 6: 79. https://doi.org/10.3390/chemistry8060079
APA StyleMironenko, R. M., Likholobov, V. A., & Lavrenov, A. V. (2026). Dynamic Behavior of Catalysts Under Operating Conditions: Part 1—Origin and Development of the Ideas. Chemistry, 8(6), 79. https://doi.org/10.3390/chemistry8060079