Abstract
The reaction mechanism of the gold-catalyzed hydrothiolation of alkenes (1) with thiols (2) has been investigated in detail. The tetranuclear gold complex, (PPh3)4Au4(SPh)2(NTf)2 (A), is a key intermediate in the catalytic hydrothiolation of alkenes. It forms instantaneously when PPh3AuNTf2 and PhSH are mixed in THF. Monitoring the reaction over time using 31P NMR spectroscopy revealed that gold complex A remained stable in the reaction system throughout the hydrothiolation process. In addition, we successfully observed a rapid ligand-exchange reaction between the thiolate group of gold complex A and thiols in solution. The gold-catalyzed alkene hydrothiolation reaction has been applied to the catalytic hydrothiolation of allenes, which have degenerate double bonds. Hydrothiolation of allenes proceeded regioselectively at the terminal double bond. However, the yield was lower than that observed for alkenes, and catalyst deactivation occurred. The hydrothiolation products of allenes were difficult to detach from the gold catalyst, necessitating an increase in the reaction temperature. Since high periodic transition metals such as gold and platinum are effective for hydrothiolation of alkenes and allenes, it is interesting to clarify whether iridium complexes, which belong to the same period as gold and platinum, could also catalyze alkene hydrothiolation. Through a detailed investigation of iridium ligands and reaction conditions, it was found that, in iridium systems, disulfide formation via oxidative coupling of thiols occurs preferentially over hydrothiolation reactions. This is likely due to steric hindrance around the iridium center, which inhibits alkene coordination to the iridium. Additionally, the hydrothiolation proceeding at low yields is believed to be a radical reaction involving electron transfer through the iridium complex.
1. Introduction
Organosulfur compounds readily react with transition metal compounds to form diverse complexes with organosulfur ligands. If these complexes could transfer organosulfur ligands to other organic molecules and regenerate transition metal complexes as catalysts, novel transition-metal-catalyzed thiolation reactions could be developed. However, unlike the remarkable progress in transition-metal-catalyzed reactions involving organosilicon, boron, and halogen compounds, organosulfur compounds coordinated to metals are often considered low-reactivity compounds that sometimes act as catalyst poisons [1]. Consequently, the development of transition-metal-catalyzed reactions using organosulfur compounds has lagged behind. Recently, however, progress has been made in developing substitution-type reactions of organosulfur compounds using transition metal catalysts. This approach enables the synthesis of various organosulfur compounds [2,3,4,5,6,7,8,9,10,11,12,13,14].
Addition-type reactions are expected to offer higher atom economy than substitution-type reactions. The reaction of acetylenes with thiols typically produces anti-Markovnikov adducts via a radical reaction initiated by trace amounts of oxygen molecules in the reaction system (Scheme 1).
Scheme 1.
Transition-metal-catalyzed hydrothiolation and air-mediated hydrothiolation.
Addition-type 1. However, adding palladium acetate (Pd(OAc)2) to the system reversed the regioselectivity and produced Markovnikov adducts [2,3,15,16,17,18]. Since anti-Markovnikov products were hardly produced at all, it was speculated that palladium acetate not only promotes the formation of Markovnikov adducts, but also suppresses the formation of anti-Markovnikov adducts. Additionally, Pd(OAc)2 undergoes a ligand exchange reaction with two equivalents of PhSH, yielding two equivalents of AcOH (Scheme 2).
Scheme 2.
Formation and aggregation of palladium sulfide.
The resulting palladium sulfide (Pd(SPh)2) might coordinate with acetylene, leading to an addition reaction. However, the initially generated Pd(SPh)2 complexes gradually aggregate each other to form clusters [Pd(SPh)2]n. This makes it insoluble in organic solvents and deactivates the metal catalyst. To prevent this, it is important to first mix the metal catalyst with an alkyne and coordinate the alkyne to the catalyst before adding sulfur compounds to the reaction system. Ananikov et al. have conducted systematic research on the structures and catalytic functions of cluster molecules generated from transition metals and sulfur compounds, reporting many important findings [19,20,21,22,23,24,25].
Addition-type reactions are expected to offer higher atom economy than substitution-type reactions. The reaction of acetylenes with thiols typically produces anti-Markovnikov adducts via a radical reaction initiated by trace amounts of oxygen molecules in the reaction system (Scheme 1). By devising catalysts and ligands, we have developed regio-complemental hydrothiolation reactions. For instance, the RhCl(PPh3)3-catalyzed addition of thiols to alkynes affords anti-Markovnikov adducts through syn addition (Scheme 1). In contrast, the addition catalyzed by PdCl2(PhCN)2 yields the hydrothiolation product via Markovnikov addition and double bond isomerization [26].
As mentioned above, in the transition-metal-catalyzed reactions of organosulfur compounds, it is essential to protect the transition metal center with an alkyne before the sulfur compound coordinates with the metal. This prevents catalyst deactivation and allows sulfur functional groups to be introduced into organic molecules catalytically. However, this method is limited to alkynes. Under similar conditions, the reaction does not proceed with alkenes that have low coordination ability and are sterically bulky. Kondo, Mitsudo, et al. discovered the ruthenium-catalyzed double thiolation reaction of alkenes with organic disulfides [27].
On the other hand, we discovered that the double hydrothiolation of alkynes with thiols might proceed through vinyl sulfide intermediates (see the third reaction in Scheme 3) [28]. This prompted us to develop a new process that includes alkenes with metal-coordinating neighboring groups at the double bond. Using a palladium catalyst, we successfully carried out the hydrothiolation of such alkenes with thiols (Scheme 3, first and second reactions) [29].
Scheme 3.
Palladium-catalyzed hydrothiolation of vinyl ethers and amides.
Furthermore, detailed studies were carried out using various transition metal complexes to achieve transition-metal-catalyzed hydrothiolation of inactive alkenes with no adjacent directing groups. We focused on gold catalysts, which have attracted considerable attention due to their high affinity for unsaturated bonds and ability to activate double bonds. Thus, we found that anti-Markovnikov-type hydrothiolation of inactive terminal alkenes proceeded smoothly using the PPh3AuNTf2 catalyst (Scheme 4) [30].
Scheme 4.
Gold-catalyzed hydrothiolation of inactivated alkenes.
In this study, we clarify the optimal conditions for the alkene hydrothiolation depending on the substrate, the behavior of the key active species, i.e., a tetranuclear gold complex, and also report on attempts towards the hydrothiolation of allenes, which are degenerate double bond compounds. Additionally, we investigated the potential of iridium complexes as catalysts for alkene hydrothiolation.
2. Materials and Methods
Unless otherwise stated, all starting materials and catalysts were purchased from commercial sources and used without further purification. THF as solvent was used for the alkene hydrothiolation after distillation from CaH2. Super dehydrated CH3CN was purchased from commercial sources and used directly for the allene hydrothiolation. Other solvents were purchased from commercial sources and used without further purification. 1H NMR spectra were recorded on a JEOL JNM-ECS400 (400 MHz) FT NMR system, a JEOLJNM ECX400 (400 MHz) FTNMR system, or a Bruker microTOF II ESI/TOF analyzer with Me4Si as an internal standard. Chemical shifts in 1H NMR were measured relative to CDCl3 and converted to δ (Me4Si) values by using δ (CDCl3) 7.26 ppm. Chemical shifts in 13C NMR were measured relative to CDCl3 and converted to δ (Me4Si) values by using δ (CDCl3) 77.00 ppm. The 1H NMR yields of the crude mixture were determined using 1,3,5-trioxane as the internal standard. 13C NMR{1H} spectra were recorded on a JEOL JNM ECX400 (100 MHz) FT NMR, JEOL JNM-ECS400 (100 MHz) FT NMR system, or a Bruker BioSpin Ascend 400 spectrometer (100 MHz). 31P NMR spectra were recorded on a JEOL JNM-ECX400 (162 MHz) FT NMR or JNM-ECS400 (162 MHz) FT NMR system in THF- d6 (or CDCl3) with an 85% H3PO4 solution as an external standard. IR spectra were recorded on a Jasco FT/IR-410 and reported in wavenumbers (cm−1). ESI and EI mass spectra were obtained by employing double focusing mass spectrometers. High resolution mass spectroscopy (HRMS) was conducted on a Bruker microTOF II ESI/TOF analyzer.
General procedure for gold-catalyzed hydrothiolation of inactivated alkenes. In a two-necked 10 mL flask with a magnetic stirring bar under N2 atmosphere were placed PPh3AuNTf2 (7.4 mg, 0.01 mmol), freshly distilled THF (0.15 mL), alkene (0.5 mmol) and thiol (0.5 mmol). The reaction was conducted at 45 °C for 20 h, and the thiol (0.33 mmol) was additionally added in 4 h later. After the reaction, the resulting mixture was filtered through silica gel and the crude solution was concentrated in vacuo. The product was purified by recycle GPC (eluent: CHCl3) to afford the hydrothiolation product.
1-(Phenylthio)decane (3aa) [30]. [CAS: 13910-18-4]: Colorless oil (115.2 mg, 92% yield); 1H NMR (400 MHz, CDCl3, ppm) δ 0.88 (t, J = 6.8 Hz, 3H), 1.18–1.46 (m, 14H), 1.63 (quint, J = 7.8 Hz, 2H), 2.90 (t, J = 7.8 Hz, 2H), 7.11–7.16 (m, 1H), 7.22–7.32 (m, 4H); 13C{1H} NMR (100 MHz, CDCl3, ppm) δ 14.1, 22.6, 28.8, 29.1 (overlap), 29.3, 29.5 (overlap), 31.9, 33.5, 125.5, 128.7 (overlap), 137.1; IR (NaCl) 3062, 2925, 2852, 1585, 1480, 1465, 1439, 1375, 1299, 1268, 1092, 1064, 1025, 736, 690 cm−1; MS (EI) [M]+ m/z = 250.
1-(Phenylthio)-3-phenylpropane (3ba) [30]. [CAS: 30134-12-4]: Colorless oil (100.4 mg, 88% yield); 1H NMR (400 MHz, CDCl3, ppm) δ 1.97 (quint, J = 7.3 Hz, 2H), 2.76 (t, J = 7.3 Hz, 2H), 2.92 (t, J = 7.3 Hz, 2H), 7.15–7.21 (m, 4H), 7.25–7.32 (m, 6H); 13C{1H} NMR (100 MHz, CDCl3, ppm) δ 30.6, 32.8, 34.6, 125.8, 125.9, 128.4, 128.5, 128.8, 129.1, 136.5, 141.2; IR (NaCl) 3060, 3024, 2933, 2855, 1603, 1583, 1559, 1496, 1480, 1453, 1438, 1420, 1351, 1280, 1251, 1183, 1155, 1093, 1070, 1025, 1002, 968, 738, 691 cm−1; MS (EI) [M]+ m/z = 228.
Time course of the gold-catalyzed hydrothiolation of allylbenzene with benzenethiol. Four 10 mL two-neck flasks were prepared, each equipped with a stir bar and dried under a nitrogen atmosphere. PPh3AuNTf2 (2 mol%), THF (0.10 mL), allylbenzene (0.5 mmol), benzenethiol (0.5 mmol), and THF (0.05 mL) were added to each flask in this order. The mixture was stirred at 45 °C, and after 2, 4, 6, and 8 h, 0.2 mL of the mixture was removed from each flask and an internal standard (1,3,5-trioxane) was added. NMR measurements (1H, 13C) were performed without evaporating under reduced pressure.
Gold-catalyzed hydrothiolation of 1-cyclopropylstyrene 1c with benzenethiol 2a. A 10 mL two-neck flask was placed with a stir bar and dried under a nitrogen atmosphere. PPh3AuNTf2 (2 mol%), THF (0.10 mL), 1-cyclopropylstyrene 1c (0.5 mmol), benzenethiol 2a (0.5 mmol), and THF (0.05 mL) were then added in this order and heated at 45 °C with stirring for 20 h. After the reaction was complete, the reaction solution was filtered through silica gel with ethyl acetate. The solvent was removed by evaporation under reduced pressure, and the mixture was then dried and subjected to NMR measurements (1H, 13C).
2-Cyclopropyl-2-phenyl-1-(phenylthio)ethane (3ca) [30]. [CAS: 1899041-80-5]: Colorless oil; 1H NMR (400 MHz, CDCl3, ppm) δ 0.42–0.45 (m, 2H), 0.53–0.57 (m, 2H), 1.39–1.46 (m, 4H), 7.12–7.31 (m, 8H), 7.58–7.60 (m, 2H); 13C{1H} NMR (100 MHz, CDCl3, ppm) δ 2.8, 3.0, 21.6, 23.6, 55.1, 126.5, 127.6, 127.8, 128.1, 128.2, 132.8, 136.2, 145.7; IR (NaCl) 3056, 3005, 2981, 2930, 1951, 1887, 1804, 1581, 1491, 1472, 1438, 1377, 1223, 1171, 1067, 1047, 1025, 916, 906, 825, 749, 693 cm−1; HRMS (FAB) Calcd for C17H19S [M+H]+: 255.1207, Found: 255.1214.
Monitoring the gold-catalyzed alkene hydrothiolation by 31P NMR.
31P NMR of PPh3AuNTf2: The sealed NMR tube was dried with a hair dryer and purged with Ar three times. Then, PPh3AuNTf2 (0.05 mmol) and d8-THF (0.4 mL) were added and left for 10 min. Then, 31P NMR measurements were performed.
Preparation of tetranuclear gold complex A: An equivalent amount of PhSH was added dropwise to the above d8-THF solution of PPh3AuNTf2 in a sealed NMR tube under an Ar atmosphere. After 10 min, 31P NMR measurement of the solution were performed.
Preparation of tetranuclear gold complex A in the presence of AIBN: An equivalent amount of PhSH and AIBN (30 mol%) was added dropwise to the above d8-THF solution of PPh3AuNTf2 in a sealed NMR tube under an Ar atmosphere. After 10 min, 31P NMR measurement of the solution were performed.
Preparation of tetranuclear gold complex A upon irradiation with a xenon lamp: An equivalent amount of PhSH was added dropwise to the above d8-THF solution of PPh3AuNTf2 in a sealed NMR tube under an Ar atmosphere. After irradiation with a xenon lamp for 10 min, 31P NMR measurement of the solution were performed.
Tetranuclear gold complex-catalyzed hydrothiolation of 1-decene 1a: The sealed NMR tube was dried in a hair dryer and purged with Ar three times. PPh3AuNTf2 (0.01 mmol), d8-THF (0.15 mL), and PhSH (0.005 mmol) were then added and left for 10 min. 31P NMR measurement was performed, confirming the formation of tetranuclear gold complex A. Then, 1-decene (0.5 mmol) and PhSH (0.83 mmol) were added and the mixture was heated at 45 °C with stirring for 20 h. After the reaction was complete, the reaction solution was filtered through silica gel with ethyl acetate. The solvent was removed under reduced pressure, dried, and 1H and 13C NMR measurements were performed.
Ligand exchange reaction of tetranuclear gold complex A. The sealed NMR tube was dried with a hair dryer and purged with Ar three times. PPh3AuNTf2 (0.01 mmol), CD2Cl2 (0.40 mL), and PhSH (0.005 mmol) were then added and left for 10 min. 31P NMR measurement was then performed. After confirming that the peak had shifted, p-toluenethiol (0.05 mmol) was added and the mixture was left for 10 min. 31P NMR measurement was then performed.
Isolation of gold complex A. PPh3AuNTf2 was dissolved in an appropriate amount of CH2Cl2 in a 50 mL round-bottom flask. Then 0.5 equivalent of PhSH was added to the CH2Cl2 solution of PPh3AuNTf2 and the resulting mixture was stirred for 10 min under a nitrogen atmosphere. Hexane was then added to the reaction solution, causing it to become cloudy. The solvent was removed under reduced pressure to approximately 30% of the original volume. This operation of adding hexane and removing solvent under reduced pressure to about 30% of the original volume was repeated approximately three times. After complete solvent removal, vacuum drying yielded a pale purple solid. The complex was identified by 1H NMR, 13C NMR, and 31P NMR measurements.
Isolation of gold complex A’ through gold complex A. Gold complex A isolated, was dissolved in an appropriate amount of CH2Cl2 in a 50 mL round-bottom flask. An equimolar amount of p-toluenethiol was then added to the CH2Cl2 solution of gold complex A and stirred for 10 min under a N2 atmosphere. Hexane was then added to the reaction solution, resulting in a cloudy white mixture. The solvent was distilled off under reduced pressure to about 30% of the original volume. This operation of adding hexane and distilling off to about 30% of the original volume was repeated approximately three times. After completely distilling off the solvent, vacuum drying yielded a white solid. The obtained complex A’ was identified by 1H NMR, 13C NMR, and 31P NMR measurements.
Isolation of gold complex A’ from the reaction of PPh3AuNTf2 with p-toluenethiol. PPh3AuNTf2 was dissolved in an appropriate amount of CH2Cl2 in a 50 mL round-bottom flask. Then, 0.5 equivalents of p-toluenethiol to PPh3AuNTf2 was added and stirred for 10 min under a N2 atmosphere. Hexane was then added to the reaction solution, resulting in a cloudy white mixture. The solvent was distilled off under reduced pressure to about 30% of the original volume. This operation of adding hexane and distilling off to about 30% of the original volume was repeated approximately three times. After complete removal of the solvent, the mixture was vacuum dried to yield a white solid. The resulting complex was identified by 1H NMR, 13C NMR, and 31P NMR.
Hydrothiolation of 1-decene 1a with gold complex A’. Deoxygenated THF solvent (0.15 mL) was added to the Schlenk tube in a glove box. The Schlenk tube was then removed from the glove box and the isolated gold complex A’ (0.05 mmol) and 1-decene (0.05 mmol) were added under an Ar atmosphere. 31P NMR measurements were then performed to confirm a peak shift. Then, p-toluenethiol (0.05 mmol) was added and the mixture was heated at 45 °C with stirring for 20 h. After the reaction was complete, the reaction solution was filtered through silica gel with ethyl acetate. The solvent was removed under reduced pressure, dried, and NMR measurements (1H, 13C, 31P) were performed.
Gold-catalyzed hydrothiolation of cyclohexylallene 4a. In a 10 mL two-neck glass vessel was placed a stirring bar and the vessel was dried under a nitrogen atmosphere. Catalyst, PPh3AuNTf2, (10 mol%) and dehydrating solvent (0.1 mL) were added to the vessel and the catalyst was dissolved. Cyclohexylallene 4a, PhSH 2a, and dehydrating solvent (0.2 mL) were then added, in this order, and the mixture was heated with stirring under a N2 atmosphere at the indicated reaction temperature for 24 h. After the reaction was complete, the resulting solution was filtered through silica gel with AcOMe. The filtrate was evaporated under reduced pressure, dried in vacuo, and subjected to NMR measurement (1H, 13C; CDCl3) using bibenzyl as the internal standard to calculate the yield.
Time course of the gold-catalyzed allene hydrothiolation monitored by 31P NMR. In a 10 mL two-necked glass vessel was placed a stirring bar and the vessel was dried under a nitrogen atmosphere. PPh3AuNTf2 (10 mol%), CD3CN (0.1 mL), cyclohexylallene 4a (4.0 mmol), PhSH 2a (4.0 mmol), and CD3CN (0.2 mL) were added in this order. The mixture was transferred to an NMR tube and subjected to 31P NMR measurement (CD3CN). The mixture was then heated under reflux with stirring under a N2 atmosphere, and 31P NMR measurements were taken every 10 min.
Synthesis of gold complex 8, P(C6H4-R)3AuNTf2. In a 20 mL two-neck glass vessel was placed the stirring bar and the vessel was dried under a nitrogen atmosphere. Me2SAuCl (0.2 mmol), P(C6H4-R)3 6 (0.2 mmol), and dehydrating CH2Cl2 (5 mL) were added and stirred at room temperature for 30 min. After the reaction was complete, the resulting solution was evaporated under reduced pressure, and isohexane (or hexane) was added. The precipitated solid was filtered off and dried in vacuo to obtain P(C6H4-R)3AuCl 7. 31P NMR measurements (CDCl3) were performed. The isolated P(C6H4-R)3AuCl 7 was dissolved in dehydrating CH2Cl2 (5 mL) and then AgNTf2 (0.2 mmol) was added. The mixture was stirred at room temperature for 15 min, and then filtered using CH2Cl2 to remove the AgCl precipitate. The filtrate was evaporated under reduced pressure and dried in vacuo to obtain P(C6H4-R)3AuNTf2 8. Then, 31P NMR measurements (CDCl3) were performed.
Hydrothiolation of cyclohexylallene 4a using gold catalysts 8a–8d. In a 10 mL two-neck glass vessel was placed the stirring bar and the vessel was dried under a nitrogen atmosphere. The gold catalyst (10 mol%) and dehydrating CH3CN (0.1 mL) were added and the catalyst was dissolved. Cyclohexylallene 4a (0.4 mmol), PhSH 2a (0.4 mmol), and dehydrating CH3CN (0.2 mL) were then added in this order, and the mixture was heated under reflux with stirring for 24 h under a N2 atmosphere. After the reaction was complete, the resulting solution was filtered through silica gel with AcOMe. The filtrate was evaporated under reduced pressure, dried in vacuo, and subjected to NMR analysis (1H, 13C; CDCl3) using bibenzyl as the standard.
Gold-catalyzed hydrothiolation of phenylallene 4b. In a 10 mL two-neck glass vessel was placed the stirring bar and the vessel was dried under a nitrogen atmosphere. The catalyst (10 mol%) and dehydrating CH3CN (0.1 mL) were added and the catalyst was dissolved. Phenylallene 4b (0.4 mmol), PhSH 2a (0.4 mmol), and dehydrating CH3CN (0.2 mL) were then added in this order, and the mixture was heated under reflux with stirring for 24 h under a N2 atmosphere. After the reaction was complete, the reaction solution was filtered through silica gel with AcOMe. The filtrate was evaporated under reduced pressure, dried in vacuo, and subjected to NMR analysis (1H, 13C; CDCl3) using bibenzyl as the standard.
Iridium-catalyzed reaction of alkenes with benzenethiol 2a. In a 10 mL two-neck glass vessel was placed the stirring bar and the vessel was dried under a nitrogen atmosphere. The catalyst (2.5–5.0 mol%), additive (5–10 mol%), alkene (0.5–1.5 mmol), and solvent (0.15–0.3 mL), and benzenethiol 2a (0.5 mmol) were added in this order, and the mixture was heated at 60–100 °C under reflux with stirring for 20 h under a N2 atmosphere. After the reaction was complete, the reaction solution was filtered through silica gel with AcOEt. The filtrate was evaporated under reduced pressure, dried in vacuo, and subjected to NMR analysis (1H, 13C; CDCl3).
3. Results and Discussion
3.1. Gold-Catalyzed Alkene Hydrothiolation
Studies on the catalytic hydrothiolation of alkenes have been reported in recent years, and new developments are anticipated. Combining central metals and ligands has enabled control of the regioselectivity of addition reactions [31,32,33,34]. Common, inexpensive metals used include copper (Cu) [35,36,37,38], cobalt (Co) [39,40], iron (Fe) [41], and zinc (Zn) [42], though examples using rare earth complexes [43,44] have also been reported. Furthermore, reactions beyond simple alkene addition have been reported, including cyclization reactions [45,46] and hydrothiolation reactions involving double bond migration [47,48,49]. Studies on reaction mechanisms using computational science have also been reported [50,51,52].
First, we attempted to hydrothiolate 1-decene using several transition metal catalysts and compared the results with those from our previously published study using the cationic gold catalyst PPh3AuNTf2 (Table 1). The reaction proceeded smoothly when PPh3AuNTf2 was used as the catalyst, producing the anti-Markovnikov-type product 3aa in moderate yield (entry 1). Under the same conditions in the absence of a catalyst, the reaction hardly produced the addition product at all (entry 2). When Pd(OAc)2 was used as the catalyst, the reaction barely proceeded (entry 3).
Table 1.
Hydrothiolation of 1-decene using several transition-metal catalysts.
However, when a Ni catalyst was used, product 3aa was obtained in small amounts (entries 4–5). The desired product was hardly obtained when Wilkinson’s or Ru catalyst was used (entries 6–7). Using an Ir catalyst produced the addition product in a very low yield and resulted in the by-production of a disulfide (entry 8). As mentioned above, gold catalysts [53,54,55,56,57,58] are the most effective for the hydrothiolation of 1-decene. To investigate why gold catalysts are effective in this hydrothiolation, we examined the reaction conditions, experimental methods, reaction mechanism, and properties of the key intermediate, the gold complex, sequentially.
To determine the optimal conditions for gold-catalyzed hydrothiolation, we chose allylbenzene as the substrate. We then optimized the amounts of the catalyst and solvent, as well as the reaction time and temperature (Table 2). The optimization of the solvent using 1-decene as the substrate has been described in our previous paper [30] and THF is a suitable solvent for gold-catalyzed hydrothiolation of 1-decene. For example, using nonpolar toluene and polar aprotic CH3CN as solvents under the conditions of entry 1 in Table 1 yielded 3% and 2% of 3aa, respectively. Furthermore, a higher temperature (70 °C) decreased the yield of 3aa (11%), suggesting the decomposition of PPh3AuNTf2 [30]. Increasing the amounts of solvent and catalyst did not improve the yield (entries 2–3). Furthermore, extending the reaction time to 25 h also did not improve the yield (entry 4). Since the reaction was performed at 25 °C, the desired product 3ba was not obtained, suggesting that this hydrothiolation is sensitive to temperature (entry 5).
Table 2.
Gold-catalyzed hydrothiolation of allylbenzene with benzenethiol.
To understand the details of this reaction, triphenylmethane was used as an internal standard, and the time course was monitored every two hours from the start until eight hours after the reaction began (Table 3). After four hours, 23% of the initial amount of PhSH remained, indicating that excess PhSH was consumed relative to the substrate alkene.
Table 3.
Changes in gold-catalyzed hydrothiolation of allylbenzene with benzenethiol over time.
Therefore, adding additional thiol four hours after the reaction began improved the yield of the addition product, 3ba. It was found that the reaction proceeded almost quantitatively, especially when 0.33 mmol of PhSH was added (Table 4). When adding excess thiol at the beginning, the catalyst may become deactivated depending on the reaction conditions and substrate; therefore, adding thiol during the reaction is recommended.
Table 4.
Gold-catalyzed hydrothiolation of allylbenzene by adding additional PhSH.
Because the present hydrothiolation affords anti-Markovnikov-type adducts, it is believed that the reaction occurs through a radical or transition-metal-catalyzed process. To determine which pathway is involved, we conducted an experiment using a vinylcyclopropane derivative radical clock system.
The rate constant for the ring-opening reaction of the cyclopropylcarbinyl radical is very large: k = 1.3 × 108 s−1 [59,60]. If the reaction proceeds via a radical mechanism, the cyclopropane ring-opened product 3la’ will be obtained according to Scheme 5.
Scheme 5.
Hydrothiolation of vinyl cyclopropane via a radical pathway.
Table 5.
Hydrothiolation of a vinylcyclopropane 1c by gold-catalyzed/radical pathway.

We attempted gold-catalyzed hydrothiolation using a vinylcyclopropane, 1-cyclopropylstyrene 1c, as the substrate. However, only the anti-Markovnikov-type adduct 3ca was obtained without formation of ring-opened product 3ca’ (Table 5, entry 1). These results suggest that this reaction does not involve a radical mechanism.
Furthermore, to confirm that this hydrothiolation proceeds via a transition-metal-catalyzed process, we conducted a detailed study under conditions favorable for radical generation (Table 5). First, we performed a blank experiment without adding a catalyst. Only the ring-opened compound 3ca’ was produced (entry 2). When 10 mol% AIBN was added to the substrate alkene to generate radicals, 3ca’ was obtained with a 37% yield (entry 3), like entry 2. Exposing the substrate to the radiation of a xenon lamp for 20 h to generate radicals also yielded only product 3ca’ (entry 4).
Next, we investigated the reaction with the addition of a gold catalyst. Despite the possible presence of oxygen radicals under air conditions, no ring-opened product 3ca’ was produced; instead, the addition product 3ca, which retains the cyclopropane ring, was produced in a 57% yield (entry 5). Furthermore, even with the addition of AIBN or irradiation with a xenon lamp, no ring-opened product was obtained when a gold catalyst was present; only 3ca was produced (entries 6–7). These results demonstrate that gold-catalyzed hydrothiolation proceeds as a transition-metal-catalyzed reaction rather than a radical reaction. Interestingly, the presence of a gold catalyst completely suppresses the radical reaction (A possible pathway for inhibiting radical pathway by the gold catalyst is shown in Supporting Information (See, Scheme S1)).
To investigate the behavior of the gold catalyst during the hydrothiolation reaction, the reaction was monitored using 31P nuclear magnetic resonance (31P NMR). Upon addition of PhSH, the peak at 28.5 ppm of the gold complex PPh3AuNTf2 disappeared, and a new peak appeared at 35.3 ppm (Chart 1). Furthermore, the 35.3 ppm peak did not change over time. Based on these results, it can be inferred that the gold catalyst first reacts with PhSH to form a new gold complex A, which then acts as a catalyst to promote the hydrothiolation reaction.

Chart 1.
Time course of the gold-catalyzed alkene hydrothiolation monitored by 31P NMR.

We also observed the reaction under radical conditions in the presence of a gold catalyst using 31P NMR (Scheme 6). 31P NMR measurements revealed a peak for gold complex A at 35.5 ppm when the gold catalyst, two equivalents of thiol, and AIBN were added to a THF solution and stirred for 10 min. The same result occurred when the gold catalyst and thiol were mixed and irradiated with a xenon lamp for 10 min. These results suggest that the gold complex, the key species for the hydrothiolation, forms even under conditions where radicals are generated.
Scheme 6.
The reaction of gold catalyst and thiol under radical conditions.
To confirm this complex’s involvement in the reaction, we synthesized it in situ and reacted it with the substrate alkene and thiol. The reaction proceeded smoothly, yielding the desired product (Scheme 7). These results suggest that gold complex A acts as a catalyst in this reaction.
Scheme 7.
Gold complex A-catalyzed hydrothiolation of 1-decene.
Regarding the structure of gold complex A, located near 35 ppm in the 31P NMR spectrum, we obtained a pale purple single crystal through recrystallization. A previous report [30] stated that X-ray crystal structure analysis of A identified the crystal as a tetranuclear gold complex, (PPh3)4Au4(SPh)2(NTf2) (Figure 1). Four gold atoms form a ring that is bridged by sulfur atoms from above and below. PPh3 is coordinated to each gold atom (Note that counter anion, −NTf, are omitted in Figure 1; For some other examples of tetranuclear gold complexes, see refs. [61,62,63,64,65,66]). We speculate that the reason why poisoning was suppressed in the gold-catalyzed alkene hydrothiolation reaction is that the tetranuclear gold complex A is soluble and stable in organic solvents and does not undergo further association under the reaction conditions. In contrast, monomeric and oligomeric gold complexes are unstable under these conditions and barely present in the reaction system. Thus, the tetranuclear gold complex A primarily acted as the catalyst for alkene hydrothiolation.
Figure 1.
The structure of gold complex A (For details see ref. [30]). A rare four-nuclear gold complex bridged by sulfur ligands is believed to be the key species.
To examine the reactivity of tetranuclear gold complex A, an excess of another thiol was used to carry out a ligand exchange reaction. Gold complex A was prepared separately (see the reaction between PPh3AuNTf2 and PhSH shown in Chart 1), dissolved in CHCl3, and reprecipitated with an excess of p-toluenethiol to obtain the white solid A’. Analysis of this complex using 1H NMR spectroscopy revealed a peak corresponding to the tolyl methyl group at approximately d 2.3 ppm (Chart 2). This result indicates that the phenylthio group in tetranuclear gold complex A readily exchanges with other thiols present in the system.
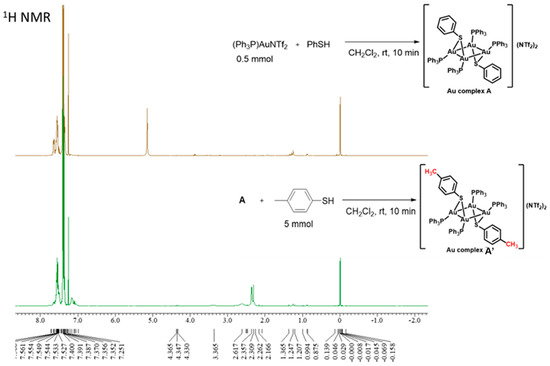
Chart 2.
Ligand exchange reaction of tetranuclear gold complex A.
Scheme 8.
Hydrothiolation of 1-decene with gold complex A’.

Furthermore, when equimolar amount of 1-decene was reacted with gold complex A’, the addition product of p-toluenethiol was obtained in a 66% yield (Scheme 8). These results revealed that the thio groups of the tetranuclear gold complex can be transferred to 1-decene.
Scheme 9.
A possible reaction pathway including four-nuclear gold complex as a key species.

Scheme 9 shows one possible reaction pathway for this alkene hydrothiolation. First, the gold complex PPh3AuNTf2 reacts with a thiol to form gold complex A. Then, an alkene coordinates with one of the gold atoms to form B. Next, a five-membered ring transition state (C) consisting of two gold atoms, a sulfur atom, and an alkene is formed. Finally, protonation occurs to produce the desired product (3), and the tetranuclear gold complex (A) is regenerated.
3.2. Gold-Catalyzed Allene Hydrothiolation
This section deals with the hydrothioation of allenes as a degenerate double bond compounds to gain further insights into the catalytic activity and regioselectivity of tetranuclear gold coordinate complexes. Allenes exhibit higher reactivity than typical C–C double bonds. In general, when benzenethiol is added to an allene, four possible regioisomers (a, b, c, and d) can form as adducts (Scheme 10).
Scheme 10.
Predicted hydrothiolation products.
In the addition reaction of benzenethiol to butylallenes under radical conditions, the thiyl radical attacks the central carbon atom at the β position or the terminal carbon atom at the γ position rather than the sterically more hindered α-position carbon atom (Scheme 11) [67].
Scheme 11.
Radical addition of benzenethiol to butylallenes.
On the other hand, we previously reported that using Pd(PPh3)4 as a catalyst enables the regioselective hydrothiolation of cyclohexylallene 4a (R = Cy: cyclohexyl) to give 5b in good yield (Table 6, entry 1) [68,69]. However, when phenylallene 4b (R = Ph) was used, which are more reactive than cyclohexylallene, the formation of regioisomers 6c and 6d was observed (entry 2). Additionally, although RuCl(PPh3)3 yields multiple regioisomers (entry 3), Pt(PPh3)4 selectively forms isomers 5c (entries 4–5).
Table 6.
Regioselectivity of the catalytic hydrothiolation of allenes a.
As mentioned above, there are only very limited examples of catalysts that promote regioselective hydrothiolation of allenes [70,71,72,73,74,75,76,77,78]. Thus, this study examined the gold-catalyzed hydrothiolation of allenes with thiols, focusing on its effect on the regioselectivity of the hydrothiolation.
The hydrothiolation of cyclohexylallene 5a with PhSH 2a was investigated using a gold catalyst, PPh3AuNTf2. When the hydrothiolation was conducted with varying amounts of the gold catalyst, the highest yield of the addition product, 5d, was obtained with 10 mol% of the catalyst, demonstrating good regioselectivity: the hydrothiolation proceeds preferentially to the terminal C–C double bond (Table 7, entries 1–3).
Table 7.
Gold-catalyzed hydrothiolation of cyclohexylallene 4a.
According to the results in entry 4 of Table 7, the target compound 5d was selectively obtained in a 16% yield when gold catalyst (10 mol%), thiol 2a (0.4 mmol), allene 5a (0.4 mmol), and CH2Cl2 (0.3 mL) were used as the solvent. A black solid was also obtained simultaneously, suggesting deactivation of the gold catalyst (entry 4). Investigating various solvents revealed that using CH3CN improved the yield of target compound 5d and enhanced selectivity (entries 4–7). A blank test was performed using CH3CN as the solvent without adding a catalyst, yielding 5b and 5d in yields of 3% and 9%, respectively. In this reaction, no black solid was observed (entry 8). To investigate the reaction at higher temperatures, 1,4-dioxane with a high boiling point was used as the solvent; however, the yield actually decreased (entry 5). It is possible that excessive temperature causes catalyst deactivation.
Further optimization of the reaction conditions was carried out using CH3CN as the optimal solvent. Lowering the reaction temperature decreased the yield of the target compound 5d (entries 9–10). In these cases, a yellow-white solid formed upon completion of the reaction. To dissolve this solid, the reaction was performed with an increased solvent volume of 1 mL. Although the amount of the yellowish-white solid formed decreased, the desired adduct was hardly obtained at all (entry 11). Assuming that this yellow-white solid is a reaction intermediate formed by the reaction of the tetranuclear gold complex with allene, it is presumed that the addition product has difficulty desorbing from the gold. Therefore, to promote elimination of the adduct, the reaction temperature was raised from room temperature to reflux. As the result, the solid dissolved completely into the reaction solution and the addition product 5d was obtained in 16% yield (entry 12).
In general, allenes have a higher coordination ability than alkenes due to their higher electron density at the unsaturated bond site. Furthermore, when sulfur-gold active species add to allenes and the gold bonds to the central carbon atom of allene, an sp2 carbon-gold bond forms. This bond has a higher bond energy than the sp3 carbon-gold bond formed when gold adds to alkenes, which makes gold more difficult to detach from an allene. Additionally, when hydrothiolation occurs to an allene, the resulting adduct retains a carbon-carbon double bond. It is presumed that, if this double bond coordinates with gold, dissociation is less likely to occur than with an alkene.
Under the reaction conditions of entry 7, a black solid was observed 12 h after the reaction began. If thiols cannot react with the coordination-unsaturated gold complex formed when the adduct leaves, gold association is likely to occur, leading to catalyst deactivation. Increasing the substrate concentration (thiols and alkenes) was found to be an effective solution. Doubling the amount of substrate yielded target 5d with a similar yield to entry 4 (entry 13). However, using only 8 mmol of thiol 2a resulted in a lower yield of 5d (entry 14). Adding cyclohexylallene 5a in two portions—at the start of the reaction and after three hours—yielded 5d in a 16% yield (entry 15).
To monitor the behavior of the gold complex during the reaction, deuterated acetonitrile was used as the solvent to observe the time-dependent changes (Chart 3): 31P NMR spectra were recorded at 10-min intervals from the start of the reaction until one hour post-initiation. The 31P NMR peak of PPh3AuNTf2 prior to the reaction was observed at 29.2 ppm. After adding allene 5a and thiol 2a, the 29.2 ppm peak disappeared, and a new peak appeared around 35 ppm. Over time, the position of the 35 ppm peak did not change significantly. Furthermore, when a gold tetranuclear complex formed in the system, a peak appeared at 34.8 ppm. This peak exhibited a nearly identical chemical shift to that observed before the reaction began. Based on these results, it is hypothesized that the gold catalyst, PPh3AuNTf2, first reacts with PhSH to form a tetranuclear gold complex A. Subsequently, the gold complex A acts as a catalyst to promote the hydrothiolation of the allenes.

Chart 3.
Time course of the gold-catalyzed allene hydrothiolation monitored by 31P NMR.

The results in Table 7 suggest that catalyst deactivation may hinder the improvement of allene hydrothiolation product yield. Therefore, we hypothesized that introducing substituents to the Ph group of the PPh3 ligand on the gold catalyst would increase the steric hindrance around the gold atom in the tetranuclear gold complex A and enhance catalyst stability. We attempted to synthesize some new gold catalysts using P(C6H4-o-Me)3 6b, P(C6H4-m-Me)3 6c, and P(C6H4-p-Me)3 6d instead of PPh3 6a (Scheme 12). 31P NMR measurements confirmed that the peak positions changed before and after both the first and second reaction steps indicated in Scheme 12.
Scheme 12.
Preparation of gold catalysts.
Table 8.
Gold-catalyzed hydrothiolation of cyclohexylallene.

Next, the hydrothiolation of cyclohexylallene was carried out using the newly synthesized gold complex catalyst 8 (Table 8). Using catalyst 8b, the target compound 5d was obtained in 33% yield while maintaining regioselectivity (entry 2). Using catalyst 8c, regioselectivity was maintained with a slightly improved yield (entry 3). Using catalyst 8d, regioselectivity was maintained with a yield of 33%, similar to entry 2 (entry 4). These results suggest that introducing a methyl group in the meta-position of the Ph group in the PPh3 ligand increases the yield of the hydrothiolation compound 5d.
As described above, we thoroughly investigated the hydrothiolation of allenes catalyzed by gold complexes using cyclohexylallene as an example. The addition occurred at the sterically unhindered terminal double bond with regioselectivity, yielding adduct 5d in which the sulfur functional group was positioned at the terminal carbon. However, the hydrothiolation of allenes yielded lower yields than the hydrothiolation of alkenes, and catalyst deactivation was observed. The adduct was difficult to detach from the gold for allenes, necessitating an increase in reaction temperature. In preliminary experiments, we attempted the hydrothiolation of phenylallene 4b using reaction conditions optimized for cyclohexylallene 4a. While the addition proceeded selectively to the terminal double bond, the addition product, 6c, with a thiolate group introduced at the central carbon, was obtained in 52% yield regioselectivity. This demonstrated that the substituent on the alkenyl group influences regioselectivity (Scheme 13).
Scheme 13.
Gold-catalyzed hydrothiolation of phenylallene 4b.
Scheme 14.
A possible pathway for the gold-catalyzed allene hydrothiolation.

Next, Scheme 14 shows a possible reaction pathway for the hydrothiolation of allenes. First, the gold catalyst forms gold complex A, and the terminal double bond of cyclohexylallene 4a coordinates to the gold atom of the gold complex A to form gold complex B. Then, the complex B, consisting of the coordinated cyclohexylallene, two gold atoms, and a sulfur atom, forms a five-membered ring transition state C. The gold-sulfur bond then breaks to form complex D. Finally, protonation by a thiol yields the desired adduct 5a, regenerating the tetranuclear gold complex A and completing the catalytic cycle.
3.3. Attempted Iridium-Catalyzed Alkene Hydrothiolation
We demonstrated the gold-catalyzed hydrothiolation of carbon-carbon double bonds. During our investigation, we discovered that the reaction either did not proceed or produced a mixture of regioisomers with lower transition metals if it did proceed. However, the hydrothiolation proceeded regioselectively with higher transition metals, such as gold and platinum. In Section 1, we also found that the iridium complex [Ir(cod)OMe]2—a period 6 element, like gold—produced hydrothiolation products of alkenes, albeit with low yields (Table 1, entry 8). Iridium forms complexes ranging from −1 to +6, but monovalent and trivalent complexes are the most common. Iridium is used less frequently as a catalyst in organic synthesis than its fellow group 9 elements, cobalt and rhodium. This is because iridium complexes have been considered too stable to be catalytically active. However, in 1979, Crabtree et al. successfully achieved the hydrogenation of tetrasubstituted alkenes in moderate yields using iridium complexes (Scheme 15) [79]. This was difficult to achieve with rhodium complexes. This hydrogenation reaction sparked increased research into iridium-catalyzed reactions. Recently, iridium-catalyzed reactions for introducing nitrogen and oxygen functional groups into alkenes have been reported [80,81].
Scheme 15.
A possible pathway for the gold-catalyzed allene hydrothiolation.
In this section, we attempted to develop an iridium-catalyzed reaction for introducing sulfur functional groups into inactivated alkenes [82,83,84,85,86,87].
Using THF as a solvent, the reaction proceeded at various temperatures, albeit with low yields (Table 9, entries 1–3). Conducting the reaction under dilute conditions did not significantly affect the yield of addition product 3aa or disulfide 9 (entry 4). Various solvents were investigated, but no favorable reaction conditions were found (entries 5–11). Disulfide was produced as a byproduct under all conditions. Disulfide was obtained in particularly high yields when aprotic polar solvents, such as DMF and DMSO, were used (entries 9–10). It is possible that the formation of Ir–S bonds contributes to the formation of disulfides. The solution turned red after the addition of a thiol, but was black initially. Additionally, a small amount of solid precipitated on the solution’s wall after the reaction. Only DMSO and CH2Cl2 produced a brown liquid without solid precipitation, though product 3aa was not obtained (entries 10–11). However, when the reaction was attempted without an iridium catalyst, product 3aa was obtained in a moderate yield (entry 12). It is presumed that the addition proceeds via a radical chain mechanism in this case. Due to the presence of a small amount of air (oxygen), (1) oxygen abstracts hydrogen from the thiol to generate a thiyl radical; (2) the thiyl radical adds to the alkene to generate a carbon radical; and (3) the carbon radical abstracts hydrogen from the thiol to produce addition product 3aa and regenerate the thiyl radical.
Table 9.
Influence of solvents on the iridium-catalyzed reaction of 1-decene with benzenethiol.
To verify the possibility that the reaction proceeds via a radical mechanism, a hydrothiolation was attempted using 2-cyclopropylstyrene as a substrate (Scheme 16). As shown in Scheme 16, in the absence of an iridium catalyst, the cyclopropane ring opens, indicating that the reaction proceeds via a radical mechanism. Based on these results, the iridium catalyst suppresses radical addition of thiols to alkenes, producing disulfides with high yields, particularly in DMF and DMSO solvents.
Scheme 16.
A mechanistic study using 1-cyclopropylstyrene.
Next, we examined the catalytic activity of iridium complexes (Table 10). When the methoxy group of the catalyst was replaced with a chloro group, the yield of the hydrothiolation product 3aa increased slightly (entries 1 and 2). IrCl3 was completely insoluble in the substrate or solvent before heating but dissolved gradually upon heating. Thus, the initial stage of the reaction is believed to occur via a radical mechanism (entry 3). Furthermore, when the Vaska complex was used as the catalyst, a large amount of disulfide was produced as a byproduct, even in toluene (entry 4).
Table 10.
Iridium-catalyzed reaction of 1-decene 1a with benzenethiol 2a.
Table 11.
Influence of the catalyst and ligand on the yields of adduct 3aa and disulfide 9.


Next, we investigated the reaction of 1-decene 1a and benzenethiol 2a using various ligands and the [Ir(cod)X]2 (X = OMe, Cl) catalyst (see Table 11). The reaction using PPh3 and BINAP as ligands with the [Ir(cod)OMe]2 catalyst was examined, but the yield of the desired product 3aa did not improve (entries 1 and 2). The yield increased slightly when 1,10-phenanthroline (phen) was used as a ligand (entry 3). However, the yield of 3aa decreased when the bulky 3,4,7,8-tmphen ligand was used (entry 4). We then investigated the reaction with various ligands and the [Ir(cod)Cl]2 catalyst. The yield of 3aa improved slightly when phen was used (entry 5). Then, we examined the use of bidentate phosphine ligands (entries 6–11). Using dppp as the ligand increased the yield of 3aa to 33% (entry 9). Conversely, no improvement in the yield of 3aa was observed with Xantphos, DPEphos, or Sphos (entries 12–14). Then, we examined bidentate amine ligands (entries 15–20). It was found that 3aa could be obtained with a yield greater than 40% when 4,4′-dmbpy or 5,5′- dmbpy was used as the ligand (entries 16 and 17).
Conversely, using highly sterically hindered bipyridyl ligands, such as the 6,6′-dimethyl-, 4,4′-diphenyl-, and 4,4′-di(t-butyl)-substituted bpys, resulted in decreased yield of the target product (entries 18–20). Furthermore, we attempted the reaction under conditions that increased the cationic nature of iridium by using AgPF6 as an additive. However, no improvement in yield was observed (entries 21 and 22). In the NHC ligand (IPr)-added system, hydrothiolation product 3aa was obtained in a 28% yield (entry 23).
Scheme 17.
A mechanistic study using 1-cyclopropylstyrene.

To gain insight into the reaction mechanism, the reaction using 2-cyclopropylstyrene as the substrate was examined (Scheme 17). In the presence of an iridium catalyst, the reaction of 1-decene with benzenethiol was carried out in toluene at 100 °C for 20 h with the addition of the dppp ligand. The cyclopropyl ring-opened product was obtained with a 40% yield. This result indicates that the iridium-catalyzed hydrothiolation occurred via a radical mechanism. However, as shown in Table 11, the yield of the hydrothiolation product 3aa varied with the iridium catalyst ligand. This suggests that the reaction is not caused by air contamination in the system but rather involves the iridium catalyst. Furthermore, the high yield of disulfides produced in the presence of an iridium catalyst suggests that disulfides might be generated via reductive elimination after forming an iridium-sulfur bond (Ir-SPh) in situ. Based on these considerations, the iridium-catalyzed (or assisted) reaction may also be a radical reaction involving the generation of a thiyl radical via a single electron transfer mechanism through the iridium catalyst. Iridium has higher ligand number than gold, so the coordination space around the iridium metal is more crowded compared with that of gold. This makes it difficult for alkenes to coordinate to iridium metal, so we believe that hydrothiolation proceeds more slowly than with a gold catalyst.
4. Conclusions
In conclusion, we investigated the reaction pathway in detail for the hydrothiolation of inactivated alkenes using the gold catalyst (Ph3P)AuNTf2. Our findings include the following:
(1) The tetranuclear gold complex, thought to be a key intermediate, exists stably in the reaction system from the beginning to the end of the reaction.
(2) The thio group of the tetranuclear gold complex undergoes ligand exchange with thiols in the solution.
(3) The addition reaction via the radical mechanism is completely suppressed in the presence of the gold complex.
(4) Because a side reaction in which thiols are consumed competitively occurs, adding thiols midway improves the yield of the hydrothiolation product.
(5) The gold-catalyzed hydrothiolation can be applied to allene hydrothiolation. Hydrothiolation proceeds regioselectively toward the terminal double bond of allenes.
(6) Compared to alkenes, the addition product is less easily eliminated from the gold catalyst and requires overheating for elimination.
(7) As with alkenes, a tetranuclear gold complex is present as a key species for the allene hydrothiolation.
(8) Unlike with alkenes, the gold catalyst becomes deactivated in the hydrothiolation of allenes, resulting in a moderate yield of the addition product.
(9) The iridium-assisted hydrothiolation of 1-decene might occur by a radical mechanism probably via a single electron transfer through the iridium catalyst.
(10) The formation of organic disulfide is observed as the major product in the iridium-catalyzed reaction with thiols.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/chemistry8040038/s1, Synthesis method of gold catalyst, hydrothiolation methods. Chart S1: 31P NMR of PPh3AuCl (CDCl3); Chart S2: 31P NMR of PPh3AuNTf2 8a (CDCl3); Chart S3: 31P NMR of [(PPh3)4Au4(SPh)2](NTf2)2), gold complex A (CDCl3); Chart S4: 31P NMR of P(C6H4-2-Me) 3AuNTf2 8b (CDCl3); Chart S5: 31P NMR of P(C6H4-3-Me) 3AuNTf2 8c (CDCl3); Chart S6: 31P NMR of P(C6H4-2-Me) 3AuNTf2 8d (CDCl3); Chart S7: 1H NMR of 1-(phenylthio)decane (3aa) (CDCl3); Chart S8: 13C NMR of 1-(phenylthio)decane (3aa) (CDCl3); Chart S9: 1H NMR (Table 11, Entry 3); Chart S10: 13C NMR (Table 11, Entry 3); Chart S11: 1H NMR (Table 11, Entry 9); Chart S12: 13C NMR (Table 11, Entry 9); Chart S13: 1H NMR (Table 11, Entry 16); Chart S14: 13C NMR (Table 11, Entry 16); Chart S15: 1H NMR (Table 11, Entry 17); Chart S16: 13C NMR (Table 11, Entry 17); Chart S17: 1H NMR (Table 11, Entry 23); Chart S18: 13C NMR (Table 11, Entry 23); Scheme S1: A possible pathway for inhibiting radical pathway.
Author Contributions
Conceptualization, A.O. and T.T.; methodology, A.O., T.T. and D.K.; Validation, A.O. and T.T.; formal analysis, A.O., K.F., R.T. and D.K.; investigation, T.T., K.F., R.T. and D.K.; resources, A.O.; writing—original draft preparation, A.O., K.F., R.T. and D.K.; writing—review and editing, A.O.; supervision, A.O., T.T. and Y.Y.; project administration, A.O.; funding acquisition, A.O. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the Grant-in-Aid for Scientific Research (24K22366, 22H02124) from the Japan Society for the Promotion of Science (JSPS).
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Materials, further inquiries can be directed to the corresponding authors.
Acknowledgments
We would like to express our gratitude to Akiko Okada (Osaka Metropolitan University) for her assistance in revising the manuscript. A.O. would like to express his sincere gratitude to Ilhyong Ryu for his assistance to this research.
Conflicts of Interest
The authors declare no conflicts of interest, either of a financial or personal nature.
References
- Yamamoto, Y.; Ogawa, A. Transition-Metal-Catalyzed Addition of Organosulfur Compounds to Alkynes and Alkenes: Catalysis and Catalyst Poisons. Chem. Eur. J. 2023, 29, e202302432. [Google Scholar] [CrossRef] [PubMed]
- Beletskaya, I.P.; Ananikov, V.P. Transition-Metal-Catalyzed C−S, C−Se, and C−Te Bond Formations via Cross-Coupling and Atom-Economic Addition Reactions. Achievements and Challenges. Chem. Rev. 2022, 122, 16110–16293. [Google Scholar] [CrossRef] [PubMed]
- Beletskaya, I.P.; Ananikov, V.P. Transition-Metal-Catalyzed C-S, C-Se, and C-Te Bond Formation via Cross-Coupling and Atom-Economic Addition Reactions. Chem. Rev. 2011, 111, 1596–1636. [Google Scholar] [CrossRef] [PubMed]
- Taj, A.; Wang, Z.; Li, L.; Yang, X.-H. Recent Advances in Transition-Metal-Catalyzed Thiolation and Sulfonylation of Alkenes. Adv. Synth. Catal. 2025, 367, e70131. [Google Scholar] [CrossRef]
- Yorimitsu, H. Catalytic Transformations of Sulfonium Salts via C-S Bond Activation. Chem. Rec. 2021, 21, 3356–3369. [Google Scholar]
- Lou, J.; Wang, Q.; Wu, P.; Wang, H.; Zhou, Y.-G.; Yu, Z. Transition-metal mediated carbon–sulfur bond activation and transformations: An update. Chem. Soc. Rev. 2020, 49, 4307–4359. [Google Scholar]
- Nogi, K.; Yorimitsu, H. Catalytic Carbonylation and Carboxylation of Organosulfur Compounds via C−S Cleavage. Chem. Asian J. 2020, 15, 441–449. [Google Scholar] [CrossRef]
- Otsuka, S.; Nogi, K.; Yorimitsu, H. C-S Bond Activation. Top. Curr. Chem. (Z) 2018, 376, 13. [Google Scholar]
- Kondo, T.; Mitsudo, T. Metal-Catalyzed Carbon−Sulfur Bond Formation. Chem. Rev. 2000, 100, 3205–3220. [Google Scholar]
- Hao, X.; Feng, D.; Huang, P.; Guo, F. A nickel-catalyzed carbon–sulfur cross-coupling reaction with disulfides enabled by mechanochemistry. Org. Chem. Front. 2024, 11, 2081–2087. [Google Scholar]
- Chen, W.; Sheng, D.; Jiang, Y.-F.; Zhu, W.-C.; Rao, W.; Shen, S.-S.; Yang, Z.-Y.; Wang, S.-Y. Nickel-Catalyzed Acid Chlorides with Tetrasulfides for the Synthesis of Thioesters and Acyl Disulfides. J. Org. Chem. 2023, 88, 15871–15880. [Google Scholar] [CrossRef] [PubMed]
- Tai, L.; Chen, L.; Shi, Y.; Chen, L.-A. Redox-active alkyl xanthate esters enable practical C–S cross-coupling by nickel catalysis. Org. Chem. Front. 2023, 10, 2505–2516. [Google Scholar]
- Ma, J.-Y.; Yao, Q.-J.; Jiang, L.-C.; Huang, F.-R.; Yue, Q.; Shi, B.-F. Copper-Mediated Enantioselective C–H Thiolation of Ferrocenes Enabled by the BINOL Ligand. J. Am. Chem. Soc. 2025, 147, 7061–7069. [Google Scholar] [PubMed]
- Tian, Y.; Li, X.-T.; Liu, J.-R.; Cheng, J.; Gao, A.; Yang, N.-Y.; Li, Z.; Guo, K.-X.; Zhang, W.; Wen, H.-T.; et al. A general copper-catalysed enantioconvergent C(sp3)–S cross-coupling via biomimetic radical homolytic substitution. Nature Chem. 2024, 16, 466–475. [Google Scholar] [CrossRef]
- Kawaguchi, S.; Yamamoto, Y.; Ogawa, A. Catalytic synthesis of sulfur and phosphorus compounds via atom-economic reactions. Mendeleev Commun. 2020, 30, 129–138. [Google Scholar] [CrossRef]
- Ogawa, A. Transition-Metal-Catalyzed S–H and Se–H Bonds Addition to Unsaturated Molecules. Top. Organomet. Chem. 2013, 43, 325–360. [Google Scholar]
- Bichler, P.; Love, J.A. Organometallic Approaches to Carbon–Sulfur Bond Formation. Top. Organomet. Chem. 2010, 31, 39–64. [Google Scholar]
- Ogawa, A. Activation and reactivity of Group 16 inter-element linkage—Transition-metal-catalyzed reactions of thiols and selenols. J. Organomet. Chem. 2000, 611, 463–474. [Google Scholar] [CrossRef]
- Ananikov, V.P.; Orlov, N.V.; Zalesskiy, S.S.; Beletskaya, I.P.; Khrustalev, V.N.; Morokuma, K.; Musaev, D.G. Catalytic Adaptive Recognition of Thiol (SH) and Selenol (SeH) Groups Toward Synthesis of Functionalized Vinyl Monomers. J. Am. Chem. Soc. 2012, 134, 6637–6649. [Google Scholar] [CrossRef]
- Ananikov, V.P.; Gayduk, K.A.; Orlov, N.V.; Beletskaya, I.P.; Khrustalev, V.N.; Antipin, M.Y. Two Distinct Mechanisms of Alkyne Insertion into the Metal–Sulfur Bond: Combined Experimental and Theoretical Study and Application in Catalysis. Chem. Eur. J. 2010, 16, 2063–2071. [Google Scholar] [CrossRef]
- Ananikov, V.P.; Orlov, N.V.; Beletskaya, I.P.; Khrustalev, V.N.; Antipin, M.Y.; Timofeeva, T. New Approach for Size- and Shape-Controlled Preparation of Pd Nanoparticles with Organic Ligands. Synthesis and Application in Catalysis. J. Am. Chem. Soc. 2007, 129, 7252–7253. [Google Scholar] [CrossRef] [PubMed]
- Beletskaya, I.P.; Ananikov, V.P. Unusual Influence of the Structures of Transition Metal Complexes on Catalytic C–S and C–Se Bond Formation Under Homogeneous and Heterogeneous Conditions. Eur. J. Org. Chem. 2007, 3431–3444. [Google Scholar] [CrossRef]
- Malyshev, D.A.; Scott, N.M.; Marion, N.; Stevens, E.D.; Ananikov, V.P.; Beletskaya, I.P.; Nolan, S.P. Homogeneous Nickel Catalysts for the Selective Transfer of a Single Arylthio Group in the Catalytic Hydrothiolation of Alkynes. Organometallics 2006, 25, 4462–4470. [Google Scholar] [CrossRef]
- Ananikov, V.P.; Malyshev, D.A.; Beletskaya, I.P.; Aleksandrov, G.G.; Eremenko, I.L. Nickel(II) Chloride-Catalyzed Regioselective Hydrothiolation of Alkynes. Adv. Synth. Catal. 2005, 347, 1993–2001. [Google Scholar]
- Ananikov, V.P.; Orlov, N.V.; Beletskaya, I.P. Efficient and Convenient Synthesis of â-Vinyl Sulfides in Nickel-Catalyzed Regioselective Addition of Thiols to Terminal Alkynes under Solvent-Free Conditions. Organometallics 2006, 25, 1970–1977. [Google Scholar]
- Ogawa, A.; Ikeda, T.; Kimura, K.; Hirao, T. Highly Regio- and Stereocontrolled Synthesis of Vinyl Sulfides via Transition-Metal-Catalyzed Hydrothiolation of Alkynes with Thiols. J. Am. Chem. Soc. 1999, 121, 5108–5114. [Google Scholar] [CrossRef]
- Kondo, T.; Uenoyama, S.Y.; Fujita, K.; Mitsudo, T. First Transition-Metal Complex Catalyzed Addition of Organic Disulfides to Alkenes Enables the Rapid Synthesis of vicinal-Dithioethers. J. Am. Chem. Soc. 1999, 121, 482–483. [Google Scholar] [CrossRef]
- Mitamura, T.; Daitou, M.; Nomoto, A.; Ogawa, A. Highly Regioselective Double Hydrothiolation of Terminal Acetylenes with Thiols Catalyzed by Palladium Diacetate. Bull. Chem. Soc. Jpn. 2011, 84, 413–415. [Google Scholar] [CrossRef]
- Tamai, T.; Ogawa, A. Regioselective Hydrothiolation of Alkenes Bearing Heteroatoms with Thiols Catalyzed by Palladium Diacetate. J. Org. Chem. 2014, 79, 5028–5035. [Google Scholar] [CrossRef]
- Tamai, T.; Fujiwara, K.; Higashimae, S.; Nomoto, A.; Ogawa, A. Gold-Catalyzed Anti-Markovnikov Selective Hydrothiolation of Unactivated Alkenes. Org. Lett. 2016, 18, 2114–2117. [Google Scholar]
- de Frémont, P. Hydrothiolation, Hydroalkoxylation, and Hydroaryloxylation. In Science of Synthesis: N-Heterocyclic Carbenes in Catalytic Organic Synthesis; Georg Thieme Verlag: Stuttgart, Germany, 2016; Volume 1, pp. 387–409. [Google Scholar]
- Kennemur, J.L.; Kortman, G.D.; Hull, K.L. Rhodium-Catalyzed Regiodivergent Hydrothiolation of Allyl Amines and Imines. J. Am. Chem Soc. 2016, 138, 11914–11919. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.; Ren, X.; Zhou, X.; Ma, C.; He, Y.; Huang, G.S. Palladium and copper co-catalyzed Markovnikov hydrothiolation of terminal olefins and alkynes. Tetrahedron Lett. 2015, 56, 6022–6029. [Google Scholar] [CrossRef]
- Jawale, D.V.; Kouatchou, J.A.T.; Fossard, F.; Miserque, F.; Geertsen, V.; Gravel, E.; Doris, E. Catalytic hydrothiolation of alkenes and alkynes using bimetallic RuRh nanoparticles on carbon nanotubes. Green Chem. 2022, 24, 1231–1237. [Google Scholar] [CrossRef]
- Delp, S.A.; Munro-Leighton, C.; Goj, L.A.; Ramirez, M.A.; Gunnoe, B.; Petersen, J.L.; Boyle, P.D. Addition of S−H Bonds across Electron-Deficient Olefins Catalyzed by Well-Defined Copper(I) Thiolate Complexes. Inorg. Chem. 2007, 46, 2365–2367. [Google Scholar] [CrossRef]
- Wang, Z.; Li, L.; Yang, X.-H. Copper-Catalyzed Transfer Markovnikov Hydrothiolation of Alkenes. Org. Lett. 2025, 27, 9637–9642. [Google Scholar] [CrossRef]
- Mondal, S.; Yashmin, S.; Khan, A.T. Synthesis of vinyl sulfides and thioethers via a hydrothiolation reaction of 4-hydroxydithiocoumarins and arylacetylenes/styrenes. Org. Biomol. Chem. 2021, 19, 9223–9230. [Google Scholar] [CrossRef]
- Xi, H.; Ma, E.; Li, Z. Copper-catalyzed selective syntheses of Markovnikov-type hydrothiolation products and thioacetals by the reactions of thiols with alkenes bearing heteroatoms. Tetrahedron 2016, 72, 4111–4116. [Google Scholar] [CrossRef]
- Liu, Y.; Du, H.-W.; Bhat, M.-u-S.; Li, Y.; Shu, W. Cobalt-Catalyzed Hydrofunctionalizations of Alkenes with sp3-Hybridized Electrophiles. Synlett 2025, 36, 1980–1994. [Google Scholar] [CrossRef]
- Li, X.-R.; Zhang, R.-J.; Xiao, Y.; Tong, Q.-X.; Zhong, J.-J. N-Sulfenyl phthalimide enabled Markovnikov hydrothiolation of unactivated alkenes via ligand promoted cobalt catalysis. Org. Chem. Front. 2024, 11, 646–653. [Google Scholar] [CrossRef]
- Greenhalgh, M.D.; Jones, A.S.; Thomas, S.P. Iron-Catalysed Hydrofunctionalisation of Alkenes and Alkynes. ChemCatChem 2015, 7, 190–222. [Google Scholar] [CrossRef]
- Taniguchi, N. Brønsted Acid-Assisted Zinc-Catalyzed Markovnikov-Type Hydrothiolation of Alkenes Using Thiols. J. Org. Chem. 2020, 85, 6528–6534. [Google Scholar] [PubMed]
- Kuciński, K.; Pawluć, P.; Marciniec, B.; Hreczycho, G. Highly Selective Hydrothiolation of Unsaturated Organosilicon Compounds Catalyzed by Scandium(III) Triflate. Chem. Eur. J. 2015, 21, 4940–4943. [Google Scholar] [PubMed]
- Kuciński, K.; Pawluć, P.; Hreczycho, G. Scandium(III) Triflate-Catalyzed anti-Markovnikov Hydrothiolation of Functionalized Olefins. Adv. Synth. Catal. 2015, 357, 3936–3942. [Google Scholar]
- Weïwer, M.; Coulombel, L.; Duñach, E. Regioselective indium(III) trifluoromethanesulfonate-catalyzed hydrothiolation of non-activated olefins. Chem. Commun. 2006, 332–334. [Google Scholar] [CrossRef]
- Weiss, C.J.; Marks, T.J. Organo-f-element catalysts for efficient and highly selective hydroalkoxylation and hydrothiolation. Dalton Trans. 2010, 39, 6576–6588. [Google Scholar]
- Zhang, Y.; Xu, X.; Zhu, S. Nickel-catalysed selective migratory hydrothiolation of alkenes and alkynes with thiols. Nature Commun. 2019, 10, 1752. [Google Scholar] [CrossRef]
- Hikida, N.; Yoshimi, Y.; Suzuki, H. Amide-Directed Rhodium-Catalyzed Chain-Walking Hydrothiolation of Internal Alkenes. Org. Lett. 2024, 26, 2500–2504. [Google Scholar] [CrossRef]
- Kathe, P.M.; Fleischer, I. Palladium-Catalyzed Tandem Isomerization/Hydrothiolation of Allylarenes. Org. Lett. 2019, 21, 2213–2217. [Google Scholar]
- Zhang, X.H.; Wang, K.T. Theoretical investigation of the mechanism of gold(i)-catalyzed hydrothiolation of alkynes and alkenes with phenthiol. RSC Adv. 2015, 5, 34439–34446. [Google Scholar]
- Ji, C.-H.; Zhang, X.H. DFT study on the mechanism of Pd(OAc)2-catalyzed hydrothiolation of alkenes bearing heteroatoms with benzenethiol. Struct. Chem. 2016, 27, 919–926. [Google Scholar]
- Feng, L.; Zhou, B.; Lu, G.-P. A DFT study on the mechanism of rhodium-catalyzed regioselective hydrothiolation of the allyl amine. Mol. Catal. 2019, 468, 62–74. [Google Scholar] [CrossRef]
- Zalesskiy, S.S.; Khrustalev, V.N.; Kostukovich, A.Y.; Ananikov, V.P. Carboxylic Group-Assisted Proton Transfer in Gold-Mediated Thiolation of Alkynes. Organometallics 2015, 34, 5214–5224. [Google Scholar]
- Kristensen, S.K.; Laursen, S.L.R.; Taarning, E.; Skrydstrup, T. Ex Situ Formation of Methanethiol: Applicationinthe Gold(I) Promoted Anti-Markovnikov Hydrothiolation of Olefins. Angew. Chem. Int. Ed. 2018, 57, 13887–13891. [Google Scholar]
- Shahzad, S.A.; Sajid, M.A.; Khan, Z.A.; Canseco-Gonzalez, D. Gold catalysis in organic transformations: A review. Synth. Comm. 2017, 47, 735–755. [Google Scholar] [CrossRef]
- Praveen, C. Carbophilic activation of π-systems via gold coordination: Towards regioselective access of intermolecular addition products. Coord. Chem. Rev. 2019, 392, 1–34. [Google Scholar]
- Murugesh, V.; Ryou, B.; Park, C.-M. Synthesis of dithioacetals via gold-catalysed hydrothiolation of vinyl sulfides. Org. Biomol. Chem. 2023, 21, 585–589. [Google Scholar]
- Brouwer, C.; Rahaman, R.; He, C. Gold(I)-Mediated Hydrothiolation of Conjugated Olefins. Synlett 2007, 1785–1789. [Google Scholar] [CrossRef]
- Griller, D.; Ingold, K.U. Free-Radical Clocks. Acc. Chem. Res. 1980, 13, 317–323. [Google Scholar]
- Newcomb, M. Competition Methods and Scales for Alkyl Radical Reaction Kinetics. Tetrahedron 1993, 49, 1151–1176. [Google Scholar] [CrossRef]
- Dau, T.M.; Chen, Y.-A.; Karttunen, A.J.; Grachova, E.V.; Tunik, S.P.; Lin, K.-T.; Hung, W.-Y.; Chou, P.-T.; Pakkanen, T.A.; Koshevoy, I.O. Tetragold(I) Complexes: Solution Isomerization and Tunable Solid-State Luminescence. Inorg. Chem. 2014, 53, 12720–12731. [Google Scholar]
- Zheng, A.-X.; Ren, Z.-G.; Li, L.-L.; Shang, H.; Li, H.-X.; Lang, J.-P. Reactions of a gold(I) thiolate complex [Au(Tab)2]2(PF6)2 (Tab = 4-(trimethylammonio)benzenethiolate) with diphosphine ligands. Dalton Trans. 2011, 40, 589–596. [Google Scholar]
- Fernández, E.J.; Laguna, A.; López-de-Luzuriaga, J.M.; Monge, M.; Sánchez-Forcada, E. Different phosphorescent excited states of tetra- and octanuclear dendritic-like phosphine gold(I) thiolate complexes: Photophysical and theoretical studies. Dalton Trans. 2011, 40, 3287–3294. [Google Scholar] [PubMed]
- Fernández, E.J.; Laguna, A.; Monge, M.; Montiel, M.; Olmos, M.E.; Pérez, J.; Sánchez-Forcada, E. Dendritic (phosphine)gold(I) thiolate complexes: Assessment of the molecular size through PGSE NMR studies. Dalton Trans. 2009, 474–480. [Google Scholar] [CrossRef]
- Ehlich, H.; Schier, A.; Schmidbaur, H. Aurophilicity-Based One-Dimensional Arrays of Gold(I) Phenylene-1,3- and -1,4-dithiolates. Inorg. Chem. 2002, 41, 3721–3727. [Google Scholar]
- Mohamed, A.A.; Chen, J.; Bruce, A.E.; Bruce, M.R.M.; Bauer, J.A.K.; Hill, D.T. Formation of a Cationic Gold(I) Complex and Disulfide by Oxidation of the Antiarthritic Gold Drug Auranofin. Inorg. Chem. 2003, 42, 2203–2205. [Google Scholar] [CrossRef]
- Pasto, D.J.; Warren, S.E.; Morrison, M.A. Aurophilicity-Based One-Dimensional Arrays of Gold(I) Phenylene-1,3 and-1,4-dithiolates. J. Org. Chem. 1981, 46, 2837–2841. [Google Scholar]
- Kodama, S.; Nomoto, A.; Kajitani, M.; Nishinaka, E.; Sonoda, M.; Ogawa, A. Transition-metal-catalyzed hydrothiolation of cyclohexylallene with benzenethiol or diphenyl disulfide. J. Sulfur Chem. 2009, 30, 309–318. [Google Scholar]
- Ogawa, A.; Kawakami, J.; Sonoda, N.; Hirao, T. Highly Regioselective Addition of Benzenethiol to Allenes Catalyzed by Palladium Acetate. J. Org. Chem. 1996, 61, 4161–4163. [Google Scholar] [CrossRef]
- Menggenbateer; Narsireddy, M.; Ferrara, G.; Nishina, N.; Jin, T.; Yamamoto, Y. Gold-catalyzed regiospecific intermolecular hydrothiolation of allenes. Tetrahedron Lett. 2010, 51, 4627–4629. [Google Scholar] [CrossRef]
- Hashmi, A.S.K.; Miriam, B. Gold-Catalyzed Addition of X-H Bonds to C-C Multiple Bonds. Aldrichimica Acta 2010, 43, 27–33. [Google Scholar]
- Prizius, A.B.; Breit, B. Z-SelectiveHydrothiolation of Racemic 1,3-Disubstituted Allenes: An Atom-Economic Rhodium-Catalyzed Dynamic Kinetic Resolution. Angew. Chem. Int. Ed. 2015, 54, 15818–15822. [Google Scholar] [CrossRef] [PubMed]
- Prizius, A.B.; Breit, B. Asymmetric Rhodium-Catalyzed Addition of Thiols to Allenes: Synthesis of Branched Allylic Thioethers and Sulfones. Angew. Chem. Int. Ed. 2015, 54, 3121–3125. [Google Scholar] [CrossRef] [PubMed]
- Ren, S.; Xue, B.; Zheng, T.; Wang, Y.; Sun, H.; Ki, X.; Fuhr, O.; Fendke, D. E-Selective Hydrothiolation of Terminal Arylallenes with Arylthiols Catalyzed by Ni (PMe3)4. Appl. Organomet. Chem. 2020, 34, e5291. [Google Scholar] [CrossRef]
- Ziyaei Halimehjani, A.; Breit, B. Rhodium-catalyzed regioselective addition of thioacids to terminal allenes: Enantioselective access to branched allylic thioesters. Chem. Commun. 2022, 58, 1704–1707. [Google Scholar] [CrossRef]
- Pages, L.; Bouquin, M.; Jaroschik, F.; Monnier, F.; Taillefer, M. Copper-Catalyzed Regio- and Stereoselective Hydrothiolation of Allenamides, Enamides, and Ynamides. J. Org. Chem. 2023, 88, 1168. [Google Scholar] [CrossRef]
- Hui, X.; Bernhard, B. Nickel-Catalyzed Regioselective Hydrothiolation of Allenes Enabled by Visible-Light Photoredox Catalysis. Org. Lett. 2024, 26, 4438–4442. [Google Scholar]
- Thakuri, R.S.; Rina, Y.A.; Schmidt, J.A.R. Regiospecific Conversion of Aliphatic Allenes to Dithioacetals via Palladium-Catalyzed Double Hydrothiolation. Eur. J. Inorg. Chem. 2024, 27, e202400339. [Google Scholar] [CrossRef]
- Crabtree, R. Iridium Compounds in Catalysis. Acc. Chem. Res. 1979, 12, 331–338. [Google Scholar] [CrossRef]
- Sevov, C.; Zhou, J.; Hartwig, J. Iridium-Catalyzed, Intermolecular Hydroamination of Unactivated Alkenes with Indoles. J. Am. Chem. Soc. 2014, 136, 3200–3207. [Google Scholar] [CrossRef]
- Haibach, M.C.; Guan, C.; Wang, D.Y.; Li, B.; Lease, N.; Steffens, A.M.; Krogh-Jespersen, K.; Goldman, A.S. Olefin Hydroaryloxylation Catalyzed by Pincer–Iridium Complexes. J. Am. Chem. Soc. 2013, 135, 15062–15070. [Google Scholar] [CrossRef]
- Burling, S.; Field, L.D.; Messerle, B.A.; Vuong, K.Q.; Turner, P. Rhodium(I) and iridium(I) complexes with bidentate N,N and P,N ligands as catalysts for the hydrothiolation of alkynes. J. Chem. Soc. Dalton Trans. 2003, 4181–4191. [Google Scholar] [CrossRef]
- Field, L.D.; Messerle, B.A.; Vuong, K.Q.; Turner, P. Rhodium(I) and iridium(I) complexes containing bidentate phosphine imidazolyl donor ligands as catalysts for the hydroamination and hydrothiolation of alkynes. Dalton Trans. 2009, 3599–3614. [Google Scholar] [CrossRef]
- Teders, M.; Henkel, C.; Anhäuser, L.; Strieth-Kalthoff, F.; Gómez-Suárez, A.; Kleinmans, R.; Kahnt, A.; Rentmeister, A.; Guldi, D.; Glorius, F. The energy-transfer-enabled biocompatible disulfide–ene reaction. Nature Chem. 2018, 10, 981–988. [Google Scholar] [CrossRef]
- Tolley, L.C.; Fernández, I.; Bezuidenhout, D.I.; Guisado-Barrios, G. Catalytic conversion of alkynes to α-vinyl sulfides mediated by carbene-linker-carbene (CXC) rhodium and iridium complexes. Catal. Sci. Technol. 2021, 11, 516–523. [Google Scholar] [CrossRef]
- Wang, X.; Li, Y.; Li, Z. Efficient visible light initiated hydrothiolations of alkenes/alkynes over Ir2S3/ZnIn2S4: Role of Ir2S3. Chinese J. Catal. 2021, 42, 409–416. [Google Scholar] [CrossRef]
- Davids, T.K.; Petersen, W.F.; Smith, G.S. Evaluation of a trinuclear heteroleptic bis-cyclometalated iridium(III) complex as a photoredox catalyst for visible light-mediated hydrothiolation reactions. Inorg. Chem. Commun. 2023, 149, 110384. [Google Scholar] [CrossRef]






































Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license.