Abstract
Benzothiazole derivatives have emerged as being highly significant in drug discovery due to their versatile biological activities and structural adaptability. Incorporating nitrogen and sulfur, this fused heterocyclic scaffold exhibits wide-ranging pharmacological properties, including anticancer, antimicrobial, anti-inflammatory, antidiabetic, neuroprotective, and diagnostic applications. A diverse set of clinically approved and investigational compounds, such as flutemetamol for Alzheimer’s diagnosis, riluzole for ALS, and quizartinib for AML, illustrates the scaffold’s therapeutic potential in varied applications. These agents act via mechanisms such as enzyme inhibition, receptor modulation, and amyloid imaging, demonstrating the scaffold’s high binding affinity and target specificity. Advances in synthetic strategies and our understanding of structure–activity relationships (SARs) continue to drive the development of novel benzothiazole-based therapeutics with improved potency, selectivity, and safety profiles. We also emphasize recent in vitro and in vivo studies, including drug candidates in clinical trials, to provide a comprehensive perspective on the therapeutic potential of benzothiazole-based compounds in modern drug discovery. This review brings together recent progress to help guide the development of new benzothiazole-based compounds for future therapeutic applications.
1. Introduction
Heterocyclic compounds are central in medicinal chemistry, offering immense structural diversity and functional versatility. Heterocyclic compounds play a vital role in medicinal chemistry due to their remarkable structural diversity and functional adaptability [1]. Found in over 90% of newly approved pharmaceuticals, these compounds are integral to modern drug discovery and development. They exhibit a broad spectrum of biological activities, including anticancer, anti-inflammatory, antifungal, antibacterial, and antiviral effects [2,3]. Nitrogen-containing heterocycles such as triazoles, tetrazoles, imidazoles, pyrimidines, and quinolines are especially prevalent in nature and hold substantial pharmacological significance [4,5,6,7,8]. Beyond the realm of medicine, heterocyclic compounds are also widely used in agrochemicals, veterinary medicine, and various industrial applications [9,10]. Their distinctive structures and multifaceted properties make them essential tools for probing and modulating biological systems, effectively bridging the disciplines of chemistry and biology [11,12,13,14,15,16].
Among them, benzothiazole (BZT) stands out as a privileged and pharmacologically potent fused heterocycle (Figure 1), consisting of a benzene ring fused with a thiazole ring (a five-membered ring containing nitrogen at the 3-position and sulfur at the 1-position). This planar bicyclic framework has emerged as a key structural motif in drug discovery, known for its ability to interact with a wide range of biological targets [17,18,19]. Benzothiazole, in particular, has emerged as a highly valuable scaffold in medicinal chemistry, exhibiting a wide array of pharmacological effects, including anticancer, antibacterial, antifungal, anti-inflammatory, and antiviral activities [20,21]. Derivatives of benzothiazole have shown significant promise as enzyme inhibitors, targeting critical biological pathways such as EGFR, VEGFR, PI3K, and topoisomerases. Some of these molecules have progressed into clinical trials, underscoring their therapeutic potential. Moreover, benzothiazole analogues have demonstrated notable efficacy in targeting neurological disorders, with applications in conditions like epilepsy, Alzheimer’s disease, and Huntington’s disease [22]. The structural versatility of the benzothiazole scaffold continues to inspire the design of new, more effective drug candidates. This ongoing research underscores the importance of benzothiazole derivatives in the development of novel therapeutic agents and points toward future opportunities for designing highly specific and potent analogues, targeting diverse biological receptors [23].
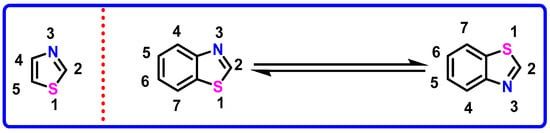
Figure 1.
Structure and tautomerism in benzothiazole.
The BZT scaffold is widely utilized for its broad spectrum of therapeutic applications, encompassing antimicrobial, antifungal, antitubercular, antimalarial, analgesic, anticonvulsant, antidiabetic, and neuroprotective properties (Figure 2) [24]. Several clinically approved drugs incorporate this motif, including riluzole, used in the treatment of amyotrophic lateral sclerosis (ALS); ethoxzolamide, a diuretic; and Zopolrestat, an aldose reductase inhibitor employed in managing diabetic complications. The success of these drugs underscores the biological relevance and drug-likeness of benzothiazole-based molecules [25]. Recent advances underscore the broad pharmacological potential of benzothiazole-based compounds, spanning approved drugs, investigational agents, and experimental probes. These compounds exhibit diverse therapeutic applications, including neuroprotection (e.g., riluzole), anticancer activity (e.g., quizartinib, Phortress), diagnostic imaging (e.g., flutemetamol), and CNS modulation (e.g., pramipexole, perospirone). While several agents have gained regulatory approval, others remain in clinical or preclinical development, highlighting the continued interest in benzothiazole scaffolds for addressing a wide array of medical needs.
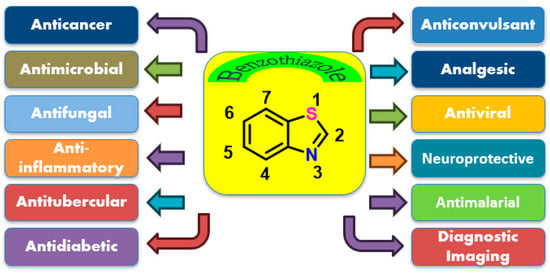
Figure 2.
Structure of benzothiazole and its multiple clinical applications.
2. Biological Importance and Mechanistic Versatility
The broad therapeutic profile of benzothiazoles can be attributed to the unique electronic and structural characteristics of the scaffold [26]. The presence of sulfur and nitrogen heteroatoms enhances the ability of BZT derivatives to engage in diverse non-covalent interactions, such as hydrogen bonding, and semi-covalent interactions, with key amino acid residues in protein binding sites [26]. This contributes to high binding affinity and target specificity. The planarity of the BZT ring system allows optimal π–π stacking interactions, which are especially important when interacting with nucleic acids or planar protein pockets. Modern medicinal chemistry increasingly emphasizes compounds with higher sp3 carbon content for improved solubility and metabolic stability [27]. However, BZT’s aromatic, sp2-rich nature remains highly valuable, especially in targeting shallow or narrow protein binding pockets, as commonly found in GPCRs, enzymes, and kinases [28].
3. Synthetic Accessibility and Structural Tunability
The benzothiazole core’s structural simplicity and synthetic feasibility make it a prime candidate for medicinal chemists developing novel drug-like molecules. Classical routes to BZT derivatives often involve the condensation of 2-aminothiophenols with carbonyl compounds, while more advanced strategies employ transition-metal-catalyzed cyclization, green chemistry techniques, and biocatalysis to access diverse analogues (Figure 3) [29,30,31]. A key site of derivatization is the C2 position, where the introduction of various functional groups such as amines (2-aminobenzothiazole), thiols (2-mercaptobenzothiazole), or aryl groups (2-arylbenzothiazole) leads to molecules with potent biological activities [32,33,34]. The 2-arylbenzothiazoles have emerged as promising anticancer agents, often displaying selective cytotoxicity against cancer cells while sparing healthy tissues. Substitution at other positions, notably C5 and C6, with electron-withdrawing groups has been shown to enhance activity, offering insights into structure–activity relationships (SARs) that guide rational drug design [32,35,36,37,38,39,40,41,42].
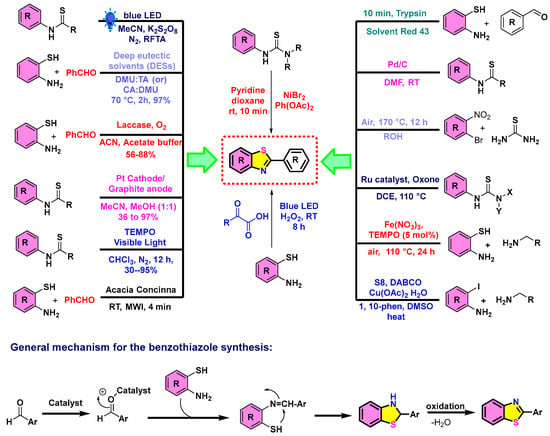
Figure 3.
Diverse strategies toward benzothiazoles via transition-metal catalysis, greener routes, and enzymatic reactions. RT = room temperature; MWI = microwave irradiation.
Figure 3 presents a detailed overview of the diverse and efficient synthetic strategies that are used for constructing benzothiazole derivatives, emphasizing both conventional and green chemistry approaches [43,44]. General synthesis involves the condensation of 2-aminothiophenols with aldehydes, followed by cyclization and oxidative aromatization to form the benzothiazole core [45]. Various catalytic systems are employed to enhance reaction efficiency and sustainability [46,47]. These include photoredox methods using blue LED light in the presence of K2S2O8 [48], laccase/O2 biocatalysis [49], and deep eutectic solvents (DESs) like DMU:TA and CA:DMU [50]. Electrochemical methods utilizing platinum cathodes and graphite anodes [51], visible-light-promoted reactions with TEMPO [52], and natural catalysts such as Acacia concinna also feature prominently [53]. Metal-catalyzed strategies using NiBr2, Ru catalysts with Oxone, Fe(NO3)3 with TEMPO, and copper-based systems further expand the synthetic versatility [54,55,56,57]. Post-synthetic modifications, including reductions, oxidations, and nucleophilic substitutions, allow for the generation of structurally diverse benzothiazole analogs [58]. Compound transformations and oxidative annulations offer opportunities for functionalization tailored to specific applications [59,60]. The adaptability and potential of benzothiazole synthesis for medicinal, biological, and materials science applications, showcasing the integration of sustainable techniques and advanced catalytic methods [30].
The introduction of a methoxy group at the C-6 position of benzothiazole significantly enhances kinase-targeted anticancer activity. For example, Ammazzalorso et al. reported that 2-substituted benzothiazole analogues bearing a C-6 methoxyphenyl moiety exhibited exceptional inhibitory potency against both wild-type and T315I-mutant Abl kinases, with IC50 values ranging from 0.03 to 0.06 nM [36]. These compounds also demonstrated strong cytotoxicity in Ba/F3 cell lines (IC50 = 0.046–0.09 µM). Additionally, hybrids of benzothiazole and pyrazole incorporating a methoxy at C-6 yielded submicromolar antiproliferative activity: one such derivative selectively inhibited A549 lung cancer cells at IC50 = 0.054 µM. These reports signify the methoxy group’s critical contribution via favorable hydrophobic and hydrogen-bonding interactions within kinase binding sites [41].
Electron-withdrawing substituents at C-5, especially fluorine, have been shown to substantially boost both anticancer efficacy and metabolic stability of benzothiazoles. Moreover, chemists introduced a fluorine at C-5 in DF-203, yielding the clinical candidate 5F-203, which maintained potent antiproliferative activity across breast, ovarian, and renal cancer cell lines while avoiding the biphasic dose–response of its parent compound. Complementary QSAR studies also support the idea that halogenizing the benzothiazole core enhances cytotoxicity; one series demonstrated superior activity in HuT78 cells with 6-Cl and 5-halogenated derivatives showing IC50 values between 3–10 µM. Additionally, a focused review on fluorinated benzazoles confirms the role of fluorine in boosting binding affinity, lipophilicity, and pharmacokinetic performance [37,38,39,40].
As well as the layering a fluorine at C-5 with a methoxy or methyl substituent at C-6 has been demonstrated to produce synergistic antiproliferative effects. In benzothiazole–pyrazole hybrids, the simultaneous presence of C-6 methoxy and ortho-fluoro on the phenyl ring yielded submicromolar inhibition (IC50 = 0.1–0.15 µM) against PC-3, 22Rv1, and MCF-7 cancer cell lines. Within this same study, variants with C-5 fluorine alone showed moderate potency (IC50 = 1.3 µM), while dual-fluorinated C-5/C-6 analogues restored high potency (IC50 = 0.23 µM). This shows how combining electron-withdrawing and electron-donating modifications at adjacent positions optimizes both electronic properties and steric complementarity to enhance biological activity [42].
4. Applications Across Therapeutic Areas
Benzothiazoles are widely investigated for their anticancer potential, with derivatives showing activity against a range of tumor types [42,61,62]. Some BZT-based compounds have progressed into clinical trials, while others serve as leads in preclinical models. In addition to oncology, BZT derivatives have demonstrated efficacy in infectious diseases, such as tuberculosis, malaria, leishmaniasis, and HIV, and in neurological disorders, including Alzheimer’s and Parkinson’s diseases [63]. In these contexts, compounds such as Thioflavin-T and Pittsburgh compound B have been developed as diagnostic agents for imaging β-amyloid plaques. The pharmacological utility of benzothiazoles extends to immunomodulation, anti-glutamatergic activity, antiparasitic action, and enzyme inhibition, targeting diverse biological pathways such as fatty acid amide hydrolase, LTD4 receptors, histamine H2 receptors, and orexin receptors. Moreover, their use is not limited to therapeutics but also employed as veterinary drugs, plant protectants, herbicides, fungicides, and rubber vulcanization accelerators (e.g., 2-mercaptobenzothiazole in tire manufacturing) [64].
Drug discovery and development remain at the forefront of innovation in medicinal, agrochemical, and pharmaceutical industries. Among the most captivating areas of this field is the synthesis of complex organic molecules, macrocycles, particularly those featuring multiple heterocyclic rings [5]. Compounds with benzo-fused nitrogen, sulfur, and oxygen-containing heterocycles such as benzothiazole, benzimidazole, indoles, and benzoxazole have garnered significant attention due to their broad spectrum of biological activities. These core structures, or pharmacophores, are integral to numerous bioactive compounds in medicine and agriculture [12]. Here, we demonstrated various benzothiazole-based marketed drugs and clinically relevant scaffolds utilized in diverse fields. Along similar lines selected pharmacological and molecular data were obtained from the DrugBank database is a comprehensive resource for approved and investigational drugs.
4.1. Flutemetamol 1
Flutemetamol (18F) is an FDA-approved radiopharmaceutical marketed under the trade name Vizamyl by GE Healthcare (Chicago, IL, USA). It is utilized in positron emission tomography (PET) imaging to assess β-amyloid plaque density in the brain, a critical pathological feature of Alzheimer’s disease (AD) [65,66,67]. This diagnostic tool aids in differentiating AD from other forms of cognitive impairment by providing in vivo visualization of amyloid burden. Structurally, flutemetamol is a fluorine-18-labeled analog of Thioflavin-T, designed to bind with high specificity and affinity to fibrillar amyloid-β aggregates (Figure 4). The benzothiazole ring system is a key pharmacophore responsible for this selective binding, facilitating the accumulation of the tracer in cortical regions enriched with amyloid deposits. After intravenous administration, flutemetamol crosses the blood–brain barrier (BBB) efficiently and distributes in the brain, with the highest retention in amyloid-positive areas. PET signal intensity typically increases for the first 30 min post-injection and then remains stable for up to 120 min, enabling a reliable imaging window. The radiolabel, fluorine-18, is a cyclotron-produced positron emitter with a physical half-life of approximately 110 min. It decays by positron emission, which upon annihilation with electrons generates pairs of 511 keV gamma photons detected by the PET scanner. Due to its short half-life, flutemetamol must be synthesized on demand, subjected to stringent quality control, and used within a narrow timeframe post-production. Pharmacokinetically, the compound is rapidly cleared from the bloodstream. During the imaging period, most circulating fluorine-18 is present as metabolites, with minimal free flutemetamol detected in plasma (Table 1). The recommended dose ranges from 1 to 10 mL, corresponding to approximately 185 MBq (5 mCi) of radioactivity, administered under the supervision of a nuclear medicine specialist [68].
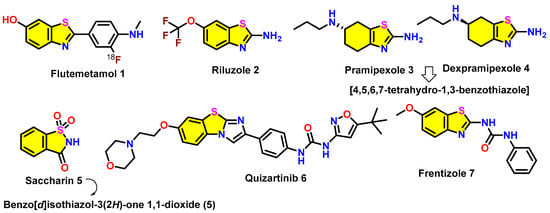
Figure 4.
Benzothiazole-based marketed drugs 1–7 (FDA-approved and those under investigation).

Table 1.
Clinical diagnosis in the 105 evaluable end-of-life patients who came to autopsy about [18F] flutemetamol PET imaging results (amyloid-positive and amyloid-negative) [66].
Figure 5 displays [18F] flutemetamol PET images interpreted as either normal (amyloid-negative) or abnormal (amyloid-positive), demonstrating key visual differences across axial, parasagittal, and coronal views. In the axial images shown in Panel a, the normal scan displays a distinct sulcal/gyral white matter pattern with prominent uptake in the frontal (f) and lateral temporal (lt) regions and visible striatal gaps (s) between the thalamus and frontal white matter. In contrast, the abnormal scan shows diffuse uptake in grey matter, loss of striatal gaps, and cortical signal extending sharply to the surface, indicative of amyloid binding. The parasagittal images in Panel b highlight increased uptake in the posterior cingulate/precuneus (pc) region and corpus callosum (cc) in the abnormal scan, while the normal scan shows lower uptake and high signal in the pons (p). In the coronal images in Panel c, the abnormal scan again demonstrates loss of the normal sulcal/gyral pattern and shows strong uptake in the posterior cingulate, inferior parietal (ip) lobes, and insula, reflecting the typical pattern of amyloid deposition. These images emphasize the utility of [18F]flutemetamol PET imaging in differentiating amyloid-positive from amyloid-negative brains, particularly in regions that are less susceptible to atrophy such as the posterior cingulate and striatum, which serve as robust indicators in visual assessment [66,67].
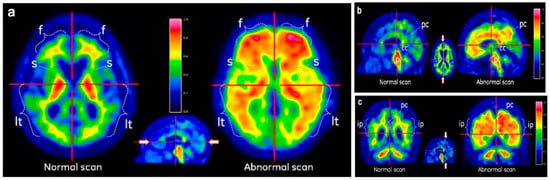
Figure 5.
Representative [18F]flutemetamol PET scans showing normal and abnormal brain uptake patterns. (a) Axial views: Left: The normal (negative) scan displays a clear sulcal/gyri pattern of white matter uptake in the frontal (f) and lateral temporal (lt) regions, along with prominent tracer accumulation in the thalamus. Right: In the abnormal (positive) scan, this pattern is lost due to increased tracer retention in the grey matter (uptake of [18F]flutemetamol), indicative of amyloid deposition. (b) Parasagittal views: Left: The normal scan shows less than 60% of the maximum intensity in the posterior cingulate/precuneus (pc) region and areas surrounding the corpus callosum (cc). Right: The abnormal scan reveals high uptake (>60% of maximum intensity) in these regions, suggestive of pathological changes. (c) Coronal views (near the level of the corpus callosum): Left: The normal scan again shows the typical white matter sulcal/gyri uptake pattern. Right: This pattern is absent in the abnormal scan, which instead shows heightened tracer uptake in the posterior cingulate (pc), extending radially to the lateral temporal lobes and insula. Especially, the inferior parietal (ip) region also exhibits strong uptake, consistent with areas less prone to atrophy and commonly involved in early amyloid deposition. Image adapted from Ref. [66]; copyright 2019, Springer.
Clinical trials have validated a strong correlation between flutemetamol PET imaging results and post-mortem histopathological assessments of β-amyloid plaque load. Additionally, the agent demonstrates high inter-reader consistency, ensuring reproducibility and reliability in image interpretation. Though flutemetamol enhances diagnostic confidence in cases of suspected Alzheimer’s disease, it is not a standalone diagnostic tool. Its use is intended to complement clinical evaluation, including neurocognitive testing and differential diagnosis. Safety and tolerability profiles are favorable, with the most commonly reported adverse effects being mild and transient. These include flushing, headache, elevated blood pressure, nausea, and dizziness. Despite its clinical utility, widespread use of flutemetamol remains limited in routine practice due to cost-related constraints [69]. As of now, Medicare coverage is restricted primarily to clinical trials, and a single scan may cost approximately USD 3000 without insurance reimbursement. In summary, flutemetamol (18F) is a highly effective amyloid PET imaging agent that plays a pivotal role in the evaluation of Alzheimer’s disease by enabling visualization of β-amyloid pathology in the living brain. Its approval represents a significant advancement in the neuroimaging of dementia and cognitive disorders [70].
4.2. Riluzole 2
Riluzole, marketed as Rilutek, is a neuroprotective agent that has been approved for the treatment of amyotrophic lateral sclerosis (ALS), a fatal neurodegenerative condition marked by progressive loss of motor neurons (Figure 4) [71]. It was the first drug approved by the FDA for ALS in 1995 and later by the European Union in 1996. Although riluzole offers only modest benefits, extending survival by 2–3 months and improving one-year survival probability by ~9%, it remains a fundamental therapy. The drug acts via multiple mechanisms, including inhibition of presynaptic glutamate release, blockade of voltage-gated sodium channels, and enhanced glutamate reuptake, collectively reducing excitotoxicity, a major contributor to motor neuron damage. It also indirectly modulates GABAA receptors and may influence intracellular events following neurotransmitter binding [72]. Structurally, riluzole is a 2-amino-benzothiazole with a trifluoromethoxy group, enhancing CNS penetration due to its lipophilicity. It is administered orally, with ~60% bioavailability, though high-fat meals reduce absorption. The drug is highly protein-bound (~96%) and extensively metabolized in the liver, primarily via CYP1A2, with additional glucuronidation. Drug interactions with CYP1A2 modulators are clinically relevant. Adverse effects are usually mild–moderate, including nausea, weakness, increased liver enzymes, and dizziness, though rare events such as neutropenia and interstitial lung disease can occur. Overdose may lead to toxic encephalopathy and methemoglobinemia, which can be treated with methylene blue [73,74,75].
In addition to ALS, riluzole has been investigated for neuropsychiatric and neurodegenerative disorders, including Huntington’s disease, Parkinson’s disease, and mood and anxiety disorders, though trials in Huntington’s and Parkinson’s have not yielded successful outcomes. Notably, riluzole has shown antioxidant and antiapoptotic activity in preclinical models of ototoxicity, such as cisplatin-induced and noise-induced hearing loss. It is also being researched concerning TDP-43 proteinopathy, a key pathological hallmark of ALS, potentially involving CK1δ signaling. Pharmaceutical innovations include sublingual formulation (BHV-0223), under development for psychiatric applications, and troriluzole, a prodrug designed for improved efficacy and broader therapeutic application. Despite limited clinical use beyond ALS, riluzole’s multifaceted mechanism of action and neuroprotective profile continue to drive interest in its repurposing and reformulation for CNS disorders [76,77,78,79].
4.3. Pramipexole 3
Pramipexole (4,5,6,7-tetrahydro-1,3-benzothiazole), marketed as Mirapex and available as a generic treatment, is a non-ergoline dopamine agonist primarily used for the treatment of Parkinson’s disease (PD) and Restless Leg Syndrome (RLS). It was first approved by the FDA in 1997 for PD and in 2006 for RLS and remains a widely prescribed agent, with over 2 million prescriptions annually in the U.S. Pramipexole exerts its therapeutic effects by selectively stimulating dopamine D2-like receptors, with a marked preference for D3 receptors, which helps restore dopaminergic function in the striatum, alleviating hallmark PD symptoms such as tremors, rigidity, and bradykinesia [80]. In RLS, the drug modulates disrupted dopaminergic pathways that underlie the characteristic leg discomfort and urges them to move. It is orally administered and is well absorbed, showing favorable bioavailability and pharmacokinetics. Structurally, pramipexole is a benzothiazole derivative containing a thiazole ring and a cyclohexylamine moiety. Compared to levodopa, pramipexole carries a lower risk of dyskinesia, making it a cornerstone in dopaminergic therapies. Importantly, only the immediate-release formulation is approved for RLS, while the extended-release version is restricted to PD treatment (Figure 4) [81].
Beyond its approved uses, pramipexole has been explored for a variety of off-label and investigational indications. In psychiatry, it has shown some potential in managing treatment-resistant depression, bipolar disorder, and SSRI-induced sexual dysfunction, likely due to its preferential D3 receptor agonism, which influences dopamine and serotonin levels in the prefrontal cortex. Although results are mixed, it has also been studied in REM sleep behavior disorder and fibromyalgia, and is under investigation for conditions like essential tremors, orthostatic tremor, and persistent genital arousal disorder. Notably, pramipexole demonstrates neuroprotective properties in preclinical models of cerebral ischemia, traumatic brain injury, and neurodegenerative conditions, acting via pathways such as Nrf2/GPX4-mediated ferroptosis inhibition and anti-inflammatory cytokine suppression. It also mitigates PD-related chronic pain through glial cell modulation and inhibition of pro-inflammatory mediators like TNF-α, IL-1β, and NF-κB. Derivatives of pramipexole, such as CJ-998 and D-264, are under study for improved selectivity and therapeutic potential. Despite its wide therapeutic scope, pramipexole’s exact mechanism remains only partially understood, especially in non-motor symptoms and mitochondrial effects, where stereoisomer-specific activity offers a pharmacological tool for distinguishing dopaminergic from non-dopaminergic pathways [82,83].
4.4. Dexpramipexole 4
Dexpramipexole (DEX, 4,5,6,7-tetrahydro-1,3-benzothiazole), also known as KNS-760704 or R-(+)-pramipexole, is an investigational oral small molecule being developed by Areteia Therapeutics as a novel therapy for eosinophilic asthma (Figure 4) [84]. Initially created for ALS treatment due to its neuroprotective effects, including lowering mitochondrial oxidative stress and enhancing neuronal resilience, its development was halted following disappointing Phase III results in ALS [85]. Nonetheless, its ability to markedly reduce eosinophil levels was discovered during these trials, prompting its repositioning for eosinophil-driven conditions like hypereosinophilic syndrome and chronic rhinosinusitis with nasal polyps. In Phase II asthma trials (e.g., EXHALE-1), dexpramipexole demonstrated significant and sustained eosinophil suppression, improved lung function metrics comparable to biologics, and a favorable safety profile with no severe adverse events or treatment discontinuations [86].
Structurally, dexpramipexole is the dextro enantiomer of pramipexole but diverges functionally due to a lack in dopamine receptor activity, allowing it to be used at much higher doses without dopaminergic side effects. Its targeted mechanism selectively impacts eosinophils while sparing other immune cells, and it has shown steroid-sparing effects in clinical studies. The drug exhibits linear dose–response behavior and excellent oral bioavailability, with evidence of tissue eosinophil depletion and bone marrow eosinophil suppression. Its pharmacokinetic simplicity and clean receptor profile make it an attractive candidate for chronic eosinophilic diseases, with three global Phase III studies currently evaluating its potential as the first approved oral eosinophil-lowering agent for asthma [87,88].
In 2020, Buonvicino reported that dexpramipexole treatment dose-dependently increased ATP content in cultured dorsal root ganglion neurons and isolated mouse optic nerves, effectively counteracting energy deficiency induced by mitochondrial uncoupling or low-glucose conditions and reducing neurotoxicity under energy-deprived or excitotoxic stress [89]. DEX, originally developed as an F1F0 ATP synthase activator for neuroprotection, has recently been shown to possess unexpected ion-channel-modulating properties. Moreover, the in vitro electrophysiological studies using patch–clamp and field recordings in rat hippocampal neurons revealed that DEX significantly enhances outward K+ currents. This effect is mediated via voltage-dependent K+ (Kv) and small-conductance Ca2+-activated K+ (SK) channels, as it is blocked by TEA and apamin, respectively. DEX also suppressed synaptic transmission and reduced excitability of CA1 pyramidal neurons. These results suggest a novel mechanism for its neuroprotective actions and position DEX as a potential lead compound for developing K+ conductance modulators [90]. Beyond its mitochondrial targeting, dexpramipexole has been shown to attenuate pyroptosis and apoptosis, improving cognitive function in sepsis-associated encephalopathy by suppressing mitochondrial-mediated cell death pathways [91]. These collective recent reports shown dexpramipexole’s multifaceted protective actions enhancing mitochondrial resilience, modulating ion channel activity, and reducing apoptosis or inflammatory cell death, signifying promising therapeutic roles in ischemic, neuroinflammatory, and cardiovascular disease models.
4.5. Saccharin 5 (Benzisothiazole-1,1-dioxide Core)
Saccharin, also known as benzosulfimide or E954 (H-1λ6,2-Benzothiazole-1,1,3(2H)-trione), is a synthetic, non-nutritive, high-intensity sweetener that is 200–700 times sweeter than sucrose while contributing no caloric value. Discovered in 1879 by Constantin Fahlberg, it was the first artificial sweetener to be commercialized and remains widely used in foods, beverages, pharmaceuticals, and oral hygiene products. Saccharin features a benzisothiazole-1,1-dioxide core, making it structurally relevant to benzothiazole-based compounds. Although its intrinsic water solubility is low, sodium and calcium saccharin salts are commonly used to enhance solubility in formulations such as tablets, syrups, and mouthwashes [92]. It is heat-stable up to at least 250 °C and remains stable across a broad pH range, making it suitable for a variety of industrial and medicinal applications. While saccharin has a known metallic or bitter aftertaste at high concentrations, it is often blended with other sweeteners to improve palatability (Figure 4). Importantly, saccharin does not affect blood glucose levels, has a glycemic index of zero, and is safe for individuals with diabetes or obesity. Despite early concerns over its carcinogenic potential that stemming from rodent studies linking it to bladder cancer, subsequent research demonstrated that humans metabolize saccharin differently, and it is now recognized as safe by the U.S. FDA, EFSA, JECFA, and other regulatory agencies in over 100 countries [93]. In addition to its sweetening properties, saccharin exhibits microbiome-modulating and bacteriostatic activities, further supporting its utility in pharmaceutical and medicinal formulations. Today, saccharin remains a trusted and extensively studied sugar substitute globally [94].
4.6. Quizartinib 6
Quizartinib, marketed as Vanflyta by Daiichi Sankyo, is a second-generation, highly potent, and selective type II FLT3 inhibitor developed specifically to target FLT3-ITD mutations; these mutations are present in approximately 25% of acute myeloid leukemia (AML) cases and are associated with poor prognosis and high relapse rates [95]. Quizartinib works by binding to the inactive conformation of the FLT3 receptor, thereby preventing autophosphorylation and downstream signaling, and effectively blocking leukemic cell proliferation and inducing apoptosis (Figure 4). Both quizartinib and its active metabolite, AC886, display nanomolar binding affinities (Kd values of 3.3 and 1.1 nM, respectively) to FLT3, showing over 10-fold selectivity compared to a broad kinase panel [96]. Quizartinib also exhibits partial inhibitory activity against c-KIT and other class III receptor tyrosine kinases, including CSF1R and PDGFRs. FDA approval was based on results from the QuANTUM-First trial, which demonstrated a 22% reduction in the risk of death in newly diagnosed FLT3-ITD-positive AML patients when quizartinib was combined with standard induction (cytarabine and anthracycline) and consolidation chemotherapy, followed by maintenance monotherapy [97].
Administered orally once daily, quizartinib is approved for adult patients with newly diagnosed FLT3-ITD-positive AML, as detected by an FDA-approved test like the LeukoStrat CDx FLT3 Mutation Assay. However, it is not indicated for maintenance monotherapy following allogeneic stem cell transplant due to a lack of demonstrated survival benefit. The drug label includes a Boxed Warning for QT prolongation, torsades de pointes, and cardiac arrest, with use restricted under the REMS program [98]. Common adverse events include neutropenia, diarrhea, mucositis, nausea, abdominal pain, and infections, while lab abnormalities may include reduced lymphocytes, electrolytes (K+, Mg2+, Ca2+), and elevated liver/muscle enzymes. QTc prolongation is dose- and concentration-dependent, necessitating ECG and electrolyte monitoring during treatment. Approved in Japan (2019), the U.S. (2023), and the EU (2023), quizartinib has completed 17 clinical trials with 11 ongoing, establishing its role as a key targeted therapy in the management of FLT3-ITD-positive AML [99,100].
4.7. Frentizole 7
Frentizole (Frentizol) is a synthetic immunosuppressive and antiviral compound originally developed for the treatment of autoimmune diseases such as rheumatoid arthritis and systemic lupus erythematosus. Structurally, it is a thiadiazole-based N-phenylurea derivative featuring a benzothiazole ring, a motif known for various bioactivities [101,102]. Pharmacologically, frentizole selectively suppresses lymphocyte proliferation in response to mitogens like Concanavalin A and pokeweed mitogen, indicating targeted immunomodulatory activity (Figure 4) [103,104,105] Notably, it does not significantly compromise host resistance to pathogens like Candida albicans or Pseudomonas aeruginosa, even at high doses, suggesting a relatively low systemic toxicity. However, despite early clinical use and promising safety in preclinical infection models, its clinical development was hindered by dose-dependent hepatotoxicity, particularly elevated liver enzyme levels, limiting its therapeutic viability [106].
More recently, frentizole has gained attention as a novel inhibitor of the interaction between amyloid beta (Aβ) peptide and mitochondrial alcohol dehydrogenase (ABAD), a pathway implicated in Alzheimer’s disease. It disrupts the Aβ–ABAD interaction with an IC50 of approximately 200 μM, reducing the potential for Aβ-mediated mitochondrial toxicity. While this suggests potential relevance in neurodegenerative disease research, the relatively high IC50 implies modest potency [107,108,109]. Despite its reported nontoxic antiviral and immunosuppressive profile in earlier studies, frentizole has not received FDA approval and remains excluded from regulatory drug databases. Currently, it is regarded as an investigational compound, primarily used in experimental settings to explore its immunosuppressive, antiviral, and neuroprotective properties [110,111].
As well as being historically recognized as a nontoxic immunosuppressant, it has recently attracted attention for its potent antitumor activity via tubulin inhibition. In 2023, Ramos et al. documented that frentizole and selected analogs such as benzamide and brominated ethyl ureas, significantly inhibited cancer cell proliferation in HeLa (IC50 = 1 µM), while also disrupting microtubule networks and inducing G2/M cell cycle arrest consistent with colchicine-site tubulin binding. In addition, it displayed activity against the glioblastoma cell line U87MG (IC50 = 7.33 µM), underscoring broader anticancer potential [110]. Beyond its anticancer role, frentizole-like derivatives have been identified as mTOR inhibitors with senomorphic properties. In 2023, Chrienova et al. [111] evaluated a series of these analogs, selecting three for in vivo studies. One of those compounds emerged as a lead candidate (dichloro derivative) with favorable drug-like properties including blood–brain barrier penetration and low acute toxicity in mice (LD50 = 560 mg/kg for males and LD50 = 576 mg/kg for female, respectively). In the context of Alzheimer’s disease, benzothiazolyl–urea derivatives inspired by frentizole have shown promising inhibition of mitochondrial ABAD/17β-HSD10 enzymatic activity [112]. Hroch et al. (2016) reported some of these as potent inhibitors as having favorable physicochemical properties, indicative of good brain penetration [108]. Later, Benek et al., (2017) identified a lead benzothiazolyl urea derivative with ABAD IC50 = 3.06 µM and promising cytotoxicity and blood–brain barrier permeability profiles [112]. Furthermore, frentizole itself was shown to reduce neuronal death and extracellular glutamate levels in vitro in cultures exposed to oligomeric Aβ1–42, indicating potential neuroprotective effects beyond immunosuppression [113].
4.8. Thioflavin-T 8
Thioflavins are benzothiazole-based fluorescent dyes primarily represented by Thioflavin-T (ThT) and Thioflavin-S (ThS), extensively used to study amyloid fibril formation associated with neurodegenerative diseases such as Alzheimer’s, Parkinson’s, Huntington’s, and prion diseases [114,115]. Thioflavin-T is a cationic, cell-permeable dye that exhibits a strong fluorescence enhancement (excitation at 440–450 nm, emission at ~482–485 nm, quantum yield ~0.43) upon binding to β-sheet-rich structures within amyloid fibrils. This binding immobilizes the dye’s two aromatic rings, restricting intramolecular rotation and enabling prolonged excited states (Figure 6) [116]. ThT’s fluorescence intensity correlates with fibril formation, making it a widely accepted tool for real-time in vitro amyloid aggregation assays and plaque visualization in brain tissues. Despite its sensitivity, ThT is not universally specific for amyloids and may occasionally bind to non-amyloid proteins or fail to detect certain fibrillar structures, leading to false positives or negatives. It also forms micelles at concentrations above 4.0 μM, which can influence binding behavior and fluorescence output, with micelle disruption at low pH further reducing signal intensity [117,118].
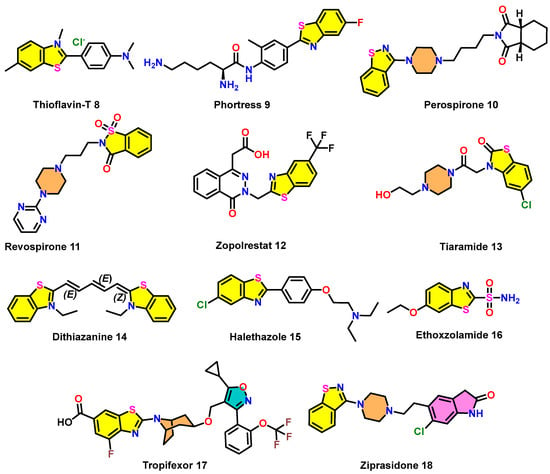
Figure 6.
Examples of clinically relevant benzothiazole scaffolds 8–18.
Turoverov team (2018) investigated α-synuclein amyloid fibrils key markers of Parkinson’s disease using Thioflavin-T (ThT) as a fluorescent probe. Reproducible fibril formation and structure were confirmed through various physicochemical methods. Binding parameters were determined via equilibrium microdialysis and spectroscopy, revealing two ThT binding modes: a high-affinity mode (K ≈ 106 M−1, stoichiometry ~1:2500) and a lower-affinity mode (K ≈ 104 M−1, stoichiometry ~1:8). Differences in binding behavior and photophysical properties supported the existence of fibril polymorphism (Figure 7) [119]. In contrast, Thioflavin-S 8 is a heterogeneous sulfonated mixture that binds to amyloid plaques similarly but lacks the characteristic spectral shift upon binding, leading to higher background fluorescence and limited utility in quantitative assays. While ThT is favored for in vitro fibrillation kinetics due to its spectral properties and quantitative potential, ThS is more commonly used in histological applications owing to its established use and compatibility with fixed tissue staining. Notably, ThT’s broader binding profile sometimes results in staining non-amyloid structures, contributing to its underuse in histopathology. Both dyes remain critical tools in amyloid research, with Congo red serving as an additional traditional dye for amyloid identification [120,121,122,123,124].
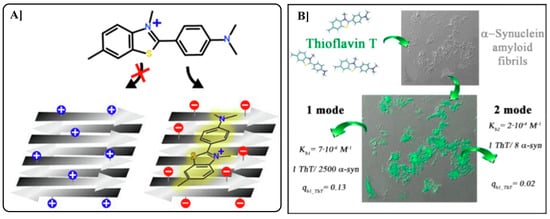
Figure 7.
The diagram illustrates how electrostatic interactions influence Thioflavin-T (ThT) 8 binding to pre-formed fibrils. On the left, electrostatic repulsion prevents ThT adsorption, while on the right, attraction to negatively charged fibril surfaces promotes ThT binding and enhanced fluorescence (A). On the right, investigation of α-synuclein amyloid fibrils using fluorescent probe Thioflavin-T 8 (B). Reproduced with permission from Refs. [119,124]; copyright 2018, MDPI, Basel, Switzerland; copyright 2020, Elsevier, Amsterdam, The Netherlands.
4.9. Phortress 9
Phortress (5F-DF-203-L-lysylamide dihydrochloride) is an experimental antitumor agent by Pharminox, the University of Nottingham, and Cancer Research UK. It is a lysylamide prodrug of 5F 203 (2-(4-amino-3-methylphenyl)-5-fluorobenzothiazole), designed for selective activity against human-derived breast, ovarian, and renal carcinomas [125,126,127]. Upon administration, Phortress is hydrolyzed to release 5F 203, which binds to the aryl hydrocarbon receptor (AhR), triggering its nuclear translocation. This induces the expression of cytochrome P450 enzymes, especially CYP1A1, which then bioactivate 5F 203 into electrophilic intermediates. These reactive species form DNA adducts selectively in sensitive tumor cells, resulting in DNA damage and apoptosis (Figure 6) [38,128,129].
Phortress is a new experimental antitumor agent with a unique mechanism distinct from current chemotherapeutics. It functions through biotransformation of its active component, 5F 203 (a fluorinated benzothiazole), by the enzyme CYP1A1. This metabolic activation produces electrophilic intermediates that bind covalently to DNA, causing lethal damage specifically in sensitive tumor cells like MCF-7. The study investigated DNA integrity and cell cycle effects, following 5F 203 exposure in sensitive (MCF-7) and resistant (MDA-MB-435) breast cancer cells using single-cell gel electrophoresis (SCGE) and flow cytometry. DNA strand breaks were observed in a dose- and time-dependent manner only in sensitive cells, both in vitro and in vivo. These findings were confirmed in mouse models, including xenografts and hollow fiber implants. Additionally, computational analyses suggested that 5F-203 impacts cell cycle-related phosphatases and kinases, supporting experimental observations of selective cell cycle disruption in responsive cells. SCGE may therefore serve as a useful pharmacodynamic tool for predicting tumor sensitivity to Phortress in clinical settings (Figure 8) [130].
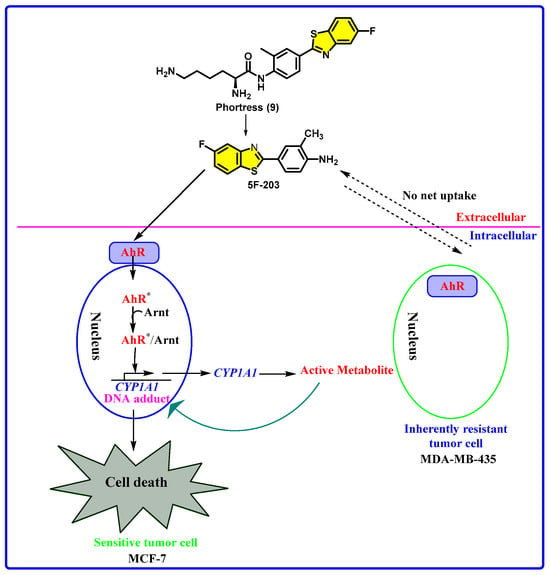
Figure 8.
Chemical structures of Phortress, 5F 203, and a summary of the mechanism of action of Phortress. (AhR* = activated AhR)
Phortress’s mechanism of action is distinct from conventional chemotherapeutics, offering a targeted approach based on tumor-specific enzymatic activation. It has demonstrated potent, selective cytotoxicity in vitro and in vivo, particularly in MCF7 (breast) and IGROV-1 (ovarian) cancer cells, and is being evaluated for pharmacokinetics, safety, and efficacy in clinical settings. Phortress exhibits high AhR affinity, and its activation is tumor-specific due to the localized expression of CYP1A1, enhancing selectivity and minimizing off-target effects. For research, it is commercially available from multiple suppliers in various quantities (Figure 8) [131,132].
4.10. Perospirone 10
Perospirone is an atypical (second-generation) antipsychotic from the azapirone family, first introduced in Japan in 2001 by Dainippon Sumitomo Pharma (brand name: Lullan). It is approved for treating schizophrenia and acute manic episodes of bipolar disorder, showing particular effectiveness against both positive and negative symptoms [133,134,135]. Pharmacologically, perospirone functions as a dopamine D2 receptor antagonist, a 5-HT2A receptor inverse agonist, and a 5-HT1A partial agonist, contributing to its antipsychotic, anxiolytic, and antidepressant properties (Figure 6). Additionally, it antagonizes α1-adrenergic, D4, and H1 histamine receptors, which accounts for its potential to cause sedation and orthostatic hypotension. It is clinically a hydrated hydrochloride salt and is primarily metabolized by CYP3A4 [136,137,138,139,140,141,142].
Perospirone shows a favorable safety profile, with a reduced risk of extrapyramidal symptoms (EPSs) compared to typical antipsychotics like haloperidol. Its D2 antagonism in the mesolimbic pathway alleviates psychotic symptoms, while 5-HT2A antagonism may enhance cognitive and negative symptoms by increasing dopamine and glutamate release in the mesocortical pathway. Moreover, 5-HT1A partial agonism helps regulate serotonergic tone. Although it is not approved outside Japan, perospirone is regarded as a unique and effective antipsychotic with receptor-targeted action that allows personalized dosing based on patient drug levels to optimize therapeutic outcomes [143,144,145,146].
4.11. Revospirone 11
Revospirone (Bay Vq 7813) is an investigational azapirone compound structurally related to buspirone and developed primarily for its anxiolytic properties. It acts as a partial agonist at serotonin 5-HT1A receptors, a mechanism associated with reduced serotonin release and anxiolytic effects [147]. Like other azapirones, Revospirone is a prodrug metabolized in vivo to 1-(2-pyrimidinyl)piperazine (1-PP), an active metabolite that serves as an α2-adrenergic receptor antagonist, potentially enhancing its central nervous system activity (Figure 6) [148].
Though it showed promise for treating generalized anxiety disorder and other neuropsychiatric conditions, Revospirone was never marketed and remains an experimental agent. Initially patented as a veterinary tranquilizer, its clinical development was discontinued, and it has no current approval or therapeutic use in humans [149,150]. Revospirone functions as a partial agonist at the serotonin 5-HT1A receptor and, via its active metabolite 1-(2-pyrimidinyl)piperazine (1-PP), also displays antagonistic activity at α2-adrenergic receptors. Although early behavioral studies demonstrated its ability to reduce 8-OH-DPAT-induced head-twitch responses in pigs after 3 to 5 days of treatment without observable side effects, the compound was never commercialized. Moreover, scientific investigation into Revospirone has remained largely inactive in recent years [148].
4.12. Zopolrestat 12
Zopolrestat is a carboxylic acid-based small molecule initially developed by Pfizer as an aldose reductase inhibitor for the treatment of diabetic complications [151]. Aldose reductase is a key enzyme in the polyol pathway, which becomes overactive under hyperglycemic conditions, contributing to oxidative stress and tissue damage. By inhibiting this enzyme, Zopolrestat was shown to normalize levels of sorbitol, fructose, and myo-inositol in diabetic rat tissues such as the sciatic nerve, lens, retina, and kidney. It also restored renal plasma flow in galactosemic rats, supporting its therapeutic potential. In addition to its primary mechanism, Zopolrestat has emerged as a potent and competitive inhibitor of glyoxalase I (GLO1), with a reported Ki of 1.2 nM. GLO1 is involved in detoxifying methylglyoxal, a cytotoxic byproduct of glycolysis implicated in diabetic neuropathy and other chronic complications (Figure 6) [152]. Structural studies revealed that Zopolrestat binds efficiently to the hydrophobic subpocket of GLO1, particularly when modified with benzothiazole groups at the C5 or C6 positions of the phthalazine ring, enhancing its potency. Despite its promising biochemical profile, Zopolrestat was discontinued during Phase III clinical trials due to concerns over toxicity and lack of clinical efficacy. Nevertheless, its dual inhibition of aldose reductase and GLO1 highlights its value as a lead scaffold for designing next-generation therapeutics targeting diabetic complications and oxidative-stress-related pathologies. Its long half-life (~30.3 h) and crystal structures in complex with GLO1 further support its utility in rational drug design [153,154,155,156].
Figure 9 illustrates the potent inhibitory activity and unique binding mechanism of Zopolrestat against human glyoxalase I (GLOI), compared to other inhibitors [152]. Figure 9a presents the chemical structure of Zopolrestat and demonstrates through a Dixon plot that it is a competitive inhibitor of recombinant human GLOI, with a low Ki value of 1.2 μM, indicating strong in vitro potency. Figure 9b shows a superposition of the crystal structures of mouse GLOI in complex with indomethacin and Zopolrestat. Unlike indomethacin, which binds weakly and does not occupy the hydrophobic subpocket or interact with Zn2+, Zopolrestat adopts a distinct V-shaped conformation and establishes multiple strong interactions, including hydrogen bonds and offset π–π stacking with key residues. These interactions occur without coordinating the Zn2+ ion, an advantage for drug development. Figure 9c compares the binding of Zopolrestat with that of a glutathione (GSH) analogue inhibitor, revealing that Zopolrestat effectively mimics critical hydrogen bonding and hydrophobic interactions typically seen in GSH-derived ligands. This suggests that Zopolrestat, although structurally unrelated to GSH, targets conserved active site features, making it a valuable non-GSH scaffold for designing novel GLOI inhibitors [152].
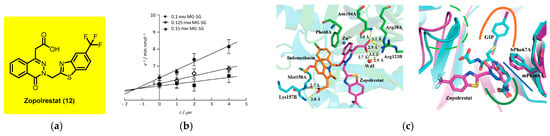
Figure 9.
(a) In vitro inhibition of recombinant human GLOI by Zopolrestat; (b) superposition of the crystal structures of mGLOI–indomethacin and mGLOI–Zopolrestat complexes; (c) comparison of binding models of Zopolrestat (magenta) with mouse GLOI and S-(N-hydroxy-N-iodophenylcarbamoyl)glutathione (GIP) (cyan) with human GLOI. Reproduced with permission from Ref. [152]; copyright 2013, Wiley-VCH, Weinheim, Germany.
4.13. Tiaramide 13
Tiaramide is a benzothiazole-derived anti-inflammatory and analgesic compound developed by Fujisawa Pharmaceutical (now Astellas Pharma, Tokyo, Japan). It functions primarily by inhibiting prostaglandin synthesis through COX-2 inhibition and suppressing the release of inflammatory mediators like histamine and thromboxane A2 [157]. These mechanisms give Tiaramide both anti-inflammatory and antiallergic properties, making it effective for conditions such as arthritis, asthma, and inflammatory bowel disease (Figure 6). It also exhibits smooth muscle relaxant and bronchodilatory effects, which were confirmed in clinical trials showing improved lung function and reduced need for bronchodilators in asthmatic patients [158].
Tiaramide offers a lower risk of gastrointestinal side effects compared to traditional NSAIDs but can still cause adverse effects such as nausea, dizziness, or GI complications with long-term use. It interacts with other NSAIDs, anticoagulants, and antihypertensives, requiring cautious use in patients with gastrointestinal, renal, or hepatic conditions. Though it was later withdrawn from the market for reasons unrelated to safety, Tiaramide remains a compound of interest due to its multitargeted action and potential to be repurposed in the treatment of inflammatory and allergic disorders [159,160].
4.14. Dithiazanine Iodide 14
Dithiazanine iodide is a benzothiazolium-based cyanine dye historically used as a broad-spectrum anthelmintic, primarily in veterinary medicine to treat nematode infections such as roundworms, whipworms, and hookworms in dogs [161,162]. Originally introduced in 1959, it was once employed for human use in treating intestinal helminth infections like trichuriasis, strongyloidiasis, and ascariasis. However, due to its significant toxicity, including severe acidosis and fatal outcomes in humans, it was withdrawn from human therapeutic use in several countries. Its toxic effects are believed to stem from interference with cellular glucose uptake, disrupting energy metabolism. Despite its withdrawal from human medicine, it remains available for veterinary use in some regions and is administered cautiously in oral doses to minimize adverse effects (Figure 6) [163,164].
Beyond its anthelmintic properties, dithiazanine has gained scientific attention for its potential applications in fields such as oncology and infectious disease due to its cytotoxic effects. Research has explored its possible roles as an anticancer agent, including studies on glioblastoma, and as an antimicrobial compound. Structurally, it is a cationic C3-cyanine dye with intense coloration and a sulfonium-substituted benzothiazole framework, contributing to its fluorescent properties. While its clinical utility is limited by toxicity, dithiazanine remains of interest in drug repurposing and chemical biology due to its bioactive profile and unique mechanism of action [165,166].
4.15. Halethazole 15
Halethazole, chemically defined as 2-(4-(5-chlorobenzothiazol-2-yl)phenoxy)-N,N-diethylethan-1-amine, is a synthetic antimicrobial agent with prominent antifungal and antiseptic properties. Its therapeutic effectiveness lies in its ability to disrupt vital cellular functions of fungal and bacterial pathogens, making it useful in the treatment and prevention of superficial skin infections. The compound’s structure, featuring a chlorinated benzothiazole core and an ether-linked diethylamino side chain, facilitates membrane penetration and interaction with microbial components, leading to cellular disruption [23,167].
Widely applied in topical formulations, Halethazole is effective against a broad spectrum of microorganisms, including fungi and bacteria. Its antiseptic action makes it valuable not only in managing dermatological infections but also as a disinfectant for minor wounds, cuts, and abrasions. The benzothiazole moiety is particularly crucial for its antimicrobial mechanism, contributing to its broad utility as a dual-action agent in clinical and over-the-counter skin care products (Figure 6) [20].
4.16. Ethoxzolamide 16
Ethoxzolamide, also known as 6-ethoxy-1,3-benzothiazole-2-sulfonamide, is a sulfonamide-based carbonic anhydrase (CA) inhibitor with a broad range of clinical applications [168]. Structurally, it features a benzothiazole ring substituted with an ethoxy group and a sulfonamide moiety, contributing to its lipophilicity and ability to cross biological membranes effectively (Figure 6 and Figure 10). Ethoxzolamide primarily acts by inhibiting carbonic anhydrase I, an enzyme that is essential for the reversible conversion of carbon dioxide and water into bicarbonate and protons. This inhibition disrupts fluid and electrolyte balance across various tissues, underlies its therapeutic efficacy [169,170]. Clinically, ethoxzolamide is used in the treatment of glaucoma, epilepsy, acute mountain sickness, duodenal ulcers, and mild diuretics. In glaucoma, it reduces intraocular pressure by decreasing the secretion of aqueous humor. In the central nervous system, its enzyme inhibition elevates the seizure threshold, making it a useful adjunctive treatment for epilepsy. In the kidneys, ethoxzolamide reduces the reabsorption of bicarbonate, sodium, and water in the proximal tubules, resulting in a diuretic effect. However, this can lead to side effects such as hypokalemia due to potassium loss. Additionally, it is used prophylactically for altitude sickness by mitigating fluid retention and associated symptoms (Figure 6 and Figure 10) [171].
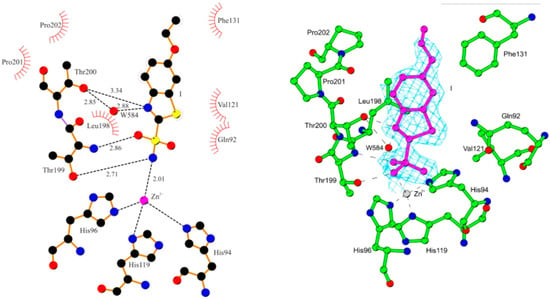
Figure 10.
Schematic representation of the active site region in the hCA II–16 complex, showing residues participating in recognition of the inhibitor molecule. Reproduced with permission from Ref. [168]; copyright 2008, Elsevier.
Beyond its approved uses, ethoxzolamide has demonstrated experimental potential in antimicrobial therapy [172,173]. Studies show that it significantly inhibits the intracellular growth of Mycobacterium tuberculosis in infected macrophages and mouse lungs, indicating possible applications in tuberculosis treatment. Despite its pharmacological benefits, ethoxzolamide has a short plasma half-life, and its effects may diminish rapidly after administration. It is generally well tolerated, but adverse effects such as dizziness, nausea, fatigue, and increased urination are possible. The drug is contraindicated by individuals with sulfonamide allergies or severe renal and hepatic dysfunction [174]. In summary, ethoxzolamide is a versatile therapeutic agent that exerts its effects by targeting carbonic anhydrase across ocular, renal, and neurological systems. Its structural features and mechanism of action enable its application in multiple medical conditions, with ongoing research exploring its potential in infectious disease management [175,176,177].
4.17. Tropifexor (LJN452) 17
Tropifexor (LJN452) is a potent, selective, non–bile-acid farnesoid X receptor (FXR) agonist developed by Novartis (Basel, Switzerland) for treating liver disorders, particularly nonalcoholic steatohepatitis (NASH) and primary biliary cholangitis (PBC). Discovered in 2017, Tropifexor incorporates a fluorinated benzothiazole moiety that enhances FXR activation with sub-nanomolar EC50 values (0.2–0.26 nM) [178]. Unlike bile-acid-based FXR agonists (e.g., obeticholic acid), Tropifexor lacks enterohepatic recirculation and exhibits improved pharmacokinetics, reducing off-target effects such as pruritus and dyslipidemia. In preclinical studies, oral Tropifexor treatment (0.01–1 mg/kg) upregulated FXR target genes SHP, BSEP, and FGF15 and suppressed CYP8B1, demonstrating effective hepatic and intestinal FXR engagement. In rodent models, it significantly reduced serum triglycerides and increased plasma FGF15, with ED50 values as low as 0.01 mg/kg.
The FLIGHT-FXR Phase 2a/b trial (NCT02855164) evaluated safety, tolerability, and efficacy in NASH patients [179]. Tropifexor (10–200 μg) led to dose-dependent reductions in ALT and hepatic fat fraction (HFF), with the 200 μg dose achieving a 39.4% reduction in HFF at week 12, sustained through 48 weeks. Improvements in body weight and biomarkers such as GGT were also observed. While pruritus was the most frequent side effect, especially at higher doses, no serious liver toxicity or drug-induced liver injury was noted [180]. Additionally, in the PBC Phase 2 trial (NCT02516605), Tropifexor dose-dependently reduced GGT levels, indicating efficacy in cholestatic liver diseases [181]. In conclusion, Tropifexor demonstrates high FXR activation potency, favorable safety, and clinically meaningful liver-targeted effects, making it a promising candidate for NASH and PBC therapy [182].
4.18. Ziprasidone 18
Ziprasidone, marketed as Geodon, is a second-generation (atypical) antipsychotic approved for the treatment of schizophrenia, bipolar mania, and acute agitation in schizophrenia. It functions primarily as a dopamine D2 and serotonin 5-HT2A receptor antagonist, with a uniquely high 5-HT2A/D2 affinity ratio [183,184,185,186]. This profile contributes to its efficacy in treating positive and negative symptoms, mood stabilization, and cognitive enhancement, while minimizing motor side effects and metabolic disturbances common with other atypical antipsychotics. Ziprasidone also inhibits serotonin and norepinephrine reuptake, adding antidepressant and anxiolytic properties. It exhibits low binding to H1, M1, and α1-adrenergic receptors, which reduces risks of sedation, orthostatic hypotension, and anticholinergic side effects [187]. Clinically, ziprasidone shows modest effectiveness compared to some peers like olanzapine but has a lower risk of weight gain and metabolic syndrome. Pharmacokinetically, oral bioavailability is ~60% when taken with food, and it is extensively metabolized in the liver, primarily by aldehyde oxidase and to a lesser extent by CYP3A4. It has a half-life of ~10 h and multiple active metabolites [188]. Studies suggest lower incidence of hyperglycemia and weight gain versus other SGAs. Though rare, ziprasidone can cause QT prolongation and, very rarely, DRESS syndrome. Clinical trials and real-world use support its favorable tolerability profile, particularly for patients concerned with weight and metabolic issues, attaining it a valuable option in long-term antipsychotic therapy [189,190,191,192]. Additionally, the demonstrated benzothiazole-based marketed drugs and clinically relevant scaffolds utilized in diverse fields are summarized briefly in the following Table 2.
Table 2.
Examples of clinically relevant benzothiazole scaffolds and their applications.
5. Privileged Scaffold and Hybrid Design
Benzothiazole has achieved the status of a “privileged structure”, defined by its high binding affinity across multiple biological receptors and its frequent appearance in active pharmaceutical ingredients [42]. This versatility enables the hybridization of the BZT core with other pharmacophores, such as indoles, pyrazoles, or thiazolidinediones, to produce multifunctional agents with enhanced activity and reduced toxicity. Hybrid molecules have shown synergistic effects, outperforming their components in terms of potency and selectivity. In this vein, computer-aided drug design (CADD), molecular docking, QSAR modeling, and pharmacophore mapping are increasingly utilized to accelerate the identification of benzothiazole-based leads with optimal ADMET (absorption, distribution, metabolism, excretion, and toxicity) profiles. This privileged core has been extensively optimized to generate derivatives with enhanced potency, selectivity, and pharmacokinetic profiles. Recent studies (2020–2025) have provided compelling in vitro and in vivo evidence for its therapeutic versatility. For instance, a recent report in 2023 by Cotman et al. demonstrates the discovery and structure-guided optimization of benzothiazole-based DNA gyrase inhibitors (Figure 11, compound 19) targeting multidrug-resistant Gram-negative pathogens, specifically Acinetobacter baumannii and Pseudomonas aeruginosa, both on the WHO’s critical priority list [193]. Starting from compound 19a, a benzyloxy-substituted benzothiazole with potent in vitro inhibition of bacterial DNA gyrase and topoisomerase IV (Topo IV), the team encountered limitations related to its poor aqueous solubility (6–12 μM), high lipophilicity (clogP = 5.8), and extremely low plasma free fraction (<0.1%). To address these issues, they introduced a methyl group at the benzylic position, yielding compound 19b, which demonstrated remarkable pharmacological improvement.
Figure 11.
Pharmacological relevance of recently reported benzothiazole scaffolds.
Compound 19b exhibited at least 10-fold lower IC50 values for bacterial Topo IV (notably 6.7 nM for A. baumannii) and a 10-fold increase in selectivity against human topoisomerase IIα (IC50 = 25,000 nM), alongside significantly enhanced solubility (>80 μM) and retained nanomolar potency against DNA gyrase. The compound also maintained excellent antibacterial activity, with MIC90 values as low as 2 μg/mL against over 60 multidrug-resistant clinical isolates and was effective even against novobiocin-resistant A. baumannii strains [193]. Structural studies (PDB codes 7PQM and 7PTG) confirmed the binding of compound 19b to the ATP-binding pocket of the GyrB subunit, forming key hydrogen bonds and cation–π interactions that rationalized its high affinity. Importantly, compound 19b exhibited no genotoxicity, mutagenicity, mitochondrial toxicity, or ion channel liabilities, and demonstrated low cytotoxicity in HepG2 cells (IC50 = 19.7 μM). These results show benzylic methylation as an appropriate strategy (Figure 11) for fine-tuning lipophilicity and enhancing pharmacokinetic and antibacterial profiles of benzothiazole-based GyrB inhibitors, aligning compound 19b as a promising preclinical candidate for Gram-negative bacterial infections [193].
Along similar lines, very recently, Ivanov et al. synthesized benzothiazole–profen hybrid amides such as compound 20a–c demonstrated notable structural and biological features relevant to anti-inflammatory drug design (Figure 11) [194]. Compound 20a, derived from ibuprofen, showed the least potent anti-inflammatory and antioxidant activities among the series; it had an IC50 of 159.94 µg/mL in the albumin denaturation assay, is significantly less effective than standard ibuprofen (IC50 = 76.05 µg/mL), and had the lowest lipophilicity (RM = 1.629), suggesting poor membrane interaction. In contrast, compound 20b, a carprofen hybrid, demonstrated superior biological activity. With an RM value of 1.755, 20b showed enhanced lipophilicity and a notable improvement in anti-inflammatory efficacy (IC50 = 65.26 µg/mL), surpassing ibuprofen. It also demonstrated moderate hydroxyl and hydrogen peroxide scavenging activity. A molecular docking analysis revealed strong binding affinities at human serum albumin (HSA) sites, particularly Sudlow II and the cleft site [194]. Meanwhile, compound 20c, incorporating flurbiprofen, emerged as one of the top candidates in terms of both biological activity and binding behavior. It exhibited an IC50 of 54.88 µg/mL in the inhibition of albumin denaturation and a respectable antioxidant profile. In docking studies, 20c (especially the S-enantiomer) showed the strongest binding at Sudlow site II (−12.3 kcal/mol) and the cleft site, forming hydrogen bonds and π–π interactions with key HSA residues such as Tyr411 and Tyr452. Molecular dynamics further confirmed the high stability of 20c-HSA complexes, supported by a low RMSD (0.15 nm) and a relatively high hydrogen bond count (0.53) in the cleft site. These findings show that aromatic-rich NSAID residues enhance both lipophilicity and biological activity, making them promising leads for anti-inflammatory drug development [194].
Recently (2024), the Tiwari group presented the design, synthesis, and evaluation of a novel benzothiazole–piperazine hybrid molecule 21 as a multitarget-directed ligand (MTDL) for Alzheimer’s disease (AD) [195]. Recognizing the multifactorial nature of AD pathogenesis, the researchers developed compound 21 to simultaneously target acetylcholinesterase (AChE) and amyloid–beta (Aβ1–42) aggregation. In silico studies, including molecular docking, molecular dynamics (MD) simulations, and MM/GBSA-free energy calculations, demonstrated strong and stable binding affinities of compound 21 to both AChE and Aβ1–42. The compound interacted effectively with the catalytic active site and peripheral anionic site of AChE, showing comparable binding free energies to standard drugs like donepezil. In vitro assays revealed that compound 21 is a potent and selective AChE inhibitor (IC50 = 0.42 μM) and suppresses Aβ aggregation more effectively than curcumin, as confirmed by Thioflavin-T assays, confocal microscopy, and TEM imaging (Figure 11). Neuroprotection studies in SH-SY5Y and Neuro2A cells showed significant rescue against H2O2 and okadaic-acid-induced toxicity. In vivo experiments in a scopolamine-induced memory impairment mouse model demonstrated that compound 21 markedly improved cognitive and memory functions in behavioral tests such as the elevated plus maze, social recognition, and passive avoidance. Histopathological studies further confirmed its neuroprotective effects in cortical and hippocampal tissues. Together, these findings establish compound 21 as a promising candidate for AD therapy, combining multitarget efficacy, favorable blood–brain barrier permeability, and neuroprotective properties [195].
Yun group reported the design, synthesis, and biological evaluation of a novel benzothiazole derivative 22 (Figure 11) as a promising antitumor agent for colorectal cancer (CRC). They demonstrated that compound 22 effectively inhibits CRC cell proliferation, migration, and invasion while inducing apoptosis via the mitochondrial intrinsic pathway (Figure 11) [196]. Mechanistic investigations revealed that compound 22 triggers the accumulation of reactive oxygen species (ROS), leading to mitochondrial membrane depolarization, cytochrome c release, and activation of proapoptotic proteins such as Bax, Bim, and Bad, while downregulating antiapoptotic proteins Bcl-2 and Bcl-x (Figure 12). This cascaded activates caspase-9 and caspase-3, culminating in apoptotic cell death. In vitro experiments, including MTT, EdU staining, flow cytometry, and wound-healing assays, confirmed the ability of compound 22 to suppress CRC cell growth and metastatic potential. RNA-seq and KEGG pathway enrichment analyses further supported apoptosis induction and disruption of cancer cell survival pathways. The in vivo studies using CT26 tumor-bearing mice showed that compound 22 significantly reduced tumor growth at tolerable doses without notable toxicity to major organs, as confirmed by histopathology and serum biochemical parameters. Immunohistochemistry analysis of tumor tissues corroborated decreased proliferation markers (Ki-67), reduced metastasis-related proteins (MMP2/MMP9), and increased cleaved caspase-3 expression. Overall, compound 22 exerts potent antitumor effects via the ROS-mediated mitochondrial apoptosis pathway and could serve as a safe and effective therapeutic candidate for CRC treatment (Figure 12).
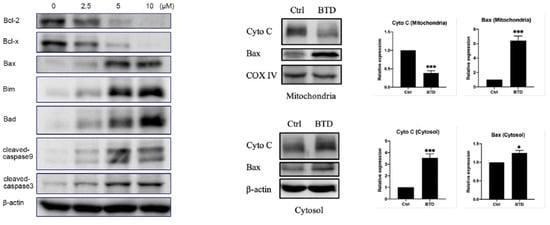
Figure 12.
Compound 22 (BTD) induced apoptosis via the mitochondrial apoptotic pathway. (* p < 0.05, ** p < 0.01, and *** p < 0.001); Reproduced with permission from Ref. [196]; copyright 2023, Frontiers.
Kumar’s group introduced (2023) the design, synthesis, and evaluation of a new series of benzothiazole derivatives bearing a 1,3,4-oxadiazole 23 moiety as promising antiepileptic agents. The pharmacophore was designed to include an aryl hydrophobic binding site, hydrogen bonding domains, and electron donor–acceptor systems critical for anticonvulsant activity (Figure 11) [197]. The in vivo antiepileptic activities were evaluated using maximal electroshock seizure (MES) and subcutaneous pentylenetetrazole (scPTZ) models in mice. Some of those derivatives showed significant protection at low doses (30 mg/kg) and prolonged duration of action, comparable or superior to standard drugs (phenytoin and phenobarbital). Neurotoxicity was assessed using a rotarod apparatus, and active compounds demonstrated minimal or no neurotoxicity. Additionally, selected compounds were evaluated for antidepressant activity, revealing negligible effects on locomotor activity, thus minimizing concerns for sedative side effects. In silico ADME predictions indicated good blood–brain barrier permeability and pharmacokinetic properties, while molecular docking studies revealed favorable binding interactions with NMDA (5IOV) and VGCC receptors, supporting their potential anticonvulsant mechanism. The structure–activity relationship (SAR) highlighted that electron-donating groups such as methoxy, nitro, and hydroxyl on the distal aryl ring enhanced activity by facilitating hydrogen bonding and lipophilicity. Finally, these benzothiazole-oxadiazole hybrids exhibit potent antiepileptic potential with significant pharmacological profiles, necessitating further development (Figure 11).
The Srour team documented (2025) a new series of benzothiazolecarbohydrazide sulfonate conjugates were synthesized and evaluated for their antiproliferative activity against three human cancer cell lines (MCF-7 (breast), HCT-116 (colon), and PC3 (prostate)) along with the normal BJ-1 cell line (Figure 11) [198]. Among these synthesized scaffolds, 24a and 24b demonstrated the most potent cytotoxic effects. Compound 24b showed strong activity against MCF-7 cells (IC50 = 78.8 μM), while 24a was notably effective against HCT-116 and PC3 cells (IC50 = 81.4 and 90.6 μM, respectively). Both compounds exhibited improved safety profiles and displaying low toxicity toward normal BJ-1 cells (Figure 11). Further mechanistic studies on compound 24b revealed that it effectively inhibited tubulin polymerization in MCF-7 cells, reducing microtubule protein levels from 632.9 to 210.3 pg/mL. Additionally, it significantly increased intracellular ROS, induced an 8.3-fold increase in DNA fragmentation, and caused G2/M phase arrest. Further, molecular docking analysis confirmed the strong binding of 24b to the colchicine-binding site of tubulin. These results suggest that compounds specifically 24b possess promising candidates as lead anticancer agents with selective cytotoxicity and a mechanism targeting tubulin dynamics (Figure 11).
The Abd El-Meguid team designed and synthesized a new series benzothiazole-based amino acid and ethyl ester derivatives 25 targeting dual inhibition of VEGFR-2 and EGFR, which are key kinases in breast and liver cancers [199]. Compounds such as carboxylic acids and ethyl esters exhibited the most potent cytotoxicity against MCF-7 (IC50 = 0.73–0.89 µM) and HepG-2 (IC50 = 2.54–2.80 µM), surpassing doxorubicin. Remarkably, ester derivatives were more effective against resistant MDA-MB-231 cells and showed superior EGFR inhibition (IC50 = 0.109–0.157 µM) compared to erlotinib (Figure 11). Along similar lines, some of these compounds also demonstrated strong VEGFR-2 inhibition (IC50 = 0.152–0.188 µM). QSAR analysis linked activity to molecular bulkiness and dipole distribution, while docking studies confirmed a type II kinase inhibitor binding mode. Overall, these derivatives are promising as dual-targeted anticancer agents with favorable potency and selectivity profiles.
In 2020, Mokhtar et al. reported that various kinds of benzothiazole-based derivatives were synthesized and evaluated for their in vitro antitumor potential against several cancer cell lines, including HepG2, HCT-116, MCF-7, PC-3, and HeLa (Figure 11) [200]. Among those series, some of them are demonstrated as notable cytotoxic activity with IC50 values comparable to the reference drug lapatinib. Of particular interest, hydrazone derivatives 26a (anthracenyl) and 26b (biphenyl) emerged as the most potent, exhibiting strong EGFR inhibition with IC50 values of 24.58 nM and 30.42 nM, respectively, that are closely similar lapatinib (17.38 nM). To better understand their mechanism, they conducted molecular docking studies revealing that compounds 26a and 26b engage critical interactions within the EGFR active site, notably forming hydrogen bonds with Lys745. These compounds were shown to be deeply embedded within the binding pocket, ensuring optimal fit and strong interactions. Surface mapping further confirmed their favorable occupation of the ATP-binding hydrophobic back pocket, supporting their potential as targeted EGFR inhibitors (Figure 11).
Rao and his team introduced a series of chalcone-conjugated imidazo [2,1-b]thiazole derivatives 27; these compounds were evaluated for their anticancer potential against a panel of human cancer cell lines, including PC-3 (prostate), A549 (lung), MCF-7 (breast), and A2780 (ovarian), using the MTT assay (Figure 11) [201]. Etoposide served as the reference standard and the entire library demonstrated a range of cytotoxic effects, with IC50 values spanning from 0.012 ± 0.0076 to 21.9 ± 8.24 µM, while etoposide exhibited IC50 values between 1.38 ± 0.56 and 3.08 ± 0.135 µM. Notably, trimethoxy derivative displayed superior activity as the most potent candidate across the tested cell lines.
Hamdi and co-workers reported various amino-benzothiazole derivatives as potential dual BRAF/VEGFR-2 inhibitors, inspired by the pharmacophoric elements of known multitarget agents. Among them, compounds 28a, 28b, 28c, and 28d emerged as the most active, exhibiting notable cytotoxicity against HepG2, HCT-116, and MCF-7 cell lines, with IC50 values ranging from 3.58 to 15.36 µM (Figure 11) [202]. These compounds also showed favorable selectivity, displaying significantly lower toxicity toward normal cells compared to sorafenib. Structure–activity relationship (SAR) analysis indicated that unsubstituted benzothiazole cores and incorporation of a hydrophobic sorafenib-like moiety enhanced anticancer activity. Especially, compound 28b demonstrated potent dual inhibition of BRAF and VEGFR-2, with IC50 values closely matching those of sorafenib. Further studies revealed that 28b induced cell cycle arrest at the S and G2-M phases and promoted apoptosis in MCF-7 cells. Molecular docking supported these findings, highlighting key interactions at both kinase binding sites, positioning compound 28b as a strong lead for further optimization (Figure 11).
Al-Sanea and co-workers documented three distinct series of 2-aminobenzothiazole-based hybrids incorporating thiazolidine-2,4-dione 29a–e, thiadiazole-aryl urea 30a–d, and cyanothiouracil 31a–d scaffolds were synthesized and structurally validated (Figure 4) [203]. Their anticancer potential was evaluated in vitro against HCT-116, HepG2, and MCF-7 cell lines using sorafenib as a reference (Figure 11). Among all candidates, compound 29a stood out with potent cytotoxic activity across all tested lines, achieving IC50 values of 5.61, 7.92, and 3.84 µM, respectively. Compounds 29e and 31a also showed significant antiproliferative effects, particularly against MCF-7 cells, with favorable safety profiles in normal WI-38 cells. Flow cytometry analysis revealed that 29a and 29e induced cell cycle arrest at the S phase, while 31a halted progression at the G1/S phase. In line with these findings, compound 29a exhibited strong VEGFR-2 inhibitory activity (IC50 = 91 nM), approaching the potency of sorafenib (53 nM). Molecular docking confirmed effective binding within the DFG region of VEGFR-2, while ADMET profiling suggested acceptable pharmacokinetic properties. In conclusion, these outcomes show compound 29a to be a promising lead for further optimization in anticancer drug development (Figure 11).
The Xue group demonstrated a series of flavonoid-based benzothiazole derivatives that were designed through structural optimization and evaluated for antifungal activity (Figure 11). Among them, compounds 32a, 32b, and 32c exhibited outstanding efficacy against Fusarium oxysporum f. sp. cucumerinum, with EC50 values of 1.2, 2.3, and 3.2 µg/mL, respectively, significantly outperforming the reference fungicide Kresoxim-methyl (EC50 = 37.8 µg/mL). Especially, 32c provided remarkable in vivo protection and therapeutic effects against cucumber fusarium wilt, achieving 82.4% and 60.5% efficacy at 200 µg/mL, compared to 60.2% and 48.6% with the standard treatment [10]. Mechanistic studies demonstrated that 32c disrupts fungal cell membranes, as evidenced by increased cytoplasmic leakage, elevated malondialdehyde levels, and compromised membrane integrity. Microscopic analyses, including SEM, confirmed structural damage to the fungal mycelia. Benzothiazole-linked flavonol derivatives, particularly 32c, hold great promise as next-generation eco-friendly antifungal agents (Figure 11) [10].
Aljuhani et al. employed click chemistry to synthesize a series of benzothiazole–1,2,3-triazole hybrids 33a–c bearing hydrazone/thiosemicarbazone linkers as potent EGFR inhibitors. These compounds exhibited strong submicromolar cytotoxicity against A549, MCF-7, and HCT-116 cancer cell lines (Figure 11) [204]. Among them, three compounds showed notable EGFR kinase inhibition (up to 98.5%) with IC50 values ranging from 0.69 to 4.82 μM. These triazole hybrids, containing benzothiazoles 33a–c, demonstrated superior safety toward normal fibroblasts (IC50 > 500 μM). In addition, molecular docking results confirmed a strong binding affinity to the EGFR active site, indicating their promise as selective and effective chemotherapeutic candidates (Figure 11).
Özdemir team reported that a series of thiadiazole–benzothiazole hybrids was synthesized and evaluated for their inhibitory effects on COX-1 and COX-2 using a colorimetric assay (Figure 11). Among them, compound 34 emerged as the most selective COX-1 inhibitor, showing 51.36% inhibition at 100 μM, while exhibiting minimal activity against COX-2 (11.05%) [205]. Molecular docking revealed that compound 34 forms key hydrogen bonds with Arg120 and Glu524 in the COX-1 active site, facilitated by its amide carbonyl group. Additionally, in silico ADME predictions suggested favorable oral bioavailability and drug-like properties. These results feature compound 34 as a promising lead scaffold for developing selective COX-1 inhibitors, potentially useful in conditions where targeted COX-1 inhibition is desirable (Figure 11).
Several benzothiazole derivatives bearing a 1,3,4-thiadiazole moiety were synthesized and evaluated for their ability to inhibit human monoamine oxidase enzymes (hMAO-A and hMAO-B), which are key targets in treating neurodegenerative and psychiatric disorders. Among the twelve synthesized compounds, compounds 35a and 35b emerged as the most potent and selective hMAO-A inhibitors, with IC50 values of 0.128 µM and 0.107 µM, respectively (Figure 11), surpassing the reference drug moclobemide [206]. Both compounds displayed no significant cytotoxicity in NIH3T3 cells. Molecular docking studies revealed strong interactions of compound 35b within the MAO-A active site, including hydrogen bonding and cation–π interactions, which contribute to its inhibitory potency. The structural features, particularly the presence of nitro groups and longer alkyl chains on the thiadiazole ring, appeared crucial for activity.
Carocci and colleagues (2023) designed and synthesized a new series of achiral benzothiazole–phthalimide hybrids, aiming to address the limitations of thalidomide. These compounds were evaluated for their anticancer, antimicrobial, and antioxidant properties. Among them, compound 36 emerged as the most promising candidate, showing potent cytotoxicity against MDA-MB-231 and MCF-7 breast cancer cells with IC50 values of 14.0 and 26.2 µM, respectively, and demonstrating significantly greater activity than thalidomide while exhibiting low toxicity toward non-malignant MCF-10A cells (Figure 11) [207]. Mechanistic studies revealed that compound 36 induced DNA damage, promoted apoptosis, and effectively suppressed cancer cell migration. Furthermore, 36 displayed notable antimicrobial activity against a broad spectrum of ESKAPE pathogens and Candida species, with MIC values ranging from 16 to 32 µg/mL. It also exhibited antioxidant potential by scavenging reactive oxygen species in fibroblast cells. Overall, compound 36 stands out as a strong lead for developing multifunctional agents targeting both cancer and associated opportunistic infections, warranting further preclinical exploration.
Boateng et al. (2023) [208] explores the development of benzothiazole-based analogues such as dopamine D4 receptor (D4R) modulators for the potential treatment of cocaine use disorder (CUD). Given the unique role of D4R in cognition, impulse control, and addiction, they designed two series of novel compounds using structure–activity relationship (SAR) strategies. From these, compound 37a emerged as a lead candidate, exhibiting high D4R binding affinity (Ki = 2.2 nM), strong selectivity over D2R and D3R, and low partial agonist activity (Figure 11). It was metabolically stable in liver microsomes and showed excellent brain penetration. The in vivo studies in rats demonstrated that compound 37a significantly reduced cocaine self-administration in a dose-dependent manner, supporting its efficacy in CUD models. Additionally, 37a showed minimal off-target effects, including weak antagonism at 5-HT2A and 5-HT2B receptors. Further SAR-guided design led to second-generation analogues with enhanced potency and selectivity, particularly compound 37b, which displayed improved D4R antagonist activity and no agonism across D2-like receptors. Overall, these results demonstrate that benzothiazole derivatives, especially 37a and 37b, are potential scaffolds for developing selective D4R-targeting therapeutics aimed at addressing CUD and related neuropsychiatric disorders [208].
Very recently, two 2-aminobenzothiazole derivatives based on isothiourea and guanidine 38a,b were evaluated for antidiabetic activity. Their molecular docking details disclosed strong binding to PPARγ (ΔG = −7.8 and −8.4 kcal/mol). Also, the in vivo studies in type 2 diabetes rats confirmed a significant glucose-lowering effect below 200 mg/dL and lipid-normalizing effects. The in silico studies supported strong ADMET profiles and DPP-IV/α-glucosidase inhibition. Both compounds 38a,b exhibited high safety (LD50 > 1750 mg/kg) and shown their potential as safe and effective PPARγ-targeted antidiabetic agents (Figure 11) [209].
Chen and Yang’s team (2024) reported that a series of twenty-five new benzothiazole derivatives was synthesized and structurally confirmed, aiming to identify compounds with dual anticancer and anti-inflammatory potential. Among them, compound 39 (6-chloro-N-(4-nitrobenzyl)benzo[d]thiazol-2-amine) demonstrated the most promising bioactivity. Compound 39 effectively inhibited the growth of A431, A549, and H1299 cancer cells and significantly suppressed the expression of inflammatory cytokines IL-6 and TNF-α (Figure 11) [210]. It also impaired cancer cell migration and induced apoptosis and cell cycle arrest at low micromolar concentrations, showing comparable effects to the reference compound such as trichloro benzothiazole derivative (Figure 11 and Figure 13). Mechanistic investigations revealed that 39 exerts its action by concurrently inhibiting the AKT and ERK signaling pathways, which are critical for tumor survival and proliferation. These findings suggest that 39 targets both tumor growth and the associated inflammatory microenvironment, offering a new therapeutic approach. Furthermore, ADMET predictions indicated favorable drug-like properties, reinforcing compound 39’s potential as a lead candidate for the development of dual-function anticancer agents [210].
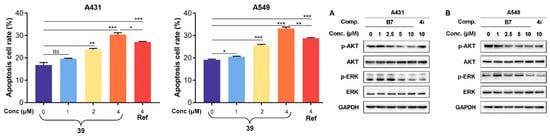
Figure 13.
Apoptosis of A431 and A549 cells treated with 39 and [210]. The protein expression levels of A431 and A549 cells are treated with 39 or [210]. (A) The protein expression levels of A431 cells treated with 39 [210] for 2 h; (B) the protein expression levels of A549 cells treated with 39 [210] for 2 h. Statistical significance differences (compared to control group) are defined as follows: p > 0.05 (not significant, ns), p ≤ 0.05 (*), p ≤ 0.01 (**) and p ≤ 0.001 (***). Reproduced with permission from Ref. [210]; copyright 2024, Frontiers.
Amoroso et al. [211] reported (2022) various phenylacetamide derivatives incorporating a benzothiazole scaffold and evaluated for antiproliferative activity against pancreatic as well as paraganglioma cancer cell lines. Most of the compounds demonstrated significant cytotoxicity at low micromolar concentrations; of these series, compound 40 emerged as the most effective, showing strong tumor selectivity and superior potency compared to the reference compound (Figure 11). Importantly, when combined with gemcitabine at submicromolar doses (0.1 µM gemcitabine and 0.5 µM of compound 40), a synergistic reduction in pancreatic cancer cell viability was observed, suggesting therapeutic potential for combination strategies. Computational target prediction further identified cannabinoid receptors (CBR1) and sentrin-specific proteases (SENP6) as likely contributors to the biological effects of 40. Docking studies confirmed favorable interactions between these targets and the synthesized compounds, indicating a promising mechanism of action. These outcomes feature the potential of benzothiazole-based phenylacetamides, particularly 40, as candidates for future anticancer therapies, warranting further biological validation and in vivo studies [211].
Moreover, several 2-arylbenzothiazole derivatives were synthesized as potential multi-angiokinase inhibitors, targeting VEGFR-2, FGFR-1, and PDGFR-β [212]. Among them, compounds based on nitro and methoxy group 41 exhibited potent enzyme inhibition with IC50 values as low as 0.13–0.19 µM for VEGFR-2 and 0.04–0.28 µM for other kinases. Compounds containing methoxy group demonstrated strong cytotoxicity against MCF-7 breast cancer cells (IC50 = 7.83 and 6.58 µM, respectively), along with significant VEGFR-2 inhibition in cell-based assays. Molecular docking confirmed type II kinase binding mode, and ADME predictions revealed favorable pharmacokinetic profiles, supporting further optimization for anticancer drug development (Figure 11) [212].
Very recently, numerous benzothiazole derivatives were synthesized to evaluate their potential as monoamine oxidase (MAO) inhibitors, targeting the treatment of neurodegenerative disorders such as Parkinson’s disease [213]. The compounds were assessed for their inhibitory activity against both MAO-A and MAO-B isoforms. Notably, compound 42b (bearing a 4-nitrobenzyl substituent) showed the strongest dual inhibition, with an IC50 of 0.336 µM for MAO-A and 0.0059 µM for MAO-B, indicating potent, broad-spectrum MAO inhibition. However, compounds 42a and 42c stood out as the most selective and potent MAO-B inhibitors, with exceptionally low IC50 values of 0.0028 µM, far surpassing the reference drug safinamide (Figure 11). Molecular docking supported these findings, revealing favorable interactions within the MAO active sites, particularly through π–π stacking and cavity-spanning orientations. Importantly, compounds like 42a and benzyl derivative also exhibited favorable physicochemical and pharmacokinetic properties, including blood–brain barrier permeability, making them strong candidates for central nervous system drug development [213].
Further, the Ahmed Kamal and Srihari’s team developed and evaluated pyrazolo–benzothiazole hybrids for anticancer and antiangiogenic activities. Among them, a halogenated derivative showed the most potent cytotoxicity across four cancer cell lines (PC-3, HT-29, A549, U87MG) with IC50 values of 3.17–6.77 µM, even superior to the reference axitinib drug [214]. It induced G0/G1 phase arrest, apoptosis, and increased ROS levels via mitochondrial dysfunction. The substituted halo compound inhibited VEGFR-2 kinase (IC50 = 97 nM) and reduced angiogenesis in zebrafish models. Molecular docking confirmed strong binding to VEGFR-2. Its high selectivity and multi-pathway effects highlight halogenated molecules as a promising lead for cancer therapy [214].
6. Conclusions
Benzothiazole and its derivatives represent a robust and versatile class of heterocyclic compounds, with extensive applications in both therapeutics and diagnostics. Their unique structural features, such as the fused benzene–thiazole ring, enable diverse chemical modifications, high target selectivity, and favorable pharmacokinetic properties. These characteristics have positioned the benzothiazole scaffold as a privileged structure in medicinal chemistry. Benzothiazole-based molecules exhibit a wide spectrum of biological activities, ranging from anticancer and antimicrobial effects to anti-inflammatory and neuroprotective actions. They have demonstrated efficacy across numerous therapeutic areas, including oncology, central nervous system (CNS) disorders, infectious diseases, and metabolic dysfunctions. Several compounds have reached clinical use, while others are under active investigation, underscoring their translational potential. As medicinal chemistry evolves, the continued integration of structure–activity relationship (SAR) studies, computational drug design, and hybrid molecule strategies will be critical in optimizing these scaffolds for next-generation drug development. The synergy between chemical innovation and biological insight will further unlock the potential of benzothiazole derivatives, enabling the discovery of safer, more effective, and precisely targeted therapeutic agents. Ultimately, benzothiazole continues to stand as a cornerstone in the ongoing quest for innovative healthcare solutions.
7. Challenges and Future Perspectives
Despite their promising biological profiles, some benzothiazole derivatives face limitations in pharmacokinetics, solubility, and toxicity, necessitating further optimization. Efforts are underway to improve bioavailability and minimize off-target effects through prodrug strategies, nanoformulations, and structural modifications. Importantly, the interdisciplinary integration of synthetic chemistry, computational tools, and biological assays is critical for overcoming these challenges. The continued advancement of synthetic methodologies, including metal-free catalysis, microwave-assisted synthesis, greener approaches, and one-pot tandem reactions, has facilitated the development of structurally diverse BZT analogues. These innovations, combined with a deeper understanding of biological mechanisms of action, are expected to drive next-generation drug discovery using a benzothiazole scaffold.
Funding
This research received no external funding.
Acknowledgments
SRC is grateful to the Brain Pool Program (2021–2023) funded by the Ministry of Science and ICT, South Korea, and Department of Pharmacology, Korea University College of Medicine, Korea University for providing financial support for the postdoctoral research.
Conflicts of Interest
The author declares no conflict of interest.
References
- Kabir, E.; Uzzaman, M. A review on biological and medicinal impact of heterocyclic compounds. Results Chem. 2022, 4, 100606. [Google Scholar] [CrossRef]
- Kotha, S.; Salman, M.; Cheekatla, S.R. Synthesis of Indenoindole Derivatives under Deep Eutectic Solvent Conditions. ChemistrySelect 2024, 9, e202402530. [Google Scholar] [CrossRef]
- Kotha, S.; Cheekatla, S.R.; Chinnam, A.K.; Jain, T. Design and synthesis of polycyclic bisindoles via Fischer indolization and ring-closing metathesis as key steps. Tetrahedron Lett. 2016, 57, 5605–5607. [Google Scholar] [CrossRef]
- Ebenezer, O.; Jordaan, M.A.; Carena, G.; Bono, T.; Shapi, M.; Tuszynski, J.A. An Overview of the Biological Evaluation of Selected Nitrogen-Containing Heterocycle Medicinal Chemistry Compounds. Int. J. Mol. Sci. 2022, 23, 8117. [Google Scholar] [CrossRef] [PubMed]
- Cheekatla, S.R.; Barik, D.; Anand, G.; Mol, K.M.R.; Porel, M. Indole-Based Macrocyclization by Metal-Catalyzed Approaches. Organics 2023, 4, 333–363. [Google Scholar] [CrossRef]
- Yu, B.; Yang, X. Why are heterocycles so special in medicinal chemistry? Chem. Biol. Drug Des. 2022, 100, 763–764. [Google Scholar] [CrossRef] [PubMed]
- Cheekatla, S.R.; Thurakkal, L.; Jose, A.; Barik, D.; Porel, M. Aza-Oxa-Triazole Based Macrocycles with Tunable Properties: Design, Synthesis, and Bioactivity. Molecules 2022, 27, 3409. [Google Scholar] [CrossRef] [PubMed]
- Kerru, N.; Gummidi, L.; Maddila, S.; Gangu, K.K.; Jonnalagadda, S.B. A Review on Recent Advances in Nitrogen-Containing Molecules and Their Biological Applications. Molecules 2020, 25, 1909. [Google Scholar] [CrossRef] [PubMed]
- Zou, Y.; Zhang, Y.; Liu, X.; Song, H.; Cai, Q.; Wang, S.; Yi, C.; Chen, J. Research Progress of Benzothiazole and Benzoxazole Derivatives in the Discovery of Agricultural Chemicals. Int. J. Mol. Sci. 2023, 24, 10807. [Google Scholar] [CrossRef] [PubMed]
- Tian, J.; Liu, Y.; Xin, H.; Hu, C.; Li, J.; Wang, Y.; Luo, X.; Qiu, Y.; Xue, W. Discovery of novel flavonoid derivatives containing benzothiazole as potential antifungal agents. Pest Manag. Sci. 2025, 81, 2288–2299. [Google Scholar] [CrossRef] [PubMed]
- Jyoti, B.; Smriti, K.; Swastika, S.; Archana, J. A Review on the Synthesis and Pharmacological Activity of Heterocyclic Compounds. Curr. Phys. Chem. 2023, 13, 2–19. [Google Scholar] [CrossRef]
- Taylor, A.P.; Robinson, R.P.; Fobian, Y.M.; Blakemore, D.C.; Jones, L.H.; Fadeyi, O. Modern advances in heterocyclic chemistry in drug discovery. Org. Biomol. Chem. 2016, 14, 6611–6637. [Google Scholar] [CrossRef] [PubMed]
- Chandra, A.; Yadav, S.C.; Cheekatla, S.R.; Kumar, A. A review on indole synthesis from nitroarenes: Classical to modern approaches. Org. Biomol. Chem. 2025, in press. [CrossRef] [PubMed]
- Kotha, S.; Salman, M.; Lal, S.; Rao Cheekatla, S.; Ansari, S. Design and Synthesis of Nitro Cage Heterocycles as Energetic Materials Derived from Pentacycloundecane (PCUD) Systems. Asian J. Org. Chem. 2023, 12, e202300060. [Google Scholar] [CrossRef]
- Kotha, S.; Cheekatla, S.R.; Fatma, A. Synthetic Approach to the ABCD Ring System of Anticancer Agent Fredericamycin A via Claisen Rearrangement and Ring-Closing Metathesis as Key Steps. ACS Omega 2019, 4, 17109–17116. [Google Scholar] [CrossRef] [PubMed]
- Kotha, S.; Cheekatla, S.R. Synthesis of Bisoxazole and Bromo-substituted Aryloxazoles. Molbank 2022, 2022, M1440. [Google Scholar] [CrossRef]
- Law, C.S.W.; Yeong, K.Y. Current trends of benzothiazoles in drug discovery: A patent review (2015–2020). Expert Opin. Ther. Pat. 2022, 32, 299–315. [Google Scholar] [CrossRef] [PubMed]
- Malik, S.; Miana, G.A.; Ata, A.; Kanwal, M.; Maqsood, S.; Malik, I.; Kazmi, Z. Synthesis, characterization, in-silico, and pharmacological evaluation of new 2-amino-6-trifluoromethoxy benzothiazole derivatives. Bioorg. Chem. 2023, 130, 106175. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Mishra, A.K. Advancement in Pharmacological Activities of Benzothiazole and its Derivatives: An Up-to-Date Review. Mini-Rev. Med. Chem. 2021, 21, 314–335. [Google Scholar] [CrossRef]
- Keri, R.S.; Patil, M.R.; Patil, S.A.; Budagumpi, S. A comprehensive review in current developments of benzothiazole-based molecules in medicinal chemistry. Eur. J. Med. Chem. 2015, 89, 207–251. [Google Scholar] [CrossRef] [PubMed]
- Colorado-Peralta, R.; Olivares-Romero, J.L.; Rosete-Luna, S.; García-Barradas, O.; Reyes-Márquez, V.; Hernández-Romero, D.; Morales-Morales, D. Copper-Coordinated Thiazoles and Benzothiazoles: A Perfect Alliance in the Search for Compounds with Antibacterial and Antifungal Activity. Inorganics 2023, 11, 185. [Google Scholar] [CrossRef]
- Yadav, R.; Meena, D.; Singh, K.; Tyagi, R.; Yadav, Y.; Sagar, R. Recent advances in the synthesis of new benzothiazole based anti-tubercular compounds. RSC Adv. 2023, 13, 21890–21925. [Google Scholar] [CrossRef] [PubMed]
- Haider, K.; Shrivastava, N.; Pathak, A.; Prasad Dewangan, R.; Yahya, S.; Shahar Yar, M. Recent advances and SAR study of 2-substituted benzothiazole scaffold based potent chemotherapeutic agents. Results Chem. 2022, 4, 100258. [Google Scholar] [CrossRef]
- Rubina, B.; Garima, K.; Dharam, P.P.; Asif, H.; Ravi, K.; Ruhi, A. A Mini Review on Recent Advancements in the Therapeutic Potentials of Benzothiazoles. Curr. Bioact. Compd. 2021, 17, 4–27. [Google Scholar] [CrossRef]
- Asiri, Y.I.; Alsayari, A.; Muhsinah, A.B.; Mabkhot, Y.N.; Hassan, M.Z. Benzothiazoles as potential antiviral agents. J. Pharm. Pharmacol. 2020, 72, 1459–1480. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Dubey, B. A Review on Emerging Benzothiazoles: Biological Aspects. J. Drug Deliv. Ther. 2022, 12 (Suppl. S4), 270–274. [Google Scholar] [CrossRef]
- Shanshan, L.; Yuan, X.; Qiancheng, S.; Xian, L.; Jing, L.; Yadong, C.; Tao, L.; Cheng, L.; Xiaomin, L.; Mingyue, Z.; et al. Non-Covalent Interactions with Aromatic Rings: Current Understanding and Implications for Rational Drug Design. Curr. Pharm. Des. 2013, 19, 6522–6533. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.C.; Alka, S.; Archana, S.; Harish, R.; Pathak, D.P. Medicinal significance of benzothiazole scaffold: An insight view. J. Enzym. Inhib. Med. Chem. 2013, 28, 240–266. [Google Scholar] [CrossRef] [PubMed]
- Kadu, V.D. Recent Advances for Synthesis of Benzothiazoles from Arylmethylamines. Asian J. Org. Chem. 2025, 14, e202400427. [Google Scholar] [CrossRef]
- Gao, X.; Liu, J.; Zuo, X.; Feng, X.; Gao, Y. Recent Advances in Synthesis of Benzothiazole Compounds Related to Green Chemistry. Molecules 2020, 25, 1675. [Google Scholar] [CrossRef] [PubMed]
- García-Báez, E.V.; Padilla-Martínez, I.I.; Tamay-Cach, F.; Cruz, A. Benzothiazoles from Condensation of o-Aminothiophenoles with Carboxylic Acids and Their Derivatives: A Review. Molecules 2021, 26, 6518. [Google Scholar] [CrossRef] [PubMed]
- Huang, G.; Cierpicki, T.; Grembecka, J. 2-Aminobenzothiazoles in anticancer drug design and discovery. Bioorg. Chem. 2023, 135, 106477. [Google Scholar] [CrossRef] [PubMed]
- Abbas, A.A.; Farghaly, T.A.; Dawood, K.M. Recent advances on anticancer and antimicrobial activities of directly-fluorinated five-membered heterocycles and their benzo-fused systems. RSC Adv. 2024, 14, 19752–19779. [Google Scholar] [CrossRef] [PubMed]
- Aayishamma, I.; Matada, G.S.P.; Pal, R.; Ghara, A.; Aishwarya, N.V.S.S.; Kumaraswamy, B.; Hosamani, K.R.; Manjushree, B.V.; Haripriya, E. Benzothiazole a privileged scaffold for Cutting-Edges anticancer agents: Exploring drug design, structure-activity relationship, and docking studies. Eur. J. Med. Chem. 2024, 279, 116831. [Google Scholar] [CrossRef]
- Nishad, R.K.; Singh, K.A.; Rahman, M.A. Synthesis and Pharmacological Activities of Benzothiazole Derivatives. Indian J. Pharm. Educ. Res. 2024, 58 (Suppl. S3), s704–s719. [Google Scholar] [CrossRef]
- Ammazzalorso, A.; Carradori, S.; Amoroso, R.; Fernández, I.F. 2-substituted benzothiazoles as antiproliferative agents: Novel insights on structure-activity relationships. Eur. J. Med. Chem. 2020, 207, 112762. [Google Scholar] [CrossRef] [PubMed]
- Brantley, E.; Trapani, V.; Alley, M.C.; Hose, C.D.; Bradshaw, T.D.; Stevens, M.F.G.; Sausville, E.A.; Stinson, S.F. Fluorinated 2-(4-amino-3-methylphenyl)Benzothiazoles Induce Cyp1a1 Expression, Become Metabolized, and Bind to Mac-romolecules in Sensitive Human Cancer Cells. Drug Metab. Dispos. 2004, 32, 1392–1401. [Google Scholar] [CrossRef] [PubMed]
- Bradshaw, T.D.; Westwell, A.D. The Development of the Antitumour Benzothiazole Prodrug, Phortress, as a Clinical Candidate. Curr. Med. Chem. 2004, 11, 1009–1021. [Google Scholar] [CrossRef] [PubMed]
- Rep Kaulić, V.; Racané, L.; Leventić, M.; Šubarić, D.; Rastija, V.; Glavaš-Obrovac, L.; Raić-Malić, S. Synthesis, Antiproliferative Evaluation and QSAR Analysis of Novel Halogen- and Amidino-Substituted Benzothiazoles and Benzimidazoles. Int. J. Mol. Sci. 2022, 23, 15843. [Google Scholar] [CrossRef] [PubMed]
- Al-Harthy, T.; Zoghaib, W.; Abdel-Jalil, R. Importance of Fluorine in Benzazole Compounds. Molecules 2020, 25, 4677. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.; Kim, J.; Yun, S.-M.; Lee, H.; Park, Y.; Hong, S.-S.; Hong, S. Discovery of New Benzothiazole-Based Inhibitors of Breakpoint Cluster Region-Abelson Kinase Including the T315I Mutant. J. Med. Chem. 2013, 56, 3531–3545. [Google Scholar] [CrossRef] [PubMed]
- Paoletti, N.; Supuran, C.T. Benzothiazole derivatives in the design of antitumor agents. Arch. Pharm. 2024, 357, 2400259. [Google Scholar] [CrossRef] [PubMed]
- Juthiga, V.V.K.V.; Boddapati, S.N.M.; Balha, M.; Tamminana, R. Comprehensive review on green methods: Synthesis of benzothiazoles. Results Chem. 2025, 15, 102212. [Google Scholar] [CrossRef]
- Shainyan, B.A.; Zhilitskaya, L.V.; Yarosh, N.O. Synthetic Approaches to Biologically Active C-2-Substituted Benzothiazoles. Molecules 2022, 27, 2598. [Google Scholar] [CrossRef] [PubMed]
- Teli, S.; Sethiya, A.; Agarwal, S. Pioneering Synthetic Strategies of 2-Substituted Benzothiazoles Using 2-Aminothiophenol. Chemistry 2024, 6, 165–206. [Google Scholar] [CrossRef]
- Yang, Z.-J.; Gong, Q.-T.; Yu, Y.; Lu, W.-F.; Wu, Z.-N.; Wang, N.; Yu, X.-Q. Fast and high-efficiency synthesis of 2-substituted benzothiazoles via combining enzyme catalysis and photoredox catalysis in one-pot. Bioorg. Chem. 2021, 107, 104607. [Google Scholar] [CrossRef] [PubMed]
- Taghizadeh, A.; Noroozi Pesyan, N. Nickel supported Fe3O4@PEG/methyl o-phenylenediamine nanosphere: An eco-friendly strategy for improved synthesis of benzothiazoles under sonication. Inorg. Chem. Commun. 2025, 175, 114127. [Google Scholar] [CrossRef]
- Bouchet, L.M.; Heredia, A.A.; Argüello, J.E.; Schmidt, L.C. Riboflavin as Photoredox Catalyst in the Cyclization of Thiobenzanilides: Synthesis of 2-Substituted Benzothiazoles. Org. Lett. 2020, 22, 610–614. [Google Scholar] [CrossRef] [PubMed]
- Maphupha, M.; Juma, W.P.; de Koning, C.B.; Brady, D. A modern and practical laccase-catalysed route suitable for the synthesis of 2-arylbenzimidazoles and 2-arylbenzothiazoles. RSC Adv. 2018, 8, 39496–39510. [Google Scholar] [CrossRef] [PubMed]
- Ahmad Wagay, S.; Hasan, A.; Ali, R. An efficient low melting mixture mediated green approach for the synthesis of 2-substituted benzothiazoles and benzimidazoles. Results Chem. 2022, 4, 100338. [Google Scholar] [CrossRef]
- Folgueiras-Amador, A.A.; Qian, X.-Y.; Xu, H.-C.; Wirth, T. Catalyst- and Supporting-Electrolyte-Free Electrosynthesis of Benzothiazoles and Thiazolopyridines in Continuous Flow. Chem. A Eur. J. 2018, 24, 487–491. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.-M.; Li, H.-X.; Young, D.J.; Zhu, D.-L.; Li, H.-Y.; Lang, J.-P. Exogenous Photosensitizer-, Metal-, and Base-Free Visible-Light-Promoted C–H Thiolation via Reverse Hydrogen Atom Transfer. Org. Lett. 2019, 21, 237–241. [Google Scholar] [CrossRef] [PubMed]
- Bhat, R.; Karhale, S.; Arde, S.; Helavi, V. Acacia concinna pod catalyzed synthesis of 2-arylbenzothia/(oxa)zole derivatives. Iran. J. Catal. 2024, 9, 173–179. [Google Scholar]
- Sharma, S.; Pathare, R.S.; Maurya, A.K.; Gopal, K.; Roy, T.K.; Sawant, D.M.; Pardasani, R.T. Ruthenium Catalyzed Intramolecular C–S Coupling Reactions: Synthetic Scope and Mechanistic Insight. Org. Lett. 2016, 18, 356–359. [Google Scholar] [CrossRef] [PubMed]
- Gao, M.-Y.; Li, J.-H.; Zhang, S.-B.; Chen, L.-J.; Li, Y.-S.; Dong, Z.-B. A Mild Synthesis of 2-Substituted Benzothiazoles via Nickel-Catalyzed Intramolecular Oxidative C–H Functionalization. J. Org. Chem. 2020, 85, 493–500. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Ding, Y.-L.; Liu, H.; Peng, S.; Ren, J.; Li, L. Copper-catalyzed multicomponent reactions of 2-iodoanilines, benzylamines, and elemental sulfur toward 2-arylbenzothiazoles. Tetrahedron Lett. 2014, 55, 945–949. [Google Scholar] [CrossRef]
- Yu, J.; Xia, Y.; Lu, M. Iron-Catalyzed Highly Efficient Aerobic Oxidative Synthesis of Benzimidazoles, Benzoxazoles, and Benzothiazoles Directly from Aromatic Primary Amines Under Solvent-Free Conditions in the Open Air. Synth. Commun. 2014, 44, 3019–3026. [Google Scholar] [CrossRef]
- Cheng, Y.; Peng, Q.; Fan, W.; Li, P. Room-Temperature Ligand-Free Pd/C-Catalyzed C–S Bond Formation: Synthesis of 2-Substituted Benzothiazoles. J. Org. Chem. 2014, 79, 5812–5819. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Yu, L.; Su, C.; Yang, Y.; Li, H.; Xu, Q. Efficient Generation of C–S Bonds via a By-Product-Promoted Selective Coupling of Alcohols, Organic Halides, and Thiourea. Adv. Synth. Catal. 2017, 359, 1649–1655. [Google Scholar] [CrossRef]
- Monga, A.; Bagchi, S.; Soni, R.K.; Sharma, A. Synthesis of Benzothiazoles via Photooxidative Decarboxylation of α-Keto Acids. Adv. Synth. Catal. 2020, 362, 2232–2237. [Google Scholar] [CrossRef]
- Yadav, K.P.; Rahman, M.A.; Nishad, S.; Maurya, S.K.; Anas, M.; Mujahid, M. Synthesis and biological activities of benzothiazole derivatives: A review. Intell. Pharm. 2023, 1, 122–132. [Google Scholar] [CrossRef]
- Hamad, H.T. Benzothiazole derivatives in cancer treatment: Synthesis and therapeutic potential: Review. MedMat 2025, 2, 17–32. [Google Scholar] [CrossRef]
- Kamal, A.; Hussaini, S.M.A.; Mohammed, S.M. Therapeutic potential of benzothiazoles: A patent review (2010–2014). Expert Opin. Ther. Pat. 2015, 25, 335–349. [Google Scholar] [CrossRef] [PubMed]
- Badgujar, N.D.; Dsouza, M.D.; Nagargoje, G.R.; Kadam, P.D.; Momin, K.I.; Bondge, A.S.; Panchgalle, S.P.; More, V.S. Recent Advances in Medicinal Chemistry with Benzothiazole-Based Compounds: An In-Depth Review. J. Chem. Rev. 2024, 6, 202–236. [Google Scholar] [CrossRef]
- Mountz, J.M.; Laymon, C.M.; Cohen, A.D.; Zhang, Z.; Price, J.C.; Boudhar, S.; McDade, E.; Aizenstein, H.J.; Klunk, W.E.; Mathis, C.A. Comparison of qualitative and quantitative imaging characteristics of [11C]PiB and [18F]flutemetamol in normal control and Alzheimer’s subjects. NeuroImage Clin. 2015, 9, 592–598. [Google Scholar] [CrossRef] [PubMed]
- Farrar, G.; Molinuevo, J.L.; Zanette, M. Is there a difference in regional read [18F]flutemetamol amyloid patterns between end-of-life subjects and those with amnestic mild cognitive impairment? Eur. J. Nucl. Med. Mol. Imaging 2019, 46, 1299–1308. [Google Scholar] [CrossRef] [PubMed]
- Buckley, C.J.; Sherwin, P.F.; Smith, A.P.L.; Wolber, J.; Weick, S.M.; Brooks, D.J. Validation of an electronic image reader training programme for interpretation of [18F]flutemetamol β-amyloid PET brain images. Nucl. Med. Commun. 2017, 38, 234–241. [Google Scholar] [CrossRef] [PubMed]
- Thal, D.R.; Beach, T.G.; Zanette, M.; Heurling, K.; Chakrabarty, A.; Ismail, A.; Smith, A.P.L.; Buckley, C. [18F]flutemetamol amyloid positron emission tomography in preclinical and symptomatic Alzheimer’s disease: Specific detection of advanced phases of amyloid-β pathology. Alzheimer’s Dement. 2015, 11, 975–985. [Google Scholar] [CrossRef] [PubMed]
- Beach, T.G.; Thal, D.R.; Zanette, M.; Smith, A.; Buckley, C. Detection of Striatal Amyloid Plaques with [18F]flutemetamol: Validation with Postmortem Histopathology. J. Alzheimer’s Dis. 2016, 52, 863–873. [Google Scholar] [CrossRef] [PubMed]
- Curtis, C.; Gamez, J.E.; Singh, U.; Sadowsky, C.H.; Villena, T.; Sabbagh, M.N.; Beach, T.G.; Duara, R.; Fleisher, A.S.; Frey, K.A.; et al. Phase 3 Trial of Flutemetamol Labeled with Radioactive Fluorine 18 Imaging and Neuritic Plaque Density. JAMA Neurol. 2015, 72, 287–294. [Google Scholar] [CrossRef] [PubMed]
- Corcia, P.; Nathalie, G.; Pierre-François, P.; Marie-Helene, S.; Annie, V.; Couratier, P. Treatment continuity of amyotrophic lateral sclerosis with available riluzole formulations: State of the art and current challenges in a ‘real-world’ setting. Amyotroph. Lateral Scler. Front. Degener. 2025, 26, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Saitoh, Y.; Takahashi, Y. Riluzole for the Treatment of Amyotrophic Lateral Sclerosis. Neurodegener. Dis. Manag. 2020, 10, 343–355. [Google Scholar] [CrossRef] [PubMed]
- Pandya, R.S.; Zhu, H.; Li, W.; Bowser, R.; Friedlander, R.M.; Wang, X. Therapeutic neuroprotective agents for amyotrophic lateral sclerosis. Cell. Mol. Life Sci. 2013, 70, 4729–4745. [Google Scholar] [CrossRef] [PubMed]
- Wagner, M.L.; Landis, B.E. Riluzole: A New Agent for Amyotrophic Lateral Sclerosis. Ann. Pharmacother. 1997, 31, 738–744. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Xin, L.; Pengjiao, A.; Zhang, B. Real-world safety profile of riluzole: A systematic analysis of data from the FAERS database and case reports. Expert Opin. Drug Saf. 2023, 22, 967–974. [Google Scholar] [CrossRef] [PubMed]
- Bissaro, M.; Federico, S.; Salmaso, V.; Sturlese, M.; Spalluto, G.; Moro, S. Targeting Protein Kinase CK1δ with Riluzole: Could It Be One of the Possible Missing Bricks to Interpret Its Effect in the Treatment of ALS from a Molecular Point of View? ChemMedChem 2018, 13, 2601–2605. [Google Scholar] [CrossRef] [PubMed]
- Cheah, B.C.; Vucic, S.; Krishnan, A.V.; Kiernan, M.C. Riluzole, Neuroprotection and Amyotrophic Lateral Sclerosis. Curr. Med. Chem. 2010, 17, 1942–1959. [Google Scholar] [CrossRef] [PubMed]
- Wymer, J.; Apple, S.; Harrison, A.; Hill, B.A. Pharmacokinetics, Bioavailability, and Swallowing Safety with Riluzole Oral Film. Clin. Pharmacol. Drug Dev. 2023, 12, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Kakuta, T.; Hirata, H.; Soda, S.; Shiobara, T.; Watanabe, M.; Tatewaki, M.; Fukushima, F.; Chibana, K.; Sugiyama, K.; Arima, M.; et al. Riluzole-induced Lung Injury in Two Patients with Amyotrophic Lateral Sclerosis. Intern. Med. 2012, 51, 1903–1907. [Google Scholar] [CrossRef] [PubMed]
- Holloway, R.G.; Biglan, K.M. A review of pramipexole and its clinical utility in Parkinson’s disease. Expert Opin. Pharmacother. 2002, 3, 197–210. [Google Scholar] [CrossRef] [PubMed]
- Wilson, S.M.; Wurst, M.G.; Whatley, M.F.; Daniels, R.N. Classics in Chemical Neuroscience: Pramipexole. ACS Chem. Neurosci. 2020, 11, 2506–2512. [Google Scholar] [CrossRef] [PubMed]
- Antonini, A.; Calandrella, D. Pharmacokinetic evaluation of pramipexole. Expert Opin. Drug Metab. Toxicol. 2011, 7, 1307–1314. [Google Scholar] [CrossRef] [PubMed]
- Varga, L.I.; Ako-Agugua, N.; Colasante, J.; Hertweck, L.; Houser, T.; Smith, J.; Watty, A.A.; Nagar, S.; Raffa, R.B. Critical review of ropinirole and pramipexole—Putative dopamine D3-receptor selective agonists—For the treatment of RLS. J. Clin. Pharm. Ther. 2009, 34, 493–505. [Google Scholar] [CrossRef] [PubMed]
- Bozik, M.E.; Mather, J.L.; Kramer, W.G.; Gribkoff, V.K.; Ingersoll, E.W. Safety, Tolerability, and Pharmacokinetics of KNS-760704 (Dexpramipexole) in Healthy Adult Subjects. J. Clin. Pharmacol. 2011, 51, 1177–1185. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui, S.; Bozik, M.; Archibald, D.; Dworetzky, S.; Mather, J.; Killingsworth, R.; Ochkur, S.; Jacobsen, E.; Panettieri, R.; Prussin, C. Late Breaking Abstract—Phase 2 trial evaluating the effects of dexpramipexole on blood eosinophils, lung function, and airway biomarkers in eosinophilic asthma. Eur. Respir. J. 2021, 58 (Suppl. S65), RCT2900. [Google Scholar] [CrossRef]
- Panch, S.R.; Bozik, M.E.; Brown, T.; Makiya, M.; Prussin, C.; Archibald, D.G.; Hebrank, G.T.; Sullivan, M.; Sun, X.; Wetzler, L.; et al. Dexpramipexole as an oral steroid-sparing agent in hypereosinophilic syndromes. Blood 2018, 132, 501–509. [Google Scholar] [CrossRef] [PubMed]
- Dworetzky, S.I.; Hebrank, G.T.; Archibald, D.G.; Reynolds, I.J.; Farwell, W.; Bozik, M.E. The targeted eosinophil-lowering effects of dexpramipexole in clinical studies. Blood Cells Mol. Dis. 2017, 63, 62–65. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui, S.; Wenzel, S.E.; Bozik, M.E.; Archibald, D.G.; Dworetzky, S.I.; Mather, J.L.; Killingsworth, R.; Ghearing, N.; Schwartz, J.T.; Ochkur, S.I.; et al. Safety and Efficacy of Dexpramipexole in Eosinophilic Asthma (EXHALE): A randomized controlled trial. J. Allergy Clin. Immunol. 2023, 152, 1121–1130.e1110. [Google Scholar] [CrossRef] [PubMed]
- Buonvicino, D.; Ranieri, G.; Pratesi, S.; Gerace, E.; Muzzi, M.; Guasti, D.; Tofani, L.; Chiarugi, A. Neuroprotection induced by dexpramipexole delays disease progression in a mouse model of progressive multiple sclerosis. Br. J. Pharmacol. 2020, 177, 3342–3356. [Google Scholar] [CrossRef] [PubMed]
- Coppi, E.; Buonvicino, D.; Ranieri, G.; Cherchi, F.; Venturini, M.; Pugliese, A.M.; Chiarugi, A. Dexpramipexole Enhances K+ Currents and Inhibits Cell Excitability in the Rat Hippocampus In Vitro. Mol. Neurobiol. 2021, 58, 2955–2962. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Fu, Q.; Ruan, J.; Shi, C.; Lu, W.; Wu, J.; Zhou, Z. Dexpramipexole ameliorates cognitive deficits in sepsis-associated encephalopathy through suppressing mitochondria-mediated pyroptosis and apoptosis. NeuroReport 2023, 34, 220–231. [Google Scholar] [CrossRef] [PubMed]
- Mahmood, A.A.R.; Al-Juboori, S.B. A Review: Saccharin Discovery, Synthesis, and Applications. Ibn Al-Haitham J. Pure Appl. Sci. 2020, 33, 43–61. [Google Scholar] [CrossRef]
- Castle, L.; Andreassen, M.; Aquilina, G.; Bastos, M.L.; Boon, P.; Fallico, B.; FitzGerald, R.; Frutos Fernandez, M.J.; Grasl-Kraupp, B.; Gundert-Remy, U. Re-evaluation of saccharin and its sodium, potassium and calcium salts (E 954) as food additives. EFSA J. 2024, 22, e9044. [Google Scholar] [CrossRef] [PubMed]
- Cohen, S.M. Saccharin: Past, present, and future. J. Am. Diet. Assoc. 1986, 86, 929–931. [Google Scholar] [CrossRef] [PubMed]
- Cortes, J. Quizartinib: A potent and selective FLT3 inhibitor for the treatment of patients with FLT3-ITD–positive AML. J. Hematol. Oncol. 2024, 17, 111. [Google Scholar] [CrossRef] [PubMed]
- Larrosa-Garcia, M.; Baer, M.R. FLT3 Inhibitors in Acute Myeloid Leukemia: Current Status and Future Directions. Mol. Cancer Ther. 2017, 16, 991–1001. [Google Scholar] [CrossRef] [PubMed]
- Moallem, F.E.; Sadra, G.C.M.; Alsadat, D.P.; Eftekhar, A.; Mehregan, S.; Sadat, D.M.; Karimi, M.A. Quizartinib: A new hope in acute myeloid leukemia, an applied comprehensive review. Future Oncol. 2024, 20, 2791–2810. [Google Scholar] [CrossRef] [PubMed]
- Levis, M.J.; Cortes, J.E.; Gammon, G.M.; Trone, D.; Kang, D.; Li, J. Laboratory and Clinical Investigations to Identify the Optimal Dosing Strategy for Quizartinib (AC220) Monotherapy in FLT3-Itd-Positive (+) Relapsed/Refractory (R/R) Acute Myeloid Leukemia (AML). Blood 2016, 128, 4042. [Google Scholar] [CrossRef]
- Erba, H.; Montesinos, P.; Vrhovac, R.; Patkowska, E.; Kim, H.J.; Zak, P.; Wang, P.N.; Mitov, T.; Hanyok, J.; Liu, L.; et al. S100: Quizartinib Prolonged Survival vs Placebo Plus Intensive Induction and Consolidation Therapy Followed by Single-Agent Continuation in Patients Aged 18–75 Years with Newly Diagnosed FLT3-ITD+ AML. HemaSphere 2022, 6, 1–2. [Google Scholar] [CrossRef]
- Qi, J.; Choi, I.; Ota, S.; Ichikawa, S.; Fujishima, N.; Iida, H.; Sugiura, I.; Sugiura, K.; Murata, Y.; Inoue, H.; et al. Safety and Pharmacokinetics of Quizartinib Combination Therapy With Standard Induction and Consolidation Chemotherapy in Patients with Newly Diagnosed Acute Myeloid Leukemia: Results from Two Phase 1 Trials in Japan and China. Clin. Pharmacol. Drug Dev. 2024, 13, 560–571. [Google Scholar] [CrossRef] [PubMed]
- Kay, D.R.; Valentine, T.V.; Walker, S.E.; Valentine, M.H.; Bole, G.G. Frentizole Therapy of Active Systemic lupus Erythematosus. Arthritis Rheum. 1980, 23, 1381–1387. [Google Scholar] [CrossRef] [PubMed]
- Scheetz, M.I.I.; Carlson, D.G.; Schinitsky, M.R. Frentizole, a novel immunosuppressive, and azathioprine: Their comparative effects on host resistance to Pseudomonas aeruginosa, Candida albicans, herpes simplex virus, and influenza (Ann Arbor) virus. Infect. Immun. 1977, 15, 145–148. [Google Scholar] [CrossRef] [PubMed]
- Walker, S.E.; Solsky, M.; Schnitzer, B. Prolonged lifespans in female nzb/nzw mice treated with the experimental immunoregulatory drug frentizole. Arthritis Rheum. 1982, 25, 1291–1297. [Google Scholar] [CrossRef] [PubMed]
- Meisel, A.D.; Maurice, B.; Ellen, G.; Diamond, H.S. Effect of Frentizole on Mitogen-Induced Blastogenesis in Human Lymphocytes. J. Immunopharmacol. 1979, 1, 483–495. [Google Scholar] [CrossRef] [PubMed]
- Shou, L.; Schwartz, S.A.; Good, R.A. Suppressor cell activity after concanavalin A treatment of lymphocytes from normal donors. J. Exp. Med. 1976, 143, 1100–1110. [Google Scholar] [CrossRef] [PubMed]
- Hatfield, S.M.; Hartley, L.W.; Schmidtke, J.R. The immunomodulatory action of frentizole, a novel immunosuppressive agent. Immunopharmacology 1982, 5, 169–179. [Google Scholar] [CrossRef] [PubMed]
- Hroch, L.; Guest, P.; Benek, O.; Soukup, O.; Janockova, J.; Dolezal, R.; Kuca, K.; Aitken, L.; Smith, T.K.; Gunn-Moore, F.; et al. Synthesis and evaluation of frentizole-based indolyl thiourea analogues as MAO/ABAD inhibitors for Alzheimer’s disease treatment. Biorg. Med. Chem. 2017, 25, 1143–1152. [Google Scholar] [CrossRef] [PubMed]
- Hroch, L.; Benek, O.; Guest, P.; Aitken, L.; Soukup, O.; Janockova, J.; Musil, K.; Dohnal, V.; Dolezal, R.; Kuca, K.; et al. Design, synthesis and in vitro evaluation of benzothiazole-based ureas as potential ABAD/17β-HSD10 modulators for Alzheimer’s disease treatment. Bioorganic Med. Chem. Lett. 2016, 26, 3675–3678. [Google Scholar] [CrossRef] [PubMed]
- Morsy, A.; Trippier, P.C. Amyloid-Binding Alcohol Dehydrogenase (ABAD) Inhibitors for the Treatment of Alzheimer’s Disease. J. Med. Chem. 2019, 62, 4252–4264. [Google Scholar] [CrossRef] [PubMed]
- Ramos, S.; Vicente-Blázquez, A.; López-Rubio, M.; Gallego-Yerga, L.; Álvarez, R.; Peláez, R. Frentizole, a Nontoxic Immunosuppressive Drug, and Its Analogs Display Antitumor Activity via Tubulin Inhibition. Int. J. Mol. Sci. 2023, 24, 17474. [Google Scholar] [CrossRef] [PubMed]
- Chrienova, Z.; Rysanek, D.; Novak, J.; Vasicova, P.; Oleksak, P.; Andrys, R.; Skarka, A.; Dumanovic, J.; Milovanovic, Z.; Jacevic, V.; et al. Frentizole derivatives with mTOR inhibiting and senomorphic properties. Biomed. Pharmacother. 2023, 167, 115600. [Google Scholar] [CrossRef] [PubMed]
- Ondrej, B.; Lukas, H.; Laura, A.; Rafael, D.; Patrick, G.; Marketa, B.; Ondrej, S.; Karel, M.; Kamil, K.; Terry, K.S.; et al. 6-Benzothiazolyl Ureas, Thioureas and Guanidines are Potent Inhibitors of ABAD/17&β-HSD10 and Potential Drugs for Alzheimer&s Disease Treatment: Design, Synthesis and in vitro Evaluation. Med. Chem. 2017, 13, 345–358. [Google Scholar] [CrossRef]
- Jekabsone, A.; Jankeviciute, S.; Pampuscenko, K.; Borutaite, V.; Morkuniene, R. The Role of Intracellular Ca2+ and Mitochondrial ROS in Small Aβ1-42 Oligomer-Induced Microglial Death. Int. J. Mol. Sci. 2023, 24, 12315. [Google Scholar] [CrossRef] [PubMed]
- Arad, E.; Green, H.; Jelinek, R.; Rapaport, H. Revisiting thioflavin T (ThT) fluorescence as a marker of protein fibrillation—The prominent role of electrostatic interactions. J. Colloid Interface Sci. 2020, 573, 87–95. [Google Scholar] [CrossRef] [PubMed]
- Xue, C.; Lin, T.Y.; Chang, D.; Guo, Z. Thioflavin T as an amyloid dye: Fibril quantification, optimal concentration and effect on aggregation. R. Soc. Open Sci. 2017, 4, 160696. [Google Scholar] [CrossRef] [PubMed]
- Stefansson, S.; Adams, D.L.; Tang, C.M. Common Benzothiazole and Benzoxazole Fluorescent DNA Intercalators for Studying Alzheimer Aβ1-42 and Prion Amyloid Peptides. BioTechniques 2012, 52, 290. [Google Scholar] [CrossRef] [PubMed]
- Hudson, S.A.; Ecroyd, H.; Kee, T.W.; Carver, J.A. The thioflavin T fluorescence assay for amyloid fibril detection can be biased by the presence of exogenous compounds. FEBS J. 2009, 276, 5960–5972. [Google Scholar] [CrossRef] [PubMed]
- Sulatsky, M.I.; Sulatskaya, A.I.; Povarova, O.I.; Antifeeva, I.A.; Kuznetsova, I.M.; Turoverov, K.K. Effect of the fluorescent probes ThT and ANS on the mature amyloid fibrils. Prion 2020, 14, 67–75. [Google Scholar] [CrossRef] [PubMed]
- Sulatskaya, A.I.; Rodina, N.P.; Sulatsky, M.I.; Povarova, O.I.; Antifeeva, I.A.; Kuznetsova, I.M.; Turoverov, K.K. Investigation of α-Synuclein Amyloid Fibrils Using the Fluorescent Probe Thioflavin, T. Int. J. Mol. Sci. 2018, 19, 2486. [Google Scholar] [CrossRef] [PubMed]
- Dogan, A. An Easier and More Sensitive Method for Amyloid Detection. In Amyloid and Related Disorders: Surgical Pathology and Clinical Correlations; Picken, M.M., Herrera, G.A., Dogan, A., Eds.; Humana Press: Totowa, NJ, USA, 2012; pp. 187–189. [Google Scholar]
- MacKeigan, T.P.; Morgan, M.L.; Stys, P.K. Quantitation of Tissue Amyloid via Fluorescence Spectroscopy Using Controlled Concentrations of Thioflavin-S. Molecules 2023, 28, 4483. [Google Scholar] [CrossRef] [PubMed]
- Girych, M.; Gorbenko, G.; Maliyov, I.; Trusova, V.; Mizuguchi, C.; Saito, H.; Kinnunen, P. Combined thioflavin T–Congo red fluorescence assay for amyloid fibril detection. Methods Appl. Fluoresc. 2016, 4, 034010. [Google Scholar] [CrossRef] [PubMed]
- Gade Malmos, K.; Blancas-Mejia, L.M.; Weber, B.; Buchner, J.; Ramirez-Alvarado, M.; Naiki, H.; Otzen, D. ThT 101: A primer on the use of thioflavin T to investigate amyloid formation. Amyloid 2017, 24, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Groenning, M. Binding mode of Thioflavin T and other molecular probes in the context of amyloid fibrils—Current status. J. Chem. Biol. 2010, 3, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Bradshaw, T.D.; Stevens, M.F.G.; Calvert, H.; Plummer, E.R. Abstract B59: Preliminary clinical experiences of Phortress: Putative role for c-MET inhibition in antitumor activity. Mol. Cancer Ther. 2009, 8 (Suppl. S12), B59. [Google Scholar] [CrossRef]
- Loaiza Perez, A.; Bradshaw, T. Exploring New Molecular Targets in Advanced Ovarian Cancer: The Aryl Hydrocarbon Receptor (AhR) and Antitumor Benzothiazole Ligands as Potential Therapeutic Candidates. In Current Trends in Cancer Management; Streba, L., Gheonea, D.I., Schenker, M., Eds.; IntechOpen: London, UK, 2018. [Google Scholar]
- Bradshaw, T.D.; Wren, J.E.; Bruce, M.; Barrett, D.A.; Leong, C.O.; Gaskell, M.; Wright, E.K.; Farmer, P.B.; Henderson, C.J.; Wolf, R.; et al. Preclinical Toxicokinetic Evaluation of Phortress [2-(4-Amino-3-Methylphenyl)-5-Fluorobenzothiazole Lysylamide Dihydrochloride] in Two Rodent Species. Pharmacology 2008, 83, 99–109. [Google Scholar] [CrossRef] [PubMed]
- Leong, C.O.; Gaskell, M.; Martin, E.A.; Heydon, R.T.; Farmer, P.B.; Bibby, M.C.; Cooper, P.A.; Double, J.A.; Bradshaw, T.D.; Stevens, M.F.G. Antitumour 2-(4-aminophenyl)benzothiazoles generate DNA adducts in sensitive tumour cells in vitro and in vivo. Br. J. Cancer 2003, 88, 470–477. [Google Scholar] [CrossRef] [PubMed]
- Hutchinson, I.; Jennings, S.A.; Vishnuvajjala, B.R.; Westwell, A.D.; Stevens, M.F.G. Antitumor Benzothiazoles. 16. Synthesis and Pharmaceutical Properties of Antitumor 2-(4-Aminophenyl)benzothiazole Amino Acid Prodrugs. J. Med. Chem. 2002, 45, 744–747. [Google Scholar] [CrossRef] [PubMed]
- Leong, C.O.; Suggitt, M.; Swaine, D.J.; Bibby, M.C.; Stevens, M.F.G.; Bradshaw, T.D. In vitro, in vivo, and in silico analyses of the antitumor activity of 2-(4-amino-3-methylphenyl)-5-fluorobenzothiazoles. Mol. Cancer Ther. 2005, 3, 1565–1575. [Google Scholar] [CrossRef]
- Bradshaw, T.D.; Mortimer, C.G.; Westwell, A.D. Update to: The Aryl Hydrocarbon Receptor in Anticancer Drug Discovery: Friend or Foe? Med. Chem. Rev. Online 2005, 2, 153–161. [Google Scholar] [CrossRef]
- Stone, E.L.; Citossi, F.; Singh, R.; Kaur, B.; Gaskell, M.; Farmer, P.B.; Monks, A.; Hose, C.; Stevens, M.F.G.; Leong, C.-O.; et al. Antitumour benzothiazoles. Part 32: DNA adducts and double strand breaks correlate with activity; synthesis of 5F203 hydrogels for local delivery. Biorg. Med. Chem. 2015, 23, 6891–6899. [Google Scholar] [CrossRef] [PubMed]
- Newman-Tancredi, A. The importance of 5-HT1A receptor agonism in antipsychotic drug action: Rationale and perspectives. Curr. Opin. Investig. Drugs 2010, 11, 802–812. [Google Scholar] [PubMed]
- Takekita, Y.; Kato, M.; Wakeno, M.; Sakai, S.; Suwa, A.; Nishida, K.; Okugawa, G.; Kinoshita, T. A 12-week randomized, open-label study of perospirone versus aripiprazole in the treatment of Japanese schizophrenia patients. Prog. Neuro Psychopharmacol. Biol. Psychiatry 2013, 40, 110–114. [Google Scholar] [CrossRef] [PubMed]
- Shiwa, T.; Amano, T.; Matsubayashi, H.; Seki, T.; Sasa, M.; Sakai, N. Perospirone, a Novel Antipsychotic Agent, Hyperpolarizes Rat Dorsal Raphe Neurons via 5-HT1A Receptor. J. Pharmacol. Sci. 2003, 93, 114–117. [Google Scholar] [CrossRef] [PubMed]
- Sumiyoshi, T.; Higuchi, Y.; Itoh, T.; Matsui, M.; Arai, H.; Suzuki, M.; Kurachi, M.; Sumiyoshi, C.; Kawasaki, Y. Effect of perospirone on P300 electrophysiological activity and social cognition in schizophrenia: A three-dimensional analysis with sLORETA. Psychiatry Res. Neuroimaging 2009, 172, 180–183. [Google Scholar] [CrossRef] [PubMed]
- Yoshino, T.; Nisijima, K.; Shioda, K.; Yui, K.; Katoh, S. Perospirone, a novel atypical antipsychotic drug, potentiates fluoxetine-induced increases in dopamine levels via multireceptor actions in the rat medial prefrontal cortex. Neurosci. Lett. 2004, 364, 16–21. [Google Scholar] [CrossRef] [PubMed]
- SM 9018. Perospirone. Drugs R&D 1999, 2, 142–143. [Google Scholar] [CrossRef] [PubMed]
- Hongjing, Y.; Kumatoshi, I.; Hiroaki, M.; Taku, A.; Masashi, S. Effects of Perospirone, a Novel Antipsychotic Agent, on the Dopaminergic Neurons in the Rat Ventral Tegmental Area. Jpn. J. Pharmacol. 1997, 75, 179–185. [Google Scholar] [CrossRef]
- Matsushita, M.; Egashira, N.; Harada, S.; Okuno, R.; Mishima, K.; Iwasaki, K.; Nishimura, R.; Fujiwara, M. Perospirone, a Novel Antipsychotic Drug, Inhibits Marble-Burying Behavior via 5-HT1A Receptor in Mice: Implications for Obsessive-Compulsive Disorder. J. Pharmacol. Sci. 2005, 99, 154–159. [Google Scholar] [CrossRef] [PubMed]
- Ohno, Y.; Ishida-Tokuda, K.; Ishibashi, T.; Sakamoto, H.; Tagashira, R.; Horisawa, T.; Yabuuti, K.; Matsumoto, K.; Kawabe, A.; Nakamura, M. Potential role of 5-HT2 and D2 receptor interaction in the atypical antipsychotic action of the novel succimide derivative, perospirone. Pol. J. Pharmacol. 1997, 49, 213–219. [Google Scholar] [PubMed]
- Ishida-ToKuda, K.; Ohno, Y.; Sakamoto, H.; Ishibashi, T.; Wakabayashi, J.; Tojima, R.; Morita, T.; Nakamura, M. Evaluation of Perospirone (SM-9018), a Novel Serotonin-2 and Dopamine-2 Receptor Antagonist, and Other Antipsychotics in the Conditioned Fear Stress-Induced Freezing Behavior Model in Rats. Jpn. J. Pharmacol. 1996, 72, 119–126. [Google Scholar] [CrossRef] [PubMed]
- Tsujino, N.; Nemoto, T.; Morita, K.; Katagiri, N.; Ito, S.; Mizuno, M. Long-term Efficacy and Tolerability of Perospirone for Young Help-seeking People at Clinical High Risk: A Preliminary Open Trial. Clin. Psychopharmacol. Neurosci. 2013, 11, 132–136. [Google Scholar] [CrossRef] [PubMed]
- Caccia, S. Pharmacokinetics and metabolism update for some recent antipsychotics. Expert Opin. Drug Metab. Toxicol. 2011, 7, 829–846. [Google Scholar] [CrossRef] [PubMed]
- Araki, T.; Yamasue, H.; Sumiyoshi, T.; Kuwabara, H.; Suga, M.; Iwanami, A.; Kato, N.; Kasai, K. Perospirone in the treatment of schizophrenia: Effect on verbal memory organization. Prog. Neuro Psychopharmacol. Biol. Psychiatry 2006, 30, 204–208. [Google Scholar] [CrossRef] [PubMed]
- Worrel, J.A.; Marken, P.A.; Beckman, S.E.; Ruehter, V.L. Atypical antipsychotic agents: A critical review. Am. J. Health Syst. Pharm. 2000, 57, 238–255. [Google Scholar] [CrossRef] [PubMed]
- Ganellin, C.R.; Triggle, D.J. Dictionary of Pharmacological Agents; Taylor & Francis: Oxfordshire, UK, 1996. [Google Scholar]
- Löscher, W.; Witte, U.; Fredow, G.; Traber, J.; Glaser, T. The behavioural responses to 8-OH-DPAT, ipsapirone and the novel 5-HT1A receptor agonist Bay Vq 7813 in the pig. Naunyn-Schmiedeberg’s Arch. Pharmacol. 1990, 342, 271–277. [Google Scholar] [CrossRef] [PubMed]
- Eison, A.S. Azapirones: History of Development. J. Clin. Psychopharmacol. 1990, 10, 2S–5S. [Google Scholar] [CrossRef] [PubMed]
- Traber, J.R.; Glaser, T. 5-HT1A receptor-related anxiolytics. Trends Pharmacol. Sci. 1987, 8, 432–437. [Google Scholar] [CrossRef]
- Mylari, B.L.; Larson, E.R.; Beyer, T.A.; Zembrowski, W.J.; Aldinger, C.E.; Dee, M.F.; Siegel, T.W.; Singleton, D.H. Novel, potent aldose reductase inhibitors: 3,4-dihydro-4-oxo-3-[[5-(trifluoromethyl)-2-benzothiazolyl]methyl]-1-phthalazineacetic acid (zopolrestat) and congeners. J. Med. Chem. 1991, 34, 108–122. [Google Scholar] [CrossRef] [PubMed]
- Zhai, J.; Zhang, H.; Zhang, L.; Zhao, Y.; Chen, S.; Chen, Y.; Peng, X.; Li, Q.; Yuan, M.; Hu, X. Zopolrestat as a Human Glyoxalase I Inhibitor and Its Structural Basis. ChemMedChem 2013, 8, 1462–1464. [Google Scholar] [CrossRef] [PubMed]
- Wilson, D.K.; Tarle, I.; Petrash, J.M.; Quiocho, F.A. Refined 1.8 A structure of human aldose reductase complexed with the potent inhibitor zopolrestat. Proc. Natl. Acad. Sci. USA 1993, 90, 9847–9851. [Google Scholar] [CrossRef] [PubMed]
- Inskeep, P.B.; Ronfeld, R.A.; Peterson, M.J.; Gerber, N. Pharmacokinetics of the Aldose Reductase Inhibitor, Zopolrestat, in Humans. J. Clin. Pharmacol. 1994, 34, 760–766. [Google Scholar] [CrossRef] [PubMed]
- Ajmer Singh, G.; Shashikant, B.; Deepti, P.; Viney, L.; Bhupinder Singh, S. Updates on Aldose Reductase Inhibitors for Management of Diabetic Complications and Non-diabetic Diseases. Mini Rev. Med. Chem. 2016, 16, 120–162. [Google Scholar] [CrossRef] [PubMed]
- Oates, P.J.; Mylari, B.L. Aldose reductase inhibitors: Therapeutic implications for diabetic complications. Expert Opin. Investig. Drugs 1999, 8, 2095–2119. [Google Scholar] [CrossRef] [PubMed]
- Berkin, K.E.; Kerr, J.W. Tiaramide–a new oral drug for the treatment of asthma. Br. J. Clin. Pharmacol. 1982, 14, 505–508. [Google Scholar] [CrossRef] [PubMed]
- Khandwala, A.; Coutts, S.; Weinryb, I. Antiallergic Activity of Tiaramide (RHC 2592-A). Int. Arch. Allergy Appl. Immunol. 2009, 69, 159–168. [Google Scholar] [CrossRef] [PubMed]
- Harvey, J.E.; Jones, P.; Tanser, A.R. A Trial of Tiaramide in Asthma. Allergy 1985, 40, 198–201. [Google Scholar] [CrossRef] [PubMed]
- Folco, G.C.; Viganò, T.; Sautebin, L.; Malandrino, S.; Omini, C.; Berti, F. The mode of action of tiaramide hydrochloride: A new antiasthmatic drug. Pharmacol. Res. Commun. 1979, 11, 703–718. [Google Scholar] [CrossRef] [PubMed]
- Aguilar, F.J. The Polyanthelmintic Action of Dithiazanine Iodide: Verification of its Effectiveness in the Most Common Helminthiases in Guatemala. Am. J. Trop. Med. Hyg. 1959, 8, 305–306. [Google Scholar] [CrossRef] [PubMed]
- Swartzwelder, J.C.; Frye, W.W.; Muhleisen, J.P.; Miller, J.H.; Lampert, R.; Chavarria, A.P.; Abadie, S.H.; Anthony, S.O.; Sappenfield, R.W. Dithiazanine, an Effective Broad-Spectrum Anthelmintic: Results of Therapy of Trichuriasis, Strongyloidiasis, Enterobiasis, Ascariasis, and Hookworm Infection. J. Am. Med. Assoc. 1957, 165, 2063–2067. [Google Scholar] [CrossRef] [PubMed]
- Abadie, S.H.; Samuels, M. A Fatality Associated with Dithiazanine Iodide Therapy. JAMA 1965, 192, 326–327. [Google Scholar] [CrossRef] [PubMed]
- Akusawa, M.; Shimizu, S. ANTHELMINTIC EFFECT OF DITHIAZANINE IODIDE ON SWINE LUNGWORMS. Kurume Med. J. 1968, 15, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Swartzwelder, J.C.; Muhleisen, J.P.; Abadie, S.H.; Frye, W.W.; Jones, C.A.; Robertson, P.E.; Hebert, J.F. Therapy of Strongyloidiasis with Dithiazanine. A.M.A. Arch. Intern. Med. 1958, 101, 658–661. [Google Scholar] [CrossRef] [PubMed]
- Vinke, B.; Sar, V.D. Dithiazanine, a new anthelminthic. Trop Geogr Med. 1959, 11, 335–338. [Google Scholar] [PubMed]
- Shikha, A.; Divyani, G.; Priyanka, K. Benzothiazole: A Versatile and Multitargeted Pharmacophore in the Field of Medicinal Chemistry. Lett. Org. Chem. 2017, 14, 729–742. [Google Scholar] [CrossRef]
- Di Fiore, A.; Pedone, C.; Antel, J.; Waldeck, H.; Witte, A.; Wurl, M.; Scozzafava, A.; Supuran, C.T.; De Simone, G. Carbonic anhydrase inhibitors: The X-ray crystal structure of ethoxzolamide complexed to human isoform II reveals the importance of thr200 and gln92 for obtaining tight-binding inhibitors. Bioorganic Med. Chem. Lett. 2008, 18, 2669–2674. [Google Scholar] [CrossRef] [PubMed]
- Masini, E.; Fabrizio, C.; Andrea, S.; Supuran, C.T. Antiglaucoma carbonic anhydrase inhibitors: A patent review. Expert Opin. Ther. Pat. 2013, 23, 705–716. [Google Scholar] [CrossRef] [PubMed]
- Maren, T.H.; Brechue, W.F.; Bar-Ilan, A. Relations among IOP reduction, ocular disposition and pharmacology of the carbonic anhydrase inhibitor ethoxzolamide. Exp. Eye Res. 1992, 55, 73–79. [Google Scholar] [CrossRef] [PubMed]
- Carta, F.; Di Cesare Mannelli, L.; Pinard, M.; Ghelardini, C.; Scozzafava, A.; McKenna, R.; Supuran, C.T. A class of sulfonamide carbonic anhydrase inhibitors with neuropathic pain modulating effects. Biorg. Med. Chem. 2015, 23, 1828–1840. [Google Scholar] [CrossRef] [PubMed]
- Youse, M.S.; Abutaleb, N.S.; Nocentini, A.; Abdelsattar, A.S.; Ali, F.; Supuran, C.T.; Seleem, M.N.; Flaherty, D.P. Optimization of Ethoxzolamide Analogs with Improved Pharmacokinetic Properties for In Vivo Efficacy against Neisseria gonorrhoeae. J. Med. Chem. 2024, 67, 15537–15556. [Google Scholar] [CrossRef] [PubMed]
- Rahman, M.M.; Tikhomirova, A.; Modak, J.K.; Hutton, M.L.; Supuran, C.T.; Roujeinikova, A. Antibacterial activity of ethoxzolamide against Helicobacter pylori strains SS1 and 26695. Gut Pathog. 2020, 12, 20. [Google Scholar] [CrossRef] [PubMed]
- Wistrand, P.J. The Use of Carbonic Anhydrase Inhibitors in Ophthalmology and Clinical Medicine. Ann. N.Y. Acad. Sci. 1984, 429, 609–619. [Google Scholar] [CrossRef] [PubMed]
- Schoenwald, R.D.; Eller, M.G.; Dixson, J.A.; Barfknecht, C.F. Topical carbonic anhydrase inhibitors. J. Med. Chem. 1984, 27, 810–812. [Google Scholar] [CrossRef] [PubMed]
- Modak, J.K.; Tikhomirova, A.; Gorrell, R.J.; Rahman, M.M.; Kotsanas, D.; Korman, T.M.; Garcia-Bustos, J.; Kwok, T.; Ferrero, R.L.; Supuran, C.T.; et al. Anti-Helicobacter pylori activity of ethoxzolamide. J. Enzym. Inhib. Med. Chem. 2019, 34, 1660–1667. [Google Scholar] [CrossRef] [PubMed]
- Johnson Benjamin, K.; Colvin Christopher, J.; Needle David, B.; Mba Medie, F.; Champion Patricia, A.D.; Abramovitch Robert, B. The Carbonic Anhydrase Inhibitor Ethoxzolamide Inhibits the Mycobacterium tuberculosis PhoPR Regulon and Esx-1 Secretion and Attenuates Virulence. Antimicrob. Agents Chemother. 2015, 59, 4436–4445. [Google Scholar] [CrossRef] [PubMed]
- Tully, D.C.; Rucker, P.V.; Chianelli, D.; Williams, J.; Vidal, A.; Alper, P.B.; Mutnick, D.; Bursulaya, B.; Schmeits, J.; Wu, X.; et al. Discovery of Tropifexor (LJN452), a Highly Potent Non-bile Acid FXR Agonist for the Treatment of Cholestatic Liver Diseases and Nonalcoholic Steatohepatitis (NASH). J. Med. Chem. 2017, 60, 9960–9973. [Google Scholar] [CrossRef] [PubMed]
- Sanyal, A.J.; Lopez, P.; Lawitz, E.J.; Lucas, K.J.; Loeffler, J.; Kim, W.; Goh, G.B.B.; Huang, J.-F.; Serra, C.; Andreone, P.; et al. Tropifexor for nonalcoholic steatohepatitis: An adaptive, randomized, placebo-controlled phase 2a/b trial. Nat. Med. 2023, 29, 392–400. [Google Scholar] [CrossRef] [PubMed]
- Lucas, K.J.; Lopez, P.; Lawitz, E.; Sheikh, A.; Aizenberg, D.; Hsia, S.; Bee, G.G.B.; Vierling, J.; Frias, J.; White, J.; et al. Tropifexor, a highly potent FXR agonist, produces robust and dose-dependent reductions in hepatic fat and serum alanine aminotransferase in patients with fibrotic NASH after 12 weeks of therapy: FLIGHT-FXR Part C interim results. Dig. Liver Dis. 2020, 52, e38. [Google Scholar] [CrossRef]
- Schramm, C.; Hirschfield, G.; Mason, A.L.; Wedemeyer, H.; Klickstein, L.; Neelakantham, S.; Koo, P.; Sanni, J.; Badman, M.; Jones, D. LBO-007—Early assessment of safety and efficacy of tropifexor, a potent non bile-acid FXR agonist, in patients with primary biliary cholangitis: An interim analysis of an ongoing phase 2 study. J. Hepatol. 2018, 68, S103. [Google Scholar] [CrossRef]
- Badman, M.K.; Chen, J.; Desai, S.; Vaidya, S.; Neelakantham, S.; Zhang, J.; Gan, L.; Danis, K.; Laffitte, B.; Klickstein, L.B. Safety, Tolerability, Pharmacokinetics, and Pharmacodynamics of the Novel Non–Bile Acid FXR Agonist Tropifexor (LJN452) in Healthy Volunteers. Clin. Pharmacol. Drug Dev. 2020, 9, 395–410. [Google Scholar] [CrossRef] [PubMed]
- Masand, P.S.; Nemeroff, C.B.; Newcomer, J.W.; Lieberman, J.A.; Schatzberg, A.F.; Weiden, P.J.; Kilts, C.D.; Harvey, P.D.; Daniel, D.G. From Clinical Research to Clinical Practice: A 4-Year Review of Ziprasidone. CNS Spectr. 2005, 10, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Caley, C.F.; Cooper, C.K. Ziprasidone: The Fifth Atypical Antipsychotic. Ann. Pharmacother. 2002, 36, 839–851. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, W.M.; Citrome, L. Ziprasidone for Schizophrenia and Bipolar Disorder: A Review of the Clinical Trials. CNS Drug Rev. 2007, 13, 137–177. [Google Scholar] [CrossRef] [PubMed]
- Stroup, T.S.; Jarskog, L.F. Review: Ziprasidone is marginally less effective than other atypical antipsychotics in people with schizophrenia. Evid. Based Ment. Health 2010, 13, 53. [Google Scholar] [CrossRef] [PubMed]
- Arnold, L.M.; McElroy, S.L.; Keck, P.E. Ziprasidone: A new atypical antipsychotic. Expert Opin. Pharmacother. 2001, 2, 1033–1042. [Google Scholar] [CrossRef] [PubMed]
- Wessels, A.M.; Bies, R.R.; Pollock, B.G.; Schneider, L.S.; Lieberman, J.A.; Stroup, S.; Li, C.H.; Coley, K.; Kirshner, M.M.; Marder, S.R. Population Pharmacokinetic Modeling of Ziprasidone in Patients with Schizophrenia from the CATIE Study. J. Clin. Pharmacol. 2011, 51, 1587–1591. [Google Scholar] [CrossRef] [PubMed]
- Daniel, D.G.; Copeland, L.F. Ziprasidone: Comprehensive overview and clinical use of a novel antipsychotic. Expert Opin. Investig. Drugs 2000, 9, 819–828. [Google Scholar] [CrossRef] [PubMed]
- Bernardo, M.; Rico-Villademoros, F.; García-Rizo, C.; Rojo, R.; Gómez-Huelgas, R. Real-World Data on the Adverse Metabolic Effects of Second-Generation Antipsychotics and Their Potential Determinants in Adult Patients: A Systematic Review of Population-Based Studies. Adv. Ther. 2021, 38, 2491–2512. [Google Scholar] [CrossRef] [PubMed]
- Mandrioli, R.; Protti, M.; Mercolini, L. Evaluation of the pharmacokinetics, safety and clinical efficacy of ziprasidone for the treatment of schizophrenia and bipolar disorder. Expert Opin. Drug Metab. Toxicol. 2015, 11, 149–174. [Google Scholar] [CrossRef] [PubMed]
- Citrome, L. Drug safety evaluation of ziprasidone. Expert Opin. Drug Saf. 2011, 10, 437–448. [Google Scholar] [CrossRef] [PubMed]
- Cotman, A.E.; Durcik, M.; Benedetto Tiz, D.; Fulgheri, F.; Secci, D.; Sterle, M.; Možina, Š.; Skok, Ž.; Zidar, N.; Zega, A.; et al. Discovery and Hit-to-Lead Optimization of Benzothiazole Scaffold-Based DNA Gyrase Inhibitors with Potent Activity against Acinetobacter baumannii and Pseudomonas aeruginosa. J. Med. Chem. 2023, 66, 1380–1425. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, I.; Manolov, S.; Bojilov, D.; Stremski, Y.; Marc, G.; Statkova-Abeghe, S.; Oniga, S.; Oniga, O.; Nedialkov, P. Synthesis of Novel Benzothiazole–Profen Hybrid Amides as Potential NSAID Candidates. Molecules 2025, 30, 107. [Google Scholar] [CrossRef] [PubMed]
- Mishra, C.B.; Shalini, S.; Gusain, S.; Kumar, P.; Kumari, S.; Choi, Y.-S.; Kumari, J.; Moku, B.K.; Yadav, A.K.; Prakash, A.; et al. Multitarget action of Benzothiazole-piperazine small hybrid molecule against Alzheimer’s disease: In silico, In vitro, and In vivo investigation. Biomed. Pharmacother. 2024, 174, 116484. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Zhao, R.; Zhou, H.; Yang, S.; Tao, F.; Xie, Y.; Wang, H.; Yun, J. A novel benzothiazole derivative induces apoptosis via the mitochondrial intrinsic pathway producing antitumor activity in colorectal cancer. Front. Pharmacol. 2023, 14, 1196158. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, B.; Kumar, R.; Salahuddin; Singh, H.; Afzal, O.; Altamimi, A.S.A.; Abdullah, M.M.; Yar, M.S.; Ahsan, M.J.; Kumar, N.; et al. Design, Synthesis, In Vivo, and In Silico Evaluation of Benzothiazoles Bearing a 1,3,4-Oxadiazole Moiety as New Antiepileptic Agents. ACS Omega 2023, 8, 2520–2530. [Google Scholar] [CrossRef] [PubMed]
- Altwaijry, N.A.; Omar, M.A.; Mohamed, H.S.; Mounier, M.M.; Afifi, A.H.; Srour, A.M. Design, synthesis, molecular docking and anticancer activity of benzothiazolecarbohydrazide–sulfonate conjugates: Insights into ROS-induced DNA damage and tubulin polymerization inhibition. RSC Adv. 2025, 15, 5895–5905. [Google Scholar] [CrossRef] [PubMed]
- Abd El-Meguid, E.A.; Naglah, A.M.; Moustafa, G.O.; Awad, H.M.; El Kerdawy, A.M. Novel benzothiazole-based dual VEGFR-2/EGFR inhibitors targeting breast and liver cancers: Synthesis, cytotoxic activity, QSAR and molecular docking studies. Bioorganic Med. Chem. Lett. 2022, 58, 128529. [Google Scholar] [CrossRef] [PubMed]
- Mokhtar, A.M.; El-Messery, S.M.; Ghaly, M.A.; Hassan, G.S. Targeting EGFR tyrosine kinase: Synthesis, in vitro antitumor evaluation, and molecular modeling studies of benzothiazole-based derivatives. Bioorg. Chem. 2020, 104, 104259. [Google Scholar] [CrossRef] [PubMed]
- Suma, V.R.; Prasad, K.; Chandrasekhar, C.; Sireesha, R.; Rao, K.R.M. Design, Synthesis, and Anticancer Evaluation of Chalcone Incorporated Benzothiazole-Imidazo [2,1-b]thiazole Derivatives. Russ. J. Org. Chem. 2023, 59, S48–S55. [Google Scholar] [CrossRef]
- Ewes, W.A.; Tawfik, S.S.; Almatary, A.M.; Ahmad Bhat, M.; El-Shafey, H.W.; Mohamed, A.A.B.; Haikal, A.; El-Magd, M.A.; Elgazar, A.A.; Balaha, M.; et al. Identification of Benzothiazoles Bearing 1,3,4-Thiadiazole as Antiproliferative Hybrids Targeting VEGFR-2 and BRAF Kinase: Design, Synthesis, BIO Evaluation and in Silico Study. Molecules 2024, 29, 3186. [Google Scholar] [CrossRef] [PubMed]
- Al-Sanea, M.M.; Hamdi, A.; Mohamed, A.A.B.; El-Shafey, H.W.; Moustafa, M.; Elgazar, A.A.; Eldehna, W.M.; Ur Rahman, H.; Parambi, D.G.T.; Elbargisy, R.M.; et al. New benzothiazole hybrids as potential VEGFR-2 inhibitors: Design, synthesis, anticancer evaluation, and in silico study. J. Enzym. Inhib. Med. Chem. 2023, 38, 2166036. [Google Scholar] [CrossRef] [PubMed]
- Aljuhani, A.; Nafie, M.S.; Albujuq, N.R.; Alsehli, M.; Bardaweel, S.K.; Darwish, K.M.; Alraqa, S.Y.; Aouad, M.R.; Rezki, N. Discovery of new benzothiazole-1,2,3-triazole hybrid-based hydrazone/thiosemicarbazone derivatives as potent EGFR inhibitors with cytotoxicity against cancer. RSC Adv. 2025, 15, 3570–3591. [Google Scholar] [CrossRef] [PubMed]
- Özdemir, A.; Temel, H.E. COX inhibitory profiles of a series of thiadiazole-benzothiazole hybrids. Eur. J. Life Sci. 2024, 3, 9–15. [Google Scholar] [CrossRef]
- Acar Çevik, U.; Osmaniye, D.; Sağlik, B.N.; Levent, S.; Çavuşoğlu, B.K.; Karaduman, A.B.; Özkay, Ü.D.; Özkay, Y.; Kaplancikli, Z.A.; Turan, G. Synthesis of new benzothiazole derivatives bearing thiadiazole as monoamine oxidase inhibitors. J. Heterocycl. Chem. 2020, 57, 2225–2233. [Google Scholar] [CrossRef]
- Barbarossa, A.; Ceramella, J.; Carocci, A.; Iacopetta, D.; Rosato, A.; Limongelli, F.; Carrieri, A.; Bonofiglio, D.; Sinicropi, M.S. Benzothiazole-Phthalimide Hybrids as Anti-Breast Cancer and Antimicrobial Agents. Antibiotics 2023, 12, 1651. [Google Scholar] [CrossRef] [PubMed]
- Boateng, C.A.; Nilson, A.N.; Placide, R.; Pham, M.L.; Jakobs, F.M.; Boldizsar, N.; McIntosh, S.; Stallings, L.S.; Korankyi, I.V.; Kelshikar, S.; et al. Pharmacology and Therapeutic Potential of Benzothiazole Analogues for Cocaine Use Disorder. J. Med. Chem. 2023, 66, 12141–12162. [Google Scholar] [CrossRef] [PubMed]
- Alvarado Salazar, J.A.; Valdes, M.; Cruz, A.; Moreno de Jesús, B.; Patiño González, D.; Olivares Corichi, I.M.; Tamay Cach, F.; Mendieta Wejebe, J.E. In Silico and In Vivo Evaluation of Novel 2-Aminobenzothiazole Derivative Compounds as Antidiabetic Agents. Int. J. Mol. Sci. 2025, 26, 909. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Zhu, Z.; Chen, S.; Fu, Y.; Zhang, J.; Guo, Y.; Xu, Z.; Xi, Y.; Wang, X.; Ye, F.; et al. Synthesis and biological evaluation of novel benzothiazole derivatives as potential anticancer and antiinflammatory agents. Front. Chem. 2024, 12, 1384301. [Google Scholar] [CrossRef] [PubMed]
- Amoroso, R.; De Lellis, L.; Florio, R.; Moreno, N.; Agamennone, M.; De Filippis, B.; Giampietro, L.; Maccallini, C.; Fernández, I.; Recio, R.; et al. Benzothiazole Derivatives Endowed with Antiproliferative Activity in Paraganglioma and Pancreatic Cancer Cells: Structure–Activity Relationship Studies and Target Prediction Analysis. Pharmaceuticals 2022, 15, 937. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Mohsen, H.T.; Abd El-Meguid, E.A.; El Kerdawy, A.M.; Mahmoud, A.E.E.; Ali, M.M. Design, synthesis, and molecular docking of novel 2-arylbenzothiazole multiangiokinase inhibitors targeting breast cancer. Arch. Pharm. 2020, 353, 1900340. [Google Scholar] [CrossRef] [PubMed]
- le Roux, A.; Petzer, A.; Cloete, S.J.; Petzer, J.P. An investigation of the monoamine oxidase inhibition properties of benzothiazole derivatives. Results Chem. 2025, 14, 102142. [Google Scholar] [CrossRef]
- Reddy, V.G.; Reddy, T.S.; Jadala, C.; Reddy, M.S.; Sultana, F.; Akunuri, R.; Bhargava, S.K.; Wlodkowic, D.; Srihari, P.; Kamal, A. Pyrazolo-benzothiazole hybrids: Synthesis, anticancer properties and evaluation of antiangiogenic activity using in vitro VEGFR-2 kinase and in vivo transgenic zebrafish model. Eur. J. Med. Chem. 2019, 182, 111609. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).