


Iodinated Salicylhydrazone Derivatives as Potent α-Glucosidase Inhibitors: Synthesis, Enzymatic Activity, Molecular Modeling, and ADMET Profiling


 ,
,  , ,
, ,  ,
, 
Abstract

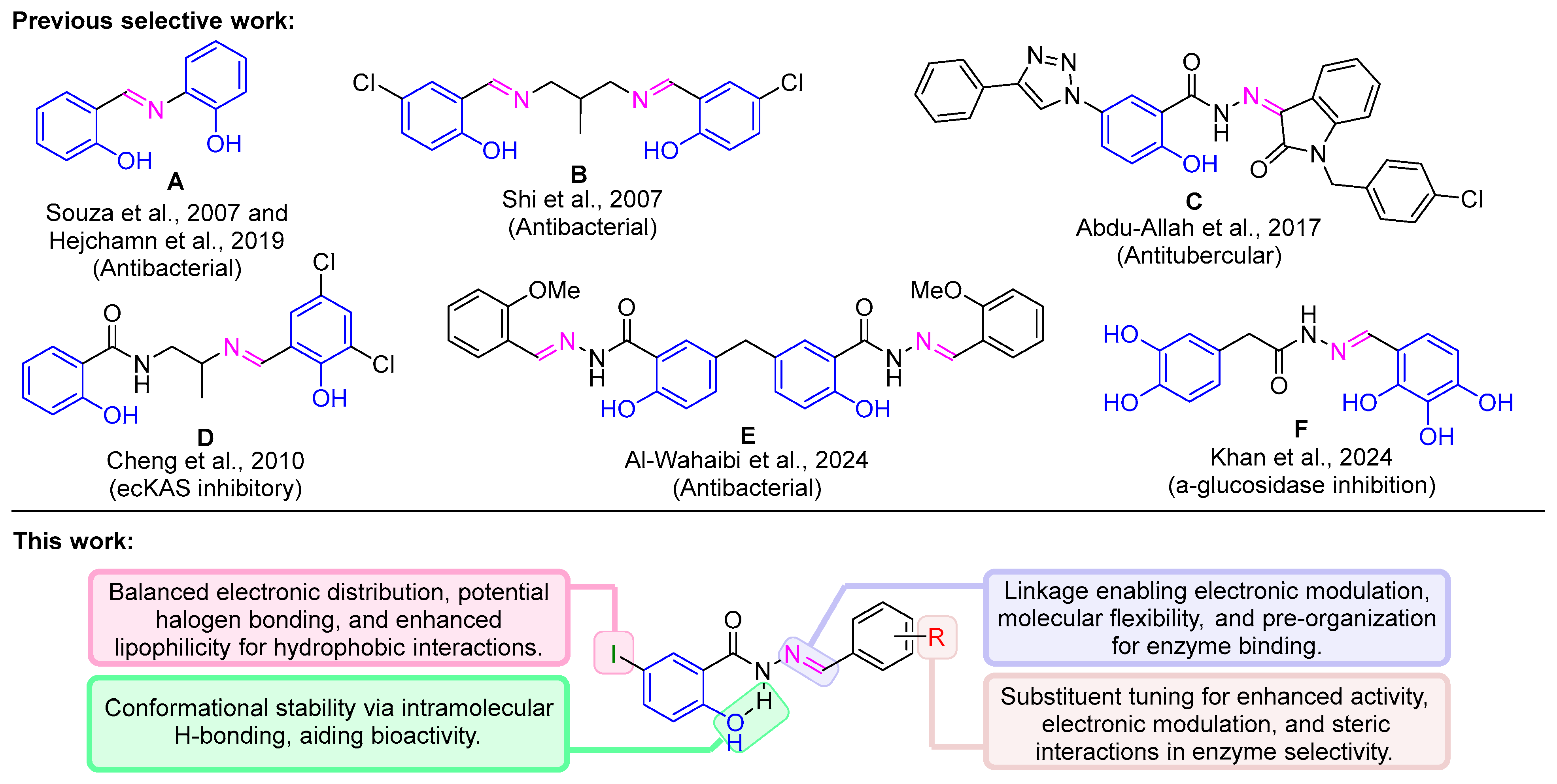
1. Introduction
2. Materials and Methods
2.1. General Method
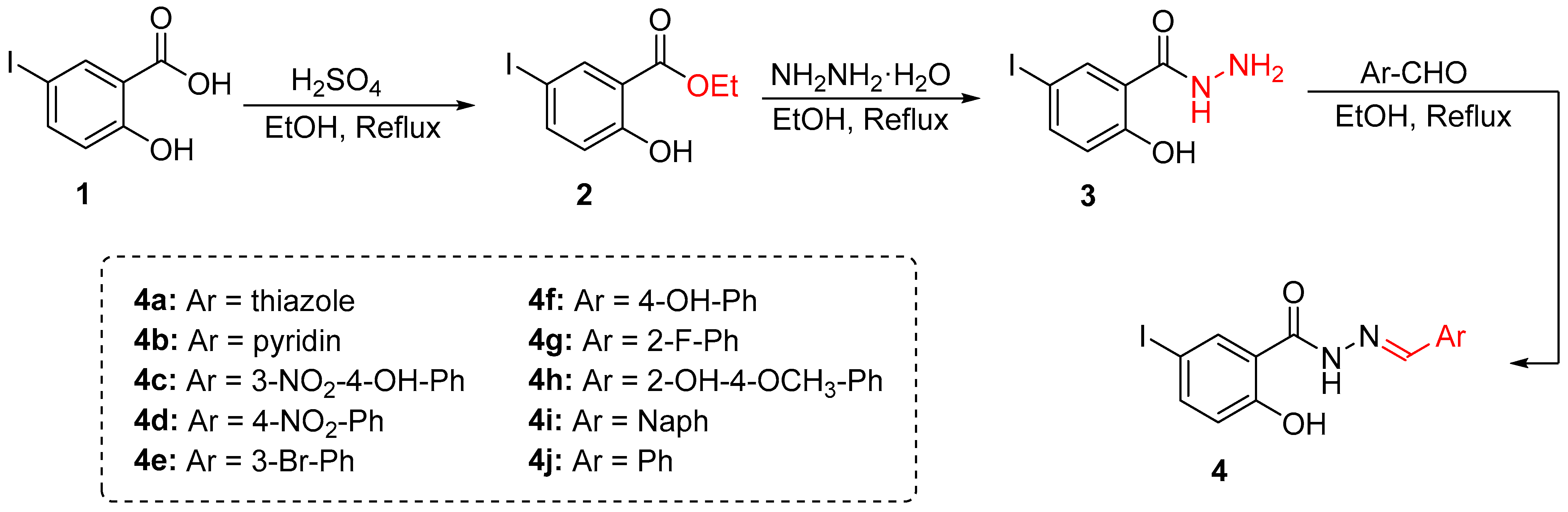
2.2. Chemistry
2.2.1. 5-Iodo-2-Salicylic Ester (2)
2.2.2. 5-Iodo-2-Hydroxybenzo-Hydrazide (3)
2.2.3. General Procedure for Synthesis
2.3. Biological Assay
2.3.1. Screening for α-Glucosidase Inhibitory Activity
2.3.2. Determination of IC50 Values
2.4. Theoretical Study
2.4.1. Density Functional Theory (DFT) and Molecular Electrostatic Potential (MEP)
2.4.2. Molecular Docking (MD) and Molecular Dynamics Simulation (MDS)
2.4.3. ADMET Predictions
2.4.4. SAR and Statistical Analysis
3. Results and Discussion
3.1. Synthesis and Characterization
3.2. Density Functional Theory (DFT) Study
3.3. ADMET Profiling
3.4. α-Glucosidase Inhibition Assay
3.5. Structure–Activity Relationship (SAR) Analysis
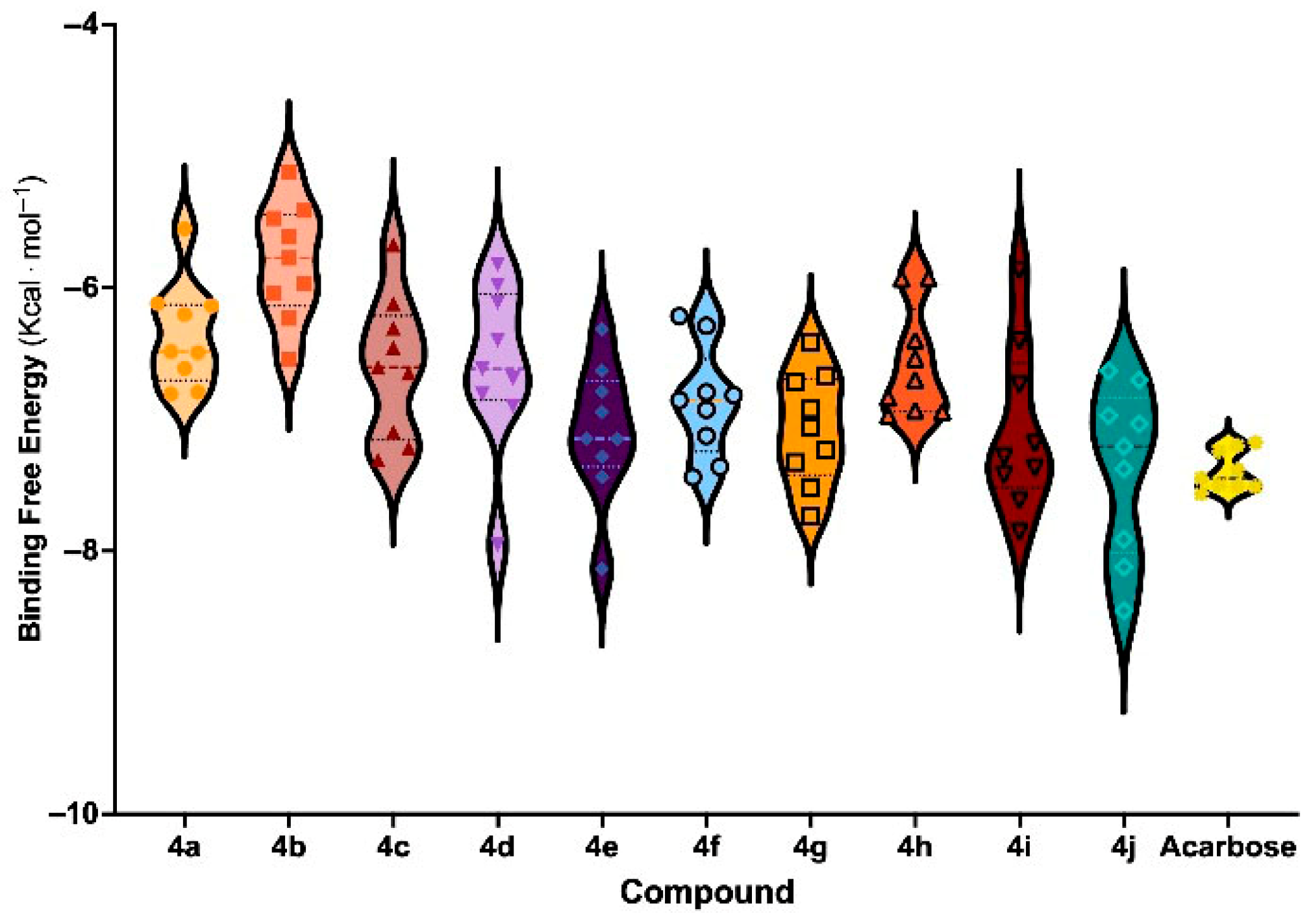
3.6. Molecular Docking and Binding Free Energy Analysis
3.7. Molecular Dynamics Simulation for 4e and 4j
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- World Health Organization. Diabetes: Fact Sheet; World Health Organization: Geneva, Switzerland, 2024; Available online: https://www.who.int/news-room/fact-sheets/detail/diabetes (accessed on 31 December 2024).
- International Diabetes Federation. IDF Diabetes Atlas, 10th ed.; International Diabetes Federation: Brussels, Belgium, 2021; Available online: https://diabetesatlas.org (accessed on 31 December 2024).
- International Diabetes Federation. Diabetes Facts and Figures; International Diabetes Federation: Brussels, Belgium, 2024; Available online: https://idf.org (accessed on 31 December 2024).
- American Diabetes Association Professional Practice Committee. Retinopathy, Neuropathy, and Foot Care: Standards of Medical Care in Diabetes—2022. Diabetes Care 2022, 45, S185–S194. [Google Scholar] [CrossRef] [PubMed]
- Ratner, R.E. Controlling Postprandial Hyperglycemia. Am. J. Cardiol. 2001, 88, 26–31. [Google Scholar] [CrossRef] [PubMed]
- Citarella, A.; Cavinato, M.; Rosini, E.; Shehi, H.; Ballabio, F.; Camilloni, C.; Fasano, V.; Silvani, A.; Passarella, D.; Pollegioni, L.; et al. Nicotinic Acid Derivatives as Novel Noncompetitive α-Amylase and α-Glucosidase Inhibitors for Type 2 Diabetes Treatment. ACS Med. Chem. Lett. 2024, 15, 1474–1481. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Liu, C.; Hu, L. Fragment-Based Dynamic Combinatorial Chemistry for Identification of Selective α-Glucosidase Inhibitors. ACS Med. Chem. Lett. 2022, 13, 1791–1796. [Google Scholar] [CrossRef] [PubMed]
- Mwakalukwa, R.; Amen, Y.; Nagata, M.; Shimizu, K. Postprandial Hyperglycemia Lowering Effect of the Isolated Compounds from Olive Mill Wastes—An Inhibitory Activity and Kinetics Studies on α-Glucosidase and α-Amylase Enzymes. ACS Omega 2020, 5, 20070–20079. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Xie, L.; Xie, J.; Liu, Y.; Chen, W. Pelargonidin-3-O-Rutinoside as a Novel α-Glucosidase Inhibitor for Improving Postprandial Hyperglycemia. Chem. Commun. 2019, 55, 39–42. [Google Scholar] [CrossRef] [PubMed]
- Hanefeld, M. The Role of Acarbose in the Treatment of Non-Insulin-Dependent Diabetes Mellitus. J. Diabetes Complicat. 1998, 12, 228–237. [Google Scholar] [CrossRef] [PubMed]
- Kageyama, S.; Nakamichi, N.; Sekino, H.; Nakano, S. Comparison of the Effects of Acarbose and Voglibose in Healthy Subjects. Clin. Ther. 1997, 19, 720–729. [Google Scholar] [CrossRef] [PubMed]
- Kashtoh, H.; Baek, K.-H. Recent Updates on Phytoconstituent Alpha-Glucosidase Inhibitors: An Approach towards the Treatment of Type Two Diabetes. Plants 2022, 11, 2722. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.-S.; Xie, H.-X.; Zhang, J.-H.; Li, Y.; Zhang, J.; Wang, K.-M.; Jiang, C.-S. An Updated Overview of Synthetic α-Glucosidase Inhibitors: Chemistry and Bioactivities. Curr. Med. Chem. 2023, 23, 2488–2526. [Google Scholar] [CrossRef] [PubMed]
- Moya-Garzón, M.D.; Martín Higueras, C.; Peñalver, P.; Romera, M.; Fernandes, M.X.; Franco-Montalbán, F.; Gómez-Vidal, J.A.; Salido, E.; Díaz-Gavilán, M. Salicylic Acid Derivatives Inhibit Oxalate Production in Mouse Hepatocytes with Primary Hyperoxaluria Type 1. J. Med. Chem. 2018, 61, 7144–7167. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Forster, E.R.; Darabedian, N.; Kim, A.T.; Pratt, M.R.; Shen, A.; Hang, H.C. Translation of Microbiota Short-Chain Fatty Acid Mechanisms Affords Anti-Infective Acyl-Salicylic Acid Derivatives. ACS Chem. Biol. 2020, 15, 1141–1147. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Lu, W.; Chen, H.; Bian, X.; Yang, G. A New Series of Salicylic Acid Derivatives as Non-Saccharide α-Glucosidase Inhibitors and Antioxidants. Biol. Pharm. Bull. 2019, 42, 231–246. [Google Scholar] [CrossRef] [PubMed]
- Aminu, K.S.; Uzairu, A.; Umar, A.B.; Ibrahim, M.T. Salicylic Acid Derivatives as Potential α-Glucosidase Inhibitors: Drug Design, Molecular Docking, and Pharmacokinetic Studies. Bull. Natl. Res. Cent. 2022, 46, 162. [Google Scholar] [CrossRef]
- Harohally, N.V.; Cherita, C.; Bhatt, P.; Appaiah, K.A. Antiaflatoxigenic and Antimicrobial Activities of Schiff Bases of 2-Hydroxy-4-methoxybenzaldehyde, Cinnamaldehyde, and Similar Aldehydes. J. Agric. Food Chem. 2017, 65, 8773–8778. [Google Scholar] [CrossRef] [PubMed]
- Thakor, P.M.; Patel, J.D.; Patel, R.J.; Chaki, S.H.; Khimani, A.J.; Vaidya, Y.H.; Chauhan, A.P.; Dholakia, A.B.; Patel, V.C.; Patel, A.J.; et al. Exploring New Schiff Bases: Synthesis, Characterization, and Multifaceted Analysis for Biomedical Applications. ACS Omega 2024, 9, 35431–35448. [Google Scholar] [CrossRef] [PubMed]
- Afzal, H.R.; Khan, N.H.; Sultana, K.; Mobashar, A.; Lareb, A.; Khan, A.; Gull, A.; Afzaal, H.; Khan, M.T.; Rizwan, M.; et al. Schiff Bases of Pioglitazone Provide Better Antidiabetic and Potent Antioxidant Effect in a Streptozotocin–Nicotinamide-Induced Diabetic Rodent Model. ACS Omega 2021, 6, 4470–4479. [Google Scholar] [CrossRef] [PubMed]
- Sarfraz, M.; Ayyaz, M.; Rauf, A.; Yaqoob, A.; Tooba; Ali, M.A.; Siddique, S.A.; Qureshi, A.M.; Sarfraz, M.H.; Aljowaie, R.M.; et al. New Pyrimidinone Bearing Aminomethylenes and Schiff Bases as Potent Antioxidant, Antibacterial, SARS-CoV-2, and COVID-19 Main Protease MPro Inhibitors: Design, Synthesis, Bioactivities, and Computational Studies. ACS Omega 2024, 9, 25730–25747. [Google Scholar] [CrossRef] [PubMed]
- Mushtaq, I.; Ahmad, M.; Saleem, M.; Ahmed, A. Pharmaceutical Significance of Schiff Bases: An Overview. Future J. Pharm. Sci. 2024, 10, 16. [Google Scholar] [CrossRef]
- Souza, A.O.; Galetti, F.; Silva, C.L.; Bicalho, B.; Parma, M.M.; Fonseca, S.F.; Marsaioli, A.J.; Trindade, A.C.L.B.; Gil, R.P.F.; Bezerra, F.S.; et al. Synthesis and Antimicrobial Activity of Novel Schiff Bases. Quim. Nova 2007, 30, 1563–1566. [Google Scholar] [CrossRef]
- Hejchman, E.; Kruszewska, H.; Maciejewska, D.; Sowirka-Taciak, B.; Tomczyk, M.; Sztokfisz-Ignasiak, A.; Jankowski, J.; Młynarczuk-Biały, I. Design, Synthesis, and Biological Activity of Schiff Bases Bearing Salicyl and 7-Hydroxycoumarinyl Moieties. Monatsh. Chem. 2019, 150, 255–266. [Google Scholar] [CrossRef]
- Shi, L.; Ge, H.-M.; Tan, S.-H.; Li, H.-Q.; Song, Y.-C.; Zhu, H.-L. Antibacterial Activity of Schiff Bases Derived from 3-Hydroxyquinoxaline-2-carboxaldehyde. Eur. J. Med. Chem. 2007, 42, 558–564. [Google Scholar] [CrossRef] [PubMed]
- Nurkenov, O.A.; Satpaeva, Z.B.; Schepetkin, I.A.; Khlebnikov, A.I.; Turdybekov, K.M.; Seilkhanov, T.M.; Fazylov, S.D. Synthesis and Biological Activity of Hydrazones of o- and p-Hydroxybenzoic Acids. Russ. J. Gen. Chem. 2017, 87, 2299–2306. [Google Scholar] [CrossRef]
- Cheng, K.; Zheng, Q.-Z.; Hou, J.; Zhou, Y.; Liu, C.-H.; Zhao, J.; Zhu, H.-L. Design, Synthesis, and Biological Evaluation of Novel Schiff Bases as Enzyme Inhibitors. Bioorg. Med. Chem. 2010, 18, 2447–2455. [Google Scholar] [CrossRef] [PubMed]
- Al-Wahaibi, L.H.; Mahmoud, M.A.; Alzahrani, H.A.; Abou-Zied, H.A.; Gomaa, H.A.M.; Youssif, B.G.M.; Bräse, S.; Rabea, S.M. Investigating Novel Chemical Scaffolds in Medicinal Chemistry. Front. Chem. 2024, 12, 1419242. [Google Scholar] [CrossRef]
- Khan, H.; Jan, F.; Shakoor, A.; Khan, A.; AlAsmari, A.F.; Alasmari, F.; Ullah, S.; Al-Harrasi, A.; Khan, M.; Ali, S. Design, Synthesis, Molecular Docking Study, and α-Glucosidase Inhibitory Evaluation of Novel Hydrazide–Hydrazone Derivatives of 3,4-Dihydroxyphenylacetic Acid. Sci. Rep. 2024, 14, 11410. [Google Scholar] [CrossRef] [PubMed]
- Guha, R. On Exploring Structure–Activity Relationships. In Methods in Molecular Biology; Ekins, S., Ed.; Humana Press: Totowa, NJ, USA, 2013; Volume 993, pp. 81–94. [Google Scholar] [CrossRef]
- Waziri, I.; Yusuf, T.L.; Kelani, M.T.; Akintemi, E.O.; Olofinsan, K.A.; Muller, A.J. Exploring the Potential of N-Benzylidenebenzohydrazide Derivatives as Antidiabetic and Antioxidant Agents: Design, Synthesis, Spectroscopic, Crystal Structure, DFT, and Molecular Docking Study. ChemistrySelect 2024, 9, e202401631. [Google Scholar] [CrossRef]
- Chai, J.-D.; Head-Gordon, M. Long-Range Corrected Hybrid Density Functionals with Damped Atom–Atom Dispersion Corrections. Phys. Chem. Chem. Phys. 2008, 10, 6615–6620. [Google Scholar] [CrossRef] [PubMed]
- Weigend, F.; Ahlrichs, R. Balanced Basis Sets of Split Valence, Triple Zeta Valence and Quadruple Zeta Valence Quality for H to Rn: Design and Assessment of Accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef] [PubMed]
- Sim, L.; Quezada-Calvillo, R.; Sterchi, E.E.; Nichols, B.L.; Rose, D.R. Human Intestinal Maltase–Glucoamylase: Crystal Structure of the N-Terminal Catalytic Subunit and Basis of Inhibition and Substrate Specificity. J. Mol. Biol. 2008, 375, 782–792. [Google Scholar] [CrossRef] [PubMed]
- Olsson, M.H.M.; Søndergaard, C.R.; Rostkowski, M.; Jensen, J.H. PROPKA3: Consistent Treatment of Internal and Surface Residues in Empirical pKa Predictions. J. Chem. Theory Comput. 2011, 7, 525–537. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, W.L.; Maxwell, D.S.; Tirado-Rives, J. Development and Testing of the OPLS All-Atom Force Field on Conformational Energetics and Properties of Organic Liquids. J. Am. Chem. Soc. 1996, 118, 11225–11236. [Google Scholar] [CrossRef]
- Xiong, G.; Wu, Z.; Yi, J.; Fu, L.; Yang, Z.; Hsieh, C.; Yin, M.; Zeng, X.; Wu, C.; Chen, X.; et al. ADMETlab 2.0: An Integrated Online Platform for Accurate and Comprehensive Predictions of ADMET Properties. Nucleic Acids Res. 2021, 49, W5–W14. [Google Scholar] [CrossRef] [PubMed]
- McNutt, A.T.; Francoeur, P.; Aggarwal, R.; Masuda, T.; Meli, R.; Ragoza, M.; Sunseri, J.; Koes, D.R. GNINA 1.0: Molecular Docking with Deep Learning. J. Cheminf. 2021, 13, 43. [Google Scholar] [CrossRef] [PubMed]
- Quiroga, R.; Villarreal, M.A. Vinardo: A Scoring Function Based on Autodock Vina Improves Scoring, Docking, and Virtual Screening. PLoS ONE 2016, 11, e0155183. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameters | 4a | 4b | 4c | 4d | 4e | 4f | 4g | 4h | 4i | 4j | Acarbose |
|---|---|---|---|---|---|---|---|---|---|---|---|
| MW (g/mol) 1 | 371.94 | 366.98 | 426.97 | 410.97 | 443.9 | 381.98 | 383.98 | 411.99 | 416 | 365.99 | 645.25 |
| logP 2 | 3.59 | 3.42 | 4.11 | 4.09 | 4.56 | 3.88 | 4.19 | 4.04 | 4.95 | 4.25 | −4.48 |
| logS 3 | −5.01 | −5.2 | −5.67 | −5.96 | −6 | −5.58 | −5.58 | −5.65 | −5.79 | −5.47 | 0.53 |
| TPSA (Å 2) 4 | 62.55 | 74.58 | 125.06 | 104.83 | 61.69 | 81.92 | 61.69 | 91.15 | 61.69 | 61.69 | 321.17 |
| Lipinski’s Rule 5 | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✗ |
| PAINS 6 | No | No | Yes | No | No | Yes | No | Yes | No | No | No |
| HIA 7 | Low | Low | Low | Low | Low | Low | Low | Low | Low | Low | Low |
| Caco-2 Perm. 8 | −4.77 | −4.71 | −4.99 | −5 | −4.93 | −4.86 | −4.59 | −5.07 | −4.74 | −4.75 | −7.29 |
| Pgp Substrate 9 | No | Yes | No | No | Yes | Yes | Yes | Yes | Yes | Yes | Yes |
| PPB (%) 10 | 98.69 | 98.12 | 99.46 | 98.92 | 99 | 98.62 | 98.67 | 98.25 | 99.15 | 98.83 | 15.22 |
| BBB Penetration 11 | No | No | No | No | No | No | No | No | No | No | No |
| CYP Inhibition 12 | 1A2, 2C9, 2C19 | 1A2, 2C9 | 1A2, 2C9 | 1A2, 2C9 | 1A2, 2C9 | 1A2, 2C9 | 1A2, 2C9 | 1A2, 2C9 | 1A2, 2C9 | 1A2, 2C9 | None |
| CL Plasma (ml/min/kg) 13 | 5.08 | 3.68 | 3.38 | 3.73 | 4.04 | 4.45 | 4.28 | 4.66 | 4.42 | 4.42 | 0.14 |
| T1/2 (h) 14 | 0.66 | 0.77 | 0.92 | 0.76 | 0.74 | 0.82 | 0.72 | 0.75 | 0.66 | 0.69 | 3.64 |
| hERG Blockade 15 | Low | Low | Low | High | Low | Low | Low | Low | High | Low | Low |
| Hepatotoxicity 15 | Low | Low | Low | Low | Low | Low | Low | Low | Low | Low | Low |
| AMES Mut. 15 | Low | Low | High | High | Low | Low | Low | Low | High | Low | High |
| Nephrotoxicity 15 | High | Low | Low | Low | Low | Low | High | High | High | Low | High |
| Entry | Compounds | Ar | % Inhibition 1 | IC50 (µM) | ΔE 2 (eV) | BFE 3 (Kcal mol−1) | Affinity 4 (pK Units) |
|---|---|---|---|---|---|---|---|
| 1 | 4a | thiazole | 66.71 ± 4.27 | nd 5 | 7.89 | −6.46 ± 0.62 | 4.74 ± 0.33 |
| 2 | 4b | pyridine | 51.65 ± 4.72 | nd | 8.09 | −5.83 ± 0.58 | 4.78 ± 0.35 |
| 3 | 4c | 3-NO2-4-OH-Ph | 45.51 ± 7.45 | nd | 7.24 | −6.62 ± 0.66 | 4.60 ± 0.27 |
| 4 | 4d | 4-NO2-Ph | 59.86 ± 2.60 | nd | 7.60 | −6.61 ± 0.78 | 4.69 ± 0.27 |
| 5 | 4e | 3-Br-Ph | 92.35 ± 2.52 * | 14.86 ± 0.24 ** | 8.03 | −7.23 ± 0.64 | 4.63 ± 0.26 |
| 6 | 4f | 4-OH-Ph | 55.75 ± 1.91 | nd | 7.73 | −6.87 ± 0.60 | 4.77 ± 0.26 |
| 7 | 4g | 2-F-Ph | 85.69 ± 1.59 | 15.58 ± 0.30 ** | 8.04 | −7.09 ± 0.63 | 4.71 ± 0.23 |
| 8 | 4h | 2-OH-4-OCH3-Ph | 14.33 ± 7.47 | nd | 7.50 | −6.59 ± 0.59 | 4.64 ± 0.24 |
| 9 | 4i | Naph | 88.64 ± 0.78 ** | 18.05 ± 0.92 ** | 7.62 | −7.10 ± 0.88 | 4.67 ± 0.20 |
| 10 | 4j | Ph | 93.84 ± 1.49 ** | 17.56 ± 0.39 ** | 8.01 | −7.38 ± 0.82 | 4.78 ± 0.31 |
| 11 | Acarbose | - | 84.66 ± 0.71 | 45.78 ± 1.95 | - | −7.41 ± 0.79 | 5.00 ± 0.22 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bhagwat, S.K.; Hernandez-Rosas, F.; Vidal-Limon, A.; Jimenez-Halla, J.O.C.; Ghotekar, B.K.; Bobade, V.D.; Delgado-Alvarado, E.; Patil, S.V.; Pawar, T.J. Iodinated Salicylhydrazone Derivatives as Potent α-Glucosidase Inhibitors: Synthesis, Enzymatic Activity, Molecular Modeling, and ADMET Profiling. Chemistry 2025, 7, 117. https://doi.org/10.3390/chemistry7040117
Bhagwat SK, Hernandez-Rosas F, Vidal-Limon A, Jimenez-Halla JOC, Ghotekar BK, Bobade VD, Delgado-Alvarado E, Patil SV, Pawar TJ. Iodinated Salicylhydrazone Derivatives as Potent α-Glucosidase Inhibitors: Synthesis, Enzymatic Activity, Molecular Modeling, and ADMET Profiling. Chemistry. 2025; 7(4):117. https://doi.org/10.3390/chemistry7040117
Chicago/Turabian StyleBhagwat, Seema K., Fabiola Hernandez-Rosas, Abraham Vidal-Limon, J. Oscar C. Jimenez-Halla, Balasaheb K. Ghotekar, Vivek D. Bobade, Enrique Delgado-Alvarado, Sachin V. Patil, and Tushar Janardan Pawar. 2025. "Iodinated Salicylhydrazone Derivatives as Potent α-Glucosidase Inhibitors: Synthesis, Enzymatic Activity, Molecular Modeling, and ADMET Profiling" Chemistry 7, no. 4: 117. https://doi.org/10.3390/chemistry7040117
APA StyleBhagwat, S. K., Hernandez-Rosas, F., Vidal-Limon, A., Jimenez-Halla, J. O. C., Ghotekar, B. K., Bobade, V. D., Delgado-Alvarado, E., Patil, S. V., & Pawar, T. J. (2025). Iodinated Salicylhydrazone Derivatives as Potent α-Glucosidase Inhibitors: Synthesis, Enzymatic Activity, Molecular Modeling, and ADMET Profiling. Chemistry, 7(4), 117. https://doi.org/10.3390/chemistry7040117