
Metal-Free C(sp3)–S Bond Cleavage of Thioethers to Selectively Access Aryl Aldehydes and Dithioacetals


Abstract
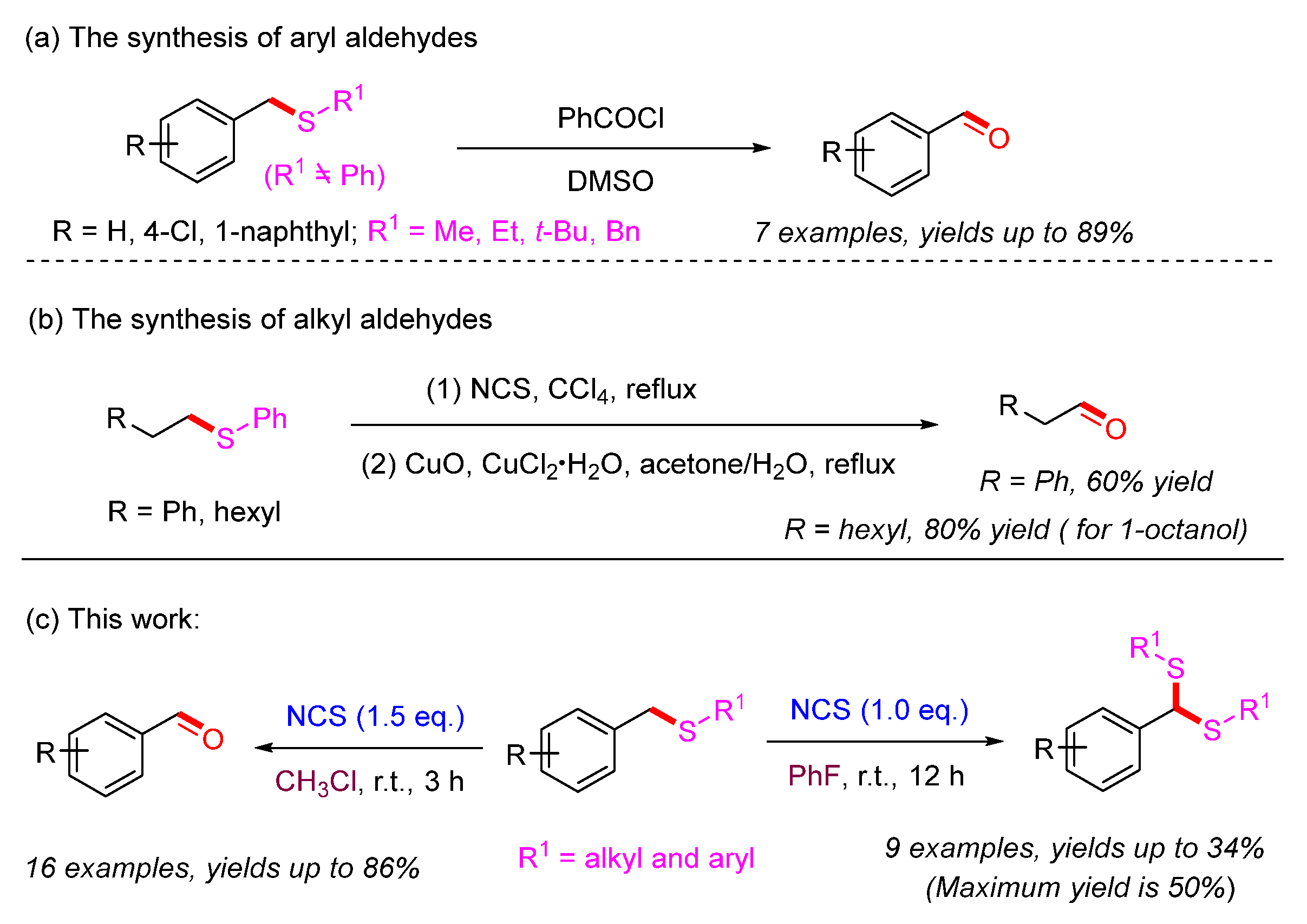
1. Introduction
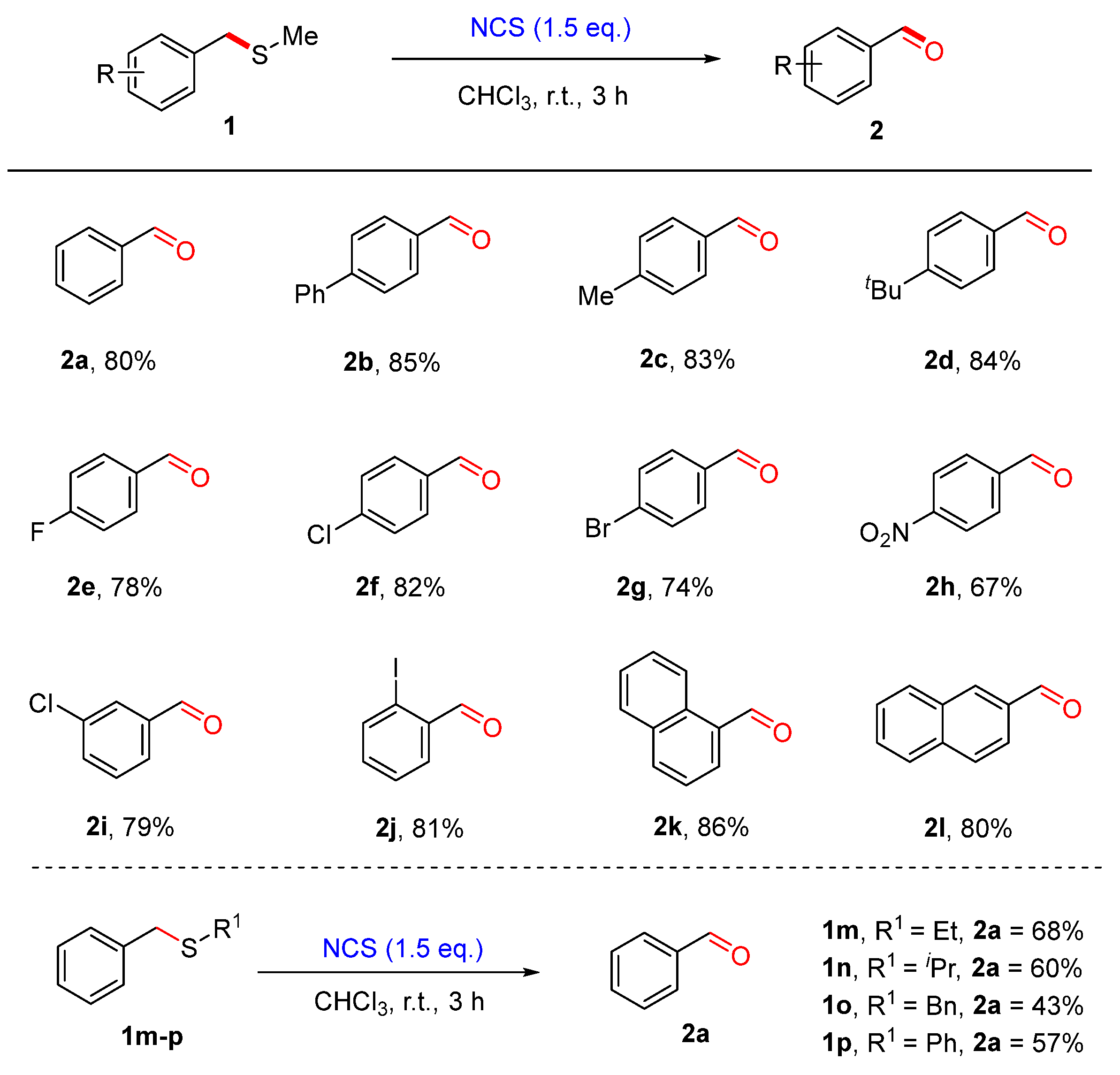
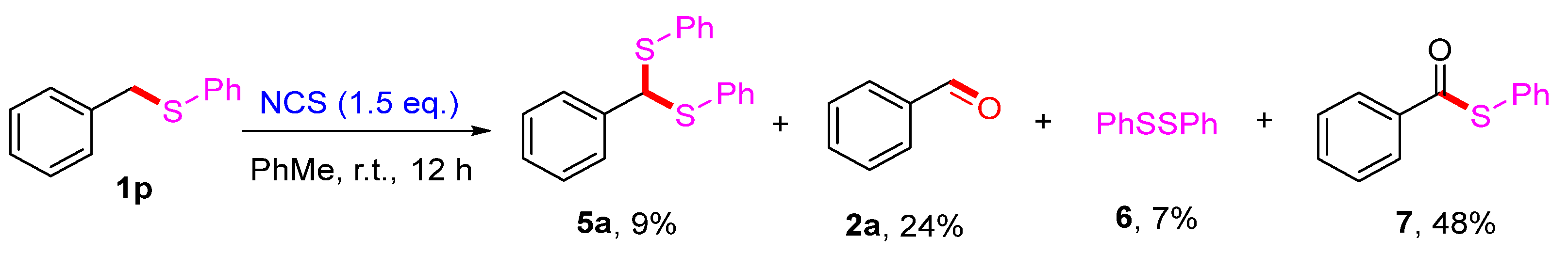
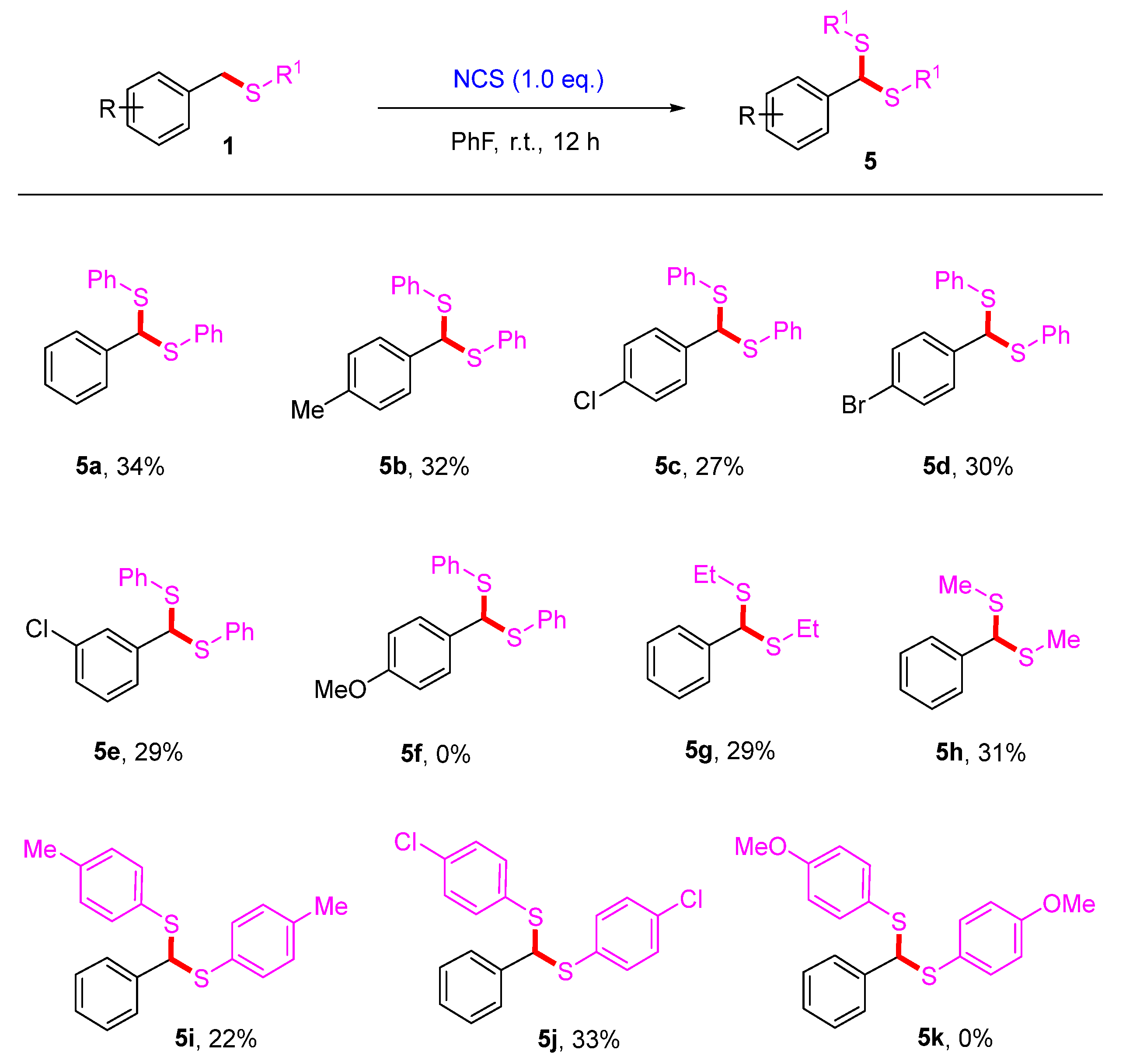
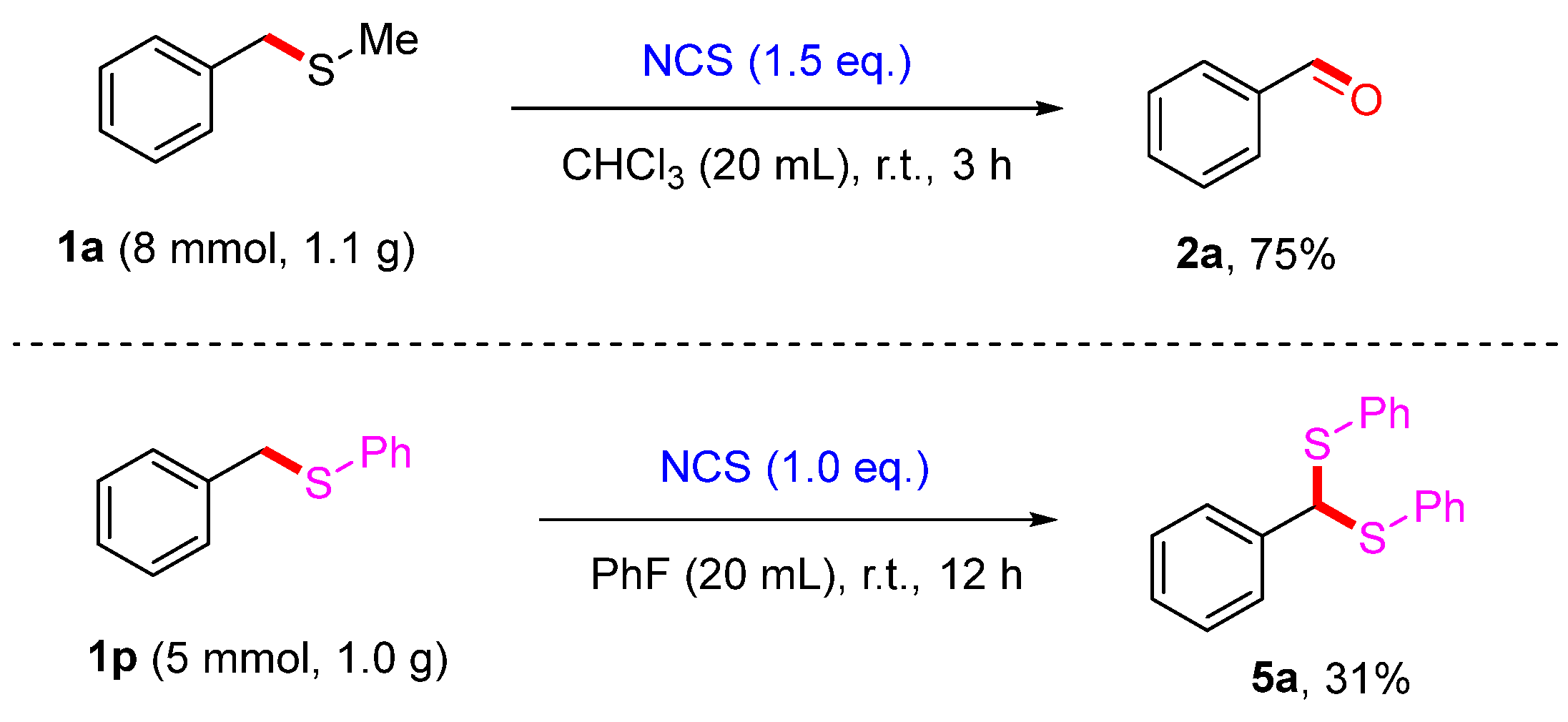
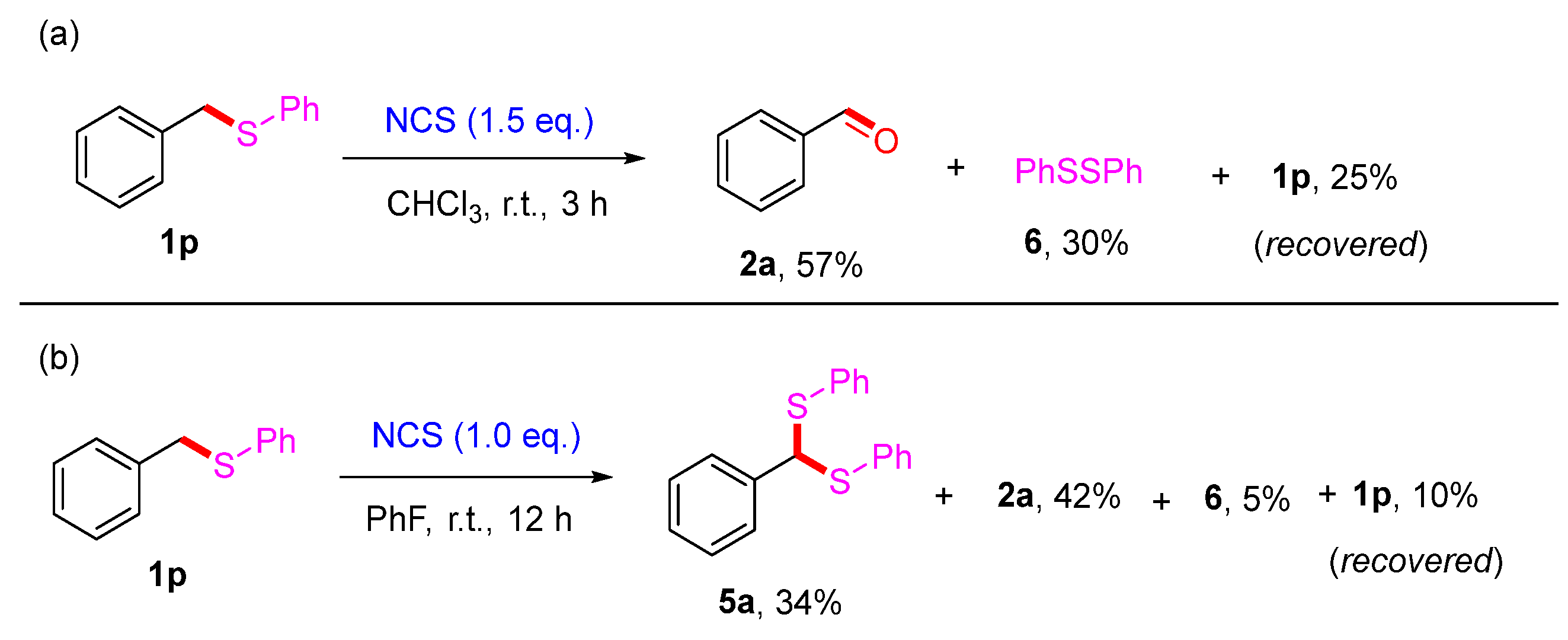
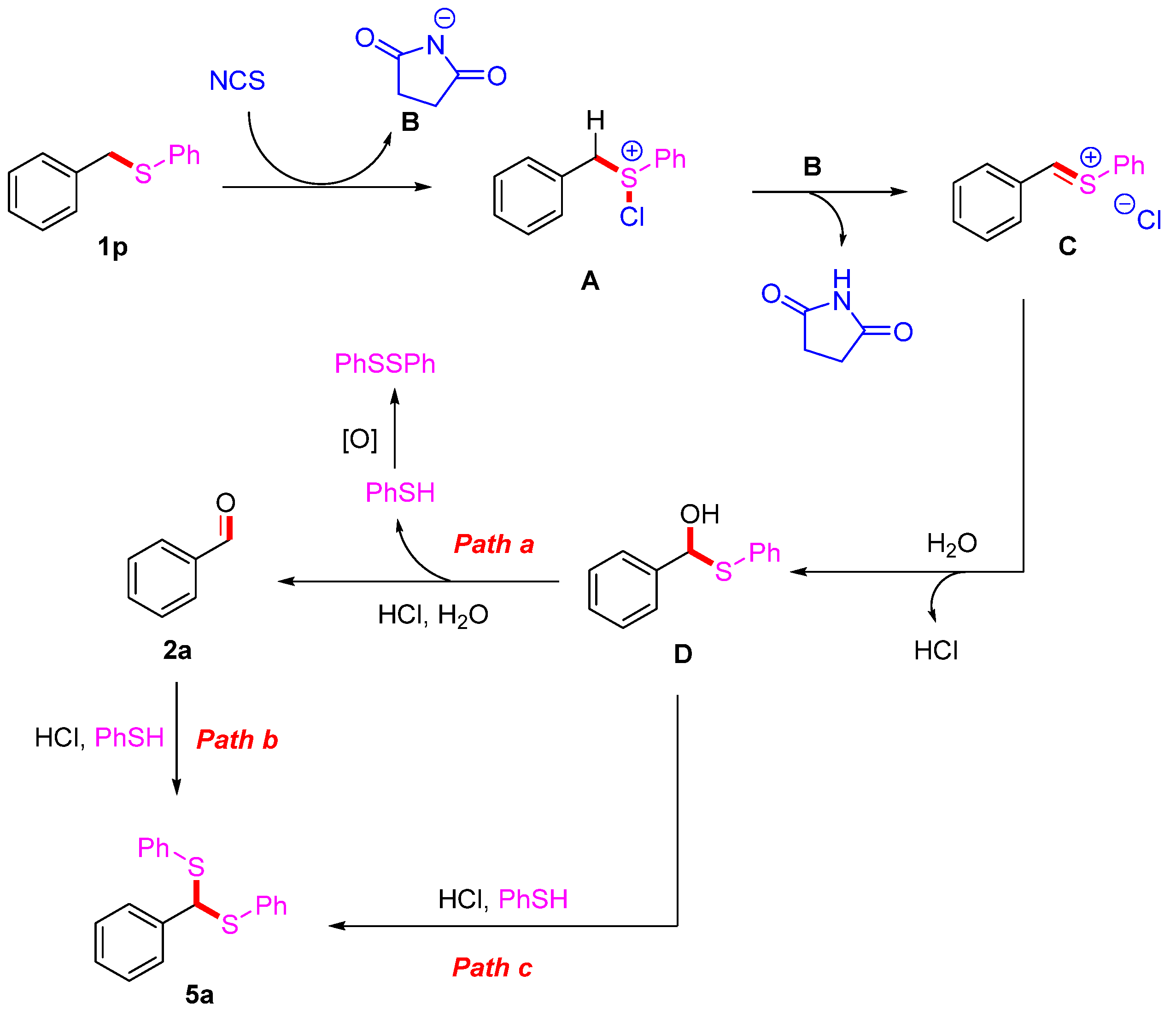
2. Results and Discussion
3. Materials and Methods
3.1. General Information
3.2. Optimization of the Reaction Conditions for Benzaldehyde 2a
3.3. Synthetic Procedures for the Synthesis of Products 2
3.4. Synthetic Procedures for the Synthesis of Products 4
3.5. Optimization of the Reaction Conditions for (Phenylmethylene)bis(phenylsulfane) (5a)
3.6. Synthetic Procedures for the Synthesis of Products 5
3.7. Synthetic Procedures for the Synthesis of (Phenylmethylene)bis(phenylsulfane) (5a), Benzaldehyde (2a), 1,2-Diphenyldisulfane (6) and S-Phenyl Benzothioate (7)
3.8. Synthetic Procedures for Gram-Scale Reactions
3.9. Control Experiments
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| NCS | N-Chlorosuccinimide |
| NCP | N-Chlorophthalimide |
| DCDMH | 1,3-Dichloro-5,5-dimethylhydantoin |
| NBS | N-Bromosuccinimide |
| NIS | N-Iodosuccinimide |
| Selectfluor | 1-Chloromethyl-4-fluoro-1,4-diazoniabicyclo[2.2.2]octane bis(tetrafluoroborate) |
| NFSI | N-Fluorobenzenesulfonamide |
| DCM | Dichloromethane |
| DCE | 1,2-Dichloroethane |
| DMSO | Dimethyl sulfoxide |
| DMF | N,N-Dimethylformamide |
References
- Kuzmin, J.; Rockl, J.; Schwarz, N.; Djossou, J.; Ahumada, G.; Ahlquist, M.; Lundberg, H. Electroreductive desulfurative transformations with thioethers as alkyl radical precursors. Angew. Chem. Int. Ed. 2023, 62, e202304272. [Google Scholar] [CrossRef] [PubMed]
- Tang, L.; Hu, Q.; Yang, K.; Elsaid, M.; Liu, C.; Ge, H. Recent advances in direct α-C(sp3)–H bond functionalization of thioethers. Green Synth. Catal. 2022, 3, 203–211. [Google Scholar] [CrossRef]
- Feng, X.; Wang, H.; Li, Z.; Tang, L.; Sun, X.; Yang, K. Transition-metal-catalyzed remote C–H functionalization of thioethers. RSC Adv. 2022, 12, 10835–10845. [Google Scholar] [CrossRef] [PubMed]
- Addison, N.D.; Jeenifer, A.L. Recent advances in well-defined, late transition metal complexes that make and/or break C–N, C–O and C–S bonds. Chem. Soc. Rev. 2017, 46, 197–238. [Google Scholar]
- David, C. Homolytic substitution at the sulfur atom as a tool for organic synthesis. Helv. Chim. Acta 2006, 89, 2167–2182. [Google Scholar]
- O’Mahony, G.E.; Ford, A.; Maguire, A.R. Asymmetric oxidation of sulfides. J. Sulfur Chem. 2013, 34, 301–341. [Google Scholar] [CrossRef]
- Wang, N.; Puli, S.; Jiang, X. Construction of sulfur-containing moieties in the total synthesis of natural products. Nat. Prod. Rep. 2020, 37, 246–275. [Google Scholar] [CrossRef]
- Sachdeva, H.; Khaturia, S.; Saquib, M.; Khatik, N.; Khandelwal, A.R.; Meena, R.; Sharma, K. Oxygen- and sulphur-containing heterocyclic compounds as potential anticancer agents. Appl. Biochem. Biotech. 2022, 194, 6438–6467. [Google Scholar] [CrossRef]
- Guo, W.; Wang, D.; Chen, Q.; Fu, Y. Advances of organosulfur materials for rechargeable metal batteries. Adv. Sci. 2022, 9, 2103989. [Google Scholar] [CrossRef]
- Wang, Y.; Li, Y.; Jiang, X. Sulfur-center-involved photocatalyzed reactions. Chem. Asian J. 2018, 13, 2208–2242. [Google Scholar] [CrossRef]
- Cinar, M.E.; Ozturk, T. Thienothiophenes, dithienothiophenes, and thienoacenes: Syntheses, oligomers, polymers, and properties. Chem. Rev. 2015, 115, 3036–3140. [Google Scholar] [CrossRef] [PubMed]
- Yorimitsu, H. Catalytic transformations of sulfonium salts via C–S Bond activation. Chem. Rec. 2021, 21, 3356–3369. [Google Scholar] [CrossRef]
- Huang, S.; Wang, M.; Jiang, X. Ni-catalyzed C–S bond construction and cleavage. Chem. Soc. Rev. 2022, 51, 8351–8377. [Google Scholar] [CrossRef] [PubMed]
- Lou, J.; Wang, Q.; Wu, P.; Wang, H.; Zhou, Y.; Yu, Z. Transition-metal mediated carbon–sulfur bond activation and transformations: An update. Chem. Soc. Rev. 2020, 49, 4307–4359. [Google Scholar] [CrossRef]
- Pan, F.; Shi, Z. Recent advances in transition-metal-catalyzed C–S activation: From thioester to (hetero)aryl thioether. ACS Catal. 2014, 4, 280–288. [Google Scholar] [CrossRef]
- Modha, S.G.; Mehta, V.P.; Eycken, E.V.V. Transition metal-catalyzed C–C bond formation via C–S bond cleavage: An overview. Chem. Soc. Rev. 2013, 42, 5042–5055. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; He, W.; Yu, Z. Transition-metal mediated carbon–sulfur bond activation and transformations. Chem. Soc. Rev. 2013, 42, 599–621. [Google Scholar] [CrossRef]
- Yang, K.; Li, Q.; Li, Z.; Sun, X. Transition-metal-free C–S bond cleavage and transformation of organosulfur compounds. Chem. Commun. 2023, 59, 5343–5364. [Google Scholar] [CrossRef]
- Fan, R.; Tan, C.; Liu, Y.; Wei, Y.; Zhao, X.; Liu, X.; Tan, J.; Yoshida, H. A leap forward in sulfonium salt and sulfur ylide chemistry. Chin. Chem. Lett. 2021, 32, 299–312. [Google Scholar] [CrossRef]
- Yang, K.; Song, M.; Ali, A.I.M.; Mudassir, S.M.; Ge, H. Recent advances in the application of Selectfluor as a “fluorine-free” functional reagent in organic synthesis. Chem. Asian J. 2020, 15, 729–741. [Google Scholar] [CrossRef]
- Li, Y.; Wang, H.; Wang, Z.; Alhumade, H.; Huang, Z.; Lei, A. Electrochemical radical-mediated selective C(sp3)–S bond activation. Chem. Sci. 2023, 14, 372–378. [Google Scholar] [CrossRef] [PubMed]
- Canestrari, D.; Lancianesi, S.; Badiola, E.; Strinna, C.; Ibrahim, H.; Adamo, M.F.A. Desulfurative chlorination of alkyl phenyl sulfides. Org. Lett. 2017, 19, 918–921. [Google Scholar] [CrossRef] [PubMed]
- Hugenberg, V.; Haufe, G. Fluoro-Pummerer rearrangement and analogous reactions. J. Fluorine Chem. 2012, 143, 238–262. [Google Scholar] [CrossRef]
- Kaiser, D.; Klose, I.; Oost, R.; Neuhaus, J.; Maulide, N. Bond-forming and -breaking reactions at sulfur(IV): Sulfoxides, sulfonium salts, sulfur ylides, and sulfinate salts. Chem. Rev. 2019, 119, 8701–8780. [Google Scholar] [CrossRef]
- Li, M.; Gu, A.; Li, J.; Liu, Y. Advanced green synthesis: Solvent-free and catalyst-free reaction. Green Synth. Catal. 2025, 6, 36–66. [Google Scholar] [CrossRef]
- Wang, F.; Wang, B.; Wang, Q.; Wang, L. Photochemical-, electrochemical-, and photoelectrochemical-catalyzed hydrogen atom transfer from aldehydes to acyl radicals and their transformations. Eur. J. Org. Chem. 2025, 28, e202401206. [Google Scholar] [CrossRef]
- Kim, H.; Walsh, P.J. Efficient approaches to the stereoselective synthesis of cyclopropyl alcohols. Acc. Chem. Res. 2012, 45, 1533–1547. [Google Scholar] [CrossRef] [PubMed]
- Parenty, A.; Moreau, X.; Campagne, J.M. Macrolactonizations in the total synthesis of natural products. Chem. Rev. 2006, 106, 911–939. [Google Scholar] [CrossRef]
- Burghardt, T.E. Developments in the deprotection of thioacetals. J. Sulfur Chem. 2005, 26, 411–427. [Google Scholar] [CrossRef]
- Wei, C.; Zhao, L.; Sun, Z.; Hu, D.; Song, B. Discovery of novel indole derivatives containing dithioacetal as potential antiviral agents for plants. Pestic. Biochem. Physiol. 2020, 166, 104568. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, J.; He, F.; Gan, X.; Song, B.; Hu, D. Design, synthesis, bioactivity and mechanism of dithioacetal derivatives containing dioxyether moiety. Bioorganic Med. Chem. Lett. 2019, 29, 2218–2223. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Liu, X.; He, W. Recent advances in the photocatalytic synthesis of aldehydes. Org. Chem. Front. 2023, 10, 4198–4210. [Google Scholar] [CrossRef]
- Roman, B.I.; Kimpe, N.D.; Stevens, C.V. Synthesis of β-, γ-, δ-, …, ω-halogenated ketones and aldehydes. Chem. Rev. 2010, 110, 5914–5988. [Google Scholar] [CrossRef] [PubMed]
- Roy, B. Recent developments in the greener approaches for the dithioacetalization of carbonyl compounds. J. Sulfur Chem. 2023, 44, 779–813. [Google Scholar] [CrossRef]
- Ryohei, O.; Hayashi, Y. Mechanism of the oxidation of α-active-methylene sulfides by dimethyl sulfoxide and benzoyl chloride. Tetrahedron Lett. 1967, 32, 3141. [Google Scholar]
- Bakuzis, P.; Bakuzis, M.L.F.; Fortes, C.C.; Santos, R. The sulfide group as an aldehyde precursor. J. Org. Chem. 1976, 41, 2769–2770. [Google Scholar] [CrossRef]
- Luo, Y.; Yuan, D.; Li, Q.; Zhou, F.; Xu, Z.; Sun, X.; Tang, L.; Yang, K. Metal-free and NBS-mediated C(sp3)–S bond cleavage of thioethers to access alkyl bromides. ChemistrySelect 2024, 9, e202401194. [Google Scholar] [CrossRef]
- Yang, K.; Luo, Y.; Hu, Q.; Song, M.; Li, Z.; Li, B.; Sun, X. Selective C(sp3)–S bond cleavage of thioethers to build up unsymmetrical disulfides. J. Org. Chem. 2023, 88, 13699–13711. [Google Scholar] [CrossRef]
- Li, Y.; Hu, Q.; Zhang, F.; Li, Z.; Sun, X.; Yang, K. Metal-free selective C–S bond cleavage of thioethers to access β-alkoxy carbonyl compounds. ChemistrySelect 2021, 6, 6268–6271. [Google Scholar] [CrossRef]
- Yang, K.; Li, Y.; Ma, Z.; Tang, L.; Yue, Y.; Zhang, H.; Li, Z.; Sun, X. Metal-free C–S bond cleavage to access N-substituted acrylamide and β-aminopropanamide. Eur. J. Org. Chem. 2019, 2019, 5812–5814. [Google Scholar] [CrossRef]
- Murphy, M.; Lynch, D.; Schaeffer, M.; Kissane, M.; Chopra, J.; O’Brien, E.; Ford, A.; Fergusonb, G.; Maguire, A.R. Investigation of the synthetic and mechanistic aspects of the highly stereoselective transformation of α-thioamides to α-thio-β-chloroacrylamides. Org. Biomol. Chem. 2007, 5, 1228–1241. [Google Scholar] [CrossRef] [PubMed]
- Foley, D.A.; Doecke, C.W.; Buser, J.Y.; Merritt, J.M.; Murphy, L.; Kissane, M.; Collins, S.G.; Maguire, A.R.; Kaerner, A. ReactNMR and reactIR as reaction monitoring and mechanistic elucidation tools: The NCS mediated cascade reaction of α-thioamides to α-thio-β-chloroacrylamides. J. Org. Chem. 2011, 76, 9630–9640. [Google Scholar] [CrossRef]
- Nimbhal, A.; Singh, R. N-Chlorosuccinimide: A versatile reagent in organic synthesis. Curr. Org. Chem. 2024, 29, 936–950. [Google Scholar] [CrossRef]
- Chauhan, P. N-Chlorosuccinimide (NCS). Synlett 2010, 8, 1285–1286. [Google Scholar] [CrossRef]
- Golebiewski, W.M.; Miroslaw, G. Applications of N-chlorosuccinimide in organic synthesis. Synthesis 2007, 23, 3599–3619. [Google Scholar] [CrossRef]
- Abraham, R.J.; Mobli, M.; Smith, R.J. 1H chemical shifts in NMR: Part 19. carbonyl anisotropies and steric effects in aromatic aldehydes and ketones. Magn. Reson. Chem. 2003, 41, 26–36. [Google Scholar] [CrossRef]
- Li, Y.; Mao, F.; Chen, T.; Zhou, Z.; Wang, Y.; Huang, J. In situ trapped and immobilized palladium nanoparticles as active and clean catalysts for Suzuki-Miyaura reaction. Adv. Synth. Catal. 2015, 357, 2827–2832. [Google Scholar] [CrossRef]
- Hong, B.; Tseng, H.; Chen, S. Synthesis of aromatic aldehydes by organocatalytic [4+2] and [3+3] cycloaddition of α,β-unsaturated aldehydes. Tetrahedron 2007, 63, 2840–2850. [Google Scholar] [CrossRef]
- Terstiege, I.; Maleczka, R.E. A new approach for the generation and reaction of organotin hydrides: The development of reactions catalytic in Tin. J. Org. Chem. 1999, 64, 342–343. [Google Scholar] [CrossRef]
- Feng, Q.; Song, Q. Aldehydes and ketones formation: Copper-catalyzed aerobic oxidative decarboxylation of phenylacetic acids and α-hydroxyphenylacetic acids. J. Org. Chem. 2014, 79, 1867–1871. [Google Scholar] [CrossRef]
- Iinuma, M.; Moriyama, K.; Togo, H. Various oxidative reactions with novel ion-supported (diacetoxyiodo)benzenes. Tetrahedron 2013, 69, 2961–2970. [Google Scholar] [CrossRef]
- Hu, P.; Tan, M.; Cheng, L.; Zhao, H.; Feng, R.; Gu, W.; Han, W. Bio-inspired iron-catalyzed oxidation of alkylarenes enables late-stage oxidation of complex methylarenes to arylaldehydes. Nat. Commun. 2019, 10, 2425. [Google Scholar] [CrossRef]
- Zhao, Y.; Snieckus, V. A practical in situ generation of the Schwartz reagent. Reduction of tertiary amides to aldehydes and hydrozirconation. Org. Lett. 2014, 16, 390–393. [Google Scholar] [CrossRef]
- Imai, S.; Togo, H. Synthetic utility of iodic acid in the oxidation of benzylic alcohols to aromatic aldehydes and ketones. Tetrahedron 2016, 72, 6948–6954. [Google Scholar] [CrossRef]
- Cabrera, S.; Arrayas, R.G.; Martın-Matute, B.; Cossıob, F.P.; Carreteroa, J.C. CuI-fesulphos complexes: Efficient chiral catalysts for asymmetric 1,3-dipolar cycloaddition of azomethine ylides. Tetrahedron 2007, 63, 6587–6602. [Google Scholar] [CrossRef]
- Vale, J.R.; Rimpilainen, T.; Sievanen, E.; Rissanen, K.; Afonso, C.A.M.; Candeias, N.R. Pot-economy autooxidative condensation of 2-aryl-2-lithio-1,3-dithianes. J. Org. Chem. 2018, 83, 1948–1958. [Google Scholar] [CrossRef] [PubMed]
- Arunprasath, D.; Sekar, G. A transition-metal-free and base-mediated carbene insertion into sulfur-sulfur and selenium-selenium bonds: An easy access to thio- and selenoacetals. Adv. Synth. Catal. 2017, 359, 698–708. [Google Scholar] [CrossRef]
- Kumar, A.; Rao, M.S.; Rao, V.K. Cerium triflate: An efficient and recyclable catalyst for chemoselective thioacetalization of carbonyl compounds under solvent-free conditions. Aust. J. Chem. 2010, 63, 135–140. [Google Scholar] [CrossRef]
- Wu, Y.C.; Zhu, J. Hafnium trifluoromethanesulfonate (hafnium triflate) as a highly efficient catalyst for chemoselective thioacetalization and transthioacetalization of carbonyl compounds. J. Org. Chem. 2008, 73, 9522–9524. [Google Scholar] [CrossRef]
- Soederstroem, M.; Matt, C.; Odell, L.R. Thioacetalation and multi-component thiomethylative friedel-crafts arylation using BF3SMe2. ACS Omega 2023, 8, 4320–4330. [Google Scholar] [CrossRef]
- Orrillo, A.G.; Escalante, A.M.; Furlan, R.L.E. Dithioacetal exchange: A new reversible reaction for dynamic combinatorial chemistry. Chem. Eur. J. 2016, 22, 6746–6749. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Li, Z.; Gao, Y.; Meng, Q.; Yu, S.; Weiss, R.G.; Tung, C.; Wu, L. Mechanistic insights into the interface-directed transformation of thiols into disulfides and molecular hydrogen by visible-light irradiation of quantum dots. Angew. Chem. Int. Ed. 2014, 53, 2085–2089. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Yu, S.; Kim, J.G.; Lee, S. Palladium-catalyzed carbonylation of thioacetates and aryl iodides for the synthesis of S-aryl thioesters. Org. Chem. Front. 2018, 5, 2447–2452. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Electrophilic Halogenation Reagents (eq.) | Solvent (mL) | Yield (2a, %) |
|---|---|---|---|
| 1 | NCS (1.5) | DCM | 61 |
| 2 | NCS (1.5) | DCE | 54 |
| 3 | NCS (1.5) | MeCN | 56 |
| 4 | NCS (1.5) | 1,4-Dioxane | 62 |
| 5 | NCS (1.5) | DMSO | trace |
| 6 | NCS (1.5) | DMF | 65 |
| 7 | NCS (1.5) | MeOH | trace |
| 8 | NCS (1.5) | PhMe | 52 |
| 9 | DCDMH (1.5) | CHCl3 | 38 |
| 10 | NCP (1.5) | CHCl3 | 75 |
| 11 | NBS (1.5) | CHCl3 | 70 |
| 12 | NIS (1.5) | CHCl3 | trace |
| 13 | Selectfluor (1.5) | CHCl3 | trace |
| 14 | NFSI (1.5) | CHCl3 | 44 |
| 15 | NCS (1.0) | CHCl3 | 51 |
| 16 | NCS (2.0) | CHCl3 | 73 |
| 17 | - | CHCl3 | 0 |
| Entry | Electrophilic Halogenation Reagents (eq.) | Solvent (mL) | Yield (5a, %) | Yield (2a, %) | Yield (6, %) | Yield (7, %) |
|---|---|---|---|---|---|---|
| 1 | NCS (1.5) | PhF | 13 | 21 | 7 | 40 |
| 2 | NCS (1.5) | PhCF3 | 6 | 22 | 10 | 47 |
| 3 | NCS (1.5) | MeCN | 0 | 43 | 25 | 0 |
| 4 | NCS (1.5) | DCE | 0 | 50 | 28 | 0 |
| 5 | NCS (1.5) | CHCl3 | 0 | 71 | 38 | 0 |
| 6 | NCS (1.5) | THF | 0 | trace | trace | 0 |
| 7 | NCS (1.5) | MeOH | 0 | trace | trace | 0 |
| 8 | NCS (1.0) | PhF | 34 | 42 | 5 | 0 |
| 9 | NCS (2.0) | PhF | trace | trace | trace | 73 |
| 10 | NBS (1.0) | PhF | 0 | trace | trace | 0 |
| 11 | NIS (1.0) | PhF | 0 | trace | trace | 0 |
| 12 | Selectfluor (1.0) | PhF | 0 | trace | trace | 0 |
| 13 | NFSI (1.0) | PhF | 0 | trace | trace | 0 |
| 14 | DCDMH (1.0) | PhF | 0 | trace | trace | 45% |
| 15 | NCP (1.0) | PhF | 12 | 30 | 10 | 0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yuan, D.; Huang, Y.; Tang, L.; Yang, K. Metal-Free C(sp3)–S Bond Cleavage of Thioethers to Selectively Access Aryl Aldehydes and Dithioacetals. Chemistry 2025, 7, 89. https://doi.org/10.3390/chemistry7030089
Yuan D, Huang Y, Tang L, Yang K. Metal-Free C(sp3)–S Bond Cleavage of Thioethers to Selectively Access Aryl Aldehydes and Dithioacetals. Chemistry. 2025; 7(3):89. https://doi.org/10.3390/chemistry7030089
Chicago/Turabian StyleYuan, Dan, Yong Huang, Long Tang, and Ke Yang. 2025. "Metal-Free C(sp3)–S Bond Cleavage of Thioethers to Selectively Access Aryl Aldehydes and Dithioacetals" Chemistry 7, no. 3: 89. https://doi.org/10.3390/chemistry7030089
APA StyleYuan, D., Huang, Y., Tang, L., & Yang, K. (2025). Metal-Free C(sp3)–S Bond Cleavage of Thioethers to Selectively Access Aryl Aldehydes and Dithioacetals. Chemistry, 7(3), 89. https://doi.org/10.3390/chemistry7030089