
Tetraanion of Tetracyclopentatetraphenylene Derivative: Global Versus Local Conjugation Modes


 , ,
, ,  and
and
Abstract
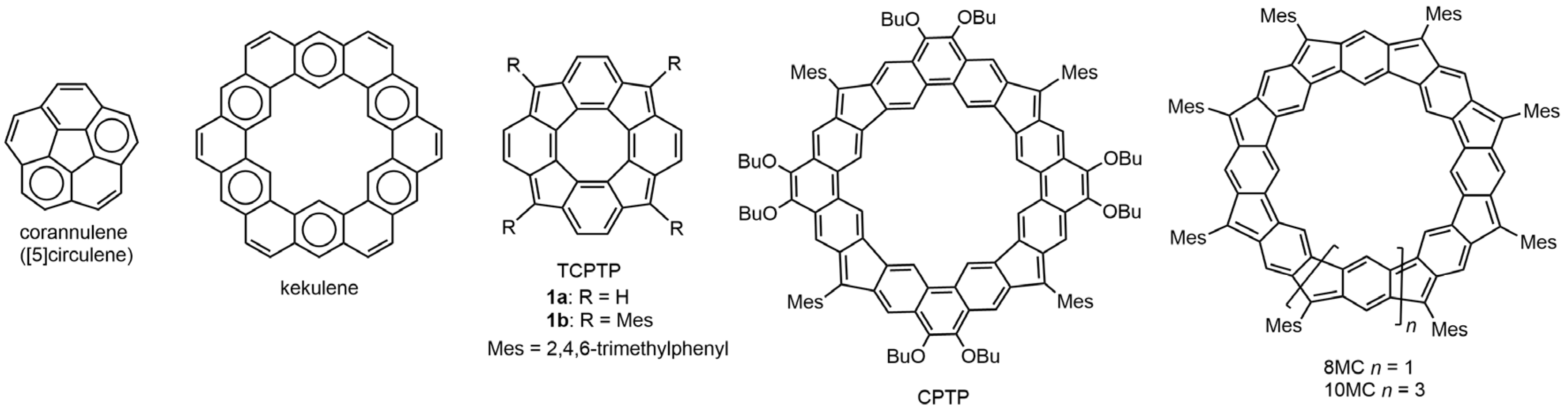
1. Introduction
2. Materials and Methods
2.1. Experimental Details
2.1.1. Reduction of 1b
2.1.2. Magnetic Circular Dichroism Measurements
2.2. Computational Details
3. Results
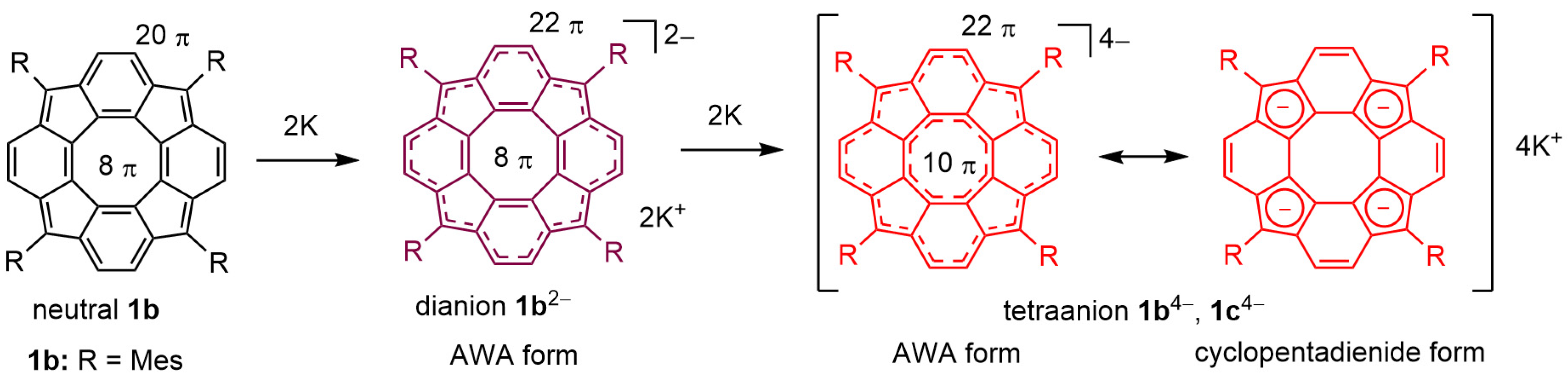
3.1. Generation and Characterization of Tetraanion 1b4−
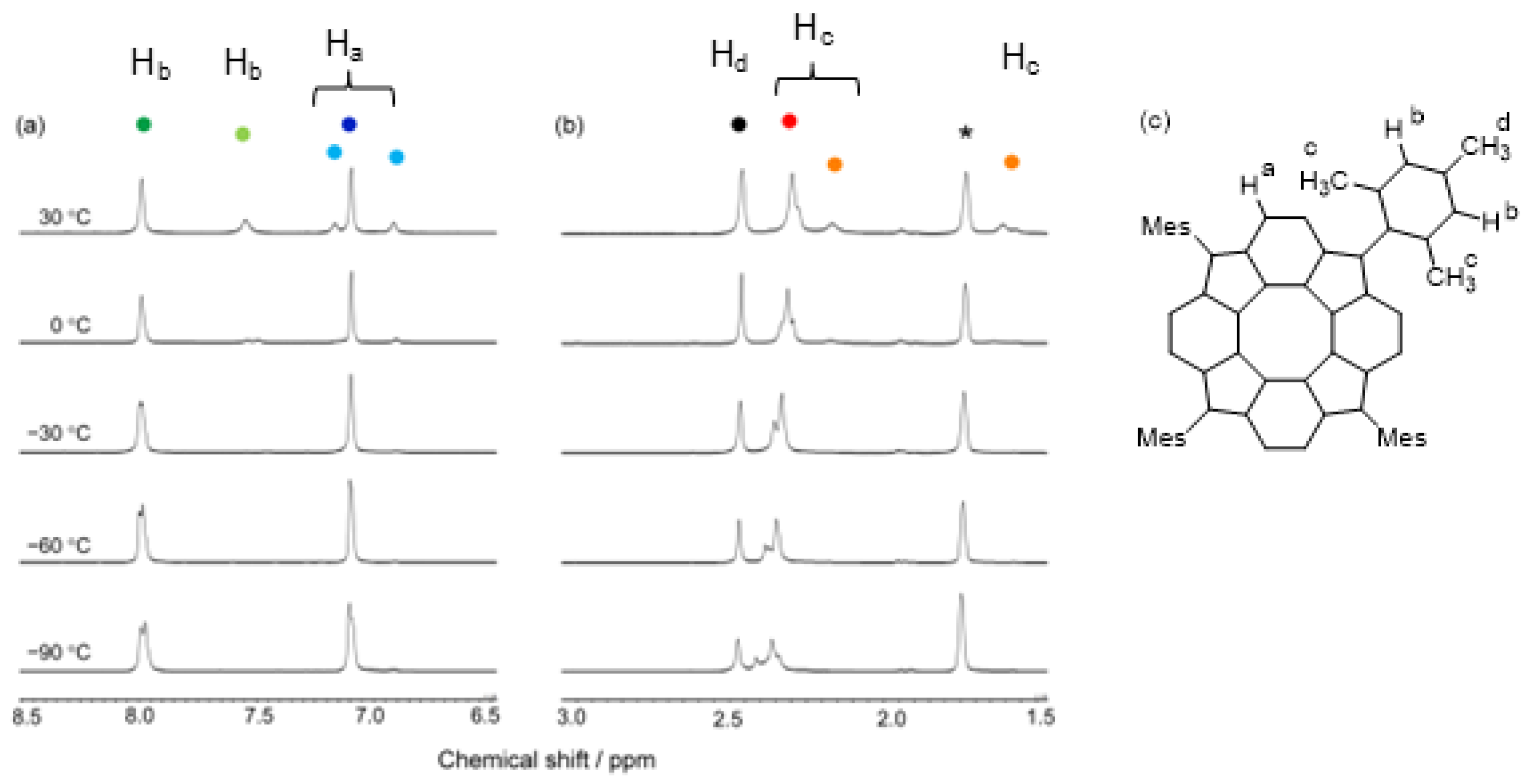
3.1.1. 1H NMR Spectroscopy
3.1.2. 13C NMR Spectroscopy
3.1.3. Magnetic Circular Dichroism Spectroscopy
3.2. Quantum Chemical Calculations for Tetraanion 1b4−
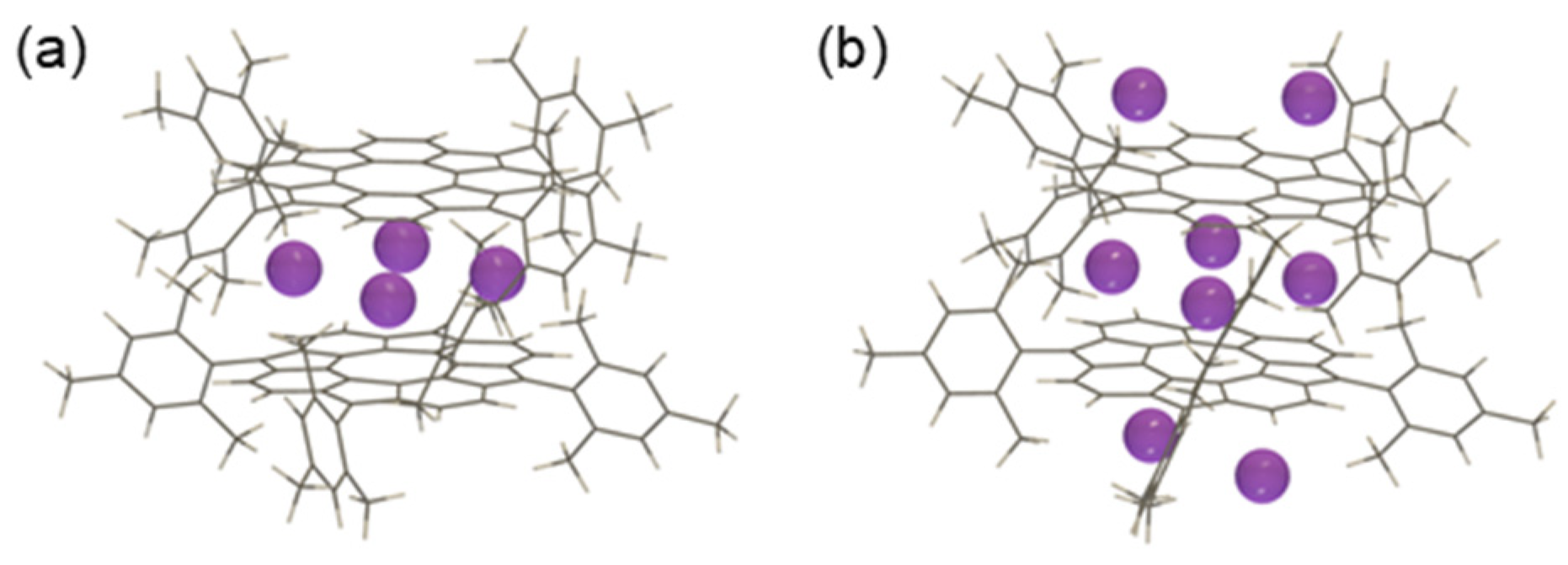
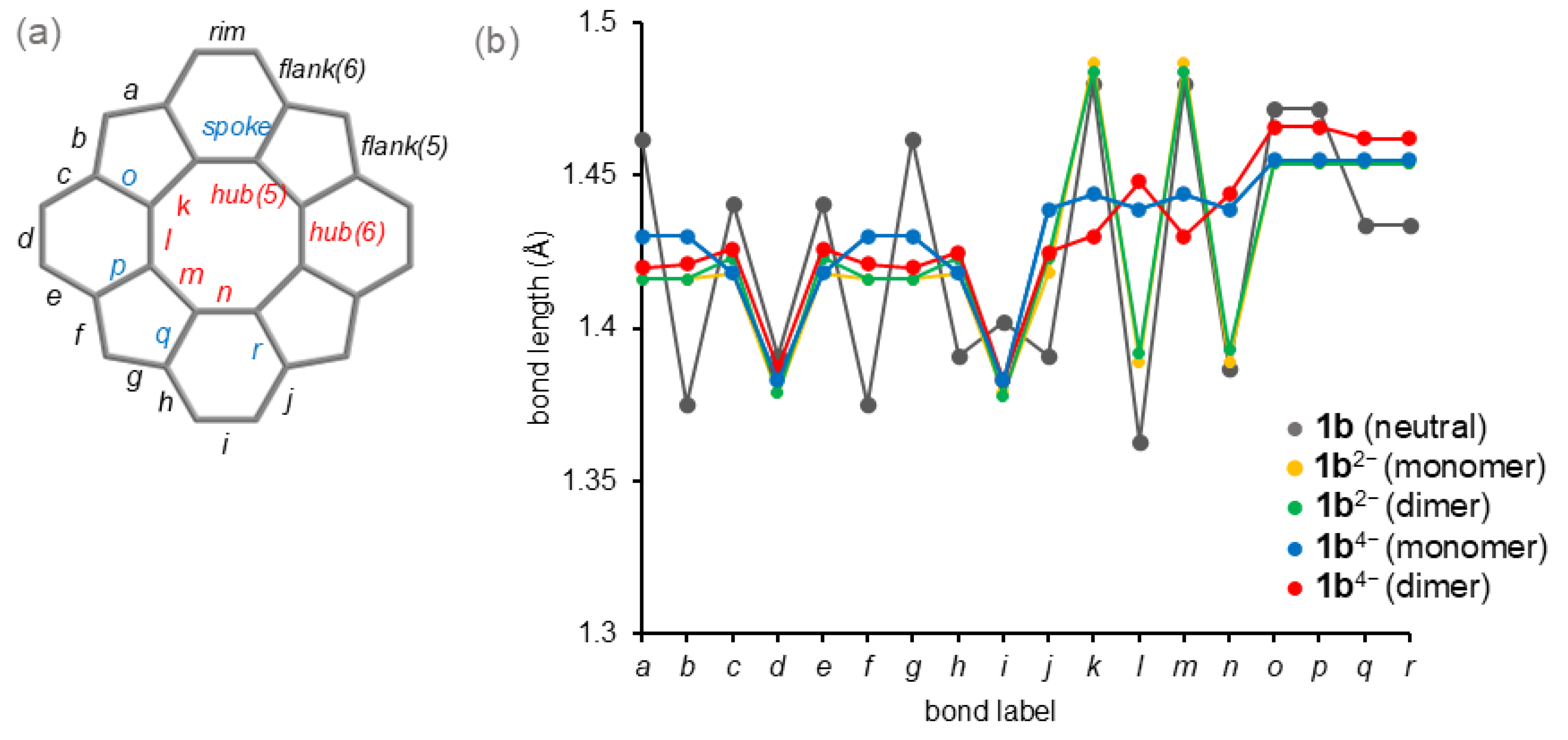
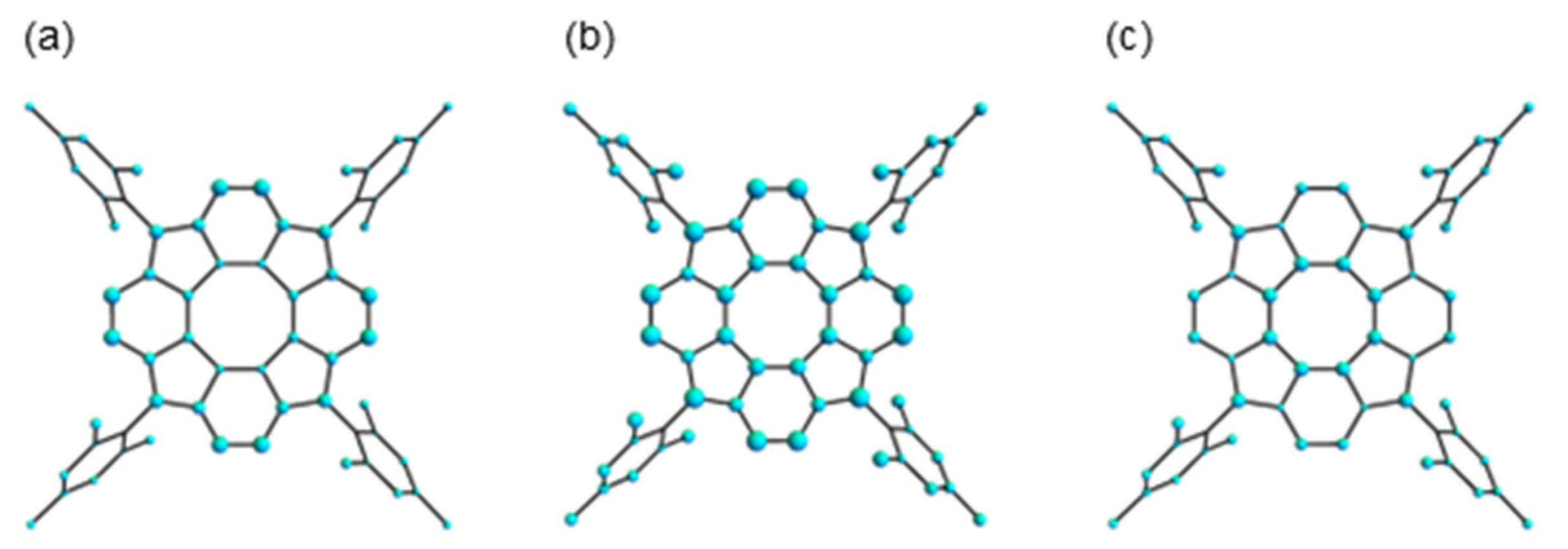
3.2.1. Structure Optimization
3.2.2. Theoretical Bond Lengths
3.2.3. Theoretical Charge Distributions
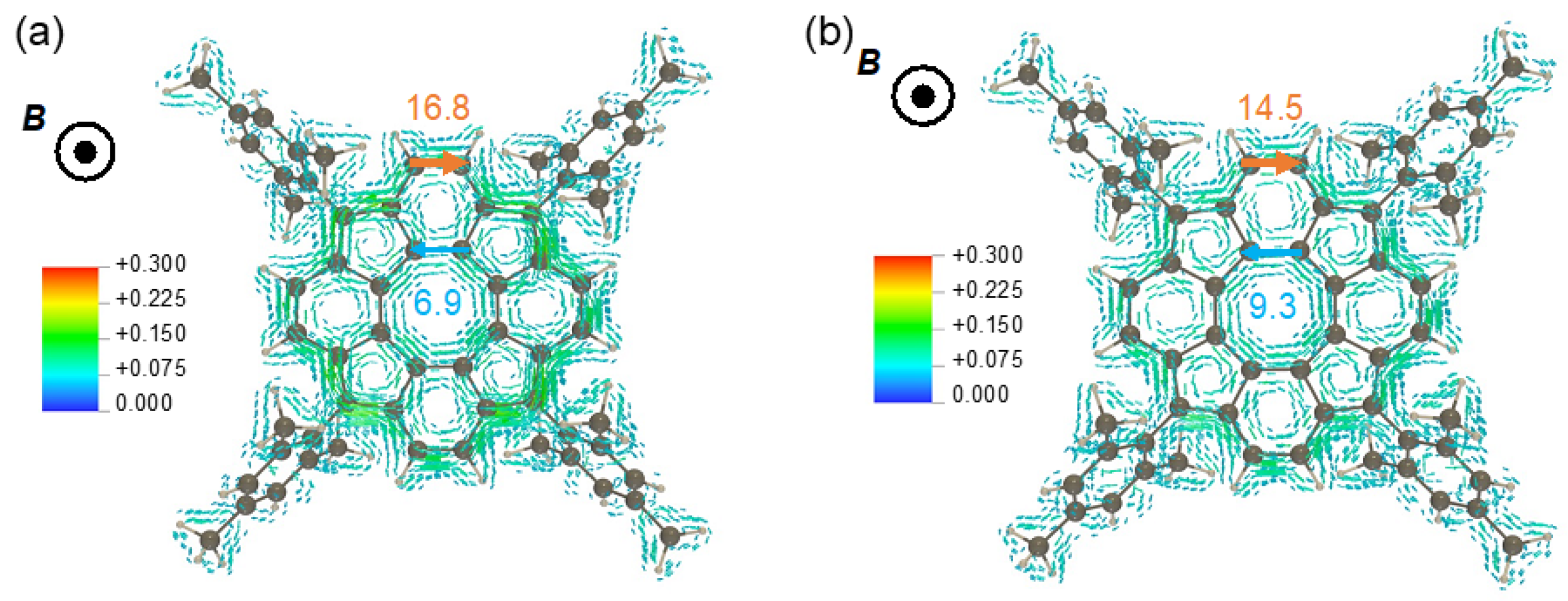
3.2.4. Magnetically Induced Ring Current Tropicities
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
| AWA | Annulene-within-an-annulene |
| BLA | Bond length alternation |
| TCPTP | Tetracyclopentatetraphenylene |
| TC | Tetracyclopentatetraphenanthlenyene |
| GIMIC | Gauge-including magnetically induced current |
| ACID | Anisotropy-induced current density |
Appendix A
References
- Müllen, K. Reduction and Oxidation of Annulenes. Chem. Rev. 1984, 84, 603–646. [Google Scholar]
- Bock, H.; Ruppert, K.; Näther, C.; Havlas, Z.; Herrmann, H.F.; Arad, C.; Göbel, I.; John, A.; Meuret, J.; Nick, S.; et al. Distorted Molecules: Perturbation Design, Preparation and Structures. Angew. Chem. Int. Ed. Engl. 2003, 31, 550–581. [Google Scholar]
- Benshafrut, R.; Shabtai, E.; Rabinovitz, M.; Scott, L.T. π-Conjugated Anions: From Carbon-Rich Anions to Charged Carbon Allotropes. Eur. J. Org. Chem. 2000, 2000, 1091–1106. [Google Scholar]
- Zabula, A.V.; Spisak, S.N.; Filatov, A.S.; Rogachev, A.Y.; Petrukhina, M.A. Record Alkali Metal Intercalation by Highly Charged Corannulene. Acc. Chem. Res. 2018, 51, 1541–1549. [Google Scholar] [PubMed]
- Katz, T.J. The Cyclooctatetraenyl Dianion. J. Am. Chem. Soc. 1960, 82, 3784–3785. [Google Scholar]
- Katz, T.J. The Chemical Interaction of the Cyclooctatetraenyl Anions. J. Am. Chem. Soc. 1960, 82, 3875–3876. [Google Scholar]
- Goldberg, S.G.; Raymond, K.N.; Harmon, C.A.; Templeton, D.H. Structure of the 10π Electron Cyclooctatetraene Dianion in Potassium Diglyme 1,3,5,7-Tetramethylcyclooctatetraene Dianion, [K((CH3OCH2CH2)2O)]2[C8H4(CH3)4]. J. Am. Chem. Soc. 1974, 96, 1348–1351. [Google Scholar]
- Müllen, K. The Dianions of Phenanthrene and 1,2,3,4-Dibenzocyclooctatetraene. Helv. Chim. Acta 1978, 61, 1296–1304. [Google Scholar]
- Paquette, L.A.; Ewing, G.D.; Traynor, E.; Gardlik, J.M. Dicyclooctatetraeno[1,2:4,5]benzene Dianion and Tetraanion. Experimental Assessment of Extended Paratropic vs. Restricted Diatropic π-Electron Delocalization. J. Am. Chem. Soc. 1977, 99, 6115–6117. [Google Scholar]
- Günther, H.; Günther, M.-E.; Mondeshka, D.; Schmickler, H. Der Benzolkern als Sonde für die π-Elektronenstruktur von Annulenen; Teil III: Dibenzoannulene und Dibenzoannulen-Ionen. Liebigs Ann. Chem. 1978, 1978, 165–175. [Google Scholar]
- Sygula, A.; Fronczek, F.R.; Rabideau, P.W. The first example of η8 coordination of lithium cations with a cyclooctatctraene dianion: Crystal structure of Li2(dibcnzo[a,c]cyclooctatetracne)(TMEDA)2. J. Organomet. Chem. 1996, 526, 389–391. [Google Scholar] [CrossRef]
- Zhou, Z.; Zhu, Y.; Wei, Z.; Bergner, J.; Neiss, C.; Doloczki, S.; Gorling, A.; Kivala, M.; Petrukhina, M.A. Reduction of π-Expanded Cyclooctatetraene with Lithium: Stabilization of the Tetra-Anion through Internal Li+ Coordination. Angew. Chem. Int. Ed. Engl. 2021, 60, 3510–3514. [Google Scholar] [CrossRef] [PubMed]
- Huber, W.; Müllen, K.; Wennerström, O. Dianion and Tetraanion of [24] Paracyclophanetetraene—New Unusual Perimeter Systems. Angew. Chem. Int. Ed. Engl. 1980, 19, 624–625. [Google Scholar] [CrossRef]
- Shabtai, E.; Segev, O.; Beust, R.; Rabinovitz, M. Charged paracyclophanes behave as annulenes with enhanced anisotropy. J. Chem. Soc. Perkin Trans. 2000, 2, 1233–1241. [Google Scholar] [CrossRef]
- Heinz, W.; Räder, H.-J.; Müllen, K. Chanzing the Size of a Cavity via an Electron-Transfer: Synthesis and Reduction of 1,5,22,26-Tetraoxa-[5,5]-(2,8)dibenzo[a,e]cyclooctatetraenophane. Tetrahedron Lett. 1989, 30, 159–162. [Google Scholar] [CrossRef]
- Tasić, M.; Ivković, J.; Carlström, G.; Melcher, M.; Bollella, P.; Bendix, J.; Gorton, L.; Persson, P.; Uhlig, J.; Strand, D. Electro-mechanically switchable hydrocarbons based on [8]annulenes. Nat. Commun. 2022, 13, 860. [Google Scholar] [CrossRef]
- Barth, W.E.; Lawton, R.G. The Synthesis of Corannulene. J. Am. Chem. Soc. 1971, 93, 1730–1745. [Google Scholar]
- Scott, L.T.; Hashemi, M.M.; Meyer, D.T.; Warren, H.B. Corannulene. A Convenient New Synthesis. J. Am. Chem. Soc. 1991, 113, 7082–7084. [Google Scholar] [CrossRef]
- Scott, L.T.; Cheng, P.-C.; Hashemi, M.M.; Bratcher, M.S.; Meyer, D.T.; Warren, H.B. Corannulene. A Three-Step Synthesis. J. Am. Chem. Soc. 1997, 119, 10963–10968. [Google Scholar] [CrossRef]
- Seiders, T.J.; Elliott, E.L.; Grube, G.H.; Siegel, J.S. Synthesis of Corannulene and Alkyl Derivatives of Corannulene. J. Am. Chem. Soc. 1999, 121, 7804–7813. [Google Scholar] [CrossRef]
- Wu, Y.-T.; Siegel, J.S. Aromatic Molecular-Bowl Hydrocarbons: Synthetic Derivatives, Their Structures, and Physical Properties. Chem. Rev. 2006, 106, 4843–4867. [Google Scholar] [CrossRef] [PubMed]
- Diederich, F.; Staab, H.A. Benzenoid versus Annulenoid Aromaticity: Synthesis and Properties of Kekulene. Angew. Chem. Int. Ed. Engl. 2003, 17, 372–374. [Google Scholar] [CrossRef]
- Staab, H.A.; Diederich, F. Synthesis of Kekulene. Chem. Ber. 1983, 116, 3487–3503. [Google Scholar] [CrossRef]
- Majewski, M.A.; Hong, Y.; Tadeusz Lis, J.; Gregoliński; Chmielewski, P.J.; Cybińska, J.; Kim, D.; Stępień, M. Octulene: A Hyperbolic Molecular Belt that Binds Chloride Anions. Angew. Chem. Int. Ed. 2016, 55, 14072–14076. [Google Scholar] [CrossRef]
- Monaco, G.; Viglione, R.G.; Zanasi, R.; Fowler, P.W. Designing Ring-Current Patterns: [10,5]-Coronene, a Circulene with Inverted Rim and Hub Currents. J. Phys. Chem. A 2006, 110, 7447–7452. [Google Scholar] [CrossRef] [PubMed]
- Monaco, G.; Zanasi, R. Investigation of the p-coronene series in the context of the ‘annulene-within-an-annulene’ model by means of ipso-centric ab initio calculations of π-electron currents. Phys. Chem. Chem. Phys. 2013, 15, 17654–17657. [Google Scholar] [CrossRef]
- Miyoshi, H.; Nobusue, S.; Shimizu, A.; Tobe, Y. Non-alternant non-benzenoid kekulenes: The birth of a new kekulene family. Chem. Soc. Rev. 2015, 44, 6560–6577. [Google Scholar] [CrossRef] [PubMed]
- Stępień, M. An Aromatic Riddle: Decoupling Annulene Conjugation in Coronoid Macrocycles. Chem 2018, 4, 1481–1483. [Google Scholar] [CrossRef]
- Buttrick, J.C.; King, B.T. Kekulenes, cycloarenes, and heterocycloarenes: Addressing electronic structure and aromaticity through experiments and calculations. Chem. Soc. Rev. 2017, 46, 7–20. [Google Scholar] [CrossRef]
- Ayalon, A.; Rabinovitz, M.; Cheng, P.C.; Scott, L.T. Corannulene Tetraanion: A Novel Species with Concentric Anionic Rings. Angew. Chem. Int. Ed. Engl. 1992, 31, 1636–1637. [Google Scholar] [CrossRef]
- Baumgarten, M.; Gherghel, L.; Wagner, M.; Weitz, A.; Rabinovitz, M.; Cheng, P.-C.; Scott, L.T. Corannulene Reduction: Spectroscopic Detection of All Anionic Oxidation States. J. Am. Chem. Soc. 1995, 117, 6254–6257. [Google Scholar]
- Ayalon, A.; Sygula, A.; Cheng, P.-C.; Rabinovitz, M.; Rabideau, P.W.; Scott, L.T. Stable High-Order Molecular Sandwiches: Hydrocarbon Polyanion Pairs with Multiple Lithium Ions Inside and Out. Science 1994, 265, 1065–1067. [Google Scholar] [PubMed]
- Zabula, A.V.; Filatov, A.S.; Spisak, S.N.; Rogachev, A.Y.; Petrukhina, M.A. A Main Group Metal Sandwich: Five Lithium Cations Jammed Between Two Corannulene Tetraanion Decks. Science 2011, 333, 1008–1011. [Google Scholar] [PubMed]
- Nobusue, S.; Miyoshi, H.; Shimizu, A.; Hisaki, I.; Fukuda, K.; Nakano, M.; Tobe, Y. Tetracyclopenta[def,jkl,pqr,vwx]tetraphenylene: A potential tetraradicaloid hydrocarbon. Angew. Chem. Int. Ed. Engl. 2015, 54, 2090–2094. [Google Scholar]
- Miyoshi, H.; Hisaki, I.; Tobe, Y. Crystal Structures of Tetramesity-Substituted Tetracyclopenta[def,jkl,pqr,vwx]tetraphenylene. Eur. J. Org. Chem. 2021, 2021, 3528–3534. [Google Scholar]
- Lu, X.; Gopalakrishna, T.Y.; Phan, H.; Herng, T.S.; Jiang, Q.; Liu, C.; Li, G.; Ding, J.; Wu, J. Global Aromaticity in Macrocyclic Cyclopenta-Fused Tetraphenanthrenylene Tetraradicaloid and Its Charged Species. Angew. Chem. Int. Ed. Engl. 2018, 57, 13052–13056. [Google Scholar]
- Liu, C.; Ni, Y.; Lu, X.; Li, G.; Wu, J. Global Aromaticity in Macrocyclic Polyradicaloids: Huckel’s Rule or Baird’s Rule? Acc. Chem. Res. 2019, 52, 2309–2321. [Google Scholar]
- Liu, C.; Sandoval-Salinas, M.E.; Hong, Y.; Gopalakrishna, T.Y.; Phan, H.; Aratani, N.; Herng, T.S.; Ding, J.; Yamada, H.; Kim, D.; et al. Macrocyclic Polyradicaloids with Unusual Super-ring Structure and Global Aromaticity. Chem 2018, 4, 1586–1595. [Google Scholar]
- Miyoshi, H.; Sugiura, R.; Kishi, R.; Spisak, S.N.; Wei, Z.; Muranaka, A.; Uchiyama, M.; Kobayashi, N.; Chatterjee, S.; Ie, Y.; et al. Dianion and Dication of Tetracyclopentatetraphenylene as Decoupled Annulene-within-an-Annulene Models. Angew. Chem. Int. Ed. Engl. 2022, 61, e202115316. [Google Scholar]
- Baryshnikov, G.V.; Valiev, R.R.; Karaush, N.N.; Sundholm, D.; Minaev, B.F. Aromaticity of the doubly charged [8]circulenes. Phys. Chem. Chem. Phys. 2016, 18, 8980–8992. [Google Scholar]
- Bladauski, D.; Broser, W.; Hecht, H.-J.; Rewicki, D.; Dietrich, H. Lithium-7bH-indeno[1,2,3-jk]fluorenide. Chem. Ber. 1979, 112, 1380–1391. [Google Scholar]
- Becker, B.C.; Huber, W.; Müllen, K. Acepleiadylene Dianion and Tetraanion. J. Am. Chem. Soc. 1980, 102, 7803–7805. [Google Scholar]
- Müllen, K.; Huber, W.; Meul, T.; Nakagawa, M.; Iyoda, M. Highly Reduced Annulenes. Novel Probes for Spectroscopy and Theory. J. Am. Chem. Soc. 1982, 104, 5403–5411. [Google Scholar]
- Shenhar, R.; Wang, H.; Hoffman, R.E.; Frish, L.; Avram, L.; Willner, I.; Rajca, A.; Rabinovitz, M. Self-Assembled, Helically Stacked Anionic Aggregates of 2,5,8,11-Tetra-tert-butylcycloocta[1,2,3,4-def;5,6,7,8-d’e’f’]bisbiphenylene, Stabilized by Electrostatic Interactions. J. Am. Chem. Soc. 2002, 124, 4685–4692. [Google Scholar]
- Iikura, H.; Tsuneda, T.; Yanai, T.; Hirao, K. A long-range correction scheme for generalized-gradientapproximation exchange functionals. J. Chem. Phys. 2001, 115, 3540–3544. [Google Scholar]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [PubMed]
- Juséllius, J.; Sundholm, D.; Gauss, J. Calculation of current densities using gauge-including atomic orbitals. J. Chem. Phys. 2004, 121, 3952–3963. [Google Scholar]
- Taubert, S.; Sundholm, D.; Juséllius, J. Calculation of spin-current densities using gauge-including atomic orbitals. J. Chem. Phys. 2011, 134, 054123. [Google Scholar]
- Fliegl, H.; Taubert, S.; Lehtonen, O.; Sundholm, D. The gauge including magnetically induced current method. Phys. Chem. Chem. Phys. 2011, 13, 20500–20518. [Google Scholar] [PubMed]
- Rauhalahti, M.; Taubert, S.; Sundholm, D.; Liélgeois, V. Calculations of current densities for neutral and doubly charged persubstituted benzenes using effective core potentials. Phys. Chem. Chem. Phys. 2017, 19, 7124–7131. [Google Scholar]
- Gaussian2gimic.py. Available online: https://github.com/qmcurrents/gimic (accessed on 10 January 2025).
- DralMol. UNamur. Available online: http://www.unamur.be/drawmol (accessed on 10 January 2025).
- Fraenkel, G.; Carter, R.E.; McLachlan, A.; Richards, J.H. Chemical Shifts in C5H5−, C6H6 and C7H7+; Chemical Shifts and π-Electron Densities. J. Am. Chem. Soc. 1960, 82, 5846–5850. [Google Scholar]
- Spiesecke, H.; Schneider, W.G. The Determination of π-Electron Densities in Azulene from Cl3 and H1 Nuclear Resonance Shifts. Tetrahedron Lett. 1961, 2, 468–472. [Google Scholar]
- Schaeffer, T.; Schneider, W.G. Proton Magnetic Resonance Shifts and the Electron Density Distribution in Aromatic Systems. Can. J. Chem. 1963, 41, 966–982. [Google Scholar]
- Olah, G.A.; Mateescu, G.D. Stable Carbonium Ions. The Tetraphenylcyclobutadiene Dication. J. Am. Chem. Soc. 1970, 92, 1430–1432. [Google Scholar]
- O’Brien, D.H.; Hart, A.J.; Russell, C.R. Carbon-13 Magnetic Resonance of Allyl, Pentadienyl, and Arylmethyl Carbanions. Empirical Calculation of π-Electron Densities. J. Am. Chem. Soc. 1975, 97, 4410–4412. [Google Scholar]
- Fliszár, S.; Cardinal, G.; Béraldin, M.-T. Charge Distributions and Chemical Effects. XXX. Relationships between Nuclear Magnetic Resonance Shifts and Atomic Charges. J. Am. Chem. Soc. 1982, 104, 5287–5292. [Google Scholar]
- Eliasson, B.; Edlund, U.; Müllen, K. Unusually Low 13C Chemical Shift Sensitivity to Charge in Cyclic 4nπ Anions. Potential Relation to Anisotropic Ring Current Effects. J. Chem. Soc. Perkin Trans. 1986, 2, 937–940. [Google Scholar]
- Du Vernet, R.; Boekelheide, V. Nuclear Magnetic Resonance Spectroscopy. Ring-Current Effects on Carbon-13 Chemical Shifts. Proc. Nat. Acad. Sci. USA 1974, 71, 2961–2964. [Google Scholar]
- Mitchell, R.H. Measuring Aromaticity by NMR. Chem. Rev. 2001, 101, 1301–1315. [Google Scholar]
- Müllen, K. The Dianions of Pyrene and Pyrene Isomers as (4n)π-Perimeter. Helv. Chim. Acta 1978, 61, 2307–2317. [Google Scholar]
- Müllen, K.; Muel, T.; Schade, P.; Schmickler, H.; Vogel, E. Dianions and Dications Derived from Bridged [4n + 2]Annulenes. Paratropism as a Function of Ring Size, Conformation, and Configuration. J. Am. Chem. Soc. 1987, 109, 4992–5003. [Google Scholar]
- Kobayashi, N.; Muranaka, A.; Mack, J. Circular Dichroism and Magnetic Circular Dichroism Spectroscopy for Organic Chemists; RSC Publishing: Cambridge, UK, 2012. [Google Scholar]
- Gorski, A.; Kohler, T.; Seidel, D.; Lee, J.T.; Orzanowska, G.; Sessler, J.L.; Waluk, J. Electronic structure, spectra, and magnetic circular dichroism of cyclohexa-, cyclohepta-, and cyclooctapyrrole. Chem. Eur. J. 2005, 11, 4179–4184. [Google Scholar]
- Matsushita, O.; Derkacheva, V.M.; Muranaka, A.; Shimizu, S.; Uchiyama, M.; Luk’yanets, E.A.; Kobayashi, N. Rectangular-shaped expanded phthalocyanines with two central metal atoms. J. Am. Chem. Soc. 2012, 134, 3411–3418. [Google Scholar] [PubMed]
- Rose, B.D.; Sumner, N.J.; Filatov, A.S.; Peters, S.J.; Zakharov, L.N.; Petrukhina, M.A.; Haley, M.M. Experimental and Computational Studies of the Neutral and Reduced States of Indeno[1,2-b]fluorene. J. Am. Chem. Soc. 2014, 136, 9181–9189. [Google Scholar]
- Zerger, R.; Rhine, W.; Stucky, G.D. π Groups in Ion Pair Bonding. The Effect of the Cation on the Structural and Spectroscopic Properties of Fluorenyl Ion Pairs. J. Am. Chem. Soc. 1974, 96, 5441–5448. [Google Scholar]
- Janiak, C. A Second Modification of Potassium Fluorenide and the Structure of Potassium 9-tert-Butylfluorenide: Effect of Crystallization Conditions and Substituents on Solid-state Contact Ion Pair Interaction. Chem. Ber. 1993, 126, 1603–1607. [Google Scholar]
- Neander, S.; Tio, F.E.; Buschmann, R.; Behrens, U.; Olbrich, F. Cyclopentadienyl, indenyl, fluorenyl, and pentamethylcyclopentadienyl complexes of potassium with 18-crown-6. J. Organomet. Chem. 1999, 582, 58–65. [Google Scholar]
- Neander, S.; Körnich, J.; Olbrich, F. Novel fluorenyl alkali metal DIGLYME complexes: Synthesis and solid state structures. J. Organomet. Chem. 2002, 656, 89–96. [Google Scholar]
- Hirshfeld, F.L. Bonded-Atom Fragments for Describing Molecular Charge Densities. Theor. Clim. Acta 1977, 44, 129–138. [Google Scholar]
- Feng, C.N.; Kuo, M.Y.; Wu, Y.T. Synthesis, Structural Analysis, and Properties of [8]Circulenes. Angew. Chem. Int. Ed. Engl. 2013, 52, 7791–7794. [Google Scholar]
- Sakamoto, Y.; Suzuki, T. Tetrabenzo[8]circulene: Aromatic Saddles from Negatively Curved Graphene. J. Am. Chem. Soc. 2013, 135, 14074–14077. [Google Scholar] [PubMed]
- Miller, R.W.; Duncan, A.K.; Schneebeli, S.T.; Gray, D.L.; Whalley, A.C. Synthesis and Structural Data of Tetrabenzo[8]circulene. Chem. Eur. J. 2014, 20, 3705–3711. [Google Scholar] [PubMed]
- Mitchell, R.H.; Khalifa, N.A.; Dingle, T.W. Synthesis of the First Large Annulene Fused to Cyclopentadienide. A Comparison of the Effective Aromaticity of Cyclopentadienide Anion with Benzene. J. Am. Chem. Soc. 1991, 113, 6696–6697. [Google Scholar]
- Jiao, H.; Schleyer, P.v.R.; Mo, Y.; McAllister, M.A.; Tidwell, T.T. Magnetic Evidence for the Aromaticity and Antiaromaticity of Charged Fluorenyl, Indenyl, and Cyclopentadienyl Systems. J. Am. Chem. Soc. 1997, 119, 7075–7083. [Google Scholar]
- Chen, Z.; Wannere, C.S.; Corminboeuf, C.M.; Puchta, R.; Schleyer, P.v.R. Nucleus-Independent Chemical Shifts (NICS) as an Aromaticity Criterion. Chem. Rev. 2005, 105, 3842–3888. [Google Scholar]
- Steiner, E.; Fowler, P.W.; Jenneskens, L.W. Counter-Rotating Ring Currents in Coronene and Corannulene. Angew. Chem. Int. Ed. Engl. 2001, 40, 362–366. [Google Scholar]
- Steiner, E.; Fowler, P.W.; Acocella, A.; Jenneskens, L.W. Visualisation of counter-rotating ring currents in kekulene. Chem. Commun. 2001, 7, 659–660. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Carbon | Method | δ 1b 2 | δ 1b2− 3 | δ 1b4− 4 | Δδ 1b/1b2− | Δδ 1b/1b4− | Δδ 1b2−/1b4− |
|---|---|---|---|---|---|---|---|
| A | exp | 123.7 | 110.5 | 108.8 | −13.2 | −14.9 | −1.7 |
| theor (monomer) | 112.9 | 99.2 | 97.7 | −13.7 | −15.2 | −1.5 | |
| theor (dimer) | – | 102.6 | 100.0 | −10.3 | −12.9 | −2.6 | |
| B | exp | 148.2 5 | 120.6 6 | 122.1 | −27.6 | −26.1 | +1.5 |
| theor (monomer) | 136.4 | 109.4 | 116.0 | −27.0 | −20.4 | +6.6 | |
| theor (dimer) | – | 109.0 | 111.1 | −27.4 | −25.3 | +2.1 | |
| C | exp | 148.2 5 | 125.5 6 | 103.4 7 | −22.7 | −44.8 | −22.1 |
| theor (monomer) | 135.4 | 115.7 | 103.4 | −19.7 | −32.0 | −12.3 | |
| theor (dimer) | – | 114.2 | 93.0 | −21.2 | −42.4 | −21.2 | |
| D | exp | 146.9 | 118.4 | 103.4 7 | −28.5 | −43.5 | −15.0 |
| theor (monomer) | 138.1 | 109.4 | 86.1 | −28.7 | −52.0 | −23.2 | |
| theor (dimer) | – | 113.3 | 92.3 | −24.8 | −45.8 | −21.0 | |
| E | exp | 127.4 | 138.0 | 142.1 | +10.6 | +14.7 | +4.1 |
| theor (monomer) | 117.3 | 129.6 | 134.6 | +12.3 | +17.3 | +5.0 | |
| theor (dimer) | – | 124.8 | 129.6 | +7.5 | +12.3 | +4.8 | |
| F | exp | 135.8 | 138.0 | 138.8 | +2.2 | +3.0 | +0.8 |
| theor (monomer) | 127.8 | 131.0 | 128.6 | +3.2 | +0.8 | −2.4 | |
| theor (dimer) | – | 129.2 | 129.6 | +1.4 | +1.8 | +0.4 | |
| G | exp | 128.4 | 127.5 | 128.1 | −0.9 | −0.3 | +0.6 |
| theor (monomer) | 117.3 | 116.8 | 115.4 | −0.5 | −1.4 | −0.9 | |
| theor (dimer) | – | 117.8 | 117.4 | +0.5 | +0.1 | −0.4 | |
| H | exp | 137.4 | 133.8 | 132.2 | −3.6 | −5.2 | −1.6 |
| theor (monomer) | 128.3 | 123.9 | 114.4 | −4.4 | −13.9 | −9.5 | |
| theor (dimer) | – | 127.4 | 123.5 | −0.9 | −4.8 | −3.9 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Miyoshi, H.; Sugiura, R.; Kishi, R.; Muranaka, A.; Uchiyama, M.; Kobayashi, N.; Ie, Y.; Nakano, M.; Tobe, Y. Tetraanion of Tetracyclopentatetraphenylene Derivative: Global Versus Local Conjugation Modes. Chemistry 2025, 7, 51. https://doi.org/10.3390/chemistry7020051
Miyoshi H, Sugiura R, Kishi R, Muranaka A, Uchiyama M, Kobayashi N, Ie Y, Nakano M, Tobe Y. Tetraanion of Tetracyclopentatetraphenylene Derivative: Global Versus Local Conjugation Modes. Chemistry. 2025; 7(2):51. https://doi.org/10.3390/chemistry7020051
Chicago/Turabian StyleMiyoshi, Hirokazu, Ryosuke Sugiura, Ryohei Kishi, Atsuya Muranaka, Masanobu Uchiyama, Nagao Kobayashi, Yutaka Ie, Masayoshi Nakano, and Yoshito Tobe. 2025. "Tetraanion of Tetracyclopentatetraphenylene Derivative: Global Versus Local Conjugation Modes" Chemistry 7, no. 2: 51. https://doi.org/10.3390/chemistry7020051
APA StyleMiyoshi, H., Sugiura, R., Kishi, R., Muranaka, A., Uchiyama, M., Kobayashi, N., Ie, Y., Nakano, M., & Tobe, Y. (2025). Tetraanion of Tetracyclopentatetraphenylene Derivative: Global Versus Local Conjugation Modes. Chemistry, 7(2), 51. https://doi.org/10.3390/chemistry7020051