

Iron-Promoted 1,5-Substitution Reaction of Endocyclic Enyne Oxiranes with MeMgBr: A Stereoselective Method for the Synthesis of Exocyclic 2,4,5-Trienol Derivatives


Abstract
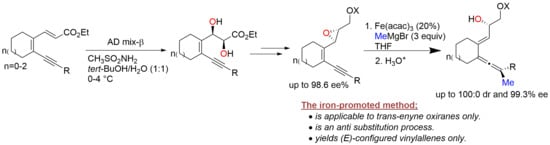
:
1. Introduction
2. Materials and Methods
2.1. General
2.2. Synthesis of Racemic Enyne Oxiranes
2.3. Synthesis of Enantiopure Trans-Enyne Oxiranes
2.4. Synthesis of Enantiopure Cis-Enyne Oxiranes
2.5. Iron-Catalyzed Reaction Protocol
3. Results and Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Qi, L.Y.; Ma, E.L.; Jia, F.; Li, Z.P. Iron-catalyzed allylic substitution reactions of allylic ethers with Grignard reagents. Tetrahedron Lett. 2016, 57, 2211–2214. [Google Scholar] [CrossRef]
- Nakamura, M.; Matsuo, K.; Inoue, T.; Nakamura, E. Iron-catalyzed regio- and stereoselective ring opening of [2.2.1]- and [3.2.1]oxabicyclic alkenes with a Grignard reagent. Org. Lett. 2003, 5, 1373–1375. [Google Scholar] [CrossRef] [PubMed]
- Kessler, S.N.; Hundemer, F.; Backvall, J.E. A Synthesis of substituted α-allenols via iron-catalyzed cross-coupling of propargyl carboxylates with Grignard reagents. ACS Catal. 2016, 6, 7448–7451. [Google Scholar] [CrossRef] [PubMed]
- Kessler, S.N.; Backvall, J.E. Iron-catalyzed cross-coupling of propargyl carboxylates and Grignard reagents: Synthesis of substituted allenes. Angew. Chem. Int. Ed. 2016, 55, 3734–3738. [Google Scholar] [CrossRef] [PubMed]
- Domingo-Legarda, P.; Soler-Yanes, R.; Quiros-Lopez, M.T.; Bunuel, E.; Cardenas, D.J. Iron-catalyzed coupling of propargyl bromides and alkyl Grignard reagents. Eur. J. Org. Chem. 2018, 2018, 4900–4904. [Google Scholar] [CrossRef]
- Sun, C.L.; Furstner, A. Formal ring-opening/cross-coupling reactions of 2-pyrones: Iron-catalyzed entry into stereodefined dienyl carboxylates. Angew. Chem. Int. Ed. 2013, 52, 13071–13075. [Google Scholar] [CrossRef]
- Hata, T.; Bannai, R.; Otsuki, M.; Urabe, H. Iron-catalyzed regio- and stereoselective substitution of γ,δ-epoxy-α,β-unsaturated Esters and Amides with Grignard Reagents. Org. Lett. 2010, 12, 1012–1014. [Google Scholar] [CrossRef]
- Furstner, A.; Mendez, M. Iron-catalyzed cross-coupling reactions: Efficient synthesis of 2,3-allenol derivatives. Angew. Chem. Int. Ed. 2003, 42, 5355–5357. [Google Scholar] [CrossRef]
- Zhang, X.B.; Qiu, Y.A.; Fu, C.L.; Ma, S.M. Highly selective 4-alkynoic acids synthesis via iron-mediated complete inversion of stereogenic carbon center. Org. Chem. Front. 2014, 1, 247–252. [Google Scholar] [CrossRef]
- Taç, D.; Aytaç, I.A.; Karatavuk, A.O.; Kuş, M.; Ziyanak, F.; Artok, L. Iron-promoted 1,5-substitution (SN2″) reactions of enyne acetates and oxiranes with Grignard reagents. Asian J. Org. Chem. 2017, 6, 1415–1420. [Google Scholar] [CrossRef]
- Sugano, G.; Kawada, K.; Shigeta, M.; Hata, T.; Urabe, H. Iron-catalyzed δ-selective conjugate addition of methyl and cyclopropyl Grignard reagents to α,β,γ,δ-unsaturated esters and amides. Tetrahedron Lett. 2019, 60, 885–890. [Google Scholar] [CrossRef]
- Sherry, B.; Furstner, D. Iron-catalyzed addition of Grignard reagents to activated vinyl cyclopropanes. Chem. Commun. 2009, 7116–7118. [Google Scholar] [CrossRef]
- Okada, S.; Arayama, K.; Murayama, R.; Ishizuka, T.; Hara, K.; Hirone, N.; Hata, T.; Urabe, H. Iron-catalyst-switched selective conjugate addition of Grignard reagents: α,β,γ,δ-Unsaturated amides as versatile templates for asymmetric three-component coupling processes. Angew. Chem. Int. Ed. 2008, 47, 6860–6864. [Google Scholar] [CrossRef]
- Lu, Z.; Chai, G.B.; Ma, S.M. Iron-catalyzed highly regio- and stereoselective conjugate addition of 2,3-allenoates with Grignard reagents. J. Am. Chem. Soc. 2007, 129, 14546–14547. [Google Scholar] [CrossRef]
- Hata, T.; Iwata, S.; Seto, S.; Urabe, H. Iron-catalyzed synthesis of allenes from 2-alken-4-ynoates and Grignard reagents. Adv. Synth. Catal. 2012, 354, 1885–1889. [Google Scholar] [CrossRef]
- Fukuhara, K.; Urabe, H. Iron-catalyzed 1,6-addition of aryl Grignard reagents to 2,4-dienoates and dienamides. Tetrahedron Lett. 2005, 46, 603–606. [Google Scholar] [CrossRef]
- Hata, T.; Nakada, T.; Oh, Y.T.; Hirone, N.; Urabe, H. Iron-catalyzed regio- and stereoselective conjugate addition of aryl-Grignard reagents to α,β,γ,δ-unsaturated sulfones and its synthetic application. Adv. Synth. Catal. 2013, 355, 1736–1740. [Google Scholar] [CrossRef]
- Tindall, D.J.; Krause, H.; Furstner, A. Iron-catalyzed cross-coupling of 1-alkynylcyclopropyl tosylates and related substrates. Adv. Synth. Catal. 2016, 358, 2398–2403. [Google Scholar] [CrossRef]
- Huang, L.; Gu, Y.T.; Furstner, A. Iron-catalyzed reactions of 2-pyridone derivatives: 1,6-addition and formal ring opening/cross coupling. Chem. Asian J. 2019, 14, 4017–4023. [Google Scholar] [CrossRef]
- Chai, G.; Zeng, B.R.; Fu, C.L.; Ma, S.M. Iron-catalyzed diastereoselective synthesis of α-(methoxycarbonyl)allylsilanes. Eur. J. Org. Chem. 2013, 2013, 148–154. [Google Scholar] [CrossRef]
- Ziyanak, F.; Kus, M.; Alkan-Karadeniz, L.; Artok, L. Palladium-catalyzed reactions of conjugated enyne oxiranes with organoborons: A diastereoselective method of the synthesis of 2,4,5-trienol derivatives. Tetrahedron 2018, 74, 3652–3662. [Google Scholar] [CrossRef]
- Zhao, J.; Yu, Y.; Ma, S. Ligand effects on the Pd-catalyzed cross-coupling reaction of 3-iodoalk-2-enoates with propargyl/1,2-allenylic metallic species: An efficient regiodivergent synthesis of 2,4,5-trienoates. Chem. Eur. J. 2010, 16, 74–80. [Google Scholar] [CrossRef]
- Yang, M.; Yokokawa, Y.; Ohmiya, N.H.; Sawamura, M. Synthesis of conjugated allenes through copper-catalyzed γ-selective and stereospecific coupling between propargylic phosphates and aryl- or alkenylboronates. Org. Lett. 2012, 14, 816–819. [Google Scholar] [CrossRef]
- Wei, X.F.; Wakaki, T.; Itoh, T.; Li, H.L.; Yoshimura, T.; Miyazaki, A.; Oisaki, K.; Hatanaka, M.; Shimizu, Y.; Kanai, M. Catalytic regio- and enantioselective proton migration from skipped Enynes to allenes. Chem 2019, 5, 585–599. [Google Scholar] [CrossRef]
- Ucuncu, M.; Karakus, E.; Kus, M.; Akpinar, G.E.O.; Aksin-Artok, Ö.; Krause, N.; Karaca, S.; Elmacı, N.; Artok, L. Rhodium- and palladium-catalyzed 1,5-substitution reactions of 2-en-4-yne acetates and carbonates with organoboronic acids. J. Org. Chem. 2011, 76, 5959–5971. [Google Scholar] [CrossRef]
- Tolstikov, G.A.; Romanova, T.Y.; Kuchin, A.V. A new method for the synthesis of vinyl-and diallenes assisted by organoaluminium compounds. J. Organomet. Chem. 1985, 285, 71–82. [Google Scholar] [CrossRef]
- Taç, D.; Artok, L. Palladium-catalyzed coupling of 2-en-4-yne carbonates with terminal alkynes. Tetrahedron Lett. 2018, 59, 895–898. [Google Scholar] [CrossRef]
- Sim, S.H.; Lee, S.I.; Seo, J.; Chung, Y.K. Mercury (II) triflate-catalyzed cycloisomerization of allenynes to allenenes. J. Org. Chem. 2007, 72, 9818–9821. [Google Scholar] [CrossRef]
- Purpura, M.; Krause, N. Regio- and stereoselective synthesis of vinylallenes by 1,5-(SN″)-substitution of enyne acetates and oxiranes with organocuprates. Eur. J. Org. Chem. 1999, 1999, 267–275. [Google Scholar] [CrossRef]
- Murakami, M.; Kadowaki, S.; Matsuda, T. Molybdenum-catalyzed ring-closing metathesis of allenynes. Org. Lett. 2005, 7, 3953–3956. [Google Scholar] [CrossRef] [PubMed]
- Molander, G.A.; Sommers, E.M.; Baker, S.R. Palladium (0)-catalyzed synthesis of chiral ene-allenes using alkenyl trifluoroborates. J. Org. Chem. 2006, 71, 1563–1568. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.I.; Sim, S.H.; Kim, S.M.; Kim, K.; Chung, Y.K. GaCl3-catalyzed allenyne cycloisomerizations to allenenes. J. Org. Chem. 2006, 71, 7120–7123. [Google Scholar] [CrossRef]
- Kuş, M.; Artok, L.; Aygün, M. Palladium-catalyzed alkoxycarbonylation of conjugated enyne oxiranes: A diastereoselective method for the synthesis of 7-hydroxy-2,3,5-trienoates. J. Org. Chem. 2015, 80, 5494–5506. [Google Scholar] [CrossRef]
- Krause, N.; Purpura, M. Remote stereocontrol in organocopper chemistry: Highly enantioselective synthesis of vinylallenes by 1, 5-substitution of enyne acetates. Angew. Chem. Int. Ed Engl. 2000, 39, 4355–4356. [Google Scholar] [CrossRef]
- Koop, U.; Handke, G.; Krause, N. Synthesis of vinylallenes by conjugate 1,6-, 1,8-, 1,10- and 1,12-addition reactions of organocuprates with acetylenic Michael acceptors and their use as dienes in intermolecular Diels-Alder reactions. Liebigs Annalen. 1996, 1996, 1487–1499. [Google Scholar]
- Keinan, E.; Bosch, E. Palladium-catalyzed propargylic vs. allylic alkylation. J. Org. Chem. 1986, 51, 4006–4016. [Google Scholar] [CrossRef]
- Karagöz, E.Ş.; Kuş, M.; Akpınar, G.E.; Artok, L. Regio-and stereoselective synthesis of 2,3,5-trienoates by palladium-catalyzed alkoxycarbonylation of conjugated enyne carbonates. J. Org. Chem. 2014, 79, 9222–9230. [Google Scholar] [CrossRef]
- Jiang, H.; Liu, X.; Zhou, L. First synthesis of 1-chlorovinyl allenes via palladium-catalyzed allenylation of alkynoates with propargyl alcohols. Chem. Eur. J. 2008, 14, 11305–11309. [Google Scholar] [CrossRef]
- Dabrowski, J.A.; Haeffner, F.; Hoveyda, A.H. Combining NHC–Cu and Brønsted base catalysis: Enantioselective allylic substitution/conjugate additions with alkynylaluminum reagents and stereospecific isomerization of the products to trisubstituted allenes. Angew. Chem. Int. Ed. 2013, 52, 7694–7699. [Google Scholar] [CrossRef]
- Cadran, N.; Cariou, K.; Herve, G.; Aubert, C.; Fensterbank, L.; Malacria, M.; Marco-Contelles, J. PtCl2-catalyzed cycloisomerizations of allenynes. J. Am. Chem. Soc. 2004, 126, 3408–3409. [Google Scholar] [CrossRef] [PubMed]
- Ben-Valid, S.; Quntar, A.A.A.; Srebnik, M. Novel vinyl phosphonates and vinyl boronates by halogenation, allylation, and propargylation of α-boryl-and α-phosphonozirconacyclopentenes. J. Org. Chem. 2005, 70, 3554–3559. [Google Scholar] [CrossRef]
- Ando, M.; Sasaki, M.; Miyashita, I.; Takeda, K. Formation of 2-cyano-2-siloxyvinylallenes via cyanide-induced brorok earrangement in γ-bromo-α,β,γ,δ-unsaturated acylsilanes. J. Org. Chem. 2015, 80, 247–255. [Google Scholar] [CrossRef]
- Akpinar, G.E.; Kus, M.; Ucuncu, M.; Karakuş, E.; Artok, L. Palladium-Catalyzed Alkoxycarbonylation of (Z)-2-En-4-yn Carbonates Leading to 2,3,5-Trienoates. Org. Lett. 2011, 13, 748–751. [Google Scholar] [CrossRef]
- Yu, S.H.; Gong, T.J.; Fu, Y. Three-component borylallenylation of alkynes: Access to densely boryl-substituted ene-allenes. Org. Lett. 2020, 22, 2941–2945. [Google Scholar] [CrossRef]
- Huang, Y.; Torker, M.S.; Li, X.H.; del Pozo, J.; Hoveyda, A.H. Racemic vinylallenes in catalytic enantioselective multicomponent processes: Rapid generation of complexity through 1,6-conjugate dditaions. Angew. Chem. Int. Ed. 2019, 58, 2685–2691. [Google Scholar] [CrossRef]
- Wang, K.K.; Andemichael, Y.W.; Dhumrongvaraporn, S. Regioselective synthesis of highly substituted arylsilanes by the reaction of the trimethylsilyl-substituted vinylallenones with enamines. Tetrahedron Lett. 1989, 30, 1311–1314. [Google Scholar] [CrossRef]
- Wu, K.M.; Midland, M.M.; Okamura, W.H. Structural effects on [1,5]-sigmatropic hydrogen shifts of vinylallenes. J. Org. Chem. 1990, 55, 4381–4392. [Google Scholar] [CrossRef]
- Murakami, M.; Amii, H.; Itami, K.; Ito, Y. A Remarkable effect of silyl substitution on electrocyclization of vinylallenes. Angew. Chem. Int. Ed. Engl. 1995, 34, 1476–1477. [Google Scholar] [CrossRef]
- López, S.; Rodríguez, J.; Rey, J.G.; de Lera, A.R. Structural effects affecting the thermal electrocyclic ring closure of vinylallenes to alkylidenecyclobutenes. J. Am. Chem. Soc. 1996, 118, 1881–1891. [Google Scholar] [CrossRef]
- Souto, J.A.; Pérez, M.; Silva López, C.; Álvarez, R.; Torrado, A.; de Lera, A.R. Competing thermal electrocyclic ring-closure reactions of (2Z)-hexa-2,4,5-trienals and their Schiff bases. Structural, kinetic, and computational studies. J. Org. Chem. 2010, 75, 4453–4462. [Google Scholar] [CrossRef] [PubMed]
- Spino, C.; Thibault, C.; Gingras, S. Stereoselective construction of tetrasubstituted exocyclic alkenes from the [4 + 2]-cycloaddition of vinylallenes. J. Org. Chem. 1998, 63, 5283–5287. [Google Scholar] [CrossRef]
- Ruiz, J.M.; Regas, D.; Afonso, M.M.; Palenzuela, J.A. Study of an unexpected rearrangement of the α-phenyl pyrane derivatives prepared via hetero-Diels–Alder reaction of acyclic vinyl allenes and aldehydes. J. Org. Chem. 2008, 73, 7246–7254. [Google Scholar] [CrossRef] [PubMed]
- Lo, V.K.Y.; Chan, Y.M.; Zhou, D.; Toy, P.H.; Che, C.M. Highly enantioselective synthesis using prolinol as a chiral auxiliary: Silver-mediated synthesis of axially chiral vinylallenes and subsequent (hetero)-Diels–Alder reactions. Org. Lett. 2019, 21, 7717–7721. [Google Scholar] [CrossRef] [PubMed]
- Murakami, M.; Itami, K.; Ito, Y. Catalytic asymmetric [4 + 1] cycloaddition of vinylallenes with carbon monoxide: Reversal of the induced chirality by the choice of metal. J. Am. Chem. Soc. 1999, 121, 4130–4135. [Google Scholar] [CrossRef]
- Murakami, M.; Itami, K.; Ito, Y. Synthesis of (vinylallene) rhodium (III) complex of planar structure: Perfect π→ σ conversion of 1,3-diene system. J. Am. Chem. Soc. 1996, 118, 11672–11673. [Google Scholar] [CrossRef]
- Mazumder, S.; Crandell, D.W.; Lord, R.L.; Baik, M.H. Switching the enantioselectivity in catalytic [4 + 1] cycloadditions by changing the metal center: Principles of inverting the stereochemical preference of an asymmetric catalysis revealed by DFT calculations. J. Am. Chem. Soc. 2014, 136, 9414–9423. [Google Scholar] [CrossRef]
- Bertrand, M.; Grimaldi, J.; Waegell, B. Diels–Alder cycloadditions of vinyl-and propenyl-allene to methyl vinyl ketone. Chem. Commun. 1968, 1141–1142. [Google Scholar] [CrossRef]
- Reich, J.; Eisenhart, E.K.; Whipple, W.L.; Kelly, M.J. Stereochemistry of vinylallene cycloadditions. J. Am. Chem. Soc. 1988, 110, 6432–6442. [Google Scholar] [CrossRef]
- Murakami, M.; Ubukata, M.; Itami, K.; Ito, Y. Rhodium-catalyzed intermolecular [4 + 2] cycloaddition of unactivated substrates. Angew. Chem. Int. Ed. 1998, 37, 2248–2250. [Google Scholar] [CrossRef]
- Murakami, M.; Ashida, S.; Matsuda, T. Dramatic effects of boryl substituents on thermal ring-closing reaction of vinylallenes. J. Am. Chem. Soc. 2004, 126, 10838–10839. [Google Scholar] [CrossRef]
- Bekele, T.; Christian, C.F.; Lipton, M.A.; Singleton, D.A. “Concerted” transition state, stepwise mechanism. Dynamics effects in C2-C6 enyne allene cyclizations. J. Am. Chem. Soc. 2005, 127, 9216–9223. [Google Scholar] [CrossRef] [PubMed]
- Bhunia, S.; Liu, R.S. Gold-catalyzed 1,3-addition of a sp3-hybridized C−H bond to alkenylcarbenoid intermediate. J. Am. Chem. Soc. 2008, 130, 16488–16489. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Toste, F.D. Gold (I)-catalyzed synthesis of functionalized cyclopentadienes. Angew. Chem. Int. Ed. 2007, 46, 912–914. [Google Scholar] [CrossRef] [PubMed]
- Funami, H.; Kusama, H.; Iwasawa, N. Preparation of substituted cyclopentadienes through platinum (II)-catalyzed cyclization of 1,2,4-trienes. Angew. Chem. Int. Ed. 2007, 46, 909–911. [Google Scholar] [CrossRef]
- Delas, C.; Urabe, H.; Sato, F. Remarkably facile, unidirectional isomerization of titanated vinylallenes to cyclobutenyltitanium compounds. A practical construction of a cyclobutene framework. J. Am. Chem. Soc. 2001, 123, 7937–7938. [Google Scholar] [CrossRef]
- Leyes, G.A.; Okamura, W.H. Studies on vitamin D (calciferol) and its analogs. 23. Effect of 3-methyl substituents on the thermal [1,5]-and [1,7]-sigmatropic hydrogen shifts of vinylallenes. J. Am. Chem. Soc. 1982, 104, 6099–6105. [Google Scholar] [CrossRef]
- Posner, G.H.; Li, Z.G.; White, M.C.; Vinader, V.; Takeuchi, K.; Guggino, S.E.; Dolan, P.; Kensler, T.W. lα,25-Dihydroxyvitamin D3 analogs featuring aromatic and heteroaromatic rings: Design, synthesis, and preliminary biological testing. J. Med. Chem. 1995, 38, 4529–4537. [Google Scholar] [CrossRef]
- Deutsch, E.A.; Snider, B.B. Synthesis of the hexahydronaphthalene moiety of (±)-compactin (ML-236B). J. Org. Chem. 1982, 47, 2682–2684. [Google Scholar] [CrossRef]
- Krause, N. Synthesis of (±)-sterpurene and hydroxylated derivatives by 1,6-addition of organocuprates to acceptor-substituted enynes. Liebigs Ann. Chem. 1993, 1993, 521–525. [Google Scholar] [CrossRef]
- Schreiber, S.L.; Kiessling, L.L. Synthesis of the bicyclic core of the esperamicin/calichemicin class of antitumor agents. J. Am. Chem. Soc. 1988, 110, 631–633. [Google Scholar] [CrossRef]
- Hoffmann-Roder, A.; Krause, N. Synthesis and properties of allenic natural products and pharmaceuticals. Angew. Chem. Int. Ed. 2004, 43, 1196–1216. [Google Scholar] [CrossRef]
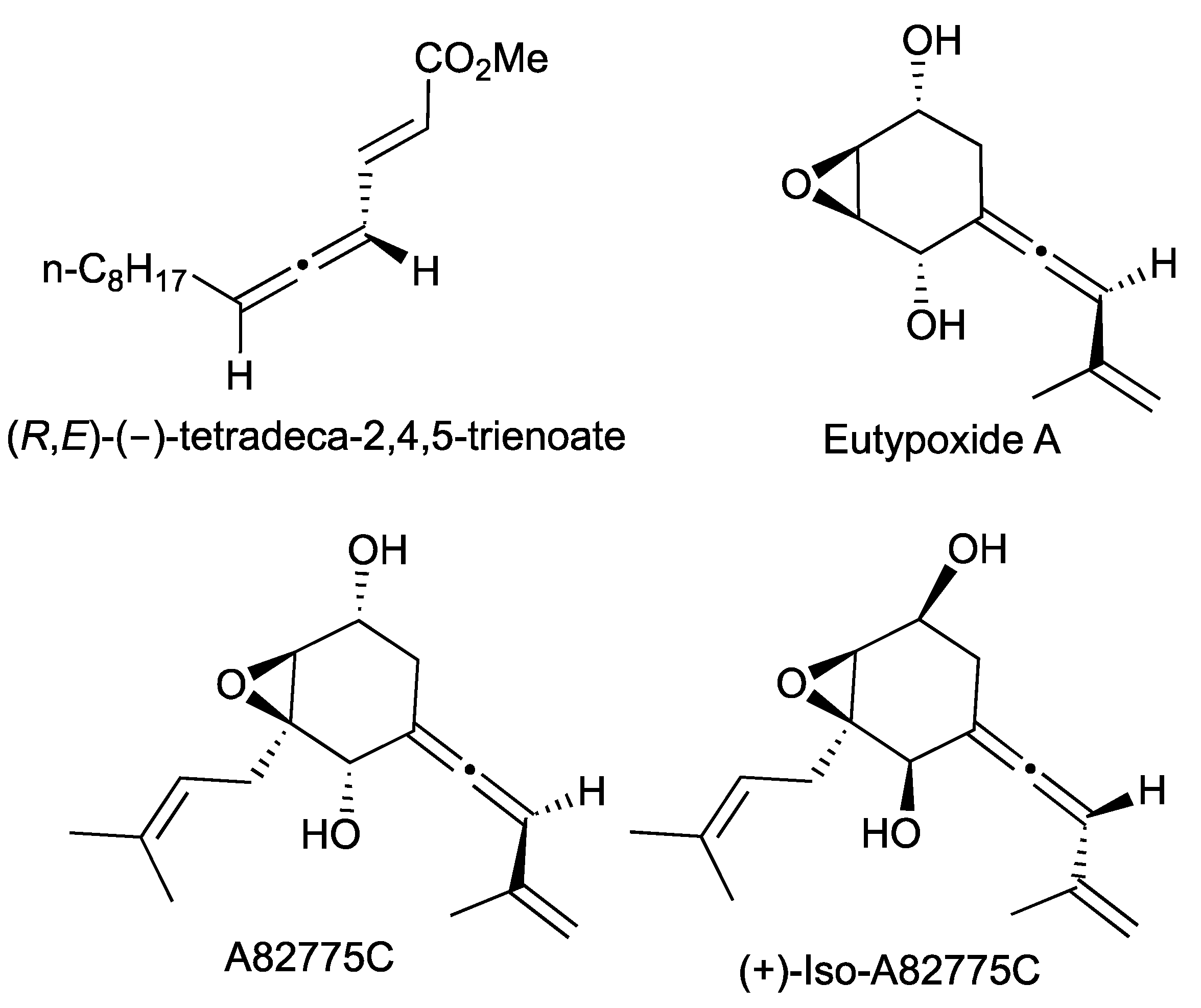
- Suzuki, T.; Watanabe, S.; Kobayashi, S.; Tanino, K. Enantioselective total synthesis of (+)-iso-A82775C, a proposed biosynthetic precursor of chloropupukeananin. Org. Lett. 2017, 19, 922–925. [Google Scholar] [CrossRef]
- Pan, Y.Y.; Liu, L.; Guan, F.F.; Li, E.W.; Jin, J.; Li, J.Y.; Che, Y.S.; Liu, G. Characterization of a prenyltransferase for iso-A82775C biosynthesis and generation of new congeners of chloropestolides. ACS Chem. Biol. 2018, 13, 703–711. [Google Scholar] [CrossRef]
- Lian, J.J.; Odedra, A.; Wu, C.J.; Liu, R.S. Ruthenium-catalyzed regioselective 1,3-methylene transfer by cleavage of two adjacent σ-carbon-carbon bonds: An easy and selective synthesis of highly subsituted benzenes. J. Am. Chem. Soc. 2005, 127, 4186–4187. [Google Scholar] [CrossRef]
- Sonogashira, K.; Tohda, Y.; Hagihara, N. A convenient synthesis of acetylenes: Catalytic substitutions of acetylenic hydrogen with bromoalkenes, iodoarenes, and bromopyridines. Tetrahedron Lett. 1975, 16, 4467–4470. [Google Scholar] [CrossRef]
- Wadsworth, W.S.; Emmons, W.D. The utility of phosphonate carbanions in olefin synthesis. J. Am. Chem. Soc. 1961, 83, 1733–1738. [Google Scholar] [CrossRef]
- Sharpless, K.B.; Amberg, W.; Bennani, Y.L.; Crispıno, G.A.; Hartung, J.; Jeong, K.S.; Kwong, H.L.; Morıkawa, K.; Wang, Z.M.; Xu, D.; et al. The osmium-catalyzed asymmetric dihydroxylation: A new ligand class and a process improvement. J. Org. Chem. 1992, 57, 2768–2771. [Google Scholar] [CrossRef]
- Kolb, H.C.; Sharpless, K.B. A simplified procedure for the stereospecific transformation of 1,2-diols into epoxides. Tetrahedron 1992, 48, 10515–10530. [Google Scholar] [CrossRef]
- Shemet, A.; Sarlah, D.; Carreira, E.M. Stereochemical studies of opening of chloro vinyl epoxides: Cyclic chloronium ions as intermediates. Org. Lett. 2015, 17, 1878–1881. [Google Scholar] [CrossRef]
- Wu, X.Y.; She, X.G.; Shi, Y.A. Highly enantioselective epoxidation of α,β-unsaturated esters by chiral dioxirane. J. Am. Chem. Soc. 2002, 124, 8792–8793. [Google Scholar] [CrossRef]
- Wang, Z.X.; Shi, Y.A. pH study on the chiral ketone catalyzed asymmetric epoxidation of hydroxyalkenes. J. Org. Chem. 1998, 63, 3099–3104. [Google Scholar] [CrossRef]
- Frohn, M.; Shi, Y. Chiral ketone-catalyzed asymmetric epoxidation of olefins. Synthesis 2000, 2000, 1979–2000. [Google Scholar] [CrossRef]
- Katsuki, T.; Sharpless, K.B. The first practical method for asymmetric epoxidation. J. Am. Chem. Soc. 1980, 102, 5974–5976. [Google Scholar] [CrossRef]
- Hanson, R.M.; Sharpless, K.B. Procedure for the catalytic asymmetric epoxidation of allylic alcohols in the presence of molecular sieves. J. Org. Chem. 1986, 51, 1922–1925. [Google Scholar] [CrossRef]
- Becket, H.; Soler, M.A.; Sharpless, K.B. Selective Asymmetric Dihydroxylation of Polyenes. Tetrahedron 1995, 51, 1345–1376. [Google Scholar] [CrossRef]
- Matsushima, Y.; Kino, J. A versatile route to 2,4,6-trideoxy-4-aminohexoses: Stereoselective syntheses of D-vicenisamine and its epimers via iodocyclization of carbamate. Tetrahedron 2017, 73, 6831–6839. [Google Scholar] [CrossRef]
- Lowe, G. The absolute configuration of allenes. Chem. Commun. 1965, 411. [Google Scholar] [CrossRef]
- Brewster, J.H. A Useful Model of Optical Activity. I. Open Chain Compounds. J. Am. Chem. Soc. 1959, 81, 5475–5483. [Google Scholar] [CrossRef]
- Brewster, J.H. Helix models of optical activity. In Topics in Stereochemistry; Allinger, N., Ernest, L., Eliel, L., Eds.; Interscience: New York, NY, USA; London, UK; Sidney, Australia, 1967; Volume 2, pp. 1–172. [Google Scholar]
- Sasaki, M.; Kondo, Y.; Moto-ishi, T.; Kawahata, M.; Yamaguchi, K.; Takeda, K. Enantioselective synthesis of allenylenol silyl ethers via chiral lithium amide mediated reduction of ynenoyl silanes and their Diels–Alder reactions. Org. Lett. 2015, 17, 1280–1283. [Google Scholar] [CrossRef]
- Li, H.; Grassi, D.; Guénée, L.; Bürgi, T.; Alexakis, A. Copper-catalyzed propargylic substitution of dichloro substrates: Enantioselective synthesis of trisubstituted allenes and formation of propargylic quaternary stereogenic centers. Chem. Eur. J. 2014, 20, 16694–16706. [Google Scholar] [CrossRef]
- Kinnel, R.; Duggan, A.J.; Eisner, T.; Meinwald, J. Panacene: An aromatic bromoallene from a sea hare. Tetrahedron Lett. 1977, 44, 3913–3916. [Google Scholar] [CrossRef]
- Baumeler, A.; Brade, W.; Haag, A.; Eugster, C. Synthese von enantiomerenreinen ‘Grasshopper’-Ketonen und verwandten Verbindungen. Helv. Chim. Acta 1990, 73, 700–715. [Google Scholar] [CrossRef]
- Kim, G.; Kim, T.; Han, S. Total synthesis of (+)-Pestalofone A and (+)-Iso-A82775C. J. Org. Chem. 2020, 85, 6815–6821. [Google Scholar] [CrossRef]
- Ye, J.; Li, S.; Chen, B.; Fan, W.; Kuang, J.; Liu, J.; Liu, Y.; Miao, B.; Wan, B.; Wang, Y.; et al. Catalytic asymmetric synthesis of optically active allenes from terminal alkynes. Org. Lett. 2012, 14, 1346–1349. [Google Scholar] [CrossRef]
- Zhang, F.H.; Guo, X.; Zeng, X.; Wang, Z. Catalytic enantioconvergent allenylation of aldehydes with propargyl halides. Angew. Chem. Int. Ed. 2022, 61, e202117114. [Google Scholar] [CrossRef]
- Chen, Y.T. Recent advances in methylation: A guide for selecting methylation reagents. Chem. Eur. J. 2019, 25, 3405–3439. [Google Scholar] [CrossRef]
- Yan, G.B.; Borah, A.J.; Wang, L.G.; Yang, M.H. Recent advances in transition metal-catalyzed methylation reactions. Adv. Synth. Catal. 2015, 357, 1333–1350. [Google Scholar] [CrossRef]
- Moulay, S. C-methylation of organic substrates: A comprehensive overview. Part I. Methane as a methylating agent. Mini-Rev. Org. Chem. 2020, 17, 805–813. [Google Scholar] [CrossRef]
- Kuntz, K.W.; Campbell, J.E.; Keilhack, H.; Pollock, R.M.; Knutson, S.K.; Porter-Scott, M.; Richon, V.M.; Sneeringer, C.J.; Wigle, T.J.; Allain, C.J.; et al. The importance of being me: Magic methyls, methyltransferase inhibitors, and the discovery of tazemetostat. J. Med. Chem. 2016, 59, 1556–1564. [Google Scholar] [CrossRef]
- Barreiro, E.J.; Kummerle, A.E.; Fraga, C.A.M. The methylation effect in medicinal chemistry. Chem. Rev. 2011, 111, 5215–5246. [Google Scholar] [CrossRef]
- Al-Afyouni, M.H.; Fillman, K.L.; Brennessel, W.W.; Neidig, M.L. Isolation and characterisation of a tetramethyliron (III) ferrate: An intermediate in the reduction pathway of ferric salts with MeMgBr. J. Am. Chem. Soc. 2014, 136, 15457–15460. [Google Scholar] [CrossRef] [PubMed]
- Furstner, A.; Martin, R.; Krause, H.; Seidel, G.; Goddard, R.; Lehmann, C.W. Preparation, structure, and reactivity of nonstabilized organoiron compounds. Implications for iron-catalyzed cross coupling reactions. J. Am. Chem. Soc. 2008, 130, 8773–8787. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Epoxide (ee%) | Product | Yield (%) | dr/ee% |
|---|---|---|---|---|
| 1 |  |  | 82 | 96:4/95.2 |
| 2 |  |  | 74 | 95:5/93.8 |
| 3 |  |  | 67 | 70:30/N.D. |
| 4 |  |  | 75 | 90:10/99.3 |
| 5 |  |  | 71 | 92:8/91.6 |
| 6 |  |  | 72 | 100:0/(±) |
| 7 |  |  | C.M. | N.D. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kuş, M.; Omur, C.; Karaca, S.; Artok, L. Iron-Promoted 1,5-Substitution Reaction of Endocyclic Enyne Oxiranes with MeMgBr: A Stereoselective Method for the Synthesis of Exocyclic 2,4,5-Trienol Derivatives. Chemistry 2023, 5, 2682-2699. https://doi.org/10.3390/chemistry5040173
Kuş M, Omur C, Karaca S, Artok L. Iron-Promoted 1,5-Substitution Reaction of Endocyclic Enyne Oxiranes with MeMgBr: A Stereoselective Method for the Synthesis of Exocyclic 2,4,5-Trienol Derivatives. Chemistry. 2023; 5(4):2682-2699. https://doi.org/10.3390/chemistry5040173
Chicago/Turabian StyleKuş, Melih, Cenk Omur, Sıla Karaca, and Levent Artok. 2023. "Iron-Promoted 1,5-Substitution Reaction of Endocyclic Enyne Oxiranes with MeMgBr: A Stereoselective Method for the Synthesis of Exocyclic 2,4,5-Trienol Derivatives" Chemistry 5, no. 4: 2682-2699. https://doi.org/10.3390/chemistry5040173
APA StyleKuş, M., Omur, C., Karaca, S., & Artok, L. (2023). Iron-Promoted 1,5-Substitution Reaction of Endocyclic Enyne Oxiranes with MeMgBr: A Stereoselective Method for the Synthesis of Exocyclic 2,4,5-Trienol Derivatives. Chemistry, 5(4), 2682-2699. https://doi.org/10.3390/chemistry5040173