
Tweaking of Peripheral Moieties in Catalytic Amyloid for Modulating Hydrogel Strength and Hydrolase Activity


Abstract
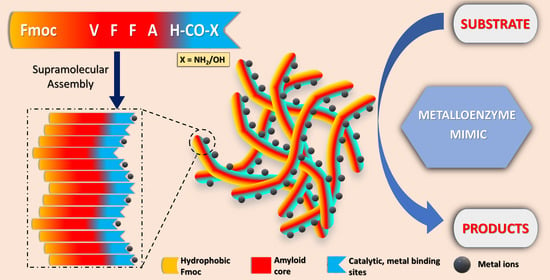
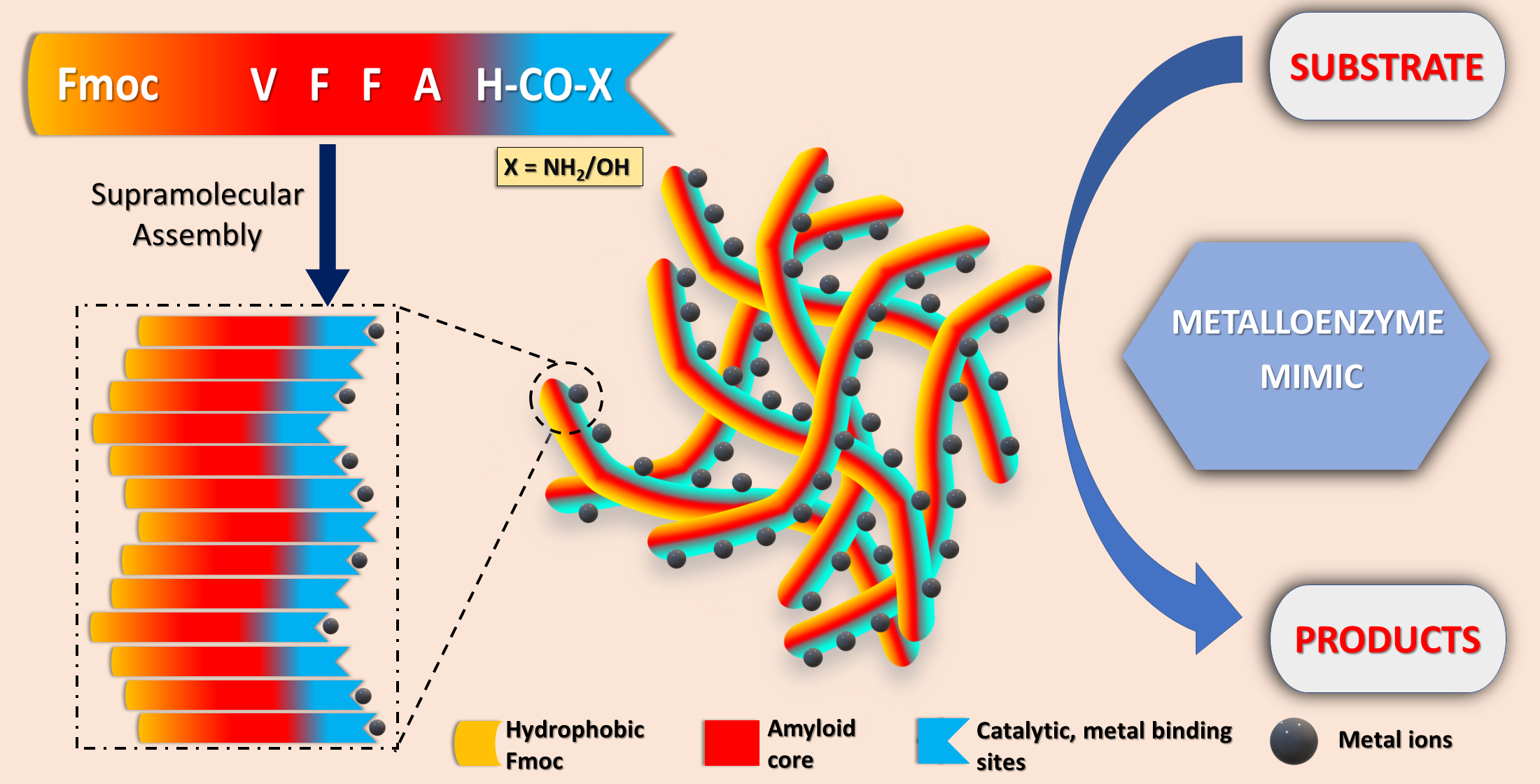
1. Introduction
2. Materials and Methods
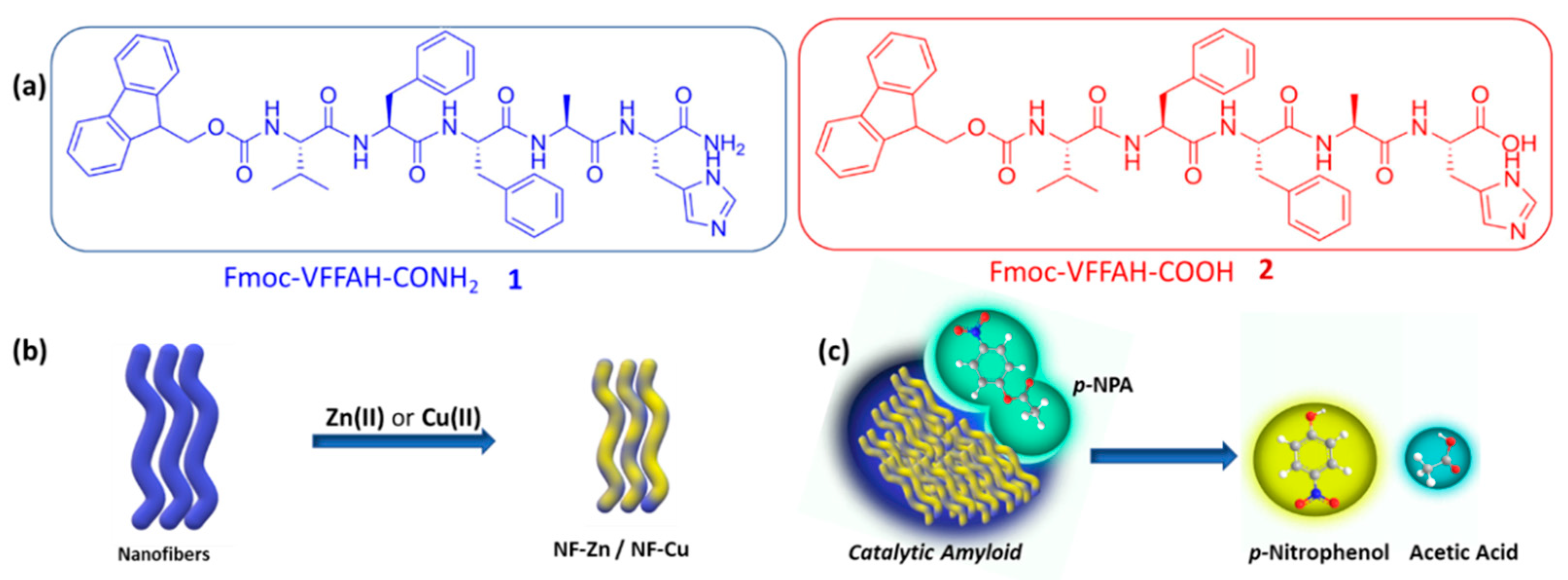
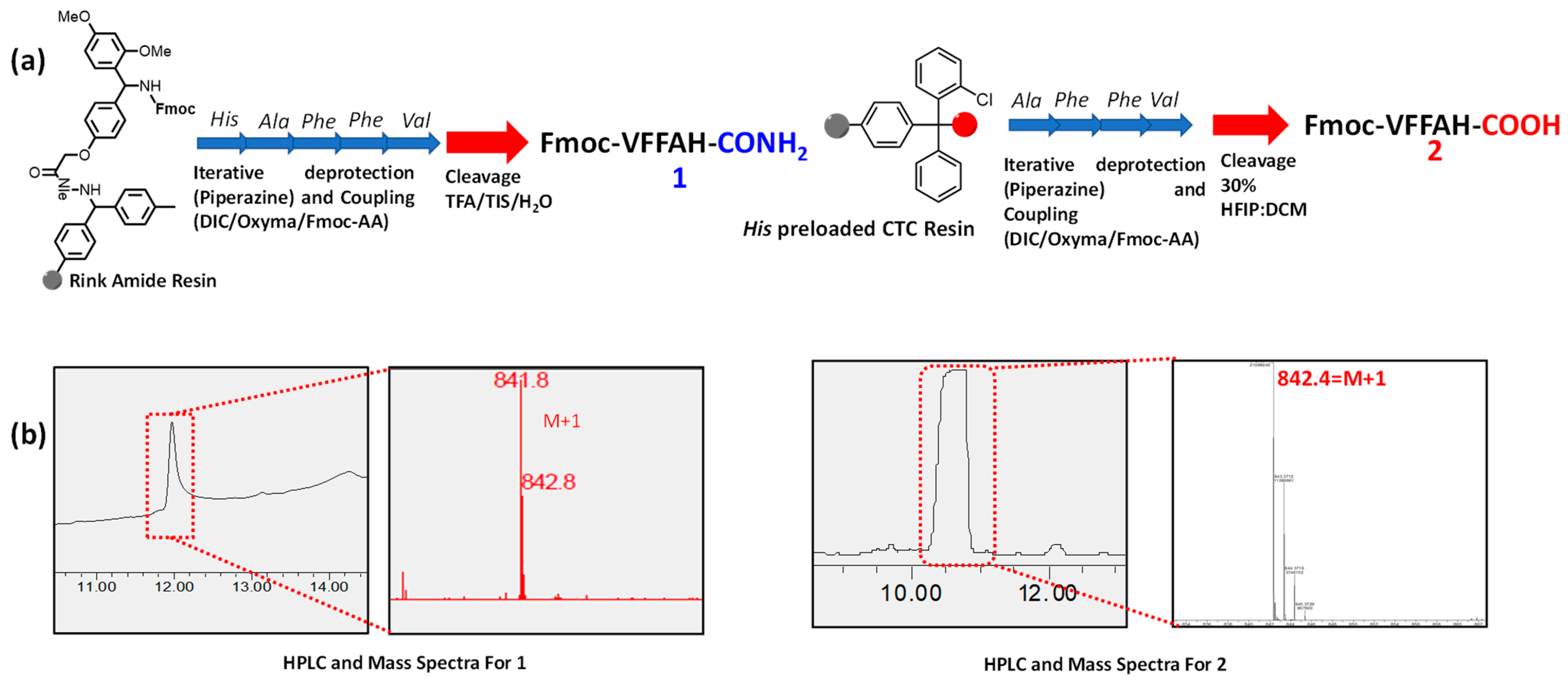
2.1. Synthesis of Peptides 1 and 2
2.2. HPLC and Mass Spectra
2.3. Self-Assembly of the Peptide Amphiphiles
2.4. Circular Dichroism Spectra
2.5. Thioflavin-T Binding Studies
2.6. Atomic Force Microscopy
2.7. Scanning Electron Microscopy
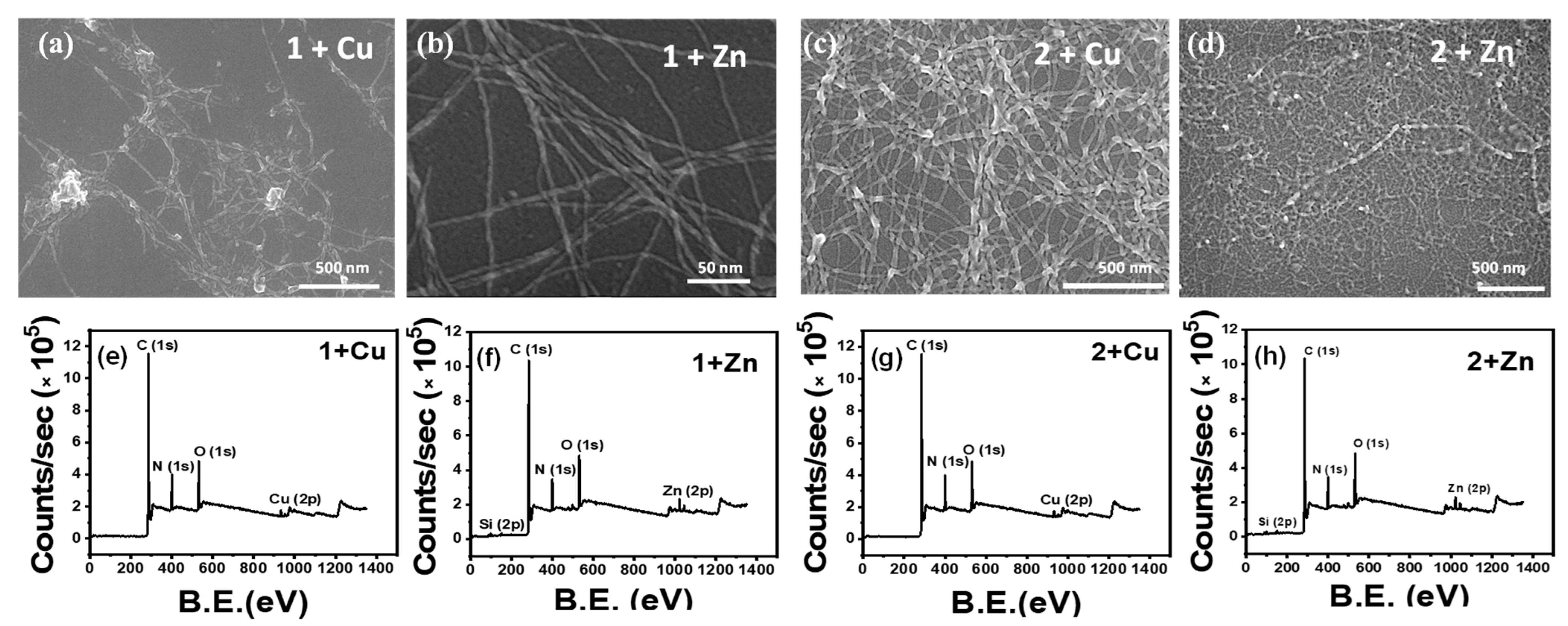
2.8. XPS Study
2.9. Fourier Transform Infrared Spectroscopy
2.10. UV–Vis Spectroscopic Studies and Titration with Metal Ions
2.11. Rheological Studies
2.12. Kinetic Assays of Para-Nitrophenyl Acetate (p-NPA) Hydrolysis
3. Results
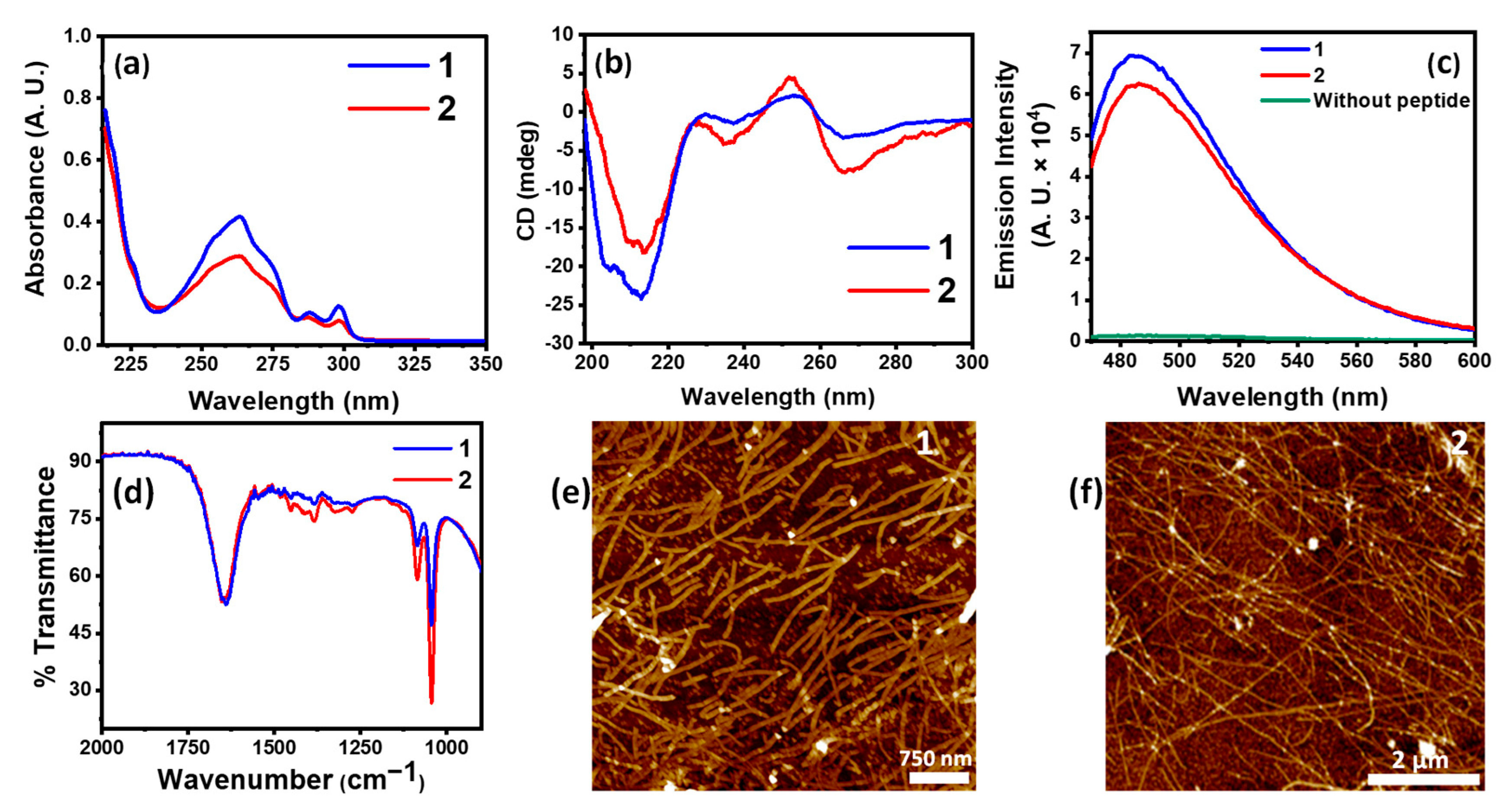
3.1. Self-Assembly Behavior of the Peptide Amphiphiles
3.2. Effect of Metal Ions
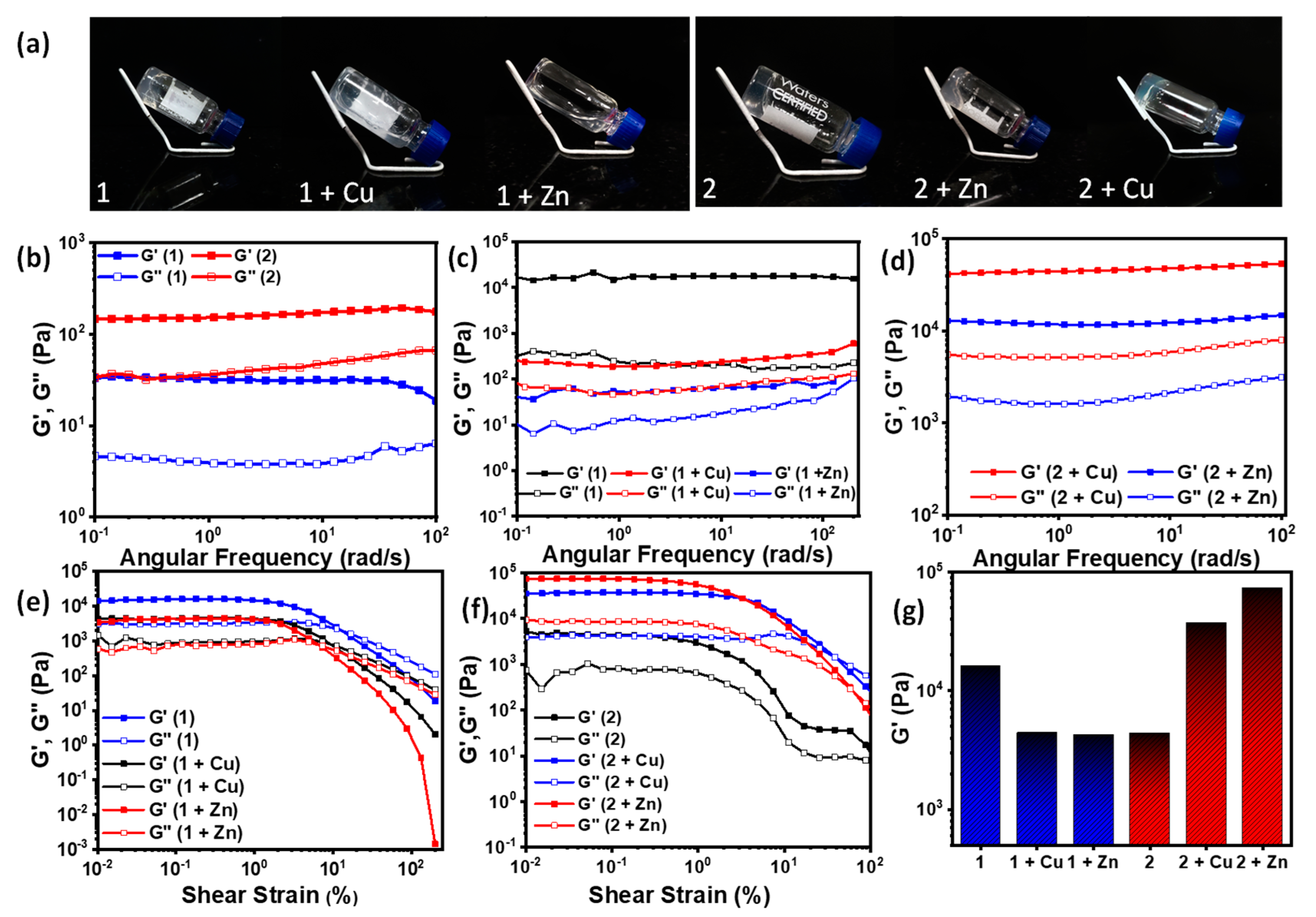
3.3. Gelation Properties
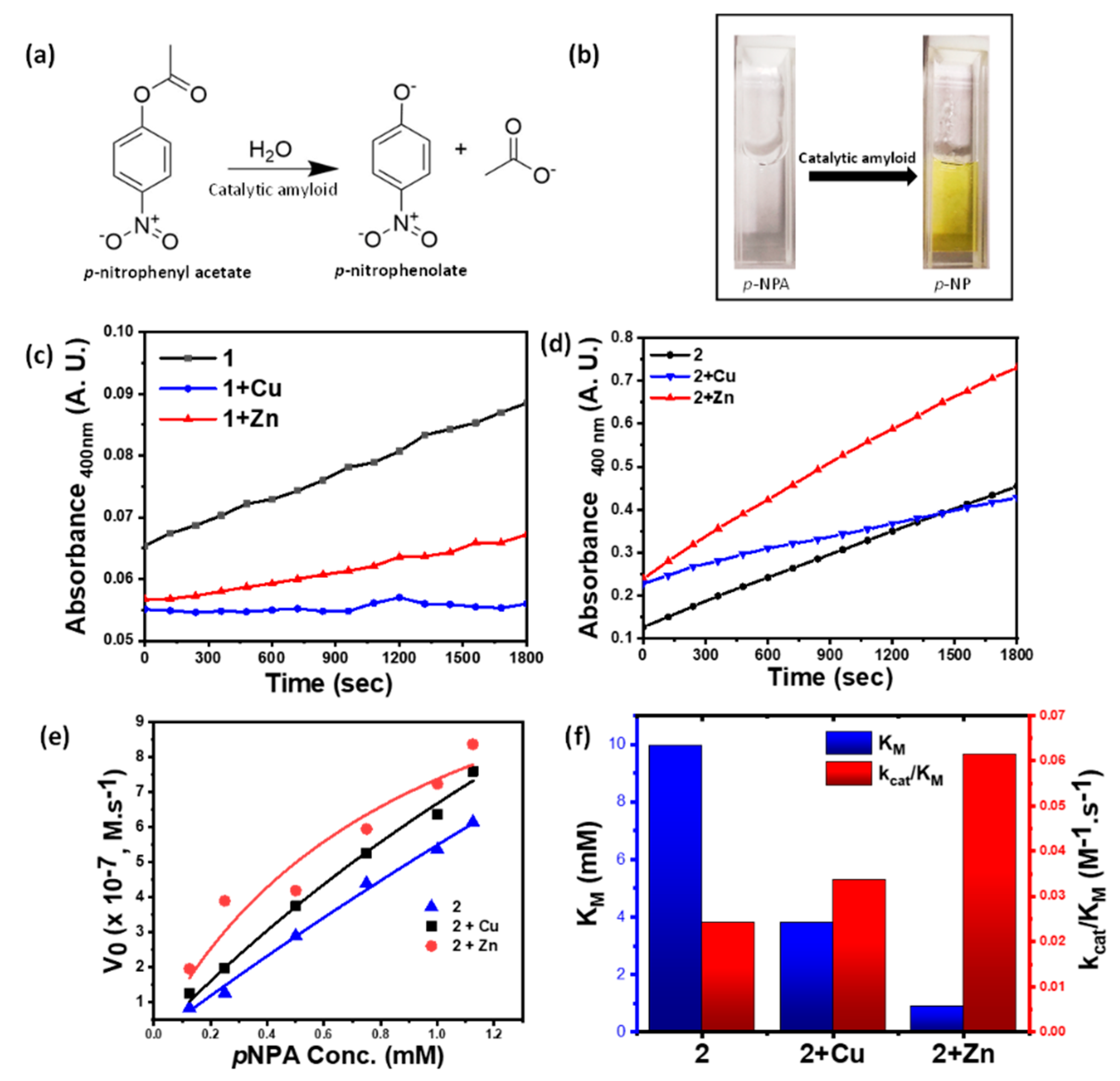
3.4. Catalysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Benkovic, S.J.; Hammes-Schiffer, S. A Perspective on Enzyme Catalysis. Science 2003, 301, 1196–1202. [Google Scholar] [CrossRef] [PubMed]
- Changeux, J.-P.; Edelstein, S.J. Allosteric Mechanisms of Signal Transduction. Science 2005, 308, 1424–1428. [Google Scholar] [CrossRef] [PubMed]
- VanEsch, J.H.; Klajn, R.; Otto, S. Chemical systems out of equilibrium. Chem. Soc. Rev. 2017, 46, 5474–5475. [Google Scholar] [CrossRef]
- Sorrenti, A.; Leira-Iglesias, J.; Markvoort, A.J.; Greef, T.F.A.d.; Hermans, T.M. Non-equilibrium supramolecular polymerization. Chem. Soc. Rev. 2017, 46, 5476–5490. [Google Scholar] [CrossRef] [PubMed]
- McManus, J.J.; Charbonneau, P.; Zaccarelli, E.; Asherie, N. The physics of protein self-assembly. Curr. Opin. Colloid Interface Sci. 2016, 22, 73–79. [Google Scholar] [CrossRef]
- Dobson, C.M. Protein folding and misfolding. Nature 2003, 426, 884–890. [Google Scholar] [CrossRef] [PubMed]
- Carny, O.; Gazit, E. A model for the role of short self-assembled peptides in the very early stages of the origin of life. FASEB J. 2005, 19, 1051–1055. [Google Scholar] [CrossRef]
- Greenwald, J.; Riek, R. On the Possible Amyloid Origin of Protein Folds. J. Mol. Biol. 2012, 421, 417–426. [Google Scholar] [CrossRef]
- Mattio, E.; Pal, A.; Leonetti, G.; Otto, S. Mechanism of building block exchange in stacks of self-replicating macrocycles. Synlett 2017, 28, 103–107. [Google Scholar] [CrossRef]
- Jaeger, K.; Eggert, T. Enantioselective biocatalysis optimized by directed evolution. Curr. Opin. Biotechnol. 2004, 15, 305–313. [Google Scholar] [CrossRef]
- Aldridge, S. Industry backs biocatalysis for greener manufacturing. Nat. Biotechnol. 2013, 31, 95–96. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Xue, X.; Luo, Q.; Li, Y.; Yang, K.; Zhuang, X.; Jiang, Y.; Zhang, J.; Liu, J.; Zou, G.; et al. Self-Assembled Peptide Nanofibers Designed as Biological Enzymes for Catalyzing Ester Hydrolysis. ACS Nano 2014, 8, 11715–11723. [Google Scholar] [CrossRef] [PubMed]
- Rufo, C.M.; Moroz, Y.S.; Moroz, O.V.; Stöhr, J.; Smith, T.A.; Hu, X.; DeGrado, W.F.; Korendovych, I.V. Short peptides self-assemble to produce catalytic amyloids. Nat. Chem. 2014, 6, 303–309. [Google Scholar] [CrossRef] [PubMed]
- Sousa, S.F.; Lopes, A.B.; Fernandes, P.A.; Ramos, M.J. The Zinc proteome: A tale of stability and functionality. Dalton Trans. 2009, 7946–7956. [Google Scholar] [CrossRef] [PubMed]
- Festa, R.A.; Thiele, D.J. Copper: An essential metal in biology. Curr. Biol. 2011, 21, R877–R883. [Google Scholar] [CrossRef]
- Frieden, E. Copper and iron metalloproteins. Trends Biochem. Sci. 1976, 1, 273–274. [Google Scholar] [CrossRef]
- Christianson, D.W.; Fierke, C.A. Carbonic Anhydrase: Evolution of the Zinc Binding Site by Nature and by Design. Acc. Chem. Res. 1996, 29, 331–339. [Google Scholar] [CrossRef]
- Zastrow, M.L.; Pecoraro, V.L. Designing Hydrolytic Zinc Metalloenzymes. Biochemistry 2014, 53, 957–978. [Google Scholar] [CrossRef]
- Singh, A.; Joo, J.-U.; Kim, D.-P. Microfluidic-driven ultrafast self-assembly of a dipeptide into stimuli-responsive 0D, 1D, and 2D nanostructures and as hydrolase mimic. Nanoscale 2022, 14, 15010–15020. [Google Scholar] [CrossRef]
- Makam, P.; Yamijala, S.S.R.K.C.; Tao, K.; Shimon, L.J.W.; Eisenberg, D.S.; Sawaya, M.R.; Wong, B.M.; Gazit, E. Non-proteinaceous hydrolase comprised of a phenylalanine metallo-supramolecular amyloid-like structure. Nat. Catal. 2019, 2, 977–985. [Google Scholar] [CrossRef]
- Lengyel, Z.; Rufo, C.M.; Moroz, Y.S.; Makhlynets, O.V.; Korendovych, I.V. Copper-Containing Catalytic Amyloids Promote Phosphoester Hydrolysis and Tandem Reactions. ACS Catal. 2018, 8, 59–62. [Google Scholar] [CrossRef] [PubMed]
- Duncan, K.L.; Ulijn, R.V. Short Peptides in Minimalistic Biocatalyst Design. Biocatalysis 2015, 1, 67–81. [Google Scholar] [CrossRef]
- Gulseren, G.; Khalily, M.A.; Tekinay, A.B.; Guler, M.O. Catalytic supramolecular self-assembled peptide nanostructures for ester hydrolysis. J. Mater. Chem. B 2016, 4, 4605–4611. [Google Scholar] [CrossRef]
- Joseph, J.P.; Singh, A.; Gupta, D.; Miglani, C.; Pal, A. Tandem Interplay of the Host–Guest Interaction and Photoresponsive Supramolecular Polymerization to 1D and 2D Functional Peptide Materials. ACS Appl. Mater. Interfaces 2019, 11, 28213–28220. [Google Scholar] [CrossRef]
- Mavlankar, N.A.; Awasthi, A.K.; Ralhan, J.; Pal, A. Amyloid-inspired Peptide Self-assembly/Disassembly as Intervened by Gold Nanoparticles and Polydopamine Coating to Dictate Spatiotemporal Organization. ChemNanoMat 2022, 8, e202200368. [Google Scholar] [CrossRef]
- Singh, A.; Joseph, J.P.; Gupta, D.; Sarkar, I.; Pal, A. Pathway driven self-assembly and living supramolecular polymerization in an amyloid-inspired peptide amphiphile. Chem. Commun. 2018, 54, 10730–10733. [Google Scholar] [CrossRef] [PubMed]
- Joseph, J.P.; Gupta, N.; Miglani, C.; Nath, D.; Singh, A.; Gupta, D.; Pal, A. Unraveling On-demand Strain-Stiffening in Nanofibrous Peptide–Polymer Conjugates to Mimic Contractility in Actinomyosin Networks. Chem. Mater. 2022, 34, 4364–4374. [Google Scholar] [CrossRef]
- Gupta, D.; Bhatt, A.; Gupta, V.; Miglani, C.; Joseph, J.P.; Ralhan, J.; Mandal, D.; Ali, M.E.; Pal, A. Photochemically Sequestered Off-Pathway Dormant States of Peptide Amphiphiles for Predictive On-Demand Piezoresponsive Nanostructures. Chem. Mater. 2022, 34, 4456–4470. [Google Scholar] [CrossRef]
- Gupta, N.; Singh, A.; Dey, N.; Chattopadhyay, S.; Joseph, J.P.; Gupta, D.; Ganguli, M.; Pal, A. Pathway-Driven Peptide–Bioglass Nanocomposites as the Dynamic and Self-Healable Matrix. Chem. Mater. 2021, 33, 589–599. [Google Scholar] [CrossRef]
- Thomas, J.; Gupta, N.; Joseph, J.P.; Chopra, V.; Pal, A.; Ghosh, D. Mechanical Integrity in a Dynamic Interpenetrating Hydrogel Network of Supramolecular Peptide–Polysaccharide Supports Enhanced Chondrogenesis. ACS Biomater. Sci. Eng. 2021, 7, 5798–5809. [Google Scholar] [CrossRef]
- Gupta, D.; Sasmal, R.; Singh, A.; Joseph, J.P.; Miglani, C.; Agasti, S.S.; Pal, A. Enzyme-responsive chiral self-sorting in amyloid-inspired minimalistic peptide amphiphiles. Nanoscale 2020, 12, 18692–18700. [Google Scholar] [CrossRef]
- Singh, A.; Joseph, J.P.; Gupta, D.; Miglani, C.; Mavlankar, N.A.; Pal, A. Photothermally switchable peptide nanostructures towards modulating catalytic hydrolase activity. Nanoscale 2021, 13, 13401–13409. [Google Scholar] [CrossRef] [PubMed]
- Hartgerink, J.D.; Beniash, E.; Stupp, S.I. Self-Assembly and Mineralization of Peptide-Amphiphile Nanofibers. Science 2001, 294, 1684–1688. [Google Scholar] [CrossRef] [PubMed]
- Al-Garawi, Z.S.; McIntosh, B.A.; Neill-Hall, D.; Hatimy, A.A.; Sweet, S.M.; Bagley, M.C.; Serpell, L.C. The amyloid architecture provides a scaffold for enzyme-like catalysts. Nanoscale 2017, 9, 10773–10783. [Google Scholar] [CrossRef] [PubMed]
- Hernick, M.; Fierke, C.A. Zinc hydrolases: The mechanisms of zinc-dependent deacetylases. Arch. Biochem. Biophys. 2005, 433, 71–84. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | 1 | 1 + Cu | 1 + Zn | 2 | 2 + Cu | 2 + Zn |
|---|---|---|---|---|---|---|
| Observation | Strong gel | Weak gel | Weak gel | Strong gel | Strong gel | Strong gel |
| Sample | Vmax (M−1 s−1) | KM (mM) | kcat/KM (mM−1 s−1) | R2 |
|---|---|---|---|---|
| 2 | 60.29 | 9.97 | 24.19 | 0.9946 |
| 2 + Cu | 32.10 | 3.80 | 33.77 | 0.9908 |
| 2 + Zn | 14.17 | 0.92 | 61.57 | 0.9114 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Patra, S.; Mavlankar, N.A.; Ramesan, L.; Singh, A.; Pal, A. Tweaking of Peripheral Moieties in Catalytic Amyloid for Modulating Hydrogel Strength and Hydrolase Activity. Chemistry 2023, 5, 1190-1202. https://doi.org/10.3390/chemistry5020080
Patra S, Mavlankar NA, Ramesan L, Singh A, Pal A. Tweaking of Peripheral Moieties in Catalytic Amyloid for Modulating Hydrogel Strength and Hydrolase Activity. Chemistry. 2023; 5(2):1190-1202. https://doi.org/10.3390/chemistry5020080
Chicago/Turabian StylePatra, Soumya, Nimisha A. Mavlankar, Lakshminarayan Ramesan, Ashmeet Singh, and Asish Pal. 2023. "Tweaking of Peripheral Moieties in Catalytic Amyloid for Modulating Hydrogel Strength and Hydrolase Activity" Chemistry 5, no. 2: 1190-1202. https://doi.org/10.3390/chemistry5020080
APA StylePatra, S., Mavlankar, N. A., Ramesan, L., Singh, A., & Pal, A. (2023). Tweaking of Peripheral Moieties in Catalytic Amyloid for Modulating Hydrogel Strength and Hydrolase Activity. Chemistry, 5(2), 1190-1202. https://doi.org/10.3390/chemistry5020080