Abstract
Linear and cyclic acenes are polycyclic aromatic hydrocarbons that can be viewed as building blocks of graphene nanoribbons and carbon nanotubes, respectively. While short linear acenes demonstrated remarkable efficiency in several optoelectronic applications, the longer members are unstable and difficult to synthesize as their cyclic counterparts. Recent progress in on-surface synthesis, a powerful tool to prepare highly reactive species, opens promising perspectives and motivates the computational investigations of these potentially functional molecules. Owing to their di- and poly-radical character, low-lying excited states dominated by doubly excited configurations are expected to become more important for longer members of both linear and cyclic molecules. In this work, we investigate the lowest-lying and the doubly excited (DE) state of linear acenes and cyclacenes, with different computational approaches, to assess the influence of the di-/poly-radical characters (increasing with the molecular dimensions) on their relative order. We show that DFT/MRCI calculations correctly reproduce the crossing of the two states for longer linear acenes, while TDUDFT calculations fail to predict the correct excitation energy trend of the DE state. The study suggests a similarity in the excited electronic state pattern of long linear and cyclic acenes leading ultimately to a lowest lying dark DE state for both.
1. Introduction
Linear acenes ([n]-acenes, Figure 1a), are polycyclic aromatic hydrocarbons (PAHs) formed by n linearly fused benzene rings and characterized by exposing two zig-zag edges. As such, linear acenes can be considered as one-dimensional strips of graphene and possible building blocks of carbon nanoribbons. Furthermore, oligoacenes are among the most efficient candidates for high-performance organic field-effect transistors (OFETs) and numerous optoelectronic applications [1,2,3,4].
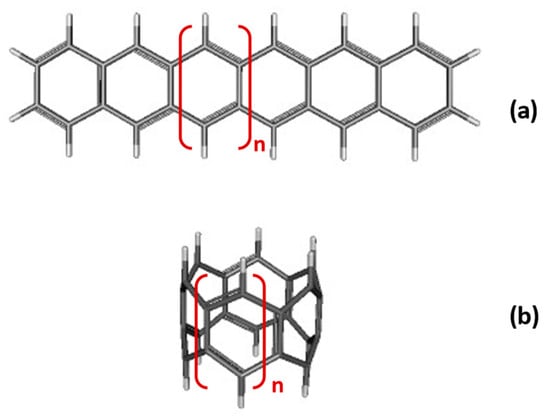
Figure 1.
Chemical structure of: (a) [n]-acenes; (b) [n]-CC carbon nanobelts. In both cases, n is the number of fused rings.
Due to the topology of their ring fusion, acenes share only one single Clar’s sextet among all six-membered rings, which implies small energy gaps between the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) compared to conjugated hydrocarbons of similar size. A small HOMO-LUMO gap is a typical feature of di-radical molecules and indeed acenes can be classified as Kekulé-type di-radicals, but their poly-radical character has also been demonstrated [5,6,7]. As other conjugated di-radicals, longer acenes also feature small singlet/triplet energy separation and characteristic absorption in the NIR region [8]. The increasing open-shell character, with the extension of the conjugation length explains the reactivity of longer acenes and their difficult synthesis. Nonetheless, several experimental studies have reported successful preparation of longer acenes in recent years, under matrix-isolation conditions or taking advantage of on-surface synthesis [4,8,9,10,11,12,13,14,15,16,17,18,19,20,21,22].
While DFT calculations carried out at restricted level indicated a triplet ground state for longer acenes [23], a singlet ground state with open-shell character was predicted by unrestricted DFT calculations [24,25]. Notably, the singlet ground state for long linear acenes is also supported by thermally assisted occupation (TAO) DFT [26,27,28] and by higher levels of theory, including both static and dynamic correlation effects [5,29,30].
Acenes possess very characteristic electronic absorption spectra dominated by three band systems known as the , , and , according to Platt’s notation [31]. These lowest lying excited states of acenes [31,32,33] are determined by excitations within an orbital space that includes two occupied (HOMO and HOMO-1) and two unoccupied (LUMO and LUMO+1) molecular orbitals (MOs). Specifically, and states are dominated by HOMO-1→LUMO and HOMO→LUMO+1 excitations, while the is dominated by the HOMO→LUMO excitation and appears at lower energies than the state except for 2-acene. However, due to their di-radical character, the simple two-electron in two-orbital (2e–2o) model [34] also predicts a low-lying, doubly excited (DE) dark state dominated by the HOMO,HOMO→LUMO,LUMO excitation, which has been identified for several conjugated di-radicaloids [35,36,37,38,39] and for heptacene [18].
The and states display a different nature (covalent and ionic) challenging time dependent DFT (TDDFT) calculations, whose quality strongly depends on the type of functional [40,41,42,43,44]. Because double excitations contribute to these low-lying excited states [33], a balanced description was reported by employing double hybrid functionals [45] and carefully assessed [46] for a set of non-empirical double-hybrid density functionals developed in recent years [47]. Low-lying excited states were also investigated at other levels of theory, including DFT combined with multi-reference configuration interaction (DFT/MRCI) [48,49], particle–particle random-phase approximation (pp-RPA) [30], complete active space self-consistent field (CASSCF) supplemented with multi-reference second-order perturbation theory (CASPT2) [50] or multi-configuration-coupled electron pair approximation (MCCEPA) [18]. Despite the impressive amount of computational investigations, the DE state of linear acenes has received comparably less attention [18,30,48].
[n]-cyclacenes, ([n]-CCs) (Figure 1b) are the cyclic analogues of acenes formed by the fusion of n benzene rings. Considered as the narrowest zig-zag carbon nanotubes, cyclacenes are expected to be highly reactive, not only due to the strain inherent in the formation of carbon nanobelts but also based on their predicted open-shell character [6,23,51,52,53]. Several attempts to synthesize cyclacenes have been reported [54,55,56,57,58,59] and there have been recent breakthroughs in the field [57,60,61,62]. In addition to conventional chemistry, on-surface synthesis, which has emerged as a powerful tool to prepare highly reactive species, opens promising perspectives [51,63].
Their open-shell electronic structure, di-radical character, singlet/triplet gap, and magnetic interactions have been determined at the DFT level and with several wavefunction-based multi-reference approaches [6,23,28,46,52,64,65,66,67,68,69]. Due to their di- and poly-radical character, excited states dominated by doubly excited configurations are expected to appear among the lowest-lying as for [n]-acenes. Notably, the energy location of the DE state in long linear acenes and [n]-CCs may influence the outcome of photoinduced events, including singlet fission processes [70,71,72,73,74]. While experimental information on [n]-CCs is not available, for linear acenes, a crossing between the DE and the state is documented to occur between 6-acene and 7-acene [18].
In this context, the objective of this study is to investigate the low-lying excited states of [n]-acenes and [n]-CCs, focusing on the relative energy order of the and DE states. To this end, low-lying excited states of [n]-acenes with n = 2–9 and [n]-CCs with n = 6–12 are investigated with several computational approaches, including a flavor of DFT-based approaches encompassing TDDFT, TDUDFT, and spin-flip TDDFT (SF-TDDFT) [75,76], along with DFT/MRCI [48,49] and second-order perturbation theory NEVPT2 [77]. The objective is to assess the quality of such different excited state approaches against the experimental data available for linear acenes and to extrapolate the results to the so-far unavailable data for [n]-CC carbon nanobelts.
2. Computational Methods
The equilibrium structures of linear acenes and cyclacenes were determined with DFT calculations employing the B3LYP functional and the 6-31G* basis set. The geometry optimization was first carried out with the restricted approach to determine a closed-shell (CS) equilibrium structure. For most of the systems investigated, a more stable open-shell broken-symmetry (BS) solution was found at the CS geometry, and therefore the equilibrium structure corresponding to the BS solution was readily determined. The overall CS-BS stability (ΔE(CS-BS)) was determined as the energy difference between the energy of the CS structure computed with restricted DFT and the energy of the BS structure computed with UDFT. At the same time, the stabilization energy only due to wavefunction relaxation at the same CS geometry was also determined and compared with the overall stabilization. Energies and shapes of frontier molecular orbitals (FMOs), both at CS and BS geometries, are collected in Tables S1–S4 and Figures S1–S4.
The excitation energies of low-lying excited states were determined with several DFT-based computational schemes, encompassing standard TDDFT based on a CS reference configuration (only to predict the state) and other flavors of the TD approach.
In recent works, we have shown that, for systems with well-localized BS frontier MOs, TDUDFT calculations can be used to predict the excitation energy of the DE state as well as the state since both excited states are described in terms of singly excited configurations at the TDUDFT level [36,38,39]. Double excitations can be recovered from TDDFT calculations also with the SF scheme [75,76]. Spin-flipping excitations enable SF-TDDFT to treat ground- and excited-state electron correlation on the same footing, while also incorporating some doubly excited configurations that are important for di-radicals. Accordingly, these two approaches were employed to investigate the excitation energy of the low lying DE state of the investigated systems.
Geometry optimization, TDDFT, and TDUDFT calculations were carried out with the Gaussian16 suite of programs [78] while SF-TDDFT calculations were carried out in the collinear approximation as implemented in the GAMESS 2016 package [79].
The and DE excitation energies were also estimated for selected molecular systems, with DFT/MRCI and CASSCF/NEVPT2 calculations. The DFT/MRCI method combines DFT (which gives information about dynamic correlation) and truncated MRCI expansions (to take the static correlation into account). In the original work [80] different parameter sets were employed for singlet and triplet state calculations. More recently, a spin-invariant parameterization has been introduced [81] which incorporates less empiricism compared to the original formulation while preserving its computational efficiency. We have carried out DFT/MRCI calculations using the DFT/MRCI software [82] with the def2-SVP basis set, and both sets of parameters are hereafter labeled as original [80] and R2018 [81].
Concerning CASSCF calculations, these were carried out with the state averaged approach, using the same weight for the lowest singlet states. The active spaces considered ranged between 8 electrons in 8 π orbitals (8,8) up to 14 electrons in 14 orbitals (14,14) (see the Supplementary Materials for further details) and were followed by second-order perturbation theory NEVPT2 [77] calculations carried out with the strongly correlated approach to include dynamical electron correlation. All the CASSCF and NEVPT2 calculations were performed using the ORCA 5.0.1 program [83] using the def2-SVP basis set. The resolution of identity approximation and the related basis sets for both Coulomb and HF exchange integrals were used (RI-JK)[84].
Two descriptors of the di-radical/multi-radical character were employed. The first is the parameter which, in the spin-unrestricted single-determinant formalism can be determined in the spin-projection scheme as [85,86]:
with calculated as:
and n is the occupation number of the frontier natural orbitals (NO). The di-radical character corresponds to while the tetraradical character is obtained for . NO occupation numbers were determined at the UHF level and UB3LYP level, namely employing the same functional used for the geometry optimization. According to the use of the spin-projection scheme, the level of theory employed to calculate the parameters will be hereafter indicated with the labels PUHF and PUB3LYP.
The second parameter considered is based on finite temperature DFT (FT-DFT) and is the NFOD value, which is the integral of the fractional orbital density (FOD) , over all space.
The is defined as [87,88]:
where and are two constants set such that only fractionally occupied orbitals are taken into account; are molecular spin orbitals; and are the fractional orbital occupancies (0 ≤ ≤ 1) determined by the Fermi–Dirac distribution. In other words, the so-defined FOD yields, for each point in real space, only the contribution of the ‘hot’ or strongly correlated electrons and is therefore an analysis tool of static correlation. The and NFOD parameters were computed at the CS and BS geometries for the entire set of di-radicals shown in Figure 1. The NFOD parameter was computed with the ORCA 5.0.1 program [83] with the default setting (TPSS/def2-TZVP level with Tel = 5000 K).
3. Results
3.1. Stabilization of Open-Shell Structures
Because geometry optimizations at the same level of theory were reported in previous works for linear acenes [24] and [n]-CCs [52,64,66], we do not discuss geometrical detail that can be found elsewhere. However, it is interesting to consider the stabilization associated with the open-shell character that develops for linear acenes only for n ≥ 6 and for all the [n]-CCs investigated. In each panel of Figure 2, the stabilization only due to the constraint relaxation in the unrestricted wavefunction (orange bars) is compared with the total stabilization including also the contribution associated with the geometrical relaxations from the CS to the BS structure (blue bars) (numerical data collected in Table S5). As previously observed, the overall stabilization increases with molecular size for both linear acenes and carbon nanobelts on account of their increasing di-/poly-radical characters. It is also commonly found that a large fraction of the total stabilization is due to relaxation of wavefunction constraints.
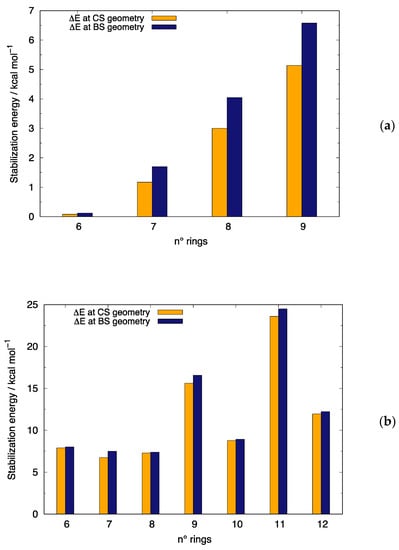
Figure 2.
Comparison between the total stabilization energy (ΔE at BS geometry) and the stabilization energy due to the relaxation of the constraint of same spatial occupation for α and β electrons in the unrestricted wavefunction (ΔE at CS geometry). From B3LYP/6-31G* calculations: (a) [n]-acenes displaying an open-shell structure and (b) even and odd [n]-CCs.
Thus, for linear acenes (Figure 2a), the total stabilization energy is mainly due to the BS wavefunction with a non-negligible contribution from geometry relaxation. However, this trend is exasperated for [n]-CCs, with an increased contribution from wavefunction relaxation (orange bars) for all [n]-CCs. A careful inspection of Figure 2b reveals a difference between even and odd [n]-CCs, with the former displaying an almost negligible stabilization due to structural relaxations, in contrast with the more relevant contribution documented for odd [n]-CCs. Notably, the large contribution of wavefunction relaxation (ca. 98% of the total stabilization) in even [n]-CCs can be rationalized by the non-bonding nature of the FMOs (Figure S2) and implies very similar CS and BS geometries.
Such different trends in stabilization energies can be traced back to the cryptoannulenic effect [89,90], which is due to the nature of peripheral circuits of cyclacenes. These peripheral circuits are formed either by 4k or 4k + 2 carbon atoms (where k is an integer), depending on the number of benzene rings in the n-cyclacene. The different character of the peripheral circuits in odd and even [n]-CCs results in several other different properties for the two sub-classes of [n]-CCs. Odd and even members of the series display rather different electronic structure, with molecules bearing an odd number of rings exhibiting a pair of degenerate HOMOs and LUMOs in contrast to even [n]-CCs [6].
3.2. Di- and Poly-Radical Characters of [n]-acenes and [n]-CCs
In previous works, the di-/poly-radical characters of linear and cyclic acenes have been thoroughly discussed [5,6,7,52,88]. Here, we consider NFOD and values and analyze them in view of their significance for the description of low-lying excited states of both linear and cyclic acenes. Our computed NFOD values confirm previous results and show a good linear correlation with the di-radical index computed at PUHF or PUB3LYP level for linear acenes (Figure S5). As previously noted [36], the values are dependent on the chosen level of theory (PUHF or PUDFT) adopted to calculate natural orbital occupation numbers, with PUB3LYP values always smaller than PUHF data (Tables S6 and S7) [36]. More importantly, the trends are similar and independent from the method chosen to calculate such descriptors.
Recalling that NFOD values correspond to the number of correlated electrons, the computed values indicate that longer acenes, featuring NFOD > 2, acquire a poly-radical character. This trend is even more marked for carbon nanobelts, where NFOD values are crucially above 2 for all odd [n]-CCs and for even [n]-CCs with (n > 8). The computed NFOD values correlate well with the tetra-radical index (Figure S6) when the two sub-classes of [n]-CCs are considered separately since assumes considerably larger values for odd [n]-CCs on account of its degenerate FMOs. Such crucial differences between even and odd [n]-CCs can be appreciated by the different slopes of the linear correlation between (computed at the PUB3LYP level) and the total stabilization energy of BS structures computed at the same level (Figure 3).
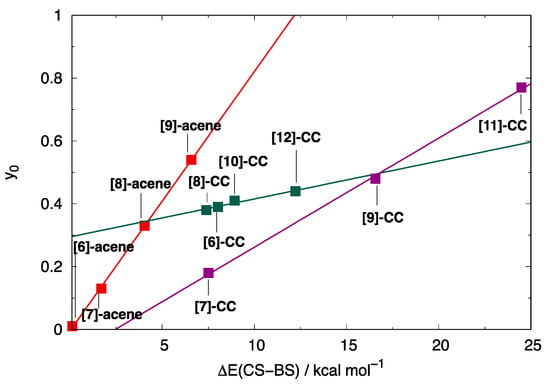
Figure 3.
Correlation between the (PUB3LYP) level and the computed stabilization of the BS structure with respect to the CS structure, both optimized at B3LYP/6-31G* level: (red squares) [n]-acenes; (green squares) even [n]-CCs; (purple squares) odd [n]-CCs.
The combination of the two descriptors (NFOD and ) discussed above gives complementing information on the reliability of the simple 2e–2o approach [34,36,38,39] to describe linear and cyclic acenes. When the NFOD value exceeds 2, it is expected that more than two electrons should be correlated for a proper description of ground and excited states. Such deviation from the ideal 2e–2o model preferentially affects odd [n]-CCs, owing to their more marked poly-radical character. Thus, in the following excited state investigations, we will restrict the attention to even [n]-CCs as well as [n]-acenes.
We now consider how the localization of computed BS orbitals is intimately related with the di-radical character . Within the 2e–2o model, the BS HOMO and LUMO orbitals of the unrestricted wavefunction can be described as linear combinations of the delocalized HOMO and LUMO obtained from the CS solution ( and ). Following previous works [85,86], we can write the BS orbitals as a function of and , with the angle of rotation with respect to the CS set of orbitals (see Table S8) and also can be recast as a function of [38,39,85]:
The linear combination of the BS frontier orbitals of each investigated molecule was determined by projecting each BS frontier orbital over the set of CS orbitals. The corresponding combination coefficients, from which the values can be determined, are collected in Figures S1 and S2.
For the set of molecules investigated, the values were determined either from the expression of the BS HOMO in terms of CS orbitals or from the expression of the BS LUMO in terms of the CS orbitals. Crosses are then placed in Figure 4 in correspondence to the computed and values, both determined at the same level of theory.
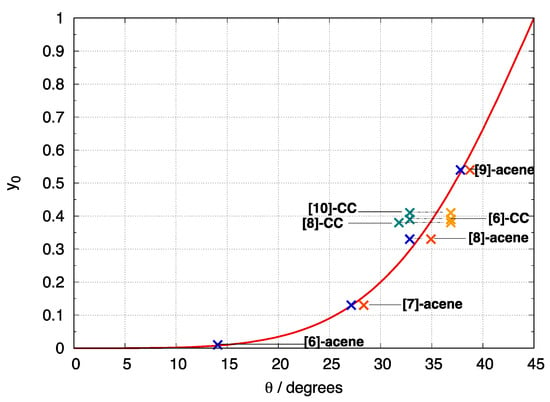
Figure 4.
Dependence of the di-radical character (PUB3LYP) as a function of BS orbital rotation angle . Analytic expression from Equation (5) (red curve); determined for the HOMO BS orbitals (orange crosses for [n]-acenes, gold crosses for [n]-CCs); determined for the LUMO BS orbitals (blue crosses for [n]-acenes, green crosses for [n]-CCs).
In Figure 4 (red curve), Equation (5) is also plotted for reference. Note that orbital rotation of 45° implies the formation of fully localized BS orbitals and corresponds to the maximum di-radical character = 1. When the rotation angle is smaller, decreases and becomes when CS and BS orbitals coincide. There is a general good agreement between the orbital rotation angle determined from the BS and CS orbitals of each molecule and the theoretical red curve. The largest deviations observed for [n]-CCs compared to [n]-acenes mainly arise from additional contributions to the BS orbitals from other CS occupied/unoccupied orbitals. Such extra contributions are in line with the generally larger tetra-radical character of [n]-CCs compared to [n]-acenes and suggest that in these cases the 2e–2o model may not fully apply.
3.3. The Excited State of [n]-acenes and [n]-CCs
Several investigations have focused on the prediction of the excitation energy of the state of [n]-acenes and in Figure 5a we collect the most relevant results taken from the literature along with our computed results and compare them with the experimental data. We should note that the acene geometries adopted in this work are not the same as those used in other studies, which can cause some differences in computed excitation energies. Inspection of the collection of data shows, clearly, the poor predictions of TDB3LYP, a trend well-known for hybrid functionals. Additionally, the SF-TDB3LYP results are unsatisfactory probably due to the use of a functional featuring an inadequate amount of exchange correlation [75] combined with the spin contamination derived by the incomplete spin-space of these calculations. Notably, an improvement over TDB3LYP is documented for longer acenes by the TDUB3LYP results. The recent TD calculations [46] carried out with non-empirical double-hybrid density functionals [47] clearly outperform the results obtained here from hybrid functionals and favorably compare with other wave-function based correlated approaches. The pp-RPA computed excitation energies [30] and our DFT/MRCI results nicely reproduce experimental data and small differences are obtained using the original or the R2018 parametrizations for the latter approach. Finally, NEVPT2 calculations, carried out for the longer acenes, are also in nice agreement with experiment. In summary, the excitation energy of the state is reliably described by several computational approaches for [n]-acenes. Extrapolating the results of [n]-acenes to [n]-CCs (Figure 5b), we can conclude that DFT/MRCI results should give a reliable prediction of the state of [n]-CCs, while the much lower TDB3LYP computed values suffer from the same limited accuracy and underestimation documented for linear acenes. In this context, we note that TDUB3LYP excitation energies are in qualitative agreement with DFT/MRCI results but show an incorrect energy trend as a function of the dimension of the [n]-CC. The NEVPT2 predictions also underestimate the excitation energy and are in contrast with the accurate results obtained for [n]-acenes, an inconsistency that may be traced back to a limited dimension of the CASSCF space [91] employed in these preliminary calculations and will require more systematic investigations.
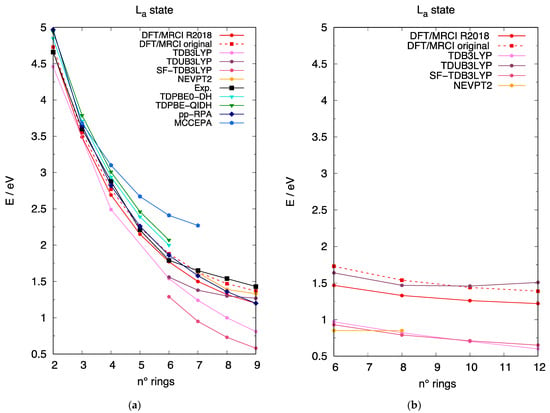
Figure 5.
The excitation energy to the state (i.e., the state dominated by the HOMO→LUMO excitation) described for different computational approaches. The excitation energy scale is the same in the two panels to allow a direct comparison between linear and cyclic acenes. (a) Excitation energy of the state for [n]-acenes as a function of the number of rings: (red circles) DFT/MRCI R2018 parametrization; (red squares, dashed line) DFT/MRCI original parametrization; (pink circles) TDB3LYP; (purple circles) TDUB3LYP; (dark-rose circles) SF-TDB3LYP; (gold circles) NEVPT2; (black squares) experimental from refs. [16,18,19,23,40]; (cyan triangles) TDPBE0-DH from ref. [46]; (green triangles) TDPBE-QIDH from ref. [46]; (blue diamonds) pp-RPA from ref. [30]; (royal-blue pentagons) MCCEPA from ref. [18]. (b) Excitation energy of the state for [n]-CCs as a function of the number of rings: (red circles) DFT/MRCI R2018 parametrization; (red squares, dashed line) DFT/MRCI original parametrization; (pink circles) TDB3LYP; (purple circles) TDUB3LYP; (dark-rose circles) SF-TDB3LYP; (gold circles) NEVPT2.
3.4. The DE Excited State of [n]-acenes and [n]-CCs
In contrast with the singly excited state, comparably fewer investigations have considered the prediction of the DE state of [n]-acenes, and in Figure 6a we collect the most relevant results taken from the literature, along with our computed results. In this case, little experimental data are available for [n]-acenes: for the smaller acenes, two-photon absorption (TPA) investigations [92,93] identified a low-lying state of Ag symmetry. Notably, while for [3]-acene the experimental data from TPA (black empty squares in Figure 6) are nicely reproduced by most calculations, for [2]-acene, the experimental excitation energy of the TPA active state is much lower compared to the predictions of the DE state. This can be reconciled in the light of our DFT/MRCI wavefunction analysis. These calculations show two low-lying excited states of Ag symmetry for naphthalene, the highest of which is the DE state while the lowest is only marginally affected by doubly excitations and its computed excitation energy (red isolated circle and square in Figure 6a, for the R2018 and original parametrizations, respectively) is in much better agreement with the experimental TPA data. On the computational side, apart from [2]-acene, the lowest Ag excited state is always the DE state for longer acenes. Overall, we note that DFT/MRCI calculations with both parametrizations are in very good agreement with the available experimental data [18,92,93] and with the results of pp-RPA calculations [30]. SF-TDB3LYP excitation energies show an acceptable trend, with a slightly too-steep energy decrease with the acene length. NEVPT2 and MCCEPA [18] appear to overestimate the excitation energy of the DE state for longer [n]-acenes. Finally, an unexpected excitation energy increase with the extension of the oligoacene is determined by TDUB3LYP calculations. Similar trends characterize the predictions of the DE state for [n]-CCs and, extrapolating from [n]-acenes, we can conclude that the quality of the DFT/MRCI results is expected to be comparable to the pp-RPA calculations also for the carbon nanobelts.
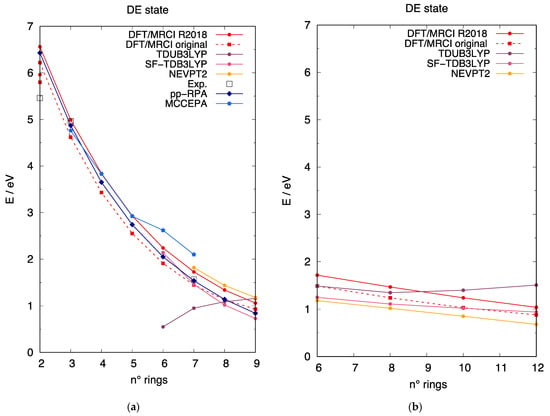
Figure 6.
The excitation energy to the DE state (i.e., the state dominated by the HOMO, HOMO→LUMO, LUMO excitation) described at different computational approaches. The excitation energy scale is the same in the two panels to allow a direct comparison between linear and cyclic acenes. (a) Excitation energy of the DE state for [n]-acenes as a function of the number of rings: (red circles) DFT/MRCI R2018 parametrization; (red squares, dashed line) DFT/MRCI original parametrization; (pink circles) TDB3LYP; (purple circles) TDUB3LYP; (dark-rose circles) SF-TDB3LYP; (gold circles) NEVPT2; (black empty squares) experimental from refs. [16,92,93]; (blue diamonds) pp-RPA from ref. [30]; (royal-blue pentagons) MCCEPA from ref. [18]. (b) Excitation energy of the DE state for [n]-CCs as a function of the number of rings: (red circles) DFT/MRCI R2018 parametrization; (red squares, dashed line) DFT/MRCI original parametrization; (pink circles) TDB3LYP; (purple circles) TDUB3LYP; (dark-rose circles) SF-TDB3LYP; (gold circles) NEVPT2.
4. Discussion
Based on the results of the previous section, we focus here on two subjects. First, we rationalize the unusual excitation energy trends predicted by TDUB3LYP calculations for the DE state, and second, we discuss the occurrence of the /DE state inversion in both [n]-acenes and [n]-CCs in view of the relevant effects on their photophysical properties.
4.1. Assessment of TDUDFT Results
The results of TDUDFT calculations can be critically analyzed by considering the nature of the wavefunction describing the DE state at the TDUDFT level [38,39], which reads:
where the first two terms correspond to singlet-spin contributions and the latter identifies the triplet-spin component. Equation (6) shows that the wavefunction of the DE state is a combination of singlet and triplet spin contributions, which implies that spin contamination can be a relevant issue. In Figure 7 we report the two spin contributions (the square of the coefficient of the triplet spin wavefunction component in blue and that of the singlet-spin component in red) for angles increasing from 0 to 45°. The graph shows that the DE state wavefunction is a pure singlet state only for fully localized BS orbitals ( = 45°), while decreasing the rotation angle, the triplet component becomes more and more important, and for = 0, the state becomes a pure triplet state. On the same figure, we also placed the investigated molecules with crosses, based on their computed rotation angles obtained from calculations.
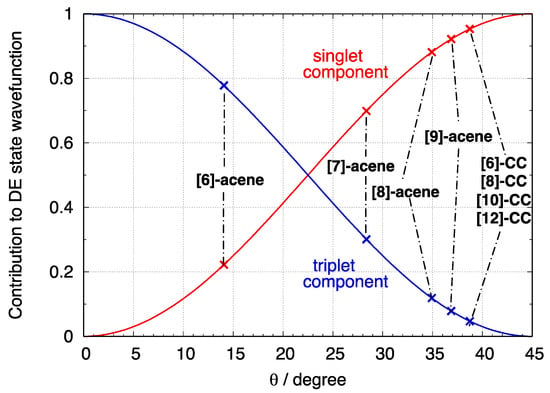
Figure 7.
Contributions of the singlet and triplet components to the DE state described at TDUDFT level according to Equation (6) as a function of BS orbital rotation angle . The investigated linear and cyclic acenes are placed on the graph with crosses based on their computed BS orbital rotation angle.
Since is related to the di-radical character as shown in a previous section, the above results demonstrate that for very small rotation angles (and di-radical character), the triplet contribution dominates and the predicted TDUB3LYP result is unreliable because of the mixed spin nature of the state, which implies a large spin contamination. As shown in Figure 7, the longer acenes and [n]-CCs display a larger than 25°. Only [6]-acene corresponds to a very small , a region in which the wavefunction of the DE state is dominated by the triplet component and is therefore unreliable. This explains the poor TDUB3LYP excitation energy of the DE state reported in Figure 6a for [6]-acene. Longer acenes and [n]-CCs fall in regions where the dominant contribution to the DE state is of singlet spin. Thus, according to the value, the di-radical character for [8]-acene and [9]-acene is large enough to guarantee a singlet-spin description of the DE state and an acceptable excitation energy at TDUB3LYP would be expected. However, at the same time the NFOD exceeds 2, indicating that more than two electrons need to be correlated. This implies that the 2e–2o model does not hold and the TDUB3LYP calculations, which provide a good description only for systems featuring large di-radical character within the 2e–2o model (only two electrons are localized, i.e., correlated), fail in such cases. Similar considerations hold for [n]-CCs featuring a large di-radical character (with close to 40°) but also a relevant tetra-radical character. Thus, the unexpected excitation energy increase of the DE state by TDUB3LYP calculations is attributed to the limits of this approach to describe poly-radicals.
4.2. Crossing of the DE and States in [n]-acenes and [n]-CCs
The relative energy location of the DE state in long linear acenes and [n]-CCs may influence the outcome of photoinduced events, such as singlet fission processes [70,71,72,73,74] TPA and luminescence efficiency. While experimental data are not available for [n]-CCs, a crossing between the DE and the states is experimentally documented to occur between [6]-acene and [7]-acene [18]. From previous sections, the method of choice to describe both excited states, among the DFT-based approaches tested, is DFT/MRCI. We can thus gauge the quality of DFT/MRCI calculations by comparing the predicted dependence of the two lowest-lying excited states as a function of the number of rings in [n]-acenes. This is shown in Figure 8a and Figure S7 for calculations carried out with the R2018 and the original parameterizations, respectively. Both DFT/MRCI parametrizations predict a crossing of the two states, with the DE state becoming the lowest excited state for longer acenes, in excellent agreement with experiment. Such crossing occurs for slightly longer acenes than experimentally observed when the R2018 parametrization is used, but we cannot rule out an influence of the chosen acene geometry on the exact crossing position. These good results for [n]-acenes let us believe that a similar quality also applies to [n]-CCs. As shown in Figure 8b, such crossing is predicted to occur between [8]-CCs and [10]-CCs when the R2018 parametrization is considered, while for the original parametrization, the DE state is the lowest excited state also for smaller carbon nanobelts. In summary, longer [n]-acenes and [n]-CCs are predicted to feature a lowest lying dark DE state.
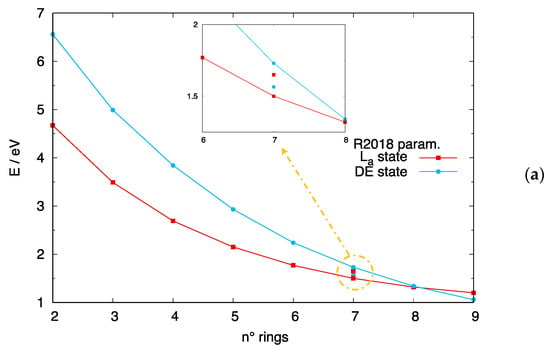
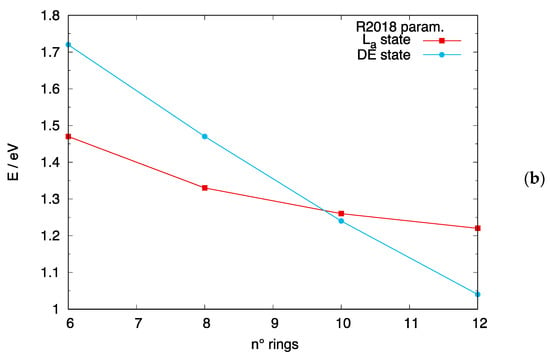
Figure 8.
Crossing between the and DE states predicted by DFT/MRCI calculations with the R2018 parametrization. (a) [n]-acenes: the isolated cyan and red data points correspond to experimental data from ref. [18]; (b) [n]-CCs.
5. Conclusions
In this work we considered the two classes of [n]-acenes and [n]-CCs and focused the attention on the influence of their increasing di-/poly-radical characters and the implications on the relative order of the two lowest-lying and DE excited states.
As in previous studies, the open-shell character of these PAHs was described by determining BS optimized geometries within the UDFT formalism. Generally, the contribution of geometry relaxation (from CS to BS) is remarkable in most conjugated di-radicals and linear acenes substantially confirmed such trend. In contrast, the total stabilization of cyclacenes was shown to be mainly due to the relaxation of constraints in the unrestricted wavefunction. Besides being a minor fraction of the total stabilization, the contribution of the geometry relaxation was shown to follow a different trend for even [n]-CCs and odd [n]-CCs, an additional manifestation of the cryptoannulenic effect which is also reflected in a larger poly-radical character of the latter sub-class of carbon nanobelts.
Several DFT based approaches encompassing TDUDFT, SF-TDDFT, and DFT/MRCI were employed to compute excitation energies of the and DE states. Among TD-based approaches, the SF-TDDFT was previously shown to capture correctly the DE state and such aptitude was confirmed here also for [n]-acenes. An unexpected excitation energy increase of the DE state as a function of molecular dimension was determined at the TDUDFT level. Such erroneous behavior was traced back to the increasing poly-radical character of longer [n]-acenes and [n]-CCs. Among the selected computational approaches, DFT/MRCI provided a balanced representation of both and DE states, while further systematic investigations will be required to assess the NEVPT2 results.
Based on the results presented here we can conclude that the electronic and optical properties of even [n]-CCs are strongly related to those of linear acenes such that the lowest excited state of both longer [n]-acenes and even [n]-CCs is the DE state, a dark state in one photon spectroscopy.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/chemistry5010044/s1, Tables S1 and S2: Frontier molecular orbital energies of [n]-acenes; Tables S3 and S4: Frontier molecular orbital energies for [n]-CCs; Table S5: Ground state energies of [n]-acenes and [n]-CCs; Tables S6 and S7: Di- and poly-radical characters of [n]-acenes and [n]-CCs; Table S8: Expression of BS frontier molecular orbitals as linear combination of CS frontier molecular orbitals; Table S9: Excitation energies of the state of [n]-acenes; Table S10: Excitation energies of the DE state of [n]-acenes; Table S11: Excitation energies of the and DE states for even [n]-CCs; Figure S1: Frontier molecular orbitals of [n]-acenes; Figure S2: Frontier molecular orbitals of even [n]-CCs; Figure S3: Frontier molecular orbitals of odd [n]-CCs; Figure S4: Energy trend of [n]-acenes’ frontier molecular orbitals; Figure S5: Relationship between and ; Figure S6: Relationship between and ; Figure S7: Crossing between the and DE state calculated at DFT/MRCI level with the original parametrization; Cartesian coordinates of all the optimized structures.
Author Contributions
Conceptualization, F.N.; investigation and formal analysis, F.N., J.-C.S.-G. and Y.D.; writing—original draft preparation, F.N.; writing—review and editing, F.N., J.-C.S.-G. and Y.D. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Data Availability Statement
The data presented in this study are available in the Supplementary Materials.
Acknowledgments
The authors thank Sofia Canola for stimulating discussions. Financial support from “Valutazione della Ricerca di Ateneo” (VRA) is acknowledged.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Chen, W.; Yu, F.; Xu, Q.; Zhou, G.; Zhang, Q. Recent Progress in High Linearly Fused Polycyclic Conjugated Hydrocarbons (PCHs, n > 6) with Well-Defined Structures. Adv. Sci. 2020, 7, 1903766. [Google Scholar] [CrossRef]
- Dong, H.; Zhu, H.; Meng, Q.; Gong, X.; Hu, W. Organic photoresponse materials and devices. Chem. Soc. Rev. 2012, 41, 1754–1808. [Google Scholar] [CrossRef]
- Wang, C.; Dong, H.; Hu, W.; Liu, Y.; Zhu, D. Semiconducting π-Conjugated Systems in Field-Effect Transistors: A Material Odyssey of Organic Electronics. Chem. Rev. 2012, 112, 2208–2267. [Google Scholar] [CrossRef]
- Watanabe, M.; Chang, Y.J.; Liu, S.-W.; Chao, T.-H.; Goto, K.; Islam, M.M.; Yuan, C.-H.; Tao, Y.-T.; Shinmyozu, T.; Chow, T.J. The synthesis, crystal structure and charge-transport properties of hexacene. Nat. Chem. 2012, 4, 574–578. [Google Scholar] [CrossRef]
- Hachmann, J.; Dorando, J.J.; Aviĺs, M.; Chan, G.K.-L.L.; Avilés, M.; Chan, G.K.-L.L. The radical character of the acenes: A density matrix renormalization group study. J. Chem. Phys. 2007, 127, 134309. [Google Scholar] [CrossRef]
- Pérez-Guardiola, A.; Sandoval-Salinas, M.E.; Casanova, D.; San-Fabián, E.; Pérez-Jiménez, A.J.; Sancho-García, J.C. The role of topology in organic molecules: Origin and comparison of the radical character in linear and cyclic oligoacenes and related oligomers. Phys. Chem. Chem. Phys. 2018, 20, 7112–7124. [Google Scholar] [CrossRef]
- Trinquier, G.; David, G.; Malrieu, J.-P. Qualitative Views on the Polyradical Character of Long Acenes. J. Phys. Chem. A 2018, 122, 6926–6933. [Google Scholar] [CrossRef]
- Sun, Z.; Wu, J. Higher Order Acenes and Fused Acenes with Near-infrared Absorption and Emission. Aust. J. Chem. 2011, 64, 519. [Google Scholar] [CrossRef]
- Zuzak, R.; Dorel, R.; Krawiec, M.; Such, B.; Kolmer, M.; Szymonski, M.; Echavarren, A.M.; Godlewski, S. Nonacene Generated by On-Surface Dehydrogenation. ACS Nano 2017, 11, 9321–9329. [Google Scholar] [CrossRef]
- Zuzak, R.; Dorel, R.; Kolmer, M.; Szymonski, M.; Godlewski, S.; Echavarren, A.M. Higher Acenes by On-Surface Dehydrogenation: From Heptacene to Undecacene. Angew. Chemie Int. Ed. 2018, 57, 10500–10505. [Google Scholar] [CrossRef]
- Krüger, J.; García, F.; Eisenhut, F.; Skidin, D.; Alonso, J.M.; Guitián, E.; Pérez, D.; Cuniberti, G.; Moresco, F.; Peña, D. Decacene: On-Surface Generation. Angew. Chem. 2017, 129, 12107–12110. [Google Scholar] [CrossRef]
- Colazzo, L.; Mohammed, M.S.G.; Dorel, R.; Nita, P.; García Fernández, C.; Abufager, P.; Lorente, N.; Echavarren, A.M.; de Oteyza, D.G. On-surface synthesis of heptacene on Ag(001) from brominated and non-brominated tetrahydroheptacene precursors. Chem. Commun. 2018, 54, 10260–10263. [Google Scholar] [CrossRef]
- Urgel, J.I.; Mishra, S.; Hayashi, H.; Wilhelm, J.; Pignedoli, C.A.; Di Giovannantonio, M.; Widmer, R.; Yamashita, M.; Hieda, N.; Ruffieux, P.; et al. On-surface light-induced generation of higher acenes and elucidation of their open-shell character. Nat. Commun. 2019, 10, 861. [Google Scholar] [CrossRef] [PubMed]
- Tönshoff, C.; Bettinger, H.F. Pushing the Limits of Acene Chemistry: The Recent Surge of Large Acenes. Chem. -A Eur. J. 2021, 27, 3193–3212. [Google Scholar] [CrossRef]
- Mondai, R.; Shah, B.K.; Neckers, D.C. Photogeneration of heptacene in a polymer matrix. J. Am. Chem. Soc. 2006, 128, 9612–9613. [Google Scholar] [CrossRef]
- Mondal, R.; Tönshoff, C.; Khon, D.; Neckers, D.C.; Bettinger, H.F. Synthesis, stability, and photochemistry of pentacene, hexacene, and heptacene: A matrix isolation study. J. Am. Chem. Soc. 2009, 131, 14281–14289. [Google Scholar] [CrossRef]
- Mondal, R.; Adhikari, R.M.; Shah, B.K.; Neckers, D.C. Revisiting the Stability of Hexacenes. Org. Lett. 2007, 9, 2505–2508. [Google Scholar] [CrossRef]
- Einholz, R.; Fang, T.; Berger, R.; Grüninger, P.; Früh, A.; Chassé, T.; Fink, R.F.; Bettinger, H.F. Heptacene: Characterization in Solution, in the Solid State, and in Films. J. Am. Chem. Soc. 2017, 139, 4435–4442. [Google Scholar] [CrossRef]
- Tönshoff, C.; Bettinger, H.F. Photogeneration of octacene and nonacene. Angew. Chem. Int. Ed. 2010, 49, 4125–4128. [Google Scholar] [CrossRef]
- Shen, B.; Tatchen, J.; Sanchez-Garcia, E.; Bettinger, H.F. Evolution of the Optical Gap in the Acene Series: Undecacene. Angew. Chem. Int. Ed. 2018, 57, 10506–10509. [Google Scholar] [CrossRef] [PubMed]
- Qu, H.; Chi, C. A stable heptacene derivative substituted with electron-deficient trifluoromethylphenyl and triisopropylsilylethynyl groups. Org. Lett. 2010, 12, 3360–3363. [Google Scholar] [CrossRef]
- Krüger, J.; Eisenhut, F.; Skidin, D.; Lehmann, T.; Ryndyk, D.A.; Cuniberti, G.; García, F.; Alonso, J.M.; Guitián, E.; Pérez, D.; et al. Electronic Resonances and Gap Stabilization of Higher Acenes on a Gold Surface. ACS Nano 2018, 12, 8506–8511. [Google Scholar] [CrossRef]
- Houk, K.N.; Lee, P.S.; Nendel, M. Polyacene and cyclacene geometries and electronic structures: Bond equalization, vanishing band gaps, and triplet ground states contrast with polyacetylene. J. Org. Chem. 2001, 66, 5517–5521. [Google Scholar] [CrossRef]
- Bendikov, M.; Duong, H.M.; Starkey, K.; Houk, K.N.; Carter, E.A.; Wudl, F. Oligoacenes: Theoretical prediction of open-shell singlet diradical ground states. J. Am. Chem. Soc. 2004, 126, 7416–7417. [Google Scholar] [CrossRef]
- Jiang, D.; Dai, S. Electronic Ground State of Higher Acenes. J. Phys. Chem. A 2008, 112, 332–335. [Google Scholar] [CrossRef]
- Chai, J.-D. Density functional theory with fractional orbital occupations. J. Chem. Phys. 2012, 136, 154104. [Google Scholar] [CrossRef]
- Chai, J.-D. Thermally-assisted-occupation density functional theory with generalized-gradient approximations. J. Chem. Phys. 2014, 140, 18A521. [Google Scholar] [CrossRef]
- Wu, C.-S.; Lee, P.-Y.; Chai, J.-D. Electronic Properties of Cyclacenes from TAO-DFT. Sci. Rep. 2016, 6, 37249. [Google Scholar] [CrossRef]
- Mostafanejad, M.; DePrince, A.E. Combining Pair-Density Functional Theory and Variational Two-Electron Reduced-Density Matrix Methods. J. Chem. Theory Comput. 2019, 15, 290–302. [Google Scholar] [CrossRef]
- Yang, Y.; Davidson, E.R.; Yang, W. Nature of ground and electronic excited states of higher acenes. Proc. Natl. Acad. Sci. USA 2016, 113, E5098–E5107. [Google Scholar] [CrossRef]
- Platt, J.R. Classification of Spectra of Cata-Condensed Hydrocarbons. J. Chem. Phys. 1949, 17, 484–495. [Google Scholar] [CrossRef]
- Negri, F.; Zgierski, M.Z. Vibronic structure of the emission spectra from single vibronic levels of the S 1 manifold in naphthalene: Theoretical simulation. J. Chem. Phys. 1996, 104, 3486–3500. [Google Scholar] [CrossRef]
- Knippenberg, S.; Starcke, J.H.; Wormit, M.; Dreuw, A. The low-lying excited states of neutral polyacenes and their radical cations: A quantum chemical study employing the algebraic diagrammatic construction scheme of second order. Mol. Phys. 2010, 108, 2801–2813. [Google Scholar] [CrossRef]
- Bonačić-Koutecký, V.; Koutecký, J.; Michl, J. Neutral and Charged Biradicals, Zwitterions, Funnels in S1, and Proton Translocation: Their Role in Photochemistry, Photophysics, and Vision. Angew. Chem. Int. Ed. Engl. 1987, 26, 170–189. [Google Scholar] [CrossRef]
- Di Motta, S.; Negri, F.; Fazzi, D.; Castiglioni, C.; Canesi, E.V. Biradicaloid and Polyenic Character of Quinoidal Oligothiophenes Revealed by the Presence of a Low-Lying Double-Exciton State. J. Phys. Chem. Lett. 2010, 1, 3334–3339. [Google Scholar] [CrossRef]
- Canola, S.; Casado, J.; Negri, F. The double exciton state of conjugated chromophores with strong diradical character: Insights from TDDFT calculations. Phys. Chem. Chem. Phys. 2018, 20, 24227–24238. [Google Scholar] [CrossRef]
- González-Cano, R.C.; Di Motta, S.; Zhu, X.; López Navarrete, J.T.; Tsuji, H.; Nakamura, E.; Negri, F.; Casado, J. Carbon-Bridged Phenylene-Vinylenes: On the Common Diradicaloid Origin of Their Photonic and Chemical Properties. J. Phys. Chem. C 2017, 121, 23141–23148. [Google Scholar] [CrossRef]
- Negri, F.; Canola, S.; Dai, Y. Spectroscopy of Open-Shell Singlet Ground-State Diradicaloids: A Computational Perspective. In Diradicaloids; Wu, J., Ed.; Jenny Stanford Publishing: New York, NY, USA, 2022; pp. 145–179. ISBN 978-981-4968-08-9. [Google Scholar]
- Canola; Dai; Negri The Low Lying Double-Exciton State of Conjugated Diradicals: Assessment of TDUDFT and Spin-Flip TDDFT Predictions. Computation 2019, 7, 68. [CrossRef]
- Grimme, S.; Parac, M. Substantial errors from time-dependent density functional theory for the calculation of excited states of large π systems. ChemPhysChem 2003, 4, 292–295. [Google Scholar] [CrossRef]
- Parac, M.; Grimme, S. A TDDFT study of the lowest excitation energies of polycyclic aromatic hydrocarbons. Chem. Phys. 2003, 292, 11–21. [Google Scholar] [CrossRef]
- Wong, B.M.; Hsieh, T.H. Optoelectronic and excitonic properties of oligoacenes: Substantial improvements from range-separated time-dependent density functional theory. J. Chem. Theory Comput. 2010, 6, 3704–3712. [Google Scholar] [CrossRef]
- Richard, R.M.; Herbert, J.M. Time-Dependent Density-Functional Description of the 1 L a State in Polycyclic Aromatic Hydrocarbons: Charge-Transfer Character in Disguise? J. Chem. Theory Comput. 2011, 7, 1296–1306. [Google Scholar] [CrossRef]
- Kuritz, N.; Stein, T.; Baer, R.; Kronik, L. Charge-transfer-like π→π* excitations in time-dependent density functional theory: A conundrum and its solution. J. Chem. Theory Comput. 2011, 7, 2408–2415. [Google Scholar] [CrossRef]
- Goerigk, L.; Grimme, S. Double-hybrid density functionals provide a balanced description of excited 1La and 1Lb states in polycyclic aromatic hydrocarbons. J. Chem. Theory Comput. 2011, 7, 3272–3277. [Google Scholar] [CrossRef]
- Sandoval-Salinas, M.E.; Brémond, E.; Pérez-Jiménez, A.J.; Adamo, C.; Sancho-García, J.C. Excitation energies of polycylic aromatic hydrocarbons by double-hybrid functionals: Assessing the PBE0-DH and PBE-QIDH models and their range-separated versions. J. Chem. Phys. 2023, 158, 044105. [Google Scholar] [CrossRef]
- Brémond, E.; Ciofini, I.; Sancho-García, J.C.; Adamo, C. Nonempirical Double-Hybrid Functionals: An Effective Tool for Chemists. Acc. Chem. Res. 2016, 49, 1503–1513. [Google Scholar] [CrossRef]
- Marian, C.M.; Gilka, N. Performance of the density functional theory/multireference configuration interaction method on electronic excitation of extended π-systems. J. Chem. Theory Comput. 2008, 4, 1501–1515. [Google Scholar] [CrossRef]
- Bettinger, H.F.; Tönshoff, C.; Doerr, M.; Sanchez-Garcia, E. Electronically Excited States of Higher Acenes up to Nonacene: A Density Functional Theory/Multireference Configuration Interaction Study. J. Chem. Theory Comput. 2016, 12, 305–312. [Google Scholar] [CrossRef]
- Bettanin, F.; Ferrão, L.F.A.; Pinheiro, M.; Aquino, A.J.A.; Lischka, H.; Machado, F.B.C.; Nachtigallova, D. Singlet La and Lb Bands for N-Acenes (N = 2-7): A CASSCF/CASPT2 Study. J. Chem. Theory Comput. 2017, 13, 4297–4306. [Google Scholar] [CrossRef]
- Schulz, F.; García, F.; Kaiser, K.; Pérez, D.; Guitián, E.; Gross, L.; Peña, D. Exploring a Route to Cyclic Acenes by On-Surface Synthesis. Angew. Chemie 2019, 131, 9136–9140. [Google Scholar] [CrossRef]
- Battaglia, S.; Faginas-Lago, N.; Andrae, D.; Evangelisti, S.; Leininger, T. Increasing Radical Character of Large [n]cyclacenes Unveiled by Wave Function Theory. J. Phys. Chem. A 2017, 121, 3746–3756. [Google Scholar] [CrossRef]
- Gleiter, R.; Esser, B.; Kornmayer, S.C. Cyclacenes: Hoop-Shaped Systems Composed of Conjugated Rings. Acc. Chem. Res. 2009, 42, 1108–1116. [Google Scholar] [CrossRef]
- Guo, Q.-H.; Qiu, Y.; Wang, M.-X.; Fraser Stoddart, J. Aromatic hydrocarbon belts. Nat. Chem. 2021, 13, 402–419. [Google Scholar] [CrossRef]
- Cheung, K.Y.; Segawa, Y.; Itami, K. Synthetic Strategies of Carbon Nanobelts and Related Belt-Shaped Polycyclic Aromatic Hydrocarbons. Chemistry 2020, 26, 14791–14801. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Kono, H.; Maekawa, T.; Segawa, Y.; Yagi, A.; Itami, K. Chemical Synthesis of Carbon Nanorings and Nanobelts. Acc. Mater. Res. 2021, 2, 681–691. [Google Scholar] [CrossRef]
- Shi, T.-H.; Wang, M.-X. Zigzag Hydrocarbon Belts. CCS Chem. 2021, 3, 916–931. [Google Scholar] [CrossRef]
- Lu, X.; Wu, J. After 60 Years of Efforts: The Chemical Synthesis of a Carbon Nanobelt. Chem 2017, 2, 619–620. [Google Scholar] [CrossRef]
- Zhang, Y.; Pun, S.H.; Miao, Q. The Scholl Reaction as a Powerful Tool for Synthesis of Curved Polycyclic Aromatics. Chem. Rev. 2022, 122, 14554–14593. [Google Scholar] [CrossRef] [PubMed]
- Shi, T.; Tong, S.; Wang, M. Construction of Hydrocarbon Nanobelts. Angew. Chemie Int. Ed. 2020, 59, 7700–7705. [Google Scholar] [CrossRef] [PubMed]
- Shi, T.-H.; Guo, Q.-H.; Tong, S.; Wang, M.-X. Toward the Synthesis of a Highly Strained Hydrocarbon Belt. J. Am. Chem. Soc. 2020, 142, 4576–4580. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Dong, S.; Shao, J.; Fan, W.; Chi, C. Synthesis of a Sidewall Fragment of a (12,0) Carbon Nanotube. Angew. Chemie Int. Ed. 2021, 60, 2658–2662. [Google Scholar] [CrossRef]
- Chen, H.; Gui, S.; Zhang, Y.; Liu, Z.; Miao, Q. Synthesis of a Hydrogenated Zigzag Carbon Nanobelt. CCS Chem. 2021, 3, 613–619. [Google Scholar] [CrossRef]
- San-Fabián, E.; Pérez-Guardiola, A.; Moral, M.; Pérez-Jiménez, A.J.; Sancho-García, J.C. Theoretical Study of Strained Carbon-based Nanobelts: Structural, Energetic, Electronic, and Magnetic Properties of [n]Cyclacenes. In Advanced Magnetic and Optical Materials; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2016; pp. 165–183. ISBN 9781119241966. [Google Scholar]
- Choi, H.S.; Kim, K.S. Structures, Magnetic Properties, and Aromaticity of Cyclacenes. Angew. Chemie Int. Ed. 1999, 38, 2256–2258. [Google Scholar] [CrossRef]
- Chen, Z.; Jiang, D.; Lu, X.; Bettinger, H.F.; Dai, S.; Schleyer, P. von R.; Houk, K.N. Open-Shell Singlet Character of Cyclacenes and Short Zigzag Nanotubes. Org. Lett. 2007, 9, 5449–5452. [Google Scholar] [CrossRef]
- Sadowsky, D.; McNeill, K.; Cramer, C.J. Electronic structures of [n]-cyclacenes (n = 6–12) and short, hydrogen-capped, carbon nanotubes. Faraday Discuss. 2010, 145, 507–521. [Google Scholar] [CrossRef]
- Pérez-Guardiola, A.; Ortiz-Cano, R.; Sandoval-Salinas, M.E.; Fernández-Rossier, J.; Casanova, D.; Pérez-Jiménez, A.J.; Sancho-García, J.C. From cyclic nanorings to single-walled carbon nanotubes: Disclosing the evolution of their electronic structure with the help of theoretical methods. Phys. Chem. Chem. Phys. 2019, 21, 2547–2557. [Google Scholar] [CrossRef]
- Ortiz, R.; Sancho-García, J.C.; Fernández-Rossier, J. Frustrated magnetic interactions in a cyclacene crystal. Phys. Rev. Mater. 2022, 6, 014406. [Google Scholar] [CrossRef]
- Casanova, D. Theoretical Modeling of Singlet Fission. Chem. Rev. 2018, 118, 7164–7207. [Google Scholar] [CrossRef]
- Zimmerman, P.M.; Zhang, Z.; Musgrave, C.B. Singlet fission in pentacene through multi-exciton quantum states. Nat. Chem. 2010, 2, 648–652. [Google Scholar] [CrossRef]
- Zimmerman, P.M.; Bell, F.; Casanova, D.; Head-Gordon, M. Mechanism for singlet fission in pentacene and tetracene: From single exciton to two triplets. J. Am. Chem. Soc. 2011, 133, 19944–19952. [Google Scholar] [CrossRef]
- Zimmerman, P.M.; Musgrave, C.B.; Head-Gordon, M. A Correlated Electron View of Singlet Fission. Acc. Chem. Res. 2013, 46, 1339–1347. [Google Scholar] [CrossRef] [PubMed]
- Zeiser, C.; Moretti, L.; Reicherter, F.; Bettinger, H.F.; Maiuri, M.; Cerullo, G.; Broch, K. Singlet Fission in Dideuterated Tetracene and Pentacene. ChemPhotoChem 2021, 5, 758–763. [Google Scholar] [CrossRef]
- Shao, Y.; Head-Gordon, M.; Krylov, A.I. The spin-flip approach within time-dependent density functional theory: Theory and applications to diradicals. J. Chem. Phys. 2003, 118, 4807–4818. [Google Scholar] [CrossRef]
- Rinkevicius, Z.; Vahtras, O.; Ågren, H. Spin-flip time dependent density functional theory applied to excited states with single, double, or mixed electron excitation character. J. Chem. Phys. 2010, 133, 114104. [Google Scholar] [CrossRef]
- Angeli, C.; Cimiraglia, R.; Evangelisti, S.; Leininger, T.; Malrieu, J.P. Introduction of n-electron valence states for multireference perturbation theory. J. Chem. Phys. 2001, 114, 10252. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Schmidt, M.W.; Baldridge, K.K.; Boatz, J.A.; Elbert, S.T.; Gordon, M.S.; Jensen, J.H.; Koseki, S.; Matsunaga, N.; Nguyen, K.A.; Su, S.; et al. General atomic and molecular electronic structure system. J. Comput. Chem. 1993, 14, 1347–1363. [Google Scholar] [CrossRef]
- Grimme, S.; Waletzke, M. A combination of Kohn-Sham density functional theory and multi-reference configuration interaction methods. J. Chem. Phys. 1999, 111, 5645–5655. [Google Scholar] [CrossRef]
- Marian, C.M.; Heil, A.; Kleinschmidt, M. The DFT/MRCI method. WIREs Comput. Mol. Sci. 2019, 9, 1–31. [Google Scholar] [CrossRef]
- Marian, C.M.; Heil, A.; Kleinschmidt, M. DFT/MRCI Software. Available online: https://www.theochem.hhu.de/software/dft/mrci (accessed on 15 October 2019).
- Neese, F.; Wennmohs, F.; Becker, U.; Riplinger, C. The ORCA quantum chemistry program package. J. Chem. Phys. 2020, 152, 224108. [Google Scholar] [CrossRef]
- Weigend, F. Hartree—Fock exchange fitting basis sets for H to Rn †. J. Comput. Chem. 2008, 29, 167–175. [Google Scholar] [CrossRef] [PubMed]
- Nakano, M. Open-Shell-Character-Based Molecular Design Principles: Applications to Nonlinear Optics and Singlet Fission. Chem. Rec. 2017, 17, 27–62. [Google Scholar] [CrossRef]
- Yamaguchi, K. The electronic structures of biradicals in the unrestricted Hartree-Fock approximation. Chem. Phys. Lett. 1975, 33, 330–335. [Google Scholar] [CrossRef]
- Grimme, S.; Hansen, A. A Practicable Real-Space Measure and Visualization of Static Electron-Correlation Effects. Angew. Chem. Int. Ed. 2015, 54, 12308–12313. [Google Scholar] [CrossRef]
- Bauer, C.A.; Hansen, A.; Grimme, S. The Fractional Occupation Number Weighted Density as a Versatile Analysis Tool for Molecules with a Complicated Electronic Structure. Chem. - A Eur. J. 2017, 23, 6150–6164. [Google Scholar] [CrossRef]
- Türker, L. Cyclacenes having dislocated rings. J. Mol. Struct. Theochem 2001, 536, 235–241. [Google Scholar] [CrossRef]
- Türker, L. Cryptoannulenic Behavior of Cyclacenes. Polycycl. Aromat. Compd. 1994, 4, 191–197. [Google Scholar] [CrossRef]
- Loos, P.-F.; Boggio-Pasqua, M.; Scemama, A.; Caffarel, M.; Jacquemin, D. Reference Energies for Double Excitations. J. Chem. Theory Comput. 2019, 15, 1939–1956. [Google Scholar] [CrossRef]
- Bergman, A.; Jortner, J. Two-photon spectroscopy utilizing dye lasers. Chem. Phys. Lett. 1972, 15, 309–315. [Google Scholar] [CrossRef]
- Bergman, A.; Jortner, J. Two-photon absorption spectra of crystalline naphthalene and of the naphthalene molecule in solution. Chem. Phys. Lett. 1974, 26, 323–326. [Google Scholar] [CrossRef]









Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).