1. Introduction
The Diels–Alder (DA) reaction [
1], which is a general class of cycloaddition reactions, is one of the most useful synthetic reactions in Organic Chemistry [
2,
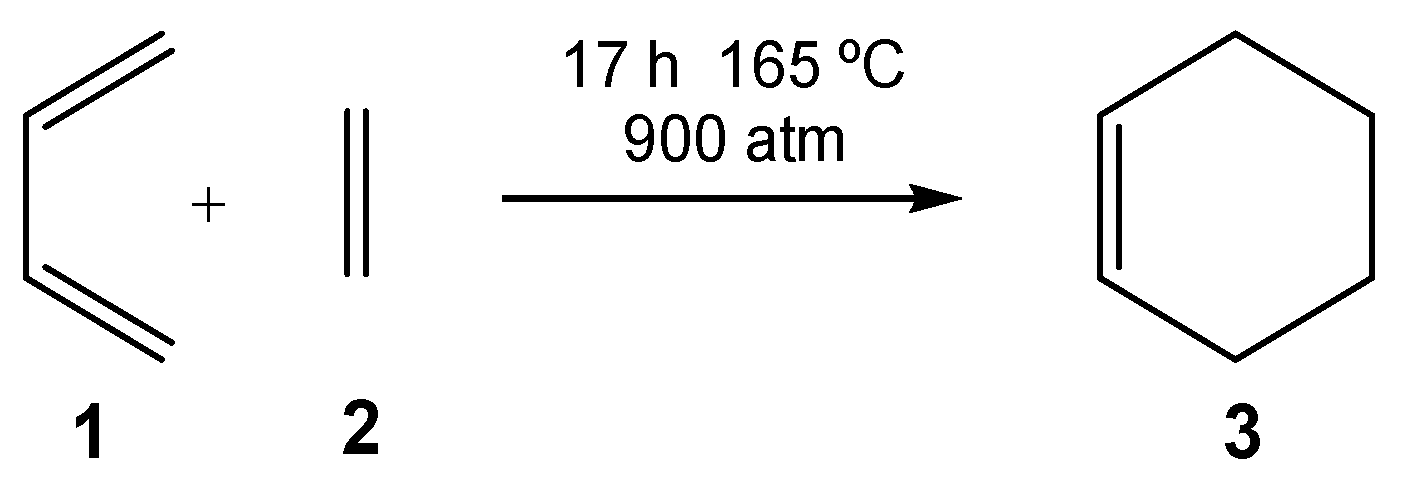
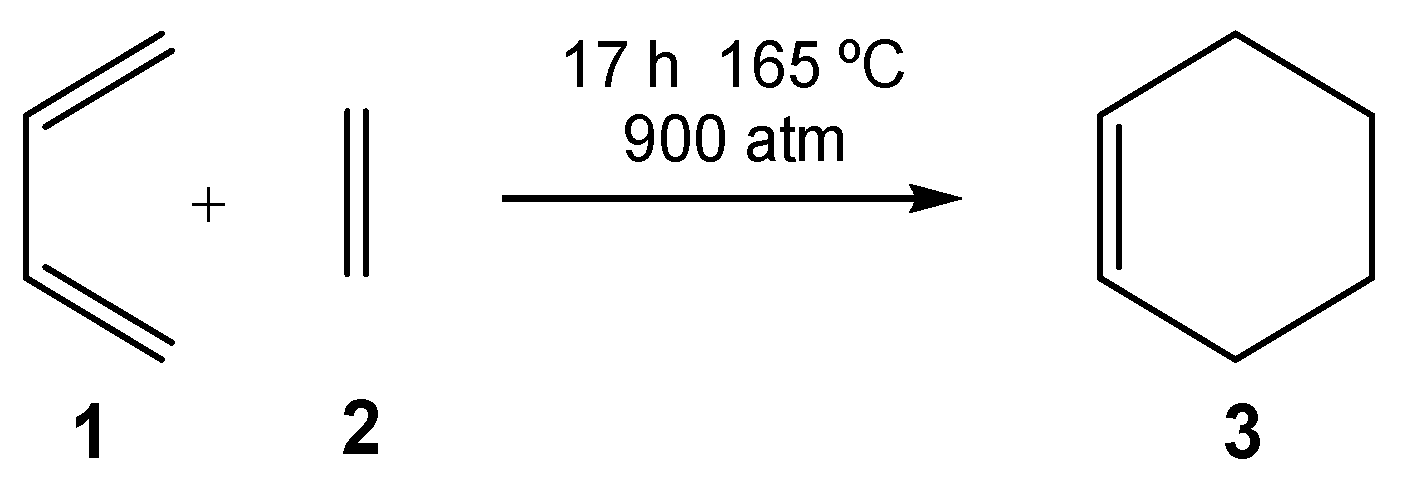
3]. By varying the nature of the diene and the ethylene, many different types of six-membered carbocyclic structures can be built up. However, not all possibilities take place easily. For instance, the DA reaction between butadiene
1 and ethylene
2, selected as the prototype of these cycloaddition reactions [
4,
5], must be forced to take place: after 17 h at 165 °C and 900 atm, a yield of 78% is obtained (see
Scheme 1) [
6].
Although this DA reaction is exothermic by 40 kcal/mol, it has a large activation energy of 27.5 kcal·mol
−1 [
6]. For DA reactions, the negative activation entropy associated with these bimolecular processes also plays an adverse role. In this way, for the DA reaction between butadiene
1 and ethylene
2, the activation entropy of the one-step process was estimated to be −40.6 eu [
7]. This unfavorable value, together with the high temperature required by the reaction, increased the activation Gibbs free energy of this DA reaction to 42.6 kcal·mol
−1.
There are two different ways to reduce this high activation Gibbs free energy: (i) to decrease the high activation enthalpy associated with the formation of the two new single bonds, and/or (ii) to decrease the high activation entropy associated with these bimolecular processes.
An exhaustive theoretical study on experimental DA reactions, allowed establishing the definitive role of the global electron density transfer [
8] (GEDT) taking place at the TSs in the feasibility of DA reactions [
9,
10]. This finding allowed establishing, in 2009, the mechanism of the polar DA (P-DA) reactions [
11], in which the favorable nucleophilic/electrophilic interactions taking place at the transition state structures (TSs) are responsible for the feasibility of the reaction.
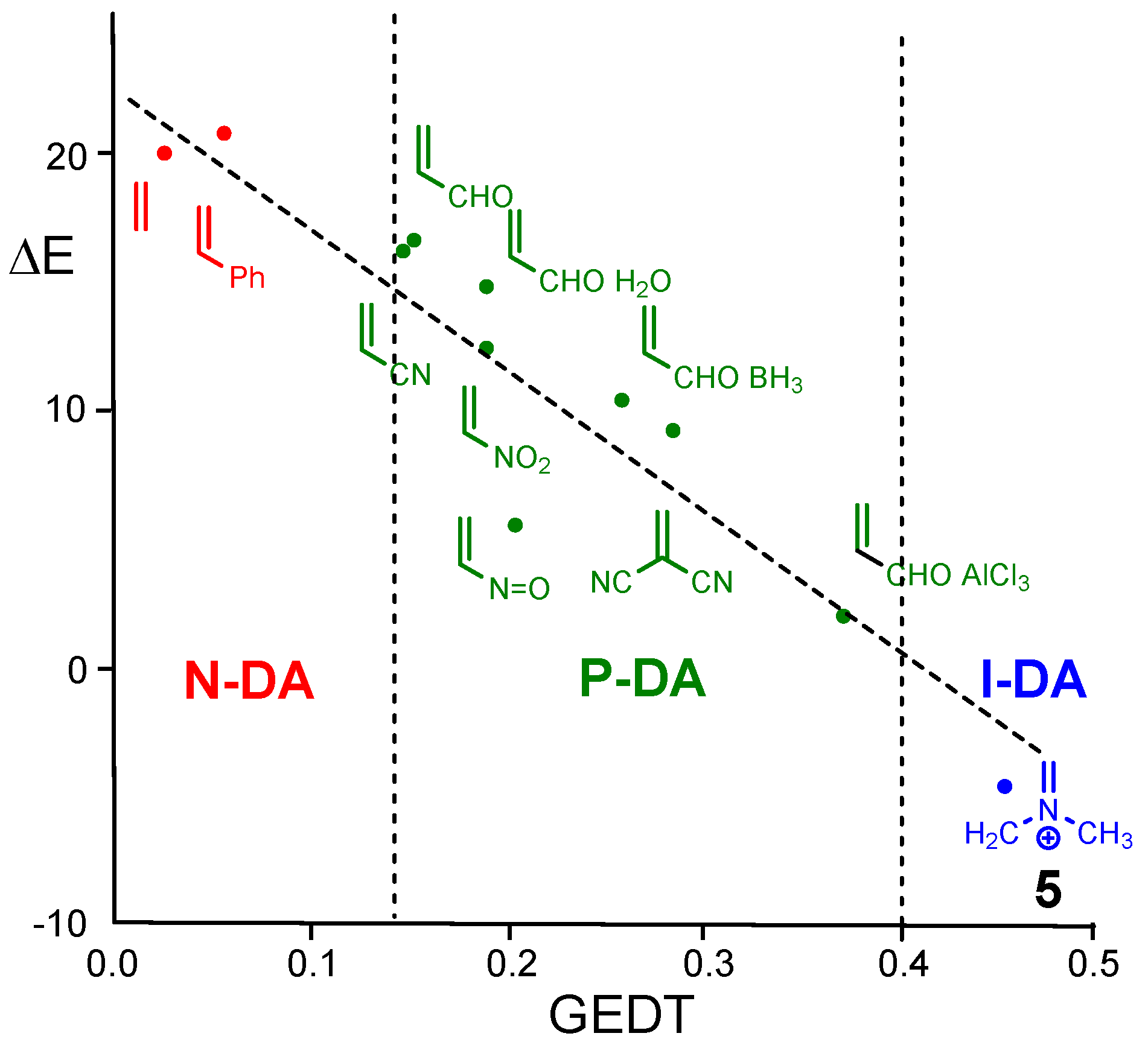
The very good correlation found between the GEDT and the activation barriers for the DA reactions of Cp
4 with a series of 12 substituted ethylenes, including iminium
5, allowed the classification of the DA reactions in non-polar DA (N-DA) (which do not take place easily experimentally), P-DA reactions, and ionic DA (I-DA) reactions, in which one of the two reagents is an ionic species (see
Figure 1) [
11]. Although, from the GEDT analysis it is not possible to stablish a clear separation between P-DA and I-DA reactions, these DA reactions have different behaviors.
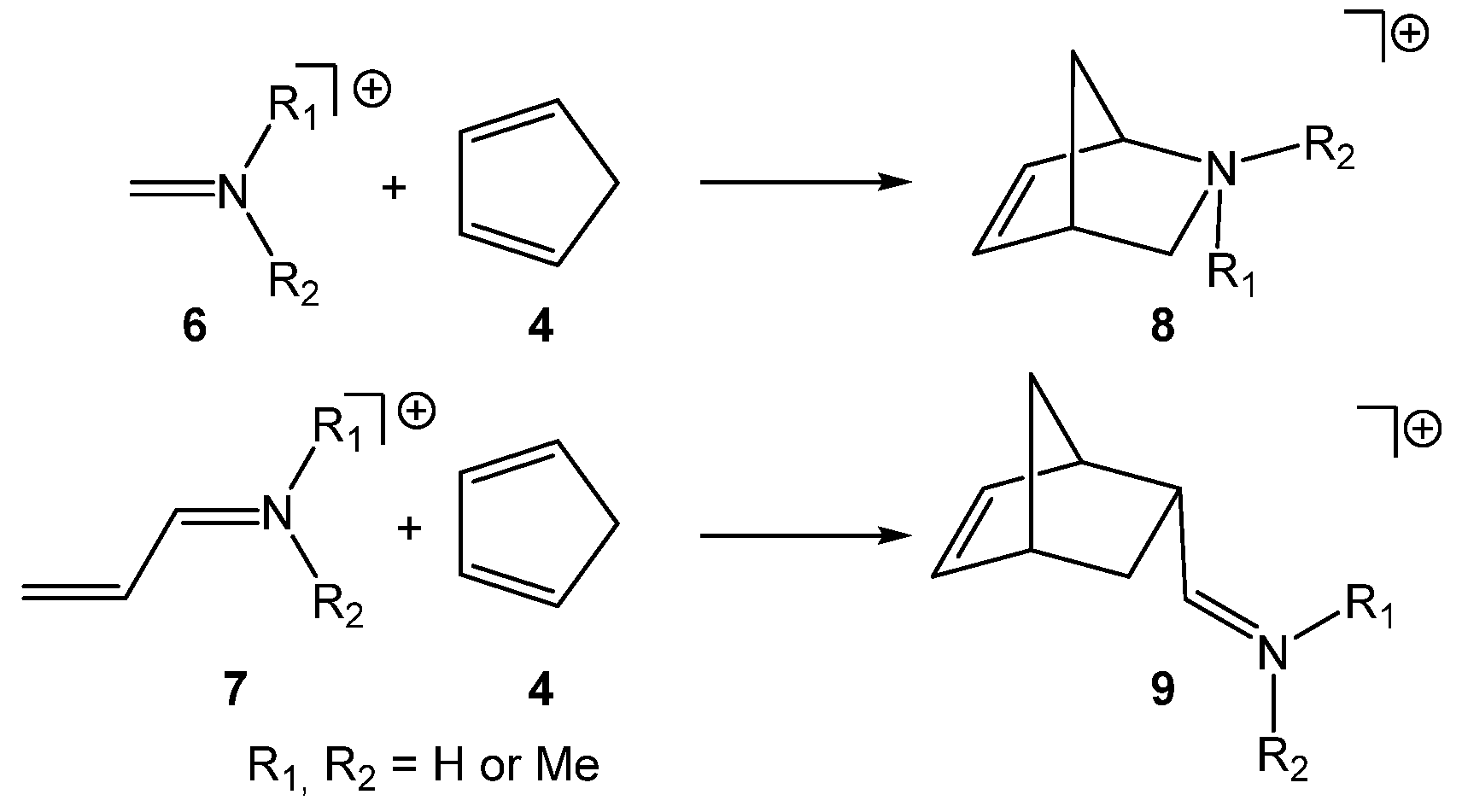
The I-DA reactions of a series of six iminiums
6 and
7 with Cp
4 have been recently studied within the Molecular Electron Density Theory [
12] (MEDT) (see
Scheme 2) [
13]. The activation energies of these I-DA reactions were found to be between 13 and 20 kcal·mol
−1 lower in energy than those associated with the corresponding P-DA reactions of neutral imines as a consequence of the superelectrophilic character of iminiums, ω > 8.20 eV. Unlike P-DA reactions, these I-DA reactions showed a low
endo stereoselectivity as a consequence of the cationic character of the TSs; however, they are highly regioselective [
13]. In addition, polar solvents have poor effects on the relative energies, and an unappreciable effect in the geometries.
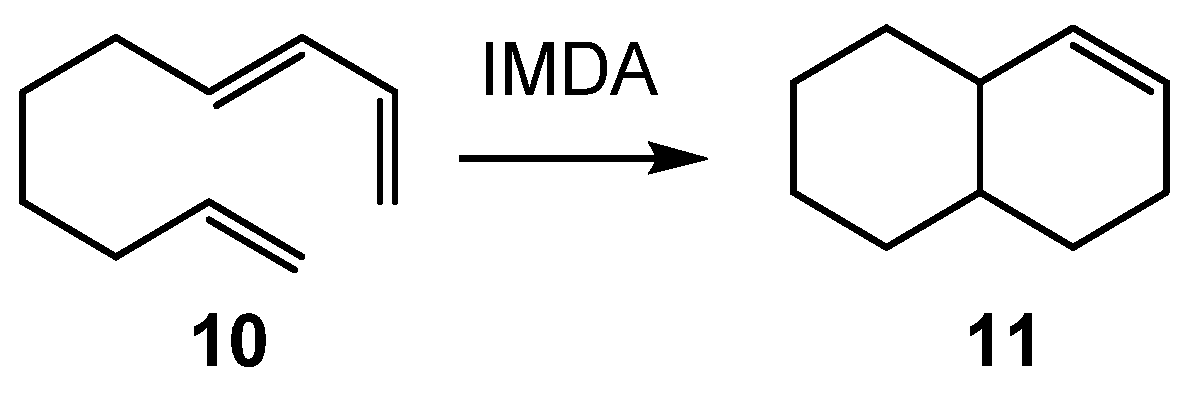
On the other hand, the high entropy cost associated with bimolecular processes may be overcome through an intramolecular process. Thus, in
Table 1, the thermodynamic data for the N-DA reaction between butadiene
1 and ethylene
2 given in
Scheme 1, and those for the intramolecular Diels–Alder (IMDA) reaction of (
E)-deca-1,3,9-triene
10 given in
Scheme 3 are gathered. The thermodynamic data of all species participating in these N-DA reactions are reported in
Table S3 in the Supplementary Materials.
The B3LYP/6-311G(d,p) activation enthalpy for the N-DA reaction between butadiene
1 and ethylene
2, ΔH
# = 25.7 kcal·mol
−1, and the activation entropy, ΔS
# = −44.8 cal·mol
−1·K, are closer to the experimental values, 27.5 kcal·mol
−1 and −40.6 cal·mol
−1·K, respectively [
6,
7]. These high values rise the activation Gibbs free energy of this intermolecular N-DA reaction computed at 165 °C to 45.4 kcal·mol
−1. Despite the fact that the activation enthalpy associated with the IMDA reaction of
10, ΔH
# = 29.4 kcal·mol
−1, is higher than that of the intermolecular one, the low activation entropy associated with this intramolecular process, ΔS
# = −19.4 cal·mol
−1·K, decreases the activation Gibbs free energy to 38.0 kcal·mol
−1. This behavior explains the feasibility of non-polar IMDA reactions.
In 1972, R. Sustmann et al. [
14] classified the cycloaddition reactions as “the normal and inverse electron-demand (NED and IED)” within the Frontier Molecular Orbital (FMO) theory [
15]. Later, in 1973, Houk renamed Sustmann’s classification, naming them as (HOMO) HO- and (LUMO) LU-controlled cycloaddition reactions [
16], emphasizing the relevance of the molecular orbital (MO) interactions in cycloaddition reactions. However, within MEDT, neither classification based on the FMO theory has any chemical significance. Note that MOs are only mathematical artefact used to construct the wave function.
Thus, for instance, the unfavorable DA reaction between butadiene
1 ethylene
2 given in
Scheme 1 is classified as NED or HO-controlled, despite that this non-polar DA reaction has a negligible GEDT at the corresponding TS [
17]. In addition, many authors have emphasized that these classifications are sometimes confusing, leading to interpretations in contract with the experimental observations [
18,
19,
20,
21]. On the order hand, in spite of the chemical similarity of the inter and intramolecular cycloaddition reactions, the later cannot be classified within these criteria as the HOMO and LUMO belong to a single molecule.
Recently, Domingo proposed the classification of the polar DA reactions as the forwards and reverse electron density flux (FEDF and REDF) [
22]. In FEDF DA reactions, the electron density always fluxes from the diene towards the ethylene. Analysis of the GEDT [
8] at the TSs unequivocally allows to classify the DA reactions. Thus, the non-polar DA reaction between butadiene
1 and ethylene
2 is classified as the null electron density flux (NEDF) [
23]. This classification has been applied recently in I-DA reactions [
13].
The GEDT measured at the TSs has proved to be a powerful tool to establish the polar character of IMDA reactions [
24,
25,
26], and consequently, it can be used to classify them depending on the course of the intramolecular electron density flux. Thus, the non-polar IMDA reaction of (
E)-deca-1,3,9-triene
10 given in
Scheme 3 is classified as NEDF.
Many theoretical studies devoted to non-polar and polar IMDA reactions can be found in the literature; however, only a small number of them have been devoted to intramolecular ionic Diels–Alder (IIDA) reactions [
27,
28].
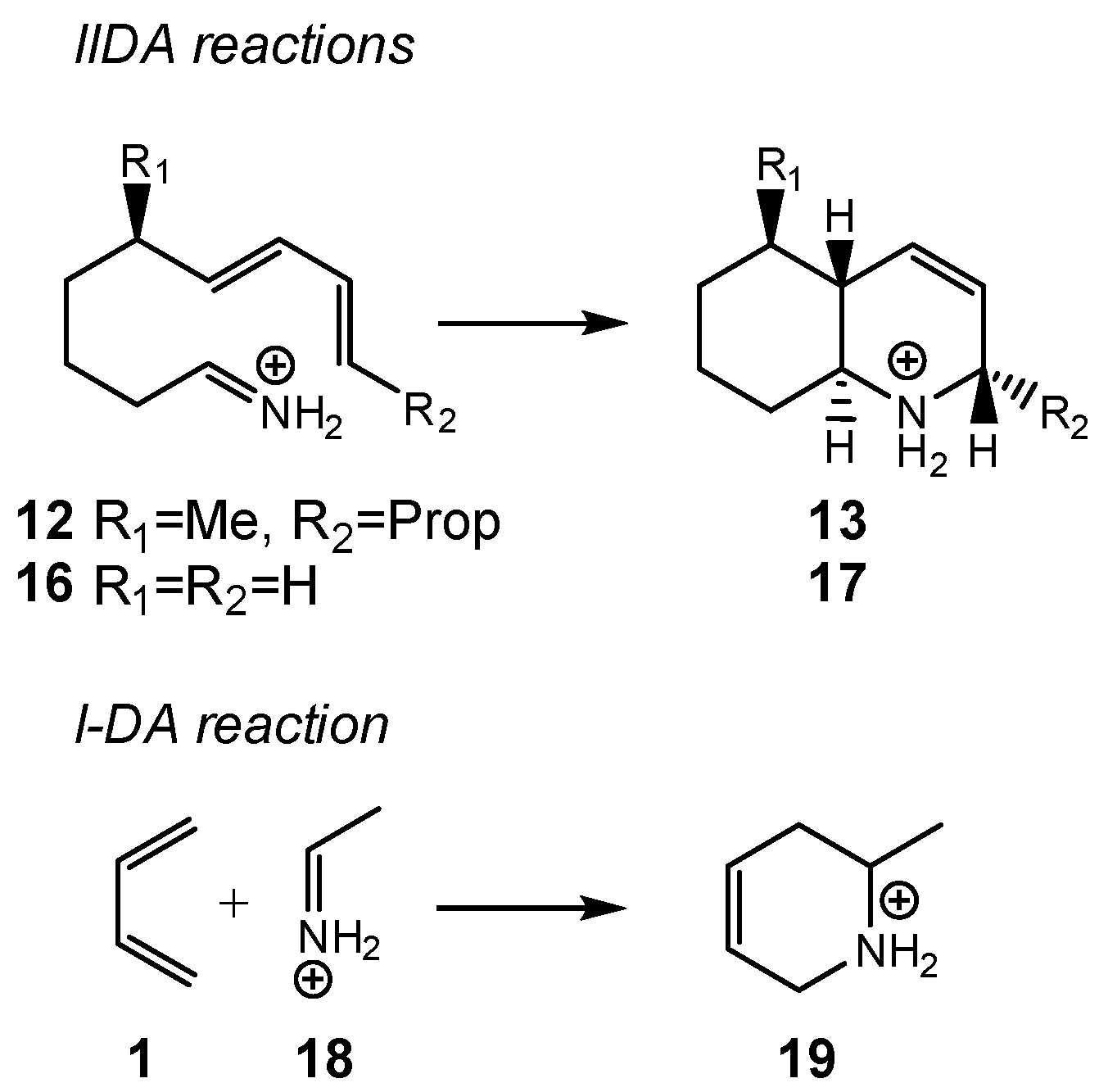
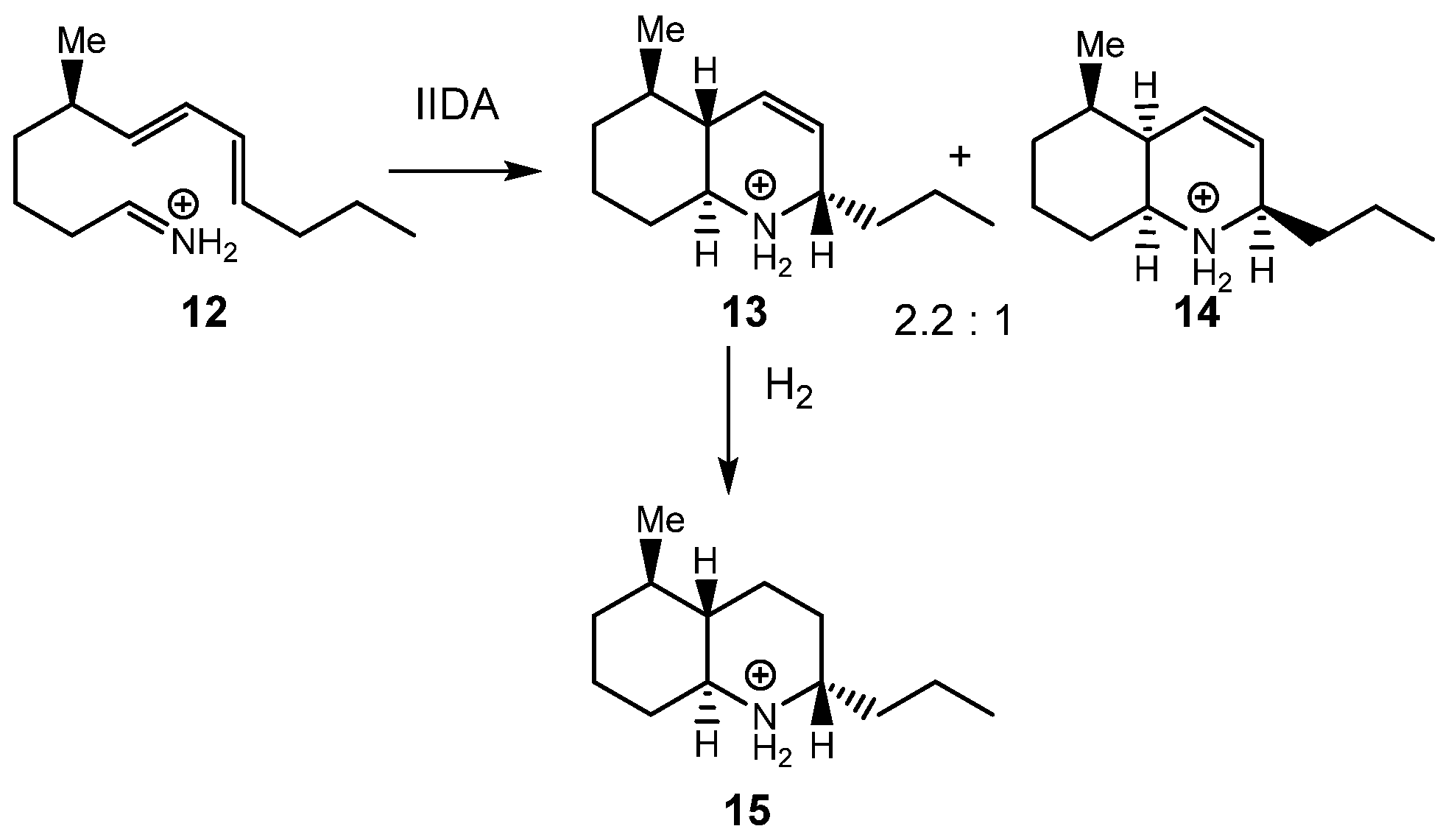
In 1988, Grieco and Parke used the IIDA reaction of dieniminium
12 in the synthesis of (−)-8a-epipumiliotoxin C
15 (see
Scheme 4) [
29]. The IIDA reaction of the generated in situ dieniminium
12 provided a mixture of the stereoisomeric quinoliniums
13 and
14 in a ratio of 2.2:1. Quinolinium
13 was further converted into
15 upon a hydrogenation reaction.
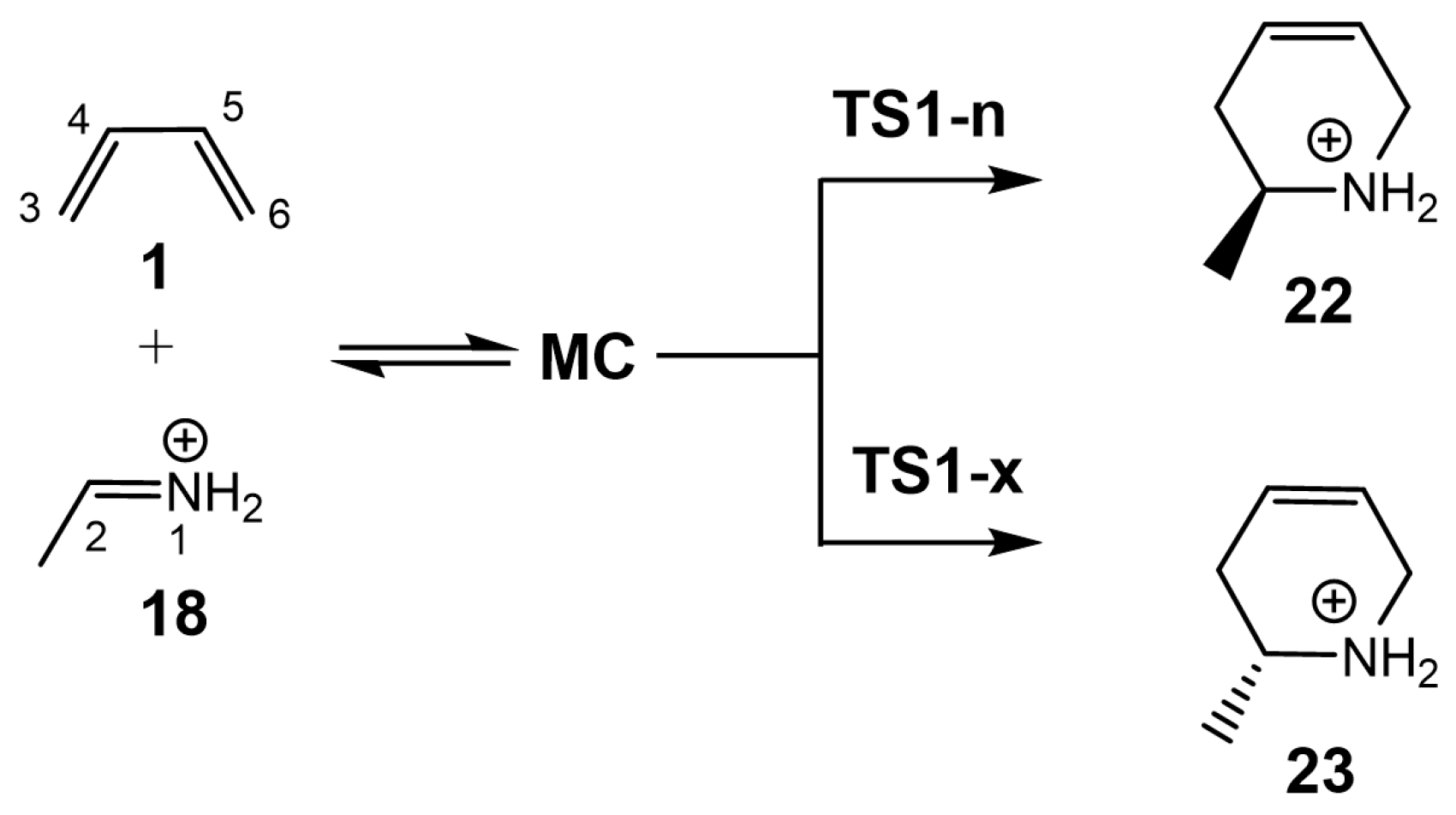
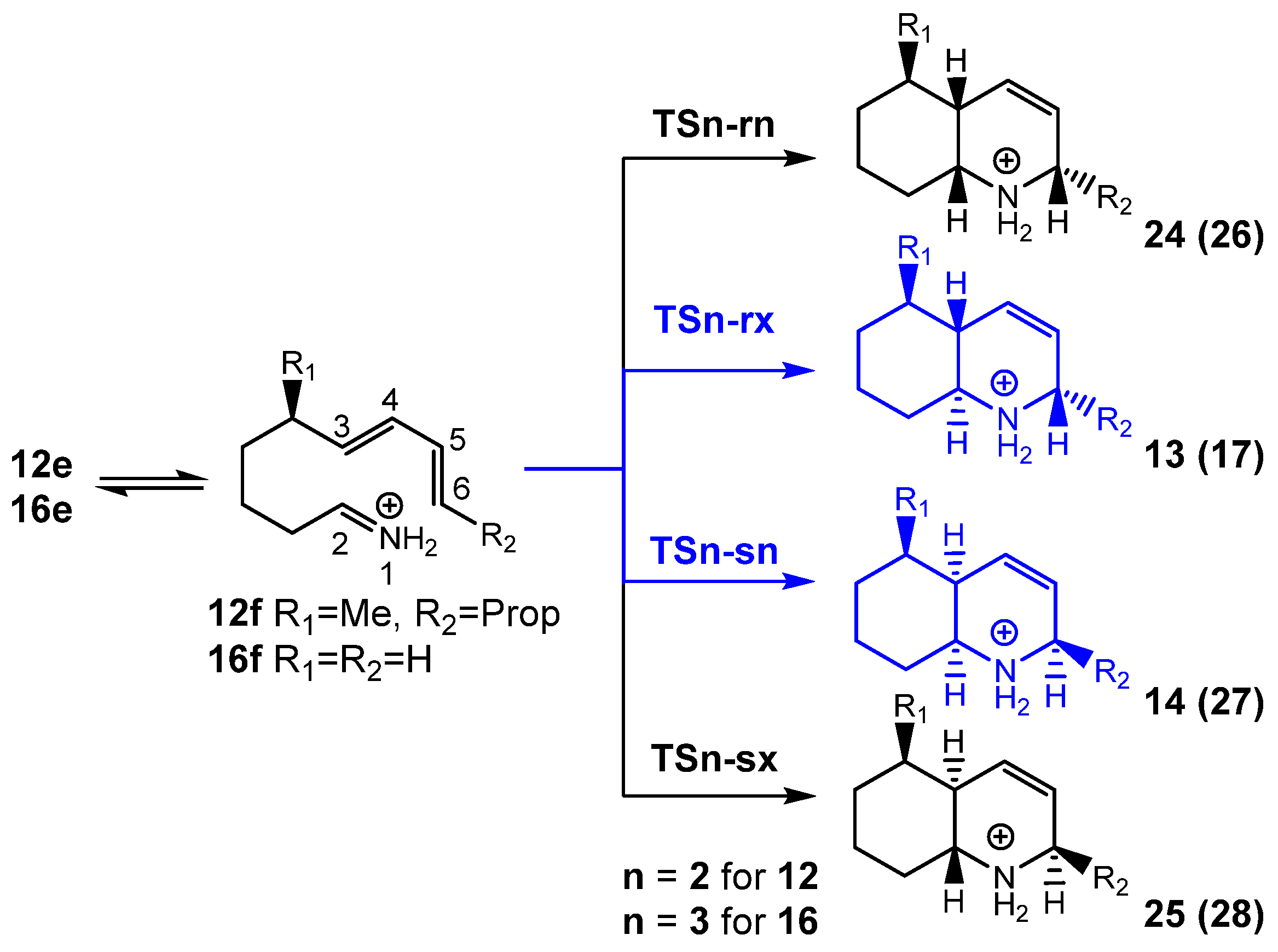
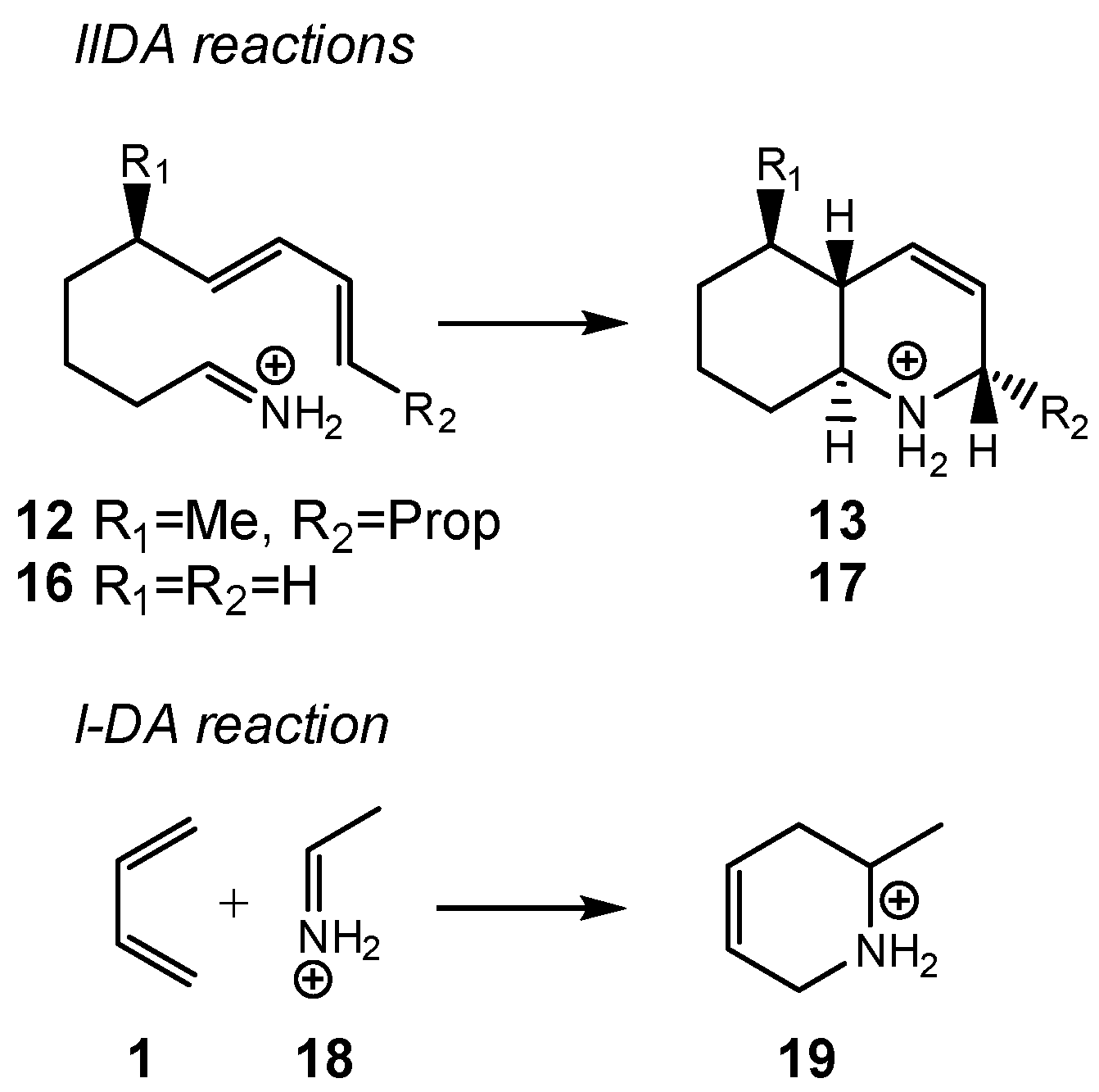
Herein, an MEDT study of the IIDA reaction of dieniminium
12, experimentally studied by Grieco and Parke [
29], is performed in order to understand the behaviors of the IIDA reactions (see
Scheme 4). To this end, the I-DA reaction of butadiene
1 with iminium
18, and the IIDA reaction of (
E)-nona-6,8-dien-1-iminium
16, as a reduced model of dieniminium
12, were also studied (see
Scheme 5). The recent classification of cycloaddition reactions based on the direction of the flux of the electron density [
22] is for the first time applied in an intramolecular cycloaddition reaction.
2. Computational Methods
DFT calculations were performed using the B3LYP [
30,
31] functional and hybrid ωB97X-D functional [
32], which includes long range exchange (denoted by X) correction as well as the semiclassical London-dispersion correction (indicated by suffix-D). The standard 6-311G(d,p) basis set was used [
33], which includes d-type polarization for second row elements and
p-type polarization functions for hydrogen atoms. The Berny method was used in optimizations [
34,
35]. Only one imaginary frequency characterized all studied TSs.
The intrinsic reaction coordinate (IRC) paths [
36] were carried out to find the unique connection given between the TSs and the minimum stationary points [
37,
38]. Solvent effects of ethanol were considered by full optimization of the gas phase structures at the same computational level using the polarizable continuum model [
39,
40] (PCM) in the framework of the self-consistent reaction field [
41,
42,
43] (SCRF).
The GEDT [
8] values were estimated by a NPA [
44,
45], using the equation
, were q are the atoms of a framework (f) at the TSs. CDFT reactivity indices [
46,
47], computed at the B3LYP/6-31G(d) level, were calculated through the equations given in reference [
47]. All calculations were carried out with the Gaussian 16 suite (Version A.03, Gaussian Inc, Wallingford, CT, USA) of programs [
48].
The topology of the electron localization function [
49] (ELF) of the B97XD/6-311G(d,p) monodeterminantal wavefunctions was carried out using the TopMod [
50] package with a cubical grid of step size of 0.1 Bohr. The Bader’s quantum theory of atoms-in-molecules [
51] (AIM) analyses were conducted using Multiwfn 3.8 software packages [
52]. GaussView program [
53] (Version 6.0, Semichem Inc., Shawnee, KS, USA) was used to visualize molecular geometries of all the systems as well as the position of the ELF basin attractors. The ELF localization domains at an isovalue of 0.75 a.u. were obtained with the Paraview software (Version 5.6.0 64-bit, Kitware inc, Clifton Park, NY, USA) [
54,
55].
4. Conclusions
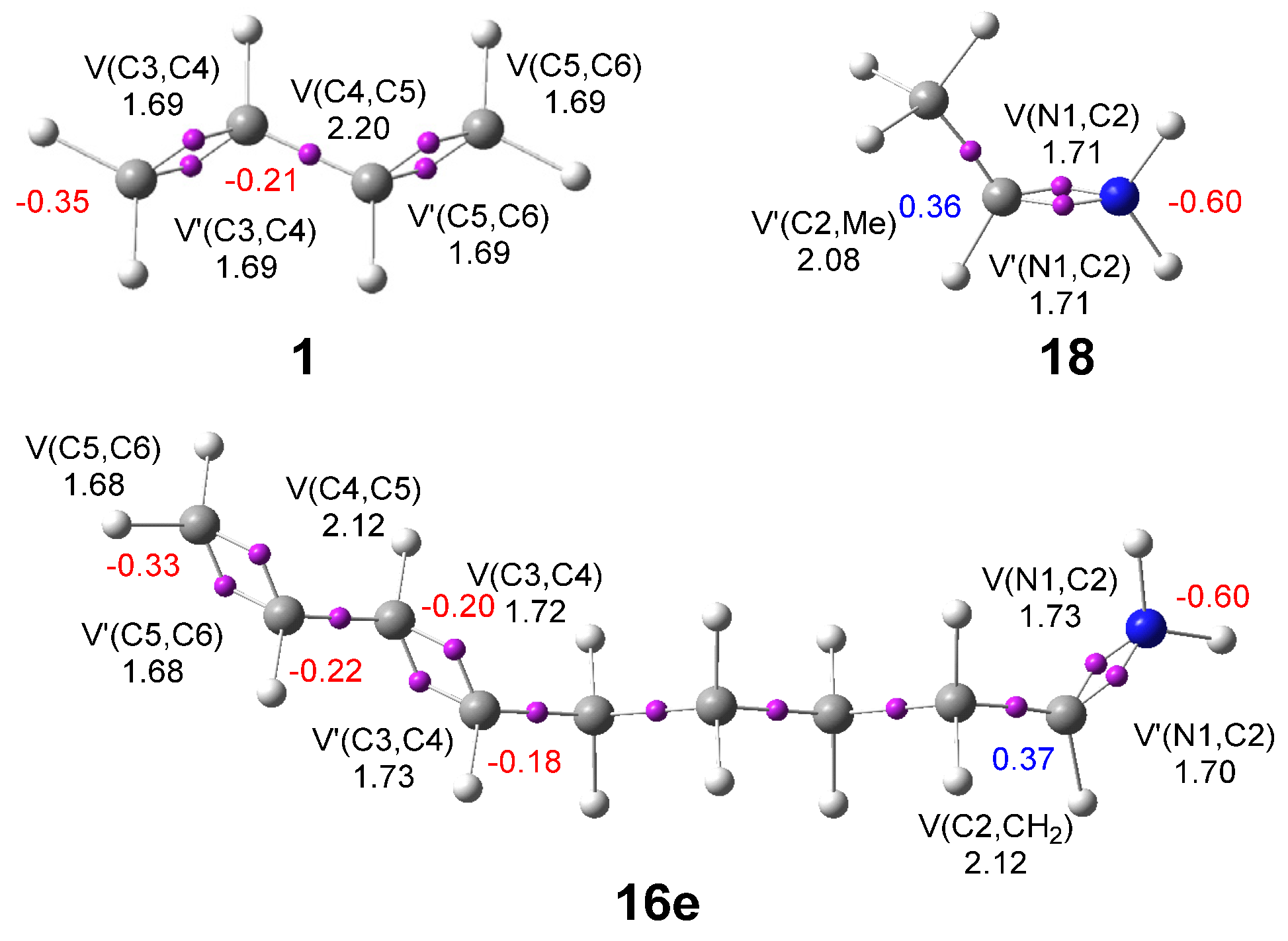
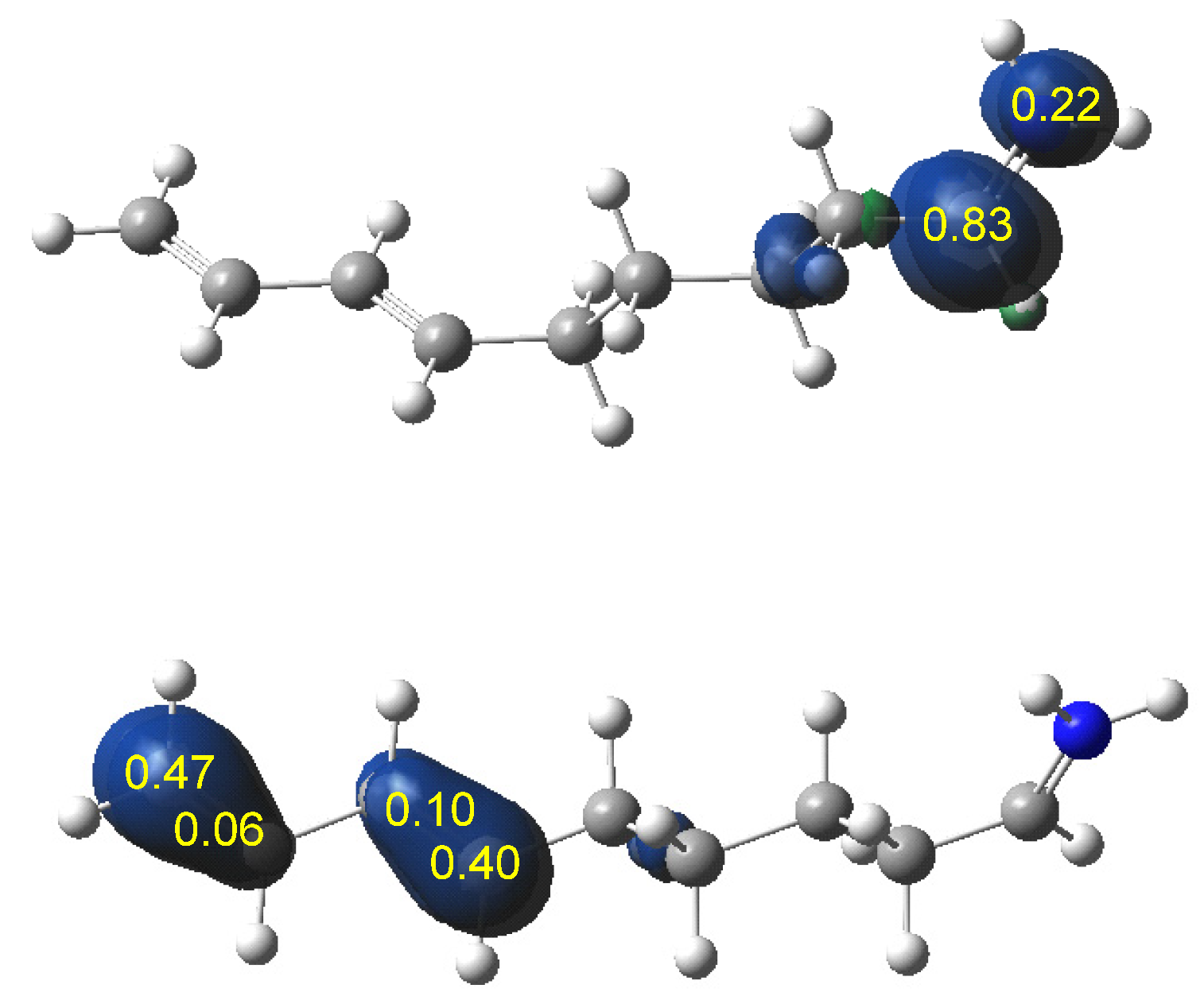
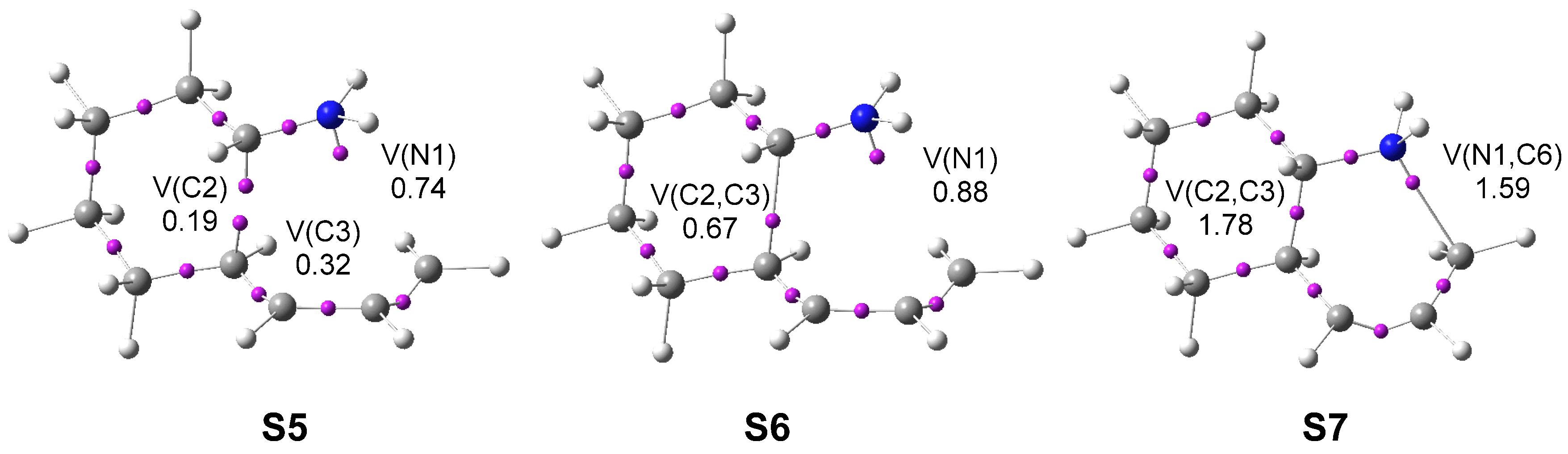
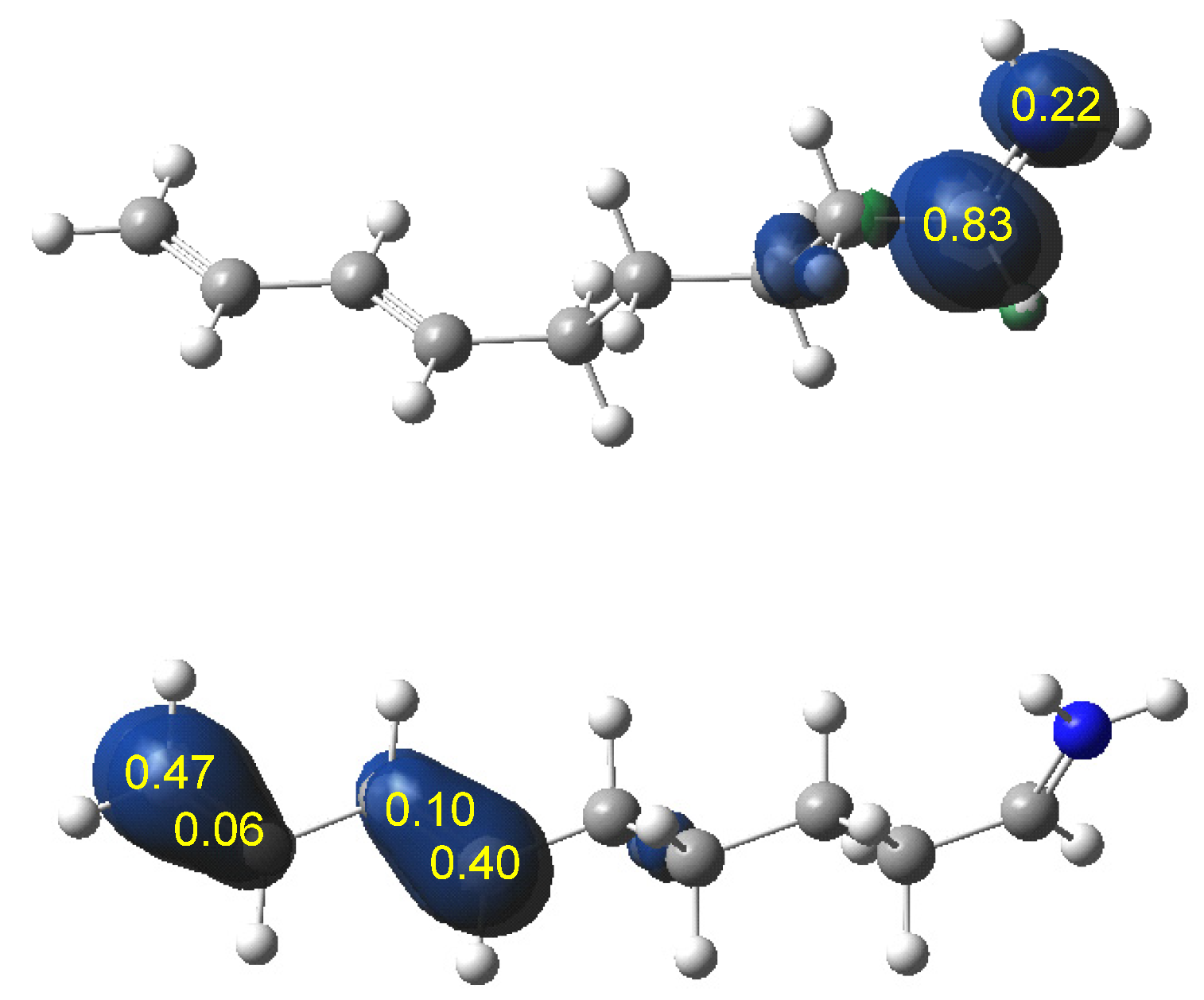
The IIDA reactions of dieniminiums 12 and 16 were studied within the MEDT at the DFT ωB97XD/6-311G(d,p) computational level. The ELF topological analysis of dieniminiums 12 and 16 showed that the electronic structure of these species can be seen as the sum of those of butadiene 1 and that of ethaniminium 18 joined by the tetramethylene chain. Analysis of the electrophilicity ω index of dieniminiums 12 and 16 pointed to the superelectrophilic character of these species. On the other hand, analysis of the Parr functions allowed characterization of the most electrophilic center, the iminium C2 carbon, and the most nucleophilic centers, the C3 and C6 carbons of the butadiene moiety, of dieniminium 16.
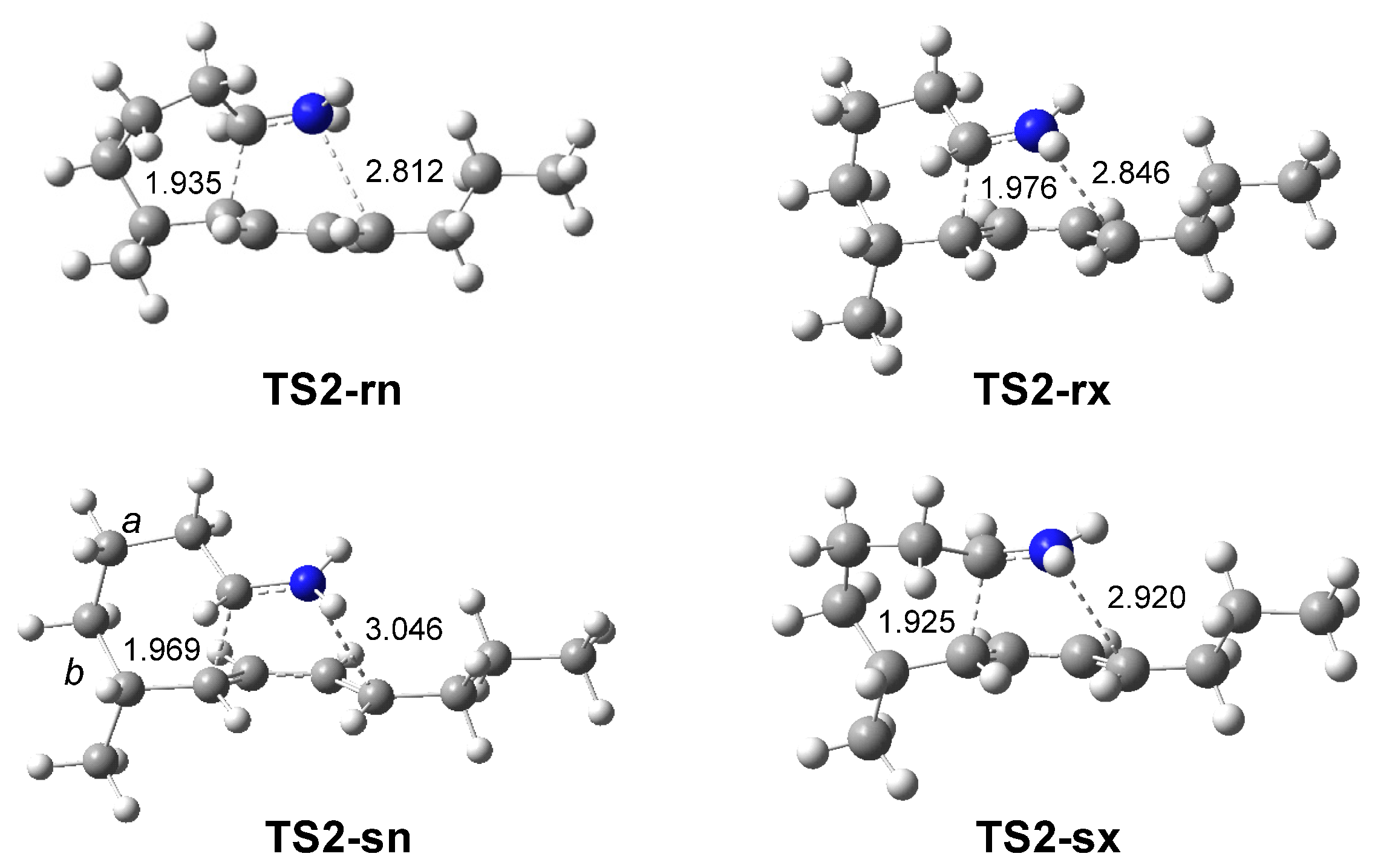
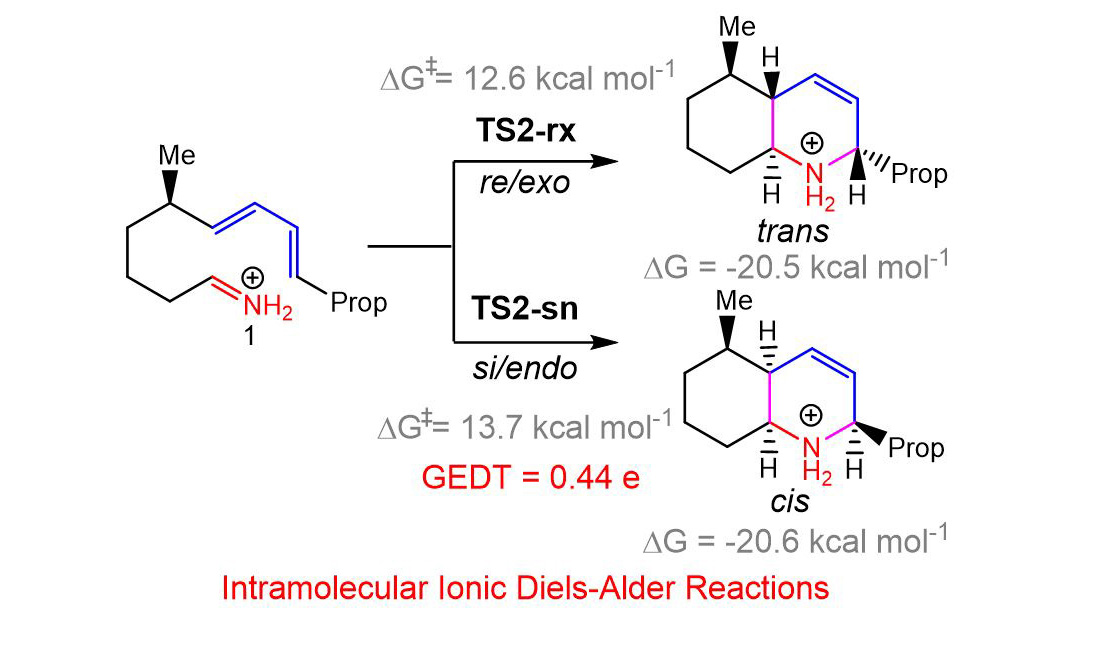
The IIDA reaction of dieniminium 12 takes place via a one-step mechanism. The activation enthalpy associated with this IIDA reaction, 8.7 kcal·mol−1, is closer to that of the I-DA reaction between butadiene 1 and ethaniminium 18, 9.3 kcal·mol−1. However, while the intermolecular I-DA reaction has an activation Gibbs free energy of 25.3 kcal·mol−1, this IIDA reaction presents an activation Gibbs free energy of only 12.6 kcal·mol−1 as a consequence of the very low activation entropy associated with this intramolecular process, −14.3 cal·mol−1·K. The strong exergonic character of the IIDA reaction, higher than 20.5 kcal·mol−1, makes the reaction irreversible.
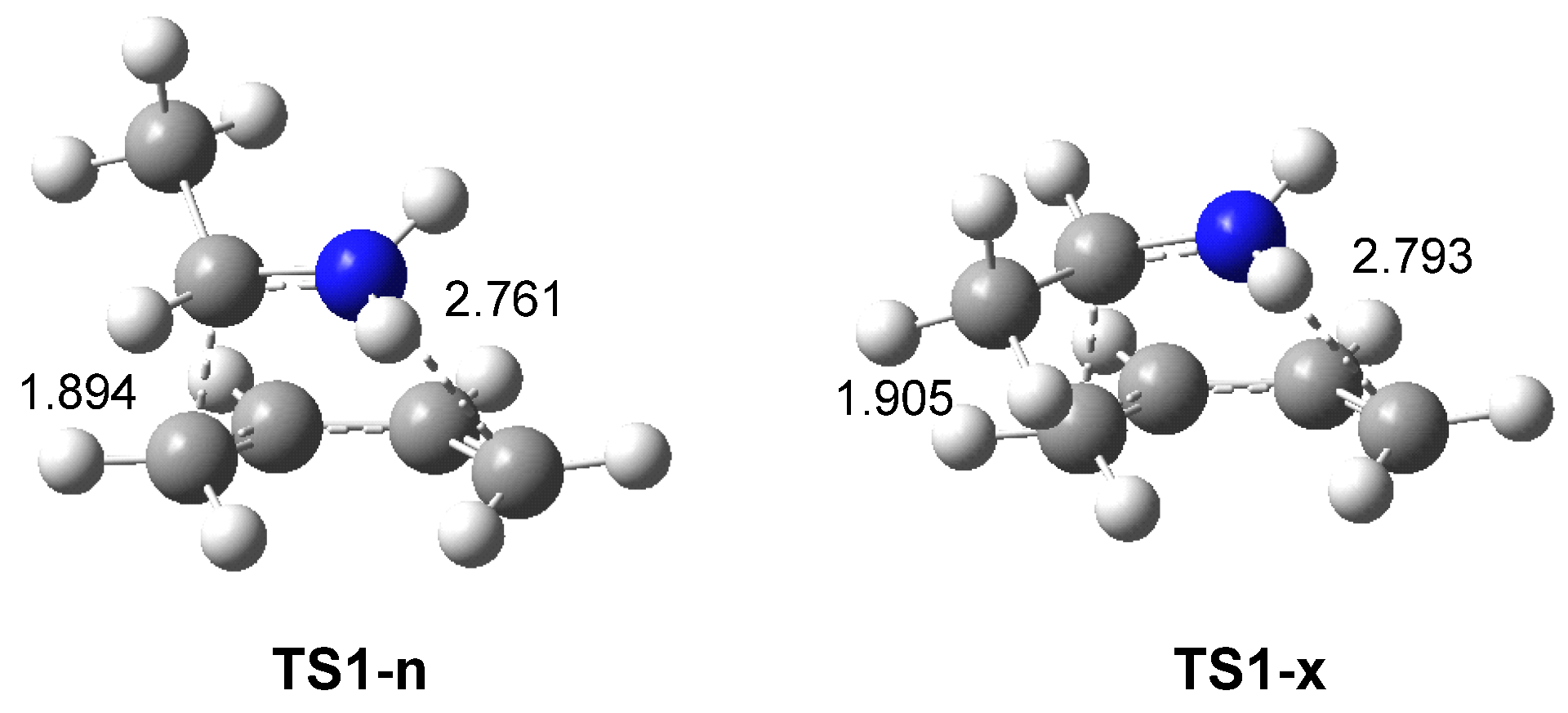
Unlike I-DA reactions, which are low
endo stereoselective [
13], these IIDA reactions present a total
re/
exo and
si/
endo diastereoselectivity, which is controlled for the most favorable chair conformations adopted by the tetramethylene chain along the
re and
si intramolecular approaches. Such as in the I-DA reactions, the IIDA reaction of dieniminium
12 presents a very high intramolecular GEDT , being classified as FEDF.
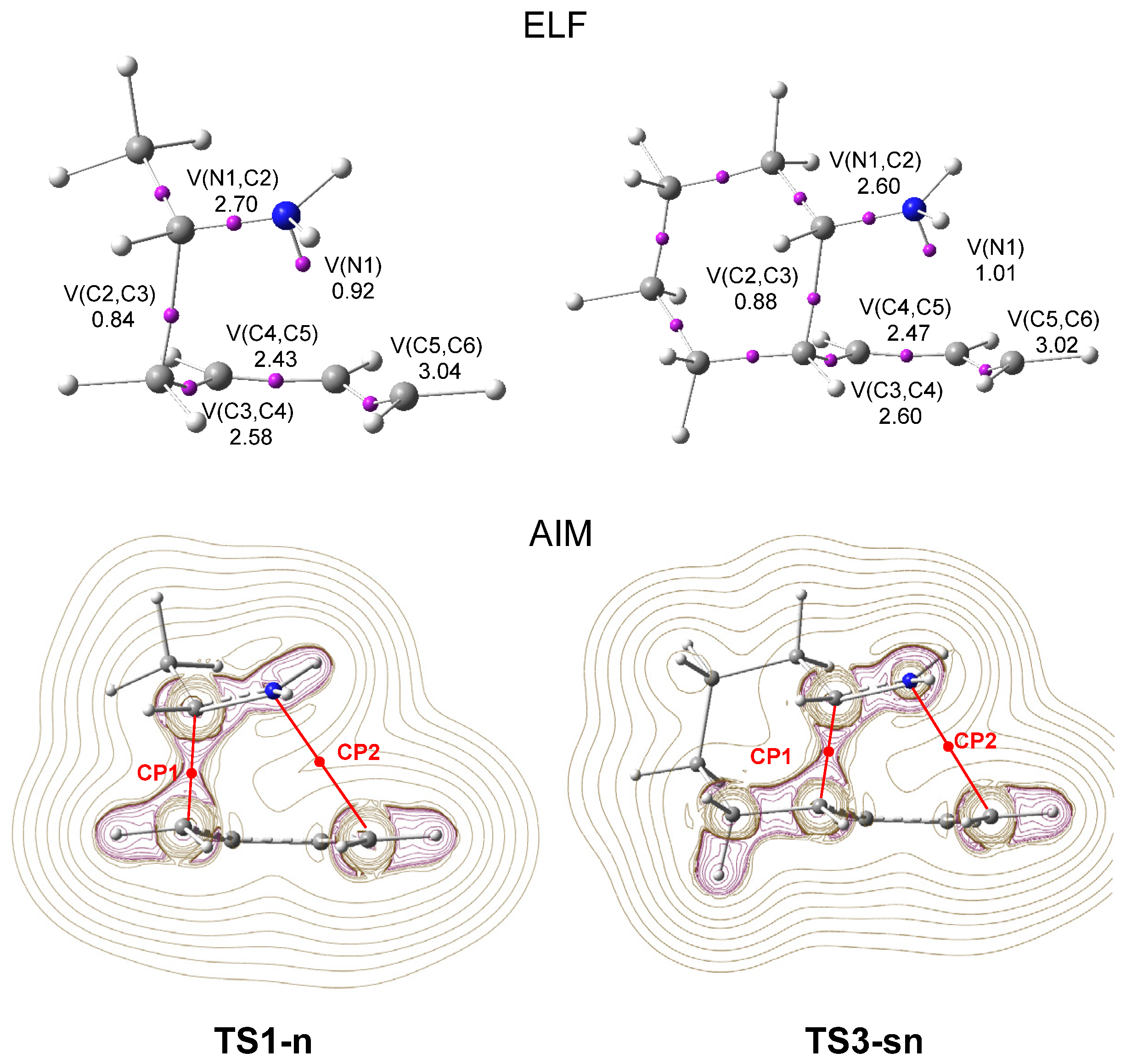
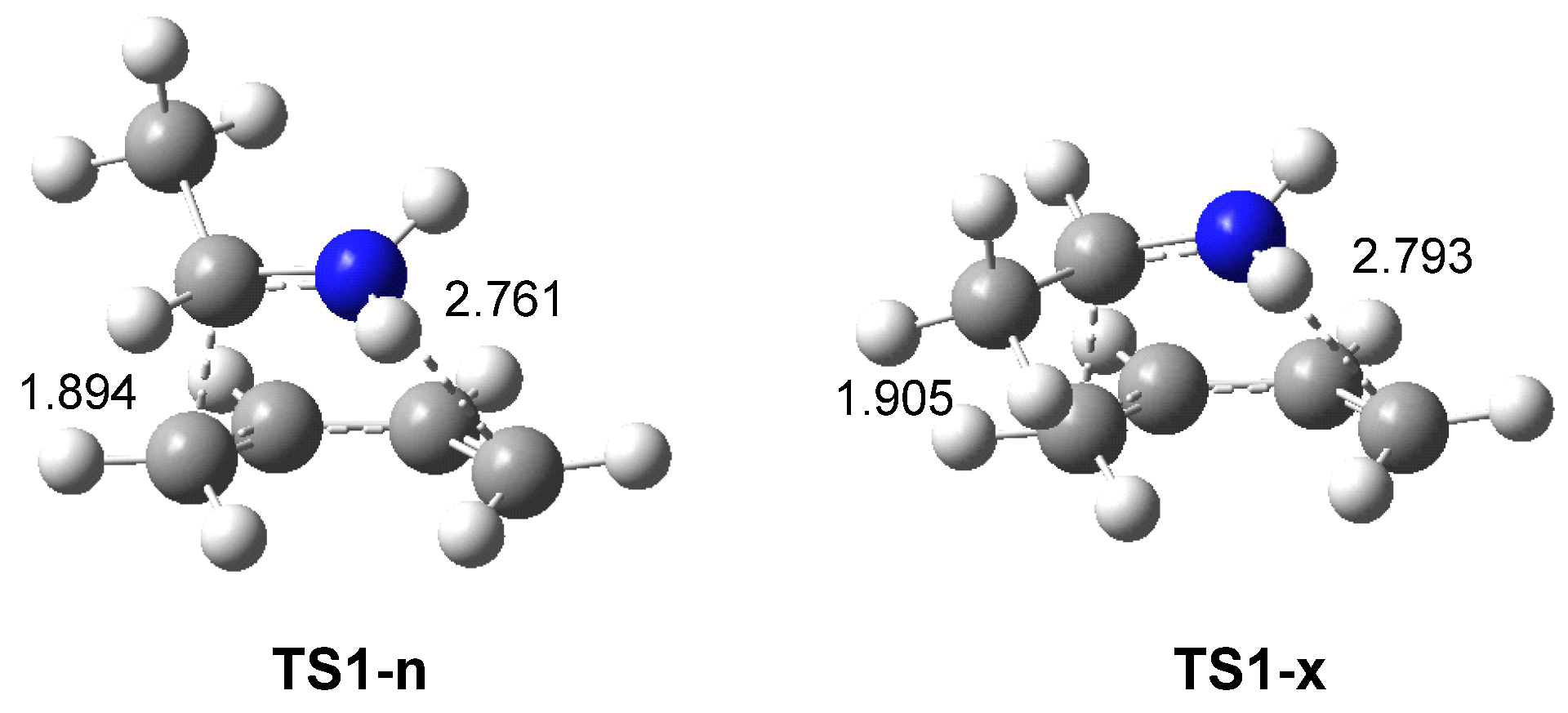
ELF topological analysis of the single bond formation along the IIDA reaction of dieniminium 16 indicated that it takes place through a non-concerted two-stage one-step mechanism. While formation of the first C2−C3 single bond begins by the merger of the non-bonding electron densities of the two pseudoradical C2 and C3 centers created along the reaction path, the formation of the second N1−C6 single bond begins mainly by the donation of the non-bonding electron density of the N1 nitrogen on the C6 carbon. Finally, ELF and AIM topological analyses of the more favorable TS associated with the I-DA reaction between butadiene 1 and ethaniminium 18 and that associated with the IIDA reaction of dieniminium 16 showed the great similarity of the two TSs.
The present MEDT study raises an interesting issue. The high GEDT taking place at the I-DA reactions is rationalized by the high Δμ values of the electronic chemical potentials μ of the two interacting species. Similar to iminiums, dieniminiums presented very low electronic chemical potentials μ values, lesser than −7.0 eV, suggesting a high GEDT at the corresponding TSs. The Sanderson’s Electronegativity Equalization Principle [
69,
70] establishes that, along a polar interaction, the GEDT fluxes from the species with the high electronic chemical potentials μ to those with a low value. However, what happens in the intramolecular processes for which the electrophilic
and nucleophilic
Parr functions recognize two distinguishable frameworks with electrophilic/nucleophilic properties similar to those present in intermolecular processes?
The answer to this question is that a regional descriptor for the electronic chemical potential μf for the different fragments of a molecule able to explain the GEDT taking place in intramolecular processes should be established.








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}