Unveiling the Unexpected Reactivity of Electrophilic Diazoalkanes in [3+2] Cycloaddition Reactions within Molecular Electron Density Theory




Abstract
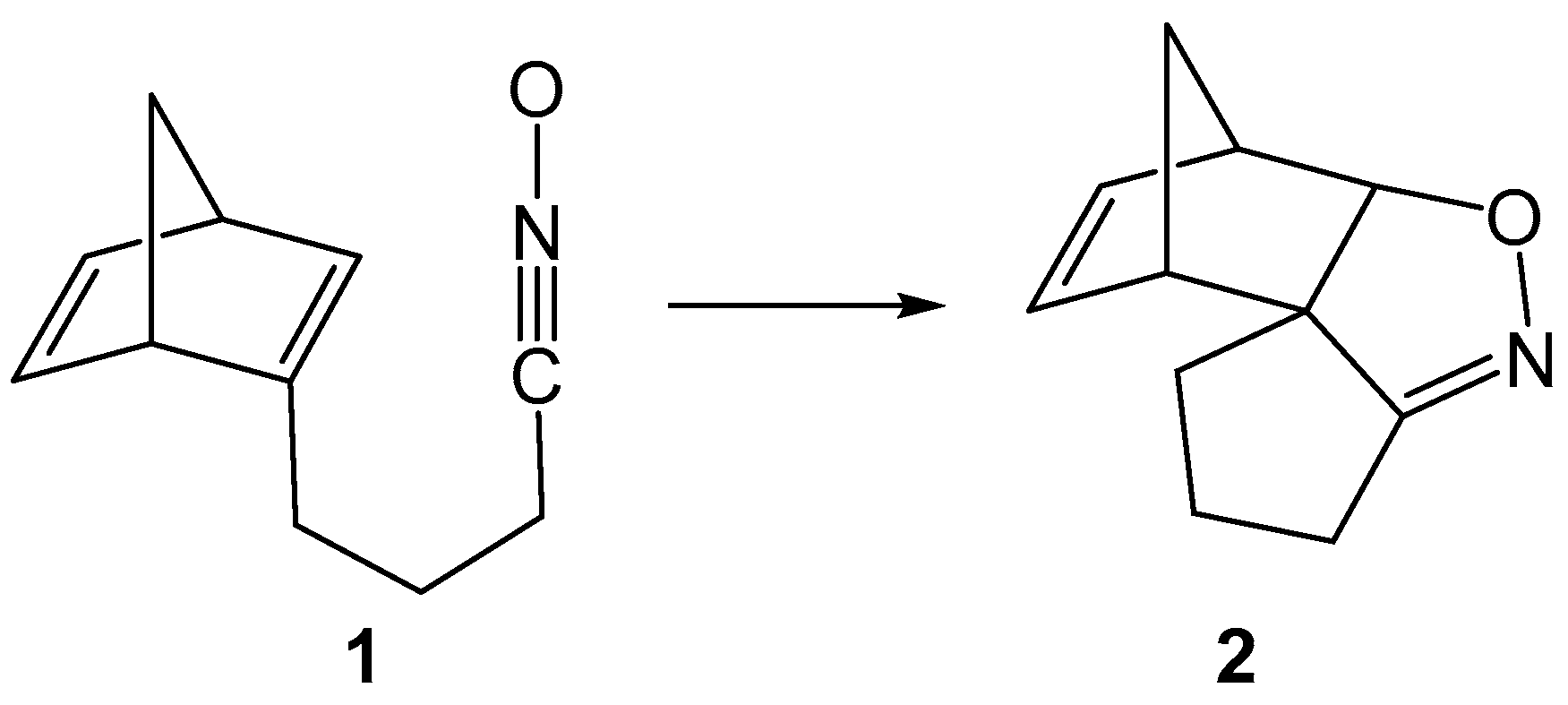
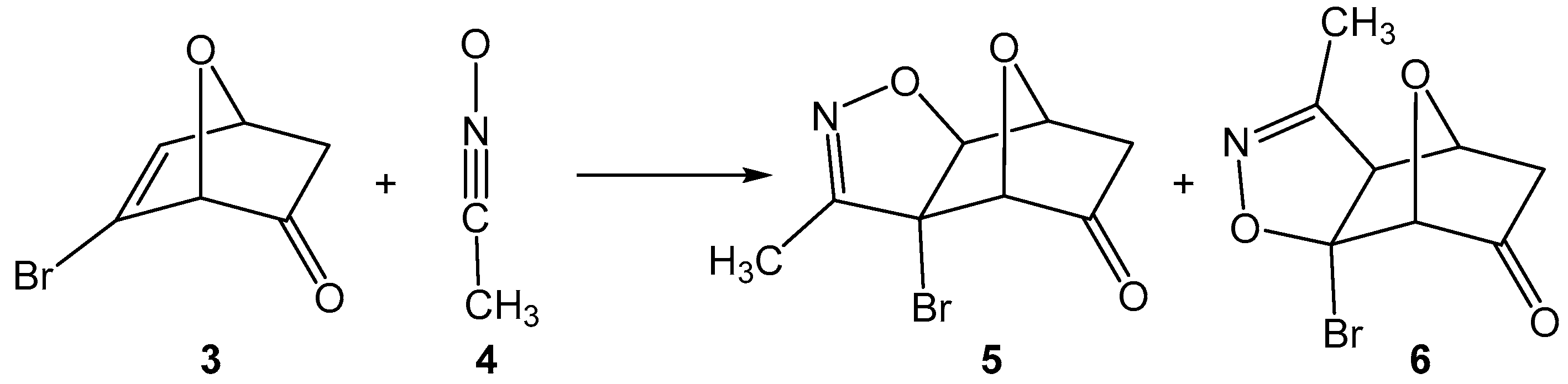
1. Introduction
2. Computational Methods
3. Results and Discussion
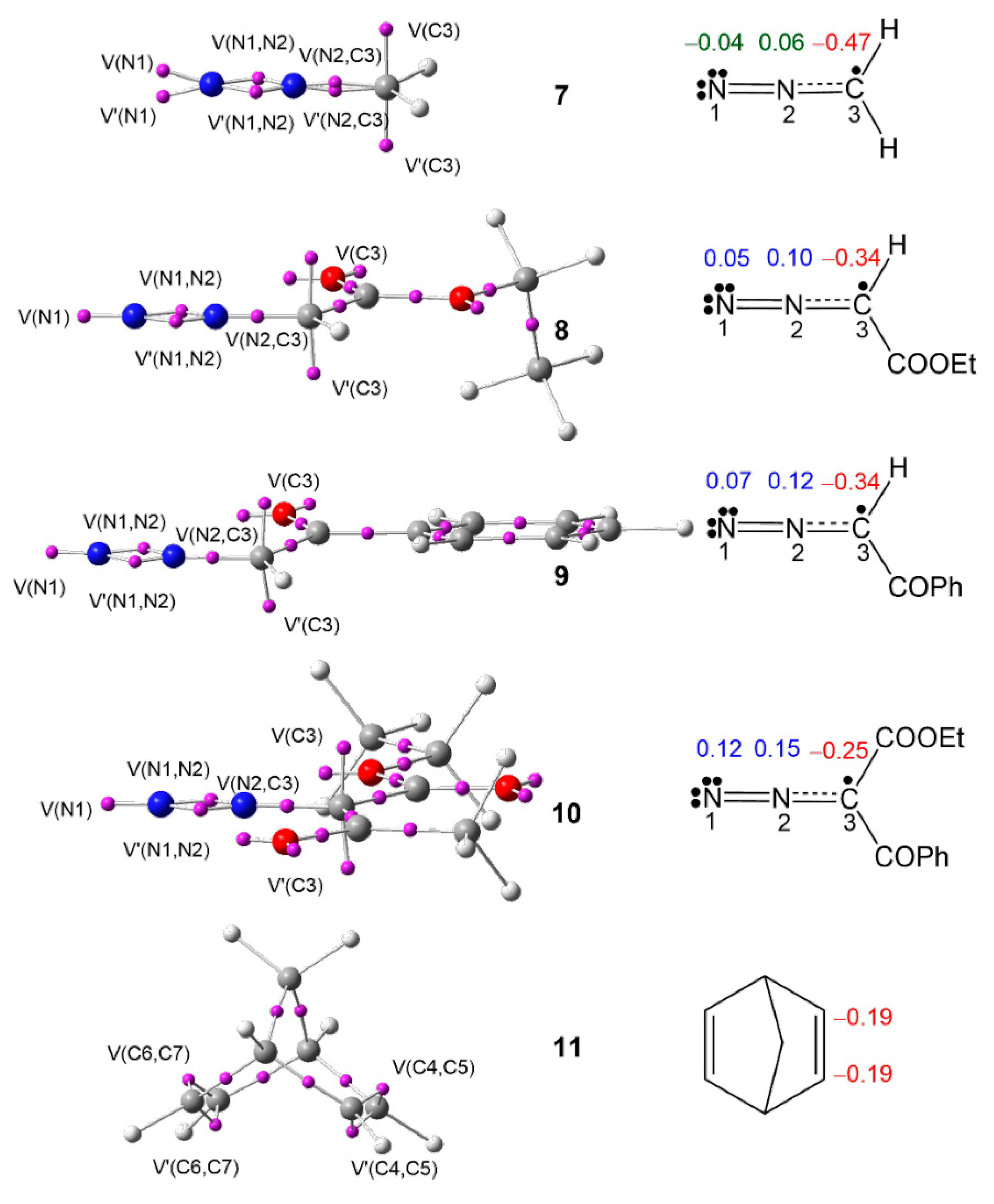
3.1. Analysis of the ELF Topology at the Ground State Electronic Structures of DAAs 7–10 and NBD 11
3.2. Analysis of the CDFT Indices of DAAs 7–10 and NBD 11
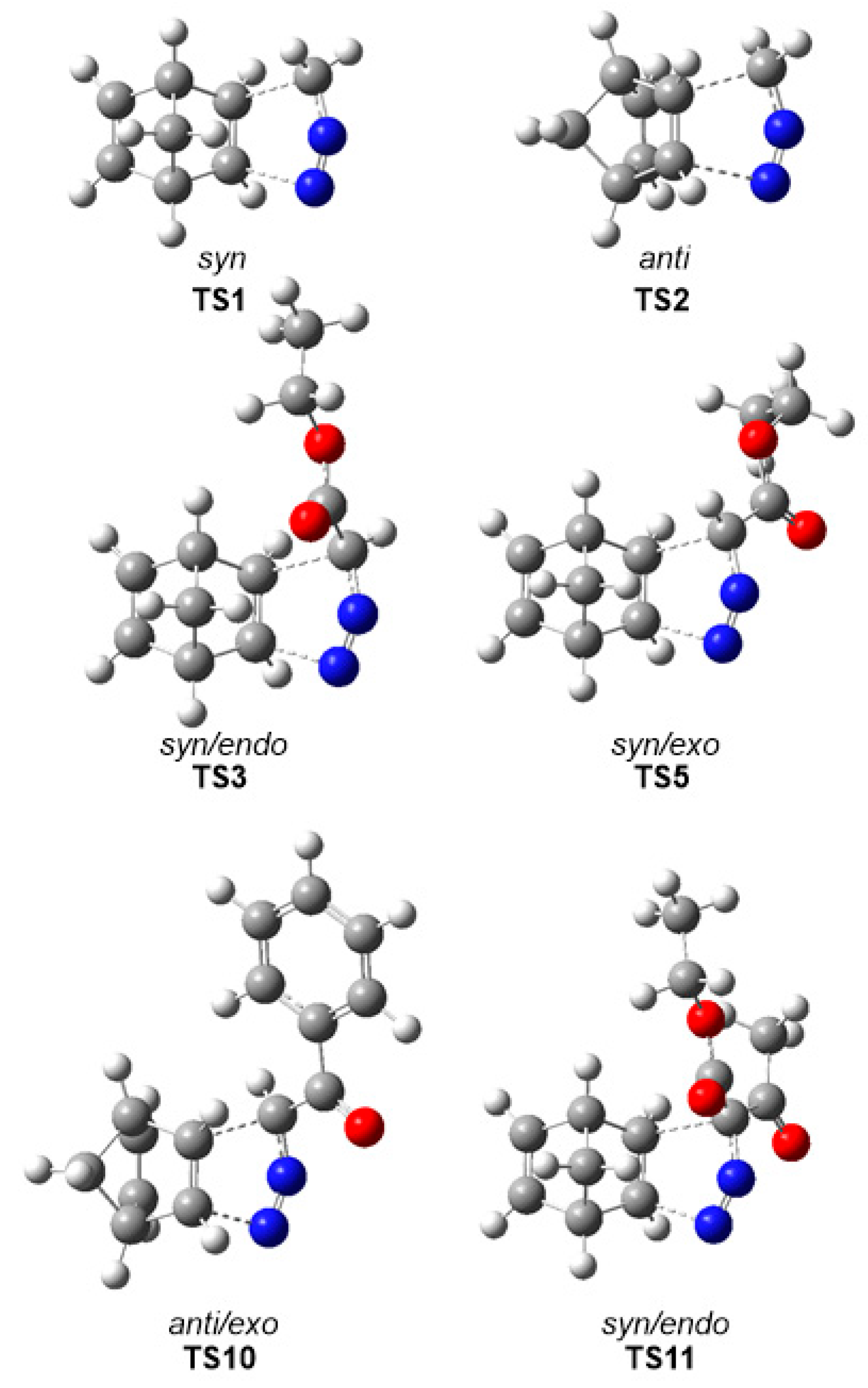
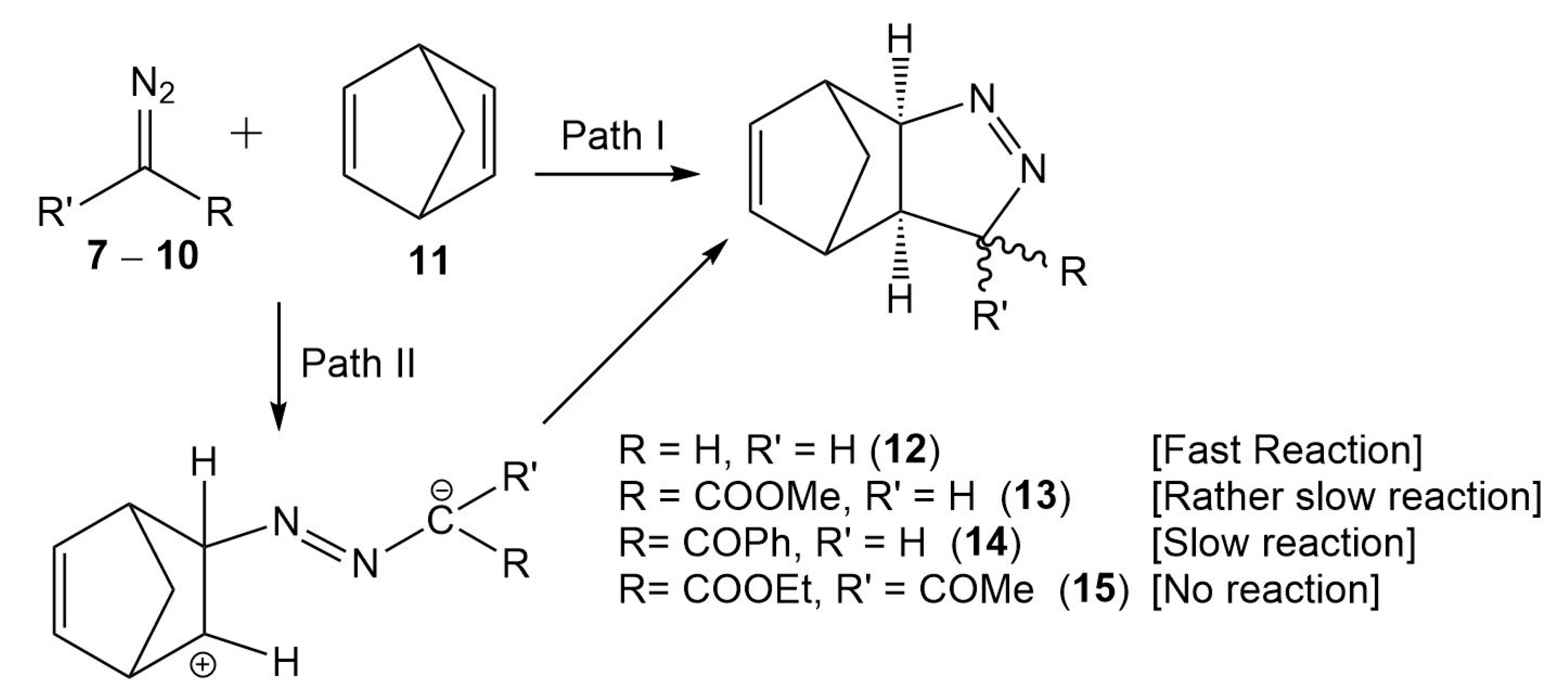
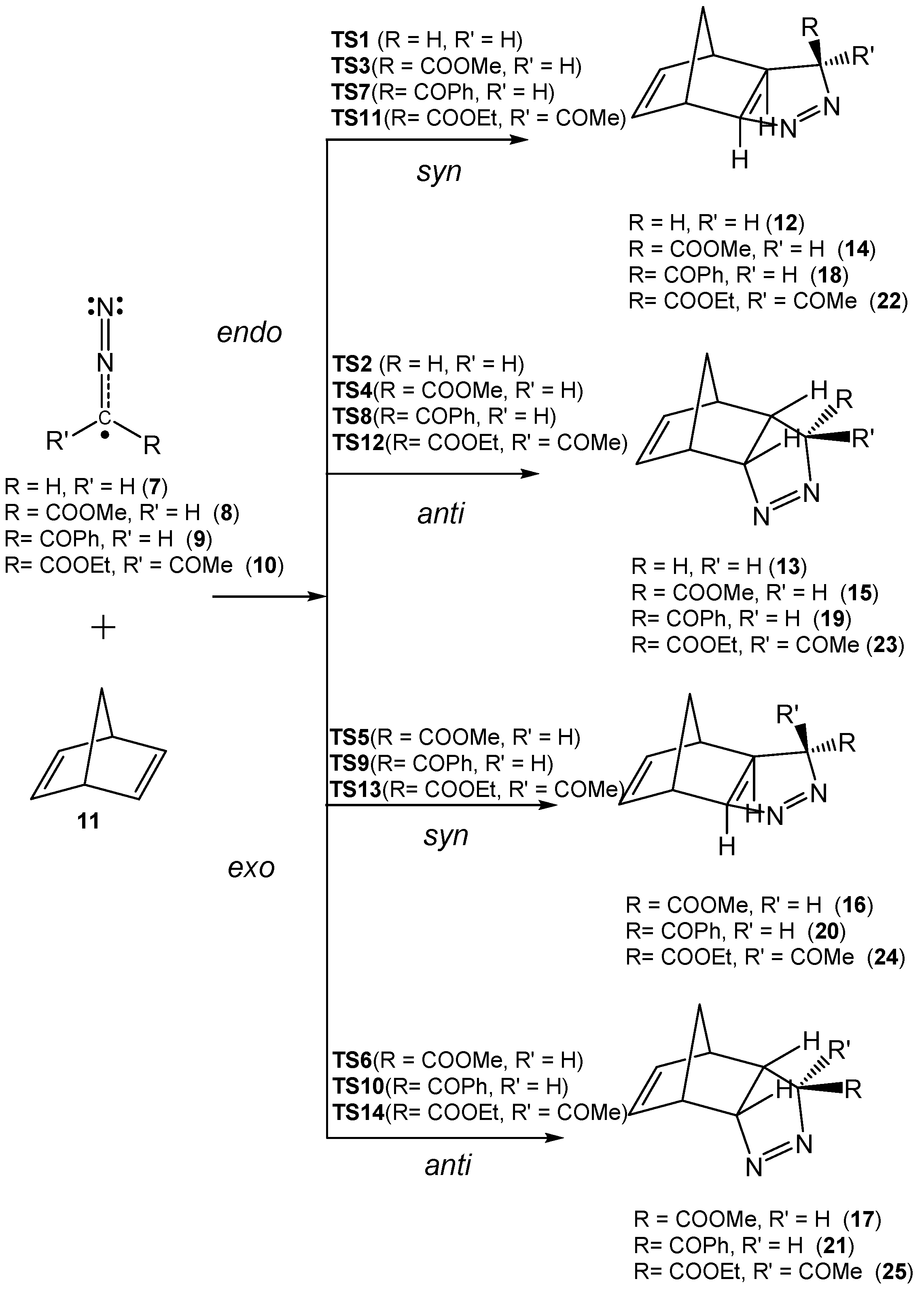
3.3. Analysis of the Reaction Paths Associated with the 32CA Reactions between DAAs 7–10 and NBD 11
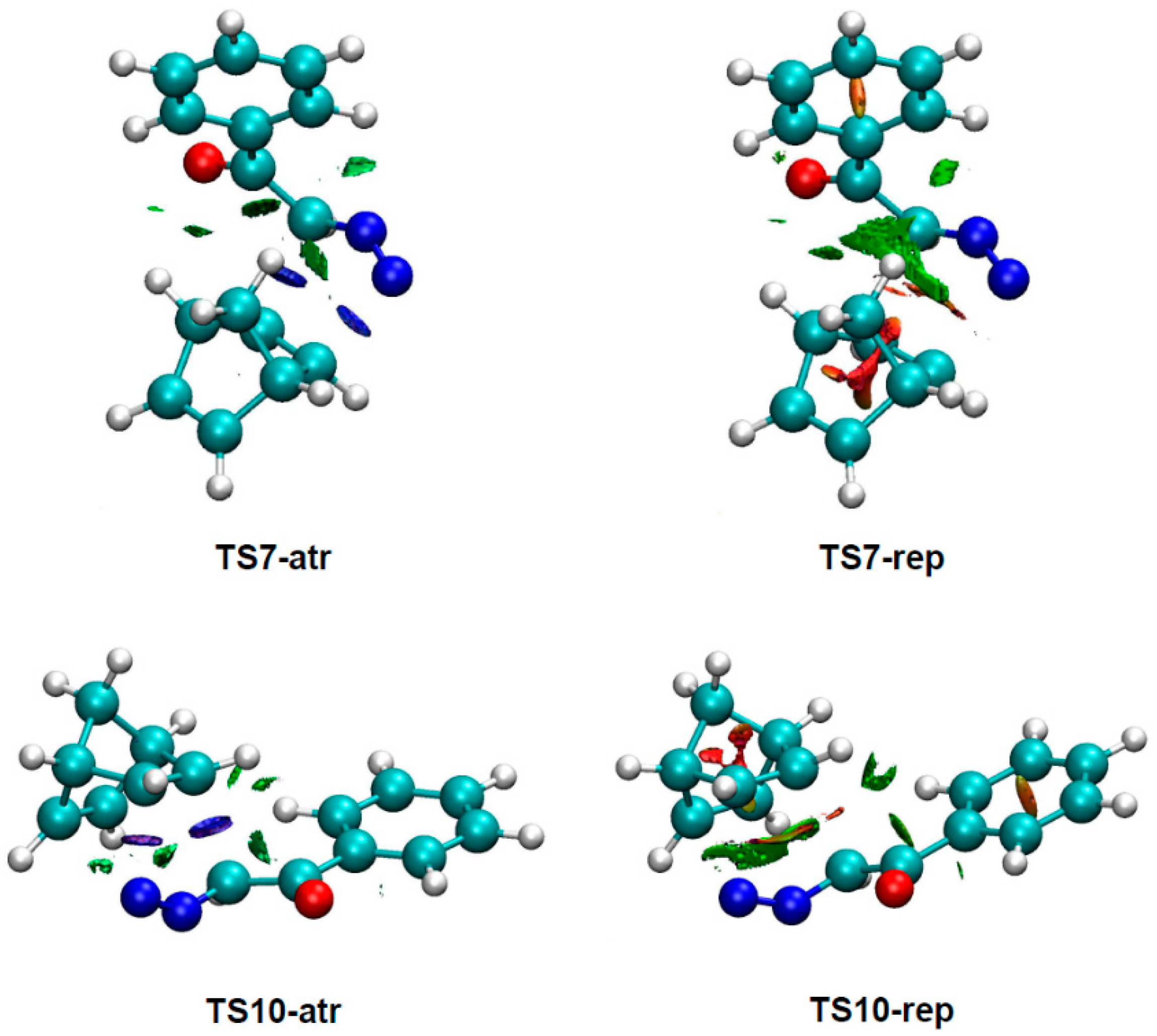
3.4. NCI Analysis of the Preferred Anti/Exo Stereoselectivity in the 32CA Reaction of DAA 9 with NBD 11
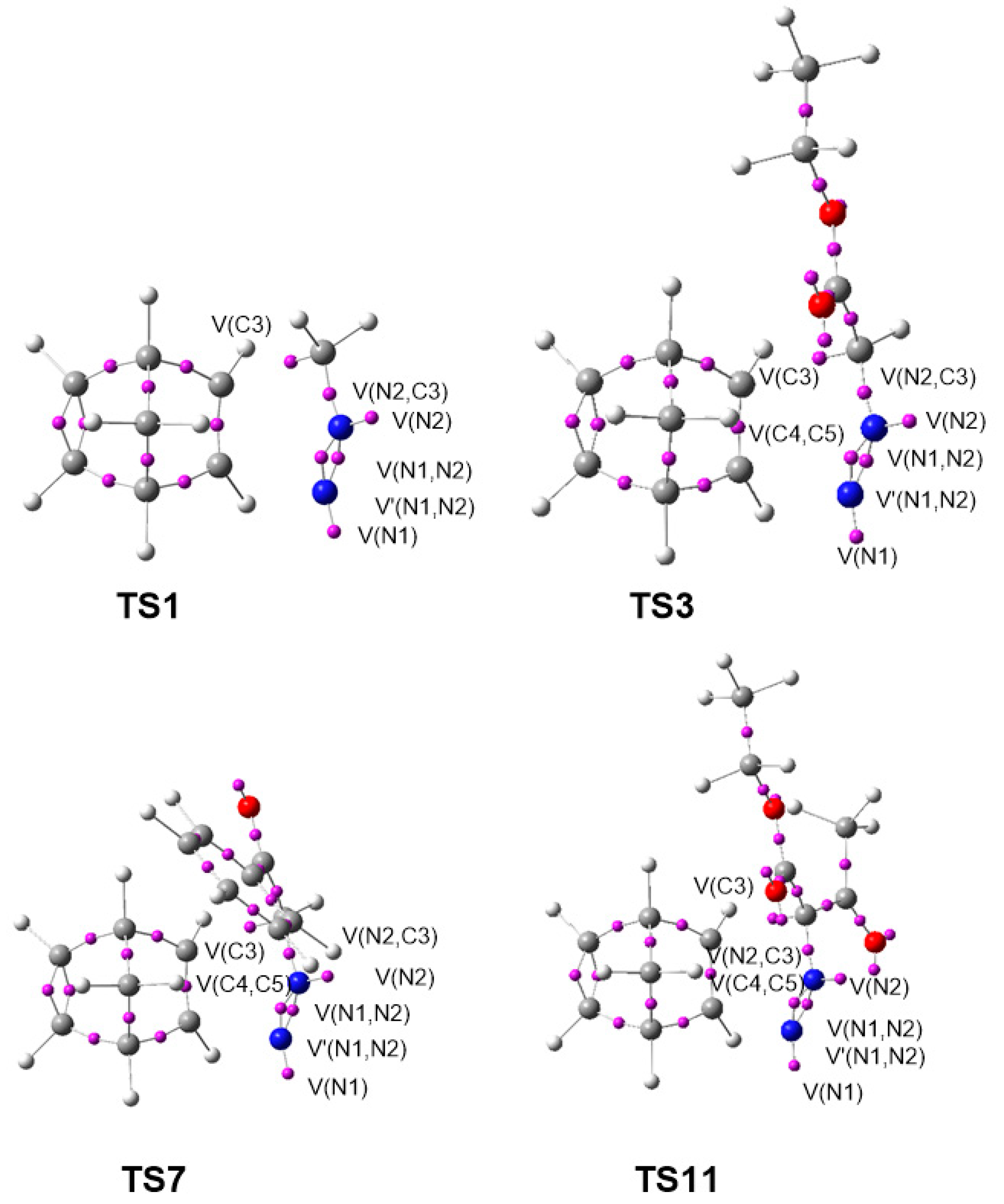
3.5. BET Study for the 32CA Reactions of DAAs 7 and 10 with NBD 11
3.6. ELF Study at the TSs Associated with the 32CA Reactions of DAAs 7–10 with NBD 11
3.7. Analysis of QTAIM Parameters at the TSs Associated with the 32CA Reactions of DAAs 7–10 with NBD 11
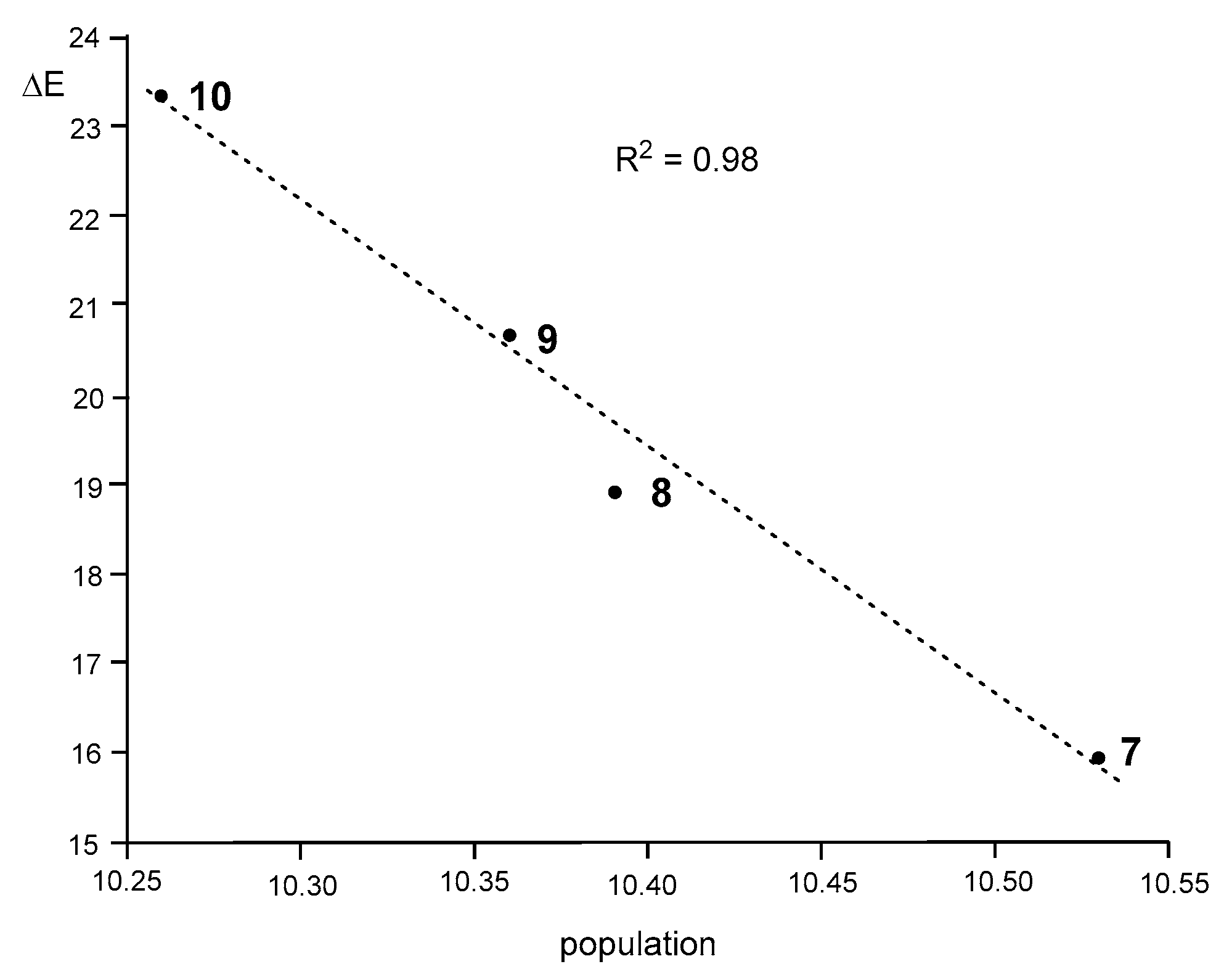
3.8. Analysis of the Origin of the Deceleration of These 32CA Reactions with the Increase of the EW Character of the Substituents Present in These DAA
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Flid, V.R.; Gringolts, M.L.; Shamsiev, R.S.; Finkelshtein, E.S. Norbornene, norbornadiene and their derivatives: Promising semi-products for organic synthesis and production of polymeric materials. Russ. Chem. Rev. 2018, 87, 1169. [Google Scholar] [CrossRef]
- Corey, E.J.; Shibasaki, M.; Nicolaou, K.C.; Malmsten, C.L.; Samuelsson, B. Simple, stereocontrolled total synthesis of a biologically active analog of the prostaglandin endoperoxides (PGH2, PGG2). Tetrahedron Lett. 1976, 17, 737–740. [Google Scholar] [CrossRef]
- Lee, M.; Ikeda, I.; Kawabe, T.; Mori, S.; Kanematsu, K. Enantioselective Total Synthesis of cis-Trikentrin B. J. Org. Chem. 1996, 61, 3406–3416. [Google Scholar] [CrossRef]
- Rosa, C.D.; Malafronte, A.; Auriemma, F.; Scoti, M.; Girolamo, R.D.; D’Alterio, M.C.; Ricci, G.; Zanchin, G.; Leone, G. Synthesis, chain conformation and crystal structure of poly(norbornadiene) having repeating 3,5-enchained nortricyclene units. Polym. Chem. 2019, 10, 4593–4603. [Google Scholar] [CrossRef]
- Petersen, A.U.; Hofmann, A.I.; Fillols, M.; Mansø, M.; Jevric, M.; Wang, Z.; Sumby, C.J.; Müller, C.; Moth-Poulsen, K. Solar Energy Storage by Molecular Norbornadiene–Quadricyclane Photoswitches: Polymer Film Devices. Adv. Sci. 2019, 6, 1900367. [Google Scholar] [CrossRef] [PubMed]
- Daştan, A.; Balci, M. High temperature bromination. Part 18: Bromination of benzonorbornadiene derivatives: Polybrominatedbenzonorbornenes and benzonorbornadienes. Tetrahedron 2005, 61, 5481–5488. [Google Scholar] [CrossRef]
- Horasan, N.; Kara, Y.; Azizoğlu, A.; Balci, M. Low and high temperature bromination of exocyclic dienes: High temperature bromination. Part 16. Tetrahedron 2003, 59, 3691–3699. [Google Scholar] [CrossRef]
- Altundas, R.; Dastan, A.; Ünaldi, N.S.; Güven, K.; Uzun, O.; Balci, M. The Di-π-methane Photorearrangement of 2,3-Disubstituted Benzobarrelenes and Benzonorbornadiene—Substituent Effects in Regioselectivity. Eur. J. Org. Chem. 2002, 526–533. [Google Scholar] [CrossRef]
- Nişancı, B.; Dalkılıç, E.; Güney, M.; Daştan, A. Synthesis and Diels–Alder cycloaddition reaction of norbornadiene and benzonorbornadiene dimers. Beilstein J. Org. Chem. 2009, 5, 39. [Google Scholar] [CrossRef]
- Khan, R.; Chen, J.; Fan, B. Versatile Catalytic Reactions of Norbornadiene Derivatives with Alkynes. Adv. Synth. Catal. 2020, 362, 1564. [Google Scholar] [CrossRef]
- Cristina, D.; Amici, M.D.; Micheli, C.D.; Gandolfi, R. Site selectivity in the reactions of 1,3-dipoles with norbornadiene derivatives. Tetrahedron 1981, 37, 1349–1357. [Google Scholar] [CrossRef]
- Aggarwal, V.K.; Roseblade, S.J.; Barrell, J.K.; Alexander, R. Highly stereoselective nitrone cycloaddition onto a chiral ketene equivalent: Asymmetric synthesis of cispentacin. Org. Lett. 2002, 4, 1227–1229. [Google Scholar] [CrossRef] [PubMed]
- Maiuolo, L.; Nino, A.D. Synthesis of isoxazolidines by 1, 3-dipolar cycloaddition: Recent advances. Targets Heterocycl. Syst. 2015, 19, 299–345. [Google Scholar]
- Rück-Braun, K.; Freysoldt, T.H.E.; Wierschem, F. 1,3-Dipolar cycloaddition on solid supports: Nitrone approach towards isoxazolidines and isoxazolines and subsequent transformations. Chem. Soc. Rev. 2005, 34, 507–516. [Google Scholar] [CrossRef] [PubMed]
- Ali, S.A.; Saeed, M.T.; Rahman, S.U. The isoxazolidines: A new class of corrosion inhibitors of mild steel in acidic medium. Corros. Sci. 2003, 45, 253–266. [Google Scholar] [CrossRef]
- Yip, C.; Handerson, S.; Tranmer, G.K.; Tam, W. Intramolecular 1,3-Dipolar Cycloadditions of Norbornadiene-Tethered Nitrile Oxides. J. Org. Chem. 2001, 66, 276–286. [Google Scholar] [CrossRef] [PubMed]
- Tranmer, G.K.; Tam, W. Intramolecular 1,3-Dipolar Cycloadditions of Norbornadiene-Tethered Nitrones. J. Org. Chem. 2001, 66, 5113–5123. [Google Scholar] [CrossRef]
- Tajabadi, J.; Bakavoli, M.; Gholizadeh, M.; Eshghi, H.; Izadyar, M. The origin of regio- and stereoselectivity in the 1,3-dipolar cycloaddition of nitrile oxides with C1-substituted 7-oxabenzonorbornadienes, a DFT study. RSC Adv. 2015, 5, 38489–38498. [Google Scholar] [CrossRef]
- Adjieufack, A.I.; Ndassa, I.M.; Mbadcam, J.K.; Ríos-Gutiérrez, M.; Domingo, L.R. Steric interactions controlling the syn stereofacial selectivity in the [3 + 2] cycloaddition reaction between acetonitrile oxide and 7-oxanorborn-5-en-2-ones: A molecular electron density theory study. J. Phys. Org. Chem. 2017, 30, e3710. [Google Scholar] [CrossRef]
- Arhin, G.; Adams, A.H.; Opoku, E.; Tia, R.; Adei, E. 1,3-Dipolar cycloaddition reactions of selected 1,3-dipoles with 7-isopropylidenenorbornadiene and follow-up thermolytic cleavage: A computational study. J. Mol. Graph. Model. 2019, 92, 267–279. [Google Scholar] [CrossRef]
- Opoku, E.; Arhin, G.; Pipim, G.B.; Adams, A.H.; Tia, R.; Adei, E. Site-, enantio- and stereo-selectivities of the 1,3-dipolar cycloaddition reactions of oxanorbornadiene with C,N-disubstituted nitrones and dimethyl nitrilimines: A DFT mechanistic study. Theor. Chem. Acc. 2020, 139, 16. [Google Scholar] [CrossRef]
- Huisgen, R. Kinetics and mechanism of 1,3-dipolar cycloadditions. Angew. Chem., Int. Ed. 1963, 2, 633–696. [Google Scholar] [CrossRef]
- Huisgen, R. Altes und Neues über aliphatische Diazoverbindungen. Angew. Chem. 1955, 67, 439–463. [Google Scholar] [CrossRef]
- Fukui, K. Molecular Orbitals in Chemistry, Physics, and Biology; Academic Press: New York, NY, USA, 1964; p. 525. [Google Scholar]
- Domingo, L.R. Molecular electron density theory. Molecules. 2016, 21, 1319. [Google Scholar] [CrossRef]
- Domingo, L.R.; Acharjee, N. Molecular Electron Density Theory: A New Theoretical Outlook on Organic Chemistry. In Frontiers in Computational Chemistry; Ul-Haq, Z., Wilson, A.K., Eds.; Bentham and Science: Singapore, 2020; Volume 5, pp. 174–227. [Google Scholar] [CrossRef]
- Domingo, L.R.; Acharjee, N. Unravelling the strain-promoted [3 + 2] cycloaddition reactions of phenyl azide with cycloalkynes from the molecular electron density theory perspective. New J. Chem. 2020, 44, 13633–13643. [Google Scholar] [CrossRef]
- Domingo, L.R.; Acharjee, N. Unveiling the high reactivity of strained dibenzocyclooctyne in [3 + 2] cycloaddition reactions with diazoalkanes through the molecular electron density theory. J. Phys. Org. Chem. 2020, 33, e4100. [Google Scholar] [CrossRef]
- Domingo, L.R.; Ríos-Gutiérrez, M.; Pérez, P. A Molecular Electron Density Theory Study of the Reactivity and Selectivities in [3 + 2] Cycloaddition Reactions of C,N-Dialkyl Nitrones with Ethylene Derivatives. J. Org. Chem. 2018, 83, 2182–2197. [Google Scholar] [CrossRef]
- Domingo, L.R.; Ríos-Gutiérrez, M.; Acharjee, N. A Molecular Electron Density Theory Study of the Chemoselectivity, Regioselectivity, and Stereofacial Selectivity in the Synthesis of an Anticancer Spiroisoxazoline derived from α-Santonin. Molecules 2019, 24, 832. [Google Scholar] [CrossRef]
- Domingo, L.R.; Ríos-Gutiérrez, M.; Adjieufack, A.I.; Ndassa, I.M.; Nouhou, C.N.; Mbadcam, J.K. Molecular Electron Density Theory Study of Fused Regioselectivity in the Intramolecular [3 + 2] Cycloaddition Reaction of Nitrones. ChemistrySelect 2018, 3, 5412–5420. [Google Scholar] [CrossRef]
- Domingo, L.R.; Acharjee, N. [3 + 2] Cycloaddition Reaction of C-Phenyl-N-methyl Nitrone to Acyclic-Olefin-Bearing-Electron-Donating Substituent: A Molecular Electron Density Theory Study. Chemistryselect 2018, 3, 8373–8380. [Google Scholar] [CrossRef]
- Gutiérrez, M.R.; Nasri, L.; Nacereddine, A.K.; Djerourou, A.; Domingo, L.R. A molecular electron density theory study of the [3 + 2] cycloaddition reaction between an azomethine imine and electron deficient ethylenes. J. Phys. Org. Chem. 2018, 31, e3830. [Google Scholar] [CrossRef]
- Domingo, L.R.; Ríos-Gutiérrez, M.; Pérez, P. A Molecular Electron Density Theory Study of the Role of the Copper-Metallation of AzomethineYlides in [3 + 2] Cycloaddition Reactions. J. Org. Chem. 2018, 83, 10959–10973. [Google Scholar] [CrossRef] [PubMed]
- Domingo, L.R.; Acharjee, N. A molecular electron density theory study of the Grignard reagent mediated regioselective direct synthesis of 1,5-disubstituted-1,2,3-triazoles. J. Phys. Org. Chem. 2020, 33, e4062. [Google Scholar] [CrossRef]
- Ríos-Gutiérrez, M.; Domingo, L.R. Unravelling the mysteries of the [3 + 2] cycloaddition reactions. Eur. J. Org. Chem. 2019, 267–282. [Google Scholar] [CrossRef]
- Domingo, L.R.; Chamorro, E.; Pérez, P. Understanding the High Reactivity of the Azomethine Ylides in [3 + 2] Cycloaddition Reactions. Lett. Org. Chem. 2010, 7, 432–439. [Google Scholar] [CrossRef]
- Domingo, L.R.; Ríos-Gutiérrez, M. A Molecular Electron Density Theory Study of the Reactivity of Azomethine Imine in [3 + 2] Cycloaddition Reactions. Molecules. 2017, 22, 750. [Google Scholar] [CrossRef]
- Ríos-Gutiérrez, M.; Domingo, L.R. The carbenoid-type reactivity of simplest nitrile imine from a molecular electron density theory perspective. Tetrahedron 2019, 75, 1961–1967. [Google Scholar] [CrossRef]
- Domingo, L.R.; Ríos-Gutiérrez, M.; Emamian, S. Understanding the domino reactions between 1-diazopropan-2-one and 1,1-dinitroethylene. A molecular electron density theory study of the [3 + 2] cycloaddition reactions of diazoalkanes with electron-deficient ethylenes. RSC Adv. 2017, 7, 15586–15595. [Google Scholar] [CrossRef]
- Becke, A.D.; Edgecombe, K.E. A simple measure of electron localization in atomic and molecular systems. J. Chem. Phys. 1990, 92, 5397–5403. [Google Scholar] [CrossRef]
- Silvi, B.; Savin, A. Classification of chemical bonds based on topological analysis of electron localization functions. Nature 1994, 371, 683–686. [Google Scholar] [CrossRef]
- Parr, R.G.; Yang, W. Density Functional Theory of Atoms and Molecules; Oxford University Press: New York, NY, USA, 1989. [Google Scholar]
- Domingo, L.R.; Ríos-Gutiérrez, M.; Pérez, P. Applications of the Conceptual Density Functional Theory Indices to Organic Chemistry Reactivity. Molecules 2016, 21, 748. [Google Scholar] [CrossRef] [PubMed]
- Krokidis, X.; Noury, S.; Silvi, B. Characterization of Elementary Chemical Processes by Catastrophe Theory. J. Phys. Chem. A 1997, 101, 7277–7282. [Google Scholar] [CrossRef]
- Bader, R.F.W. Atoms in Molecules: A Quantum Theory; Clarendon Press: Oxford, UK, 1990. [Google Scholar]
- Bader, R.F.W.; Essén, H. The characterization of atomic interactions. J. Chem. Phys. 1984, 80, 1943–1960. [Google Scholar] [CrossRef]
- Schlegel, H.B. Optimization of equilibrium geometries and transition structures. J. Comput. Chem. 1982, 3, 214–218. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. Hybrid Meta Density Functional Theory Methods for Thermochemistry, Thermochemical Kinetics, and Noncovalent Interactions: The MPW1B95 and MPWB1K Models and Comparative Assessments for Hydrogen Bonding and van der Waals Interactions. J. Phys. Chem. A 2004, 108, 6908–6918. [Google Scholar] [CrossRef]
- Hehre, W.J.; Radom, L.; Schleyer, P.V.R.; Pople, J. Ab initio Molecular Orbital Theory; Wiley: New York, NY, USA, 1986. [Google Scholar]
- Fukui, K. Formulation of the reaction coordinate. J. Phys. Chem. 1970, 74, 4161–4163. [Google Scholar] [CrossRef]
- González, C.; Schlegel, H.B. Reaction path following in mass-weighted internal coordinates. J. Phys. Chem. 1990, 94, 5523–5527. [Google Scholar] [CrossRef]
- González, C.; Schlegel, H.B. Improved algorithms for reaction path following: Higher-order implicit algorithms. Chem. Phys. 1991, 95, 5853–5860. [Google Scholar] [CrossRef]
- Tomasi, J.; Persico, M. Molecular Interactions in Solution: An Overview of Methods Based on Continuous Distributions of the Solvent. Chem. Rev. 1994, 94, 2027–2094. [Google Scholar] [CrossRef]
- Simkin, B.I.A.; Sheikhet, I.I. Quantum Chemical and Statistical Theory of Solutions: A Computational Approach; Ellis Horwood: London, UK, 1995. [Google Scholar]
- Cossi, M.; Barone, V.; Cammi, R.; Tomasi, J. Ab Initio Study of Solvated Molecules: A New Implementation of the Polarizable Continuum Model. Chem. Phys. Lett. 1996, 255, 327–335. [Google Scholar] [CrossRef]
- Cances, E.; Mennucci, B.; Tomasi, J. A new integral equation formalism for the polarizable continuum model: Theoretical background and applications to isotropic and anisotropic dielectrics. J. Chem. Phys. 1997, 107, 3032–3041. [Google Scholar] [CrossRef]
- Barone, V.; Cossi, M.; Tomasi, J. Geometry optimization of molecular structures in solution by the polarizable continuum model. J. Comput. Chem. 1998, 19, 404–417. [Google Scholar] [CrossRef]
- Domingo, L.R. A new C–C bond formation model based on the quantum chemical topology of electron density. RSC Adv. 2014, 4, 32415–32428. [Google Scholar] [CrossRef]
- Reed, A.E.; Weinstock, R.B.; Weinhold, F. Natural population analysis. J. Chem. Phys. 1985, 83, 735–746. [Google Scholar] [CrossRef]
- Reed, A.E.; Curtiss, L.A.; Weinhold, F. Intermolecular interactions from a natural bond orbital, donor-acceptor viewpoint. Chem. Rev. 1988, 88, 899–926. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comp. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef]
- Ahrens, J.; Geveci, B.; Law, C. ParaView: An End-User Tool for Large Data Visualization; Visualization Handbook; Elsevier: Amsterdam, The Netherlands, 2005. [Google Scholar]
- Ayachit, U. The ParaView Guide: A Parallel Visualization Application; Kitware: New York, NY, USA, 2015. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Johnson, E.R.; Keinan, S.; Mori-Sánchez, P.; Contreras-García, J.; Cohen, A.J.; Yang, W. Revealing noncovalent interactions. J. Am. Chem. Soc. 2010, 132, 6498–6506. [Google Scholar] [CrossRef]
- Boto, R.A.; Peccati, F.; Laplaza, R.; Quan, C.; Carbone, A.; Piquemal, J.P.; Maday, Y.; Contreras-García, J. NCIPLOT4: Fast, Robust, and Quantitative Analysis of Noncovalent Interactions. J. Chem. Theory Comput. 2020, 16, 4150–4158. [Google Scholar] [CrossRef]
- Domingo, L.R.; Aurell, M.J.; Pérez, P.; Contreras, R. Quantitative characterization of the global electrophilicity power of common diene/dienophile pairs in Diels–Alder reactions. Tetrahedron 2002, 58, 4417–4423. [Google Scholar] [CrossRef]
- Jaramillo, P.; Domingo, L.R.; Chamorro, E.; Pérez, P. A further exploration of a nucleophilicity index based on the gas-phase ionization potentials. J. Mol. Struct. Theochem. 2008, 865, 68–72. [Google Scholar] [CrossRef]
- Domingo, L.R.; Ríos-Gutiérrez, M.; Pérez., P. A molecular electron density theory study of the participation of tetrazines in aza-Diels–Alder reactions. RSC Adv. 2020, 10, 15394–15405. [Google Scholar] [CrossRef]
- Domingo, L.R.; Kula, K.; Ríos-Gutiérrez, M. Unveiling the Reactivity of Cyclic AzomethineYlides in [3 + 2] Cycloaddition Reactions within the Molecular Electron Density Theory. Eur. J. Org. Chem. 2020, 2020, 5938–5948. [Google Scholar] [CrossRef]
- Lane, J.R.; Contreras-García, J.; Piquemal, J.P.; Miller, B.; Kjaergaard, H.G. Are Bond Critical Points Really Critical for Hydrogen Bonding? J. Chem. Theory Comput. 2013, 9, 3263–3266. [Google Scholar] [CrossRef] [PubMed]
- Domingo, L.R.; Sáez, J.A.; Zaragozá, R.J.; Arnó, M. Understanding the Participation of Quadricyclane as Nucleophile in Polar [2σ + 2σ + 2π] Cycloadditions toward Electrophilic π Molecules. J. Org. Chem. 2008, 73, 8791–8799. [Google Scholar] [CrossRef] [PubMed]
- Domingo, L.R.; Ríos-Gutiérrez, M.; Pérez, P. How does the global electron density transfer diminish activation energies in polar cycloaddition reactions? A Molecular Electron Density Theory study. Tetrahedron 2017, 73, 1718–1724. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 7 | 8 | 9 | 10 | 11 | |
|---|---|---|---|---|---|
| V(N1) | 1.89 | 3.61 | 3.58 | 3.47 | |
| V′(N1) | 1.89 | ||||
| V(N1,N2) | 1.87 | 1.99 | 2.05 | 2.05 | |
| V′(N1,N2) | 1.87 | 1.89 | 1.85 | 1.92 | |
| V(C3,N2) | 3.01 | 2.90 | 2.88 | 2.82 | |
| V(C3) | 0.46 | 0.46 | 0.39 | 0.49 | |
| V′(C3) | 0.46 | 0.46 | 0.49 | 0.49 | |
| V(C4,C5) | 1.74 | ||||
| V′(C4,C5) | 1.74 | ||||
| V(C6,C7) | 1.74 | ||||
| V′(C6,C7) | 1.74 |
| μ | η | ω | N | |
|---|---|---|---|---|
| DAA 10 | −4.58 | 4.87 | 2.15 | 2.09 |
| DAA 8 | −4.27 | 4.79 | 1.90 | 2.45 |
| DAA 9 | −4.20 | 4.76 | 1.85 | 2.53 |
| DAA 7 | −3.63 | 4.70 | 1.40 | 3.13 |
| NBD 11 | −2.94 | 5.88 | 0.74 | 3.23 |
| ethylene 26 | −3.37 | 7.77 | 0.73 | 1.87 |
| Gas Phase | Acetonitrile | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| ∆E | ∆H | ∆S | ∆G | GEDT | ∆E | ∆H | ∆S | ∆G | GEDT | |
| TS1 | 15.9 | 16.8 | −42.3 | 29.4 | 0.10 a | 16.7 | 17.7 | −40.8 | 29.9 | 0.10 |
| 12 | −53.6 | −49.0 | −47.3 | −34.8 | −55.6 | −50.8 | −46.0 | −37.1 | ||
| TS2 | 16.6 | 17.4 | −42.9 | 30.2 | 0.12 a | 17.6 | 18.5 | −41.6 | 30.9 | 0.12 |
| 13 | −52.9 | −48.3 | −47.5 | −34.1 | −55.2 | −50.4 | −46.1 | −36.7 | ||
| TS3 | 18.9 | 19.4 | −42.3 | 32.0 | 0.04 | 20.0 | 20.8 | −42.5 | 33.5 | 0.03 |
| 14 | −44.0 | −40.3 | −46.5 | −26.4 | −46.3 | −42.1 | −48.2 | −27.7 | ||
| TS4 | 22.0 | 22.6 | −44.1 | 35.7 | 0.04 | 23.3 | 24.1 | −44 | 37.2 | 0.02 |
| 15 | −42.1 | −38.2 | −49.2 | −23.6 | −45.3 | −41.2 | −49.2 | −26.5 | ||
| TS5 | 19.5 | 20.1 | −42.5 | 32.8 | 0.04 | 20.9 | 21.8 | −44.1 | 35.0 | 0.03 |
| 16 | −45.3 | −41.4 | −47.4 | −27.3 | −46.3 | −42.1 | −46.4 | −28.3 | ||
| TS6 | 20.4 | 20.8 | −41.4 | 33.2 | 0.06 | 21.9 | 22.5 | −42.4 | 35.1 | 0.04 |
| 17 | −44.1 | −40.4 | −43.5 | −27.4 | −45.9 | −41.8 | −45.8 | −28.2 | ||
| TS7 | 21.4 | 22.0 | −45.9 | 35.7 | 0.02 | 23.3 | 24.1 | −47.0 | 38.1 | 0.01 |
| 18 | −40.3 | −37.2 | −55.0 | −20.8 | −39.1 | −35.0 | −42.4 | −22.4 | ||
| TS8 | 22.3 | 23.1 | −48.3 | 37.5 | 0.05 | 24.0 | 24.7 | −46.3 | 38.5 | 0.02 |
| 13 | −39.5 | −35.5 | −52.2 | −20.0 | −42.4 | −38.0 | −52.2 | −22.5 | ||
| TS9 | 21.7 | 22.5 | −47.0 | 36.5 | 0.04 | 24.0 | 24.9 | −46.8 | 38.8 | 0.02 |
| 20 | −41.3 | −37.3 | −50.5 | −22.2 | −41.2 | −37.1 | −49.5 | −22.3 | ||
| TS10 | 20.6 | 21.1 | −46.0 | 34.8 | 0.05 | 22.8 | 23.5 | −45.9 | 37.2 | 0.02 |
| 21 | −44.6 | −40.8 | −51.1 | −25.5 | −43.8 | −39.8 | −50.1 | −24.8 | ||
| TS11 | 23.3 | 24.2 | −47.7 | 38.5 | 0.04 | 23.6 | 24.7 | −47.8 | 38.9 | 0.08 b |
| 22 | −30.1 | −26.8 | −48.0 | −12.5 | −34.2 | −30.6 | −48.3 | −16.2 | ||
| TS12 | 26.2 | 27-0 | −48.5 | 41.4 | 0.04 | 26.7 | 27.6 | −47.9 | 41.9 | 0.08 b |
| 23 | −35.1 | −31.8 | −49.9 | −16.9 | −36.3 | −32.9 | −51.2 | −17.6 | ||
| TS13 | 23.9 | 24.2 | −46.0 | 37.9 | 0.04 | 23.9 | 24.8 | −46.1 | 38.6 | 0.08 b |
| 24 | −34.0 | −30.8 | −43.1 | −18.0 | −36.1 | −32.6 | −48.6 | −18.1 | ||
| TS14 | 26.5 | 27.2 | −49.5 | 41.9 | 0.03 | 26.9 | 27.9 | −50.6 | 43.0 | 0.08 b |
| 25 | −31.5 | −28.2 | −50.6 | −13.1 | −34.4 | −30.9 | −49.2 | −16.2 | ||
| C–C | C–N | C–C | C–N | |
|---|---|---|---|---|
| Gas Phase | Acetonitrile | |||
| TS1 | 2.245 | 2.333 | 2.230 | 2.357 |
| TS2 | 2.208 | 2.311 | 2.196 | 2.338 |
| TS3 | 2.250 | 2.236 | 2.251 | 2.247 |
| TS4 | 2.231 | 2.201 | 2.242 | 2.206 |
| TS5 | 2.246 | 2.220 | 2.253 | 2.229 |
| TS6 | 2.218 | 2.203 | 2.231 | 2.204 |
| TS7 | 2.255 | 2.227 | 2.251 | 2.214 |
| TS8 | 2.237 | 2.173 | 2.247 | 2.185 |
| TS9 | 2.230 | 2.239 | 2.244 | 2.226 |
| TS10 | 2.233 | 2.163 | 2.261 | 2.155 |
| TS11 | 2.296 | 2.080 | 2.337 | 2.087 |
| TS12 | 2.289 | 2.052 | 2.331 | 2.062 |
| TS13 | 2.291 | 2.088 | 2.338 | 2.084 |
| TS14 | 2.289 | 2.052 | 2.330 | 2.057 |
| V(N1) | V(N1,N2) | V′(N1,N2) | V(N2) | V(C3,N2) | V(C3) | V(C4,C5) | |
|---|---|---|---|---|---|---|---|
| TS1 | 3.64 | 1.57 | 1.58 | 1.74 | 2.04 | 0.87 | 3.32 |
| TS2 | 3.63 | 1.57 | 1.59 | 1.76 | 2.03 | 0.87 | 3.29 |
| TS3 | 3.56 | 1.64 | 1.48 | 1.83 | 2.04 | 0.81 | 3.26 |
| TS4 | 3.55 | 1.73 | 1.35 | 1.90 | 2.00 | 0.88 | 3.22 |
| TS5 | 3.56 | 1.64 | 1.47 | 1.84 | 2.04 | 0.80 | 3.26 |
| TS6 | 3.56 | 3.10 | 1.85 | 2.04 | 0.81 | 3.25 | |
| TS7 | 3.59 | 1.68 | 1.41 | 1.87 | 2.03 | 0.73 | 3.25 |
| TS8 | 3.53 | 1.64 | 1.46 | 1.88 | 1.99 | 0.88 | 3.24 |
| TS9 | 3.58 | 1.61 | 1.47 | 1.86 | 2.05 | 0.81 | 3.28 |
| TS10 | 3.53 | 1.69 | 1.40 | 1.88 | 2.04 | 0.78 | 3.23 |
| TS11 | 3.47 | 1.76 | 1.30 | 1.96 | 2.01 | 0.74 | 3.18 |
| TS12 | 3.48 | 3.01 | 2.01 | 2.00 | 0.78 | 3.16 | |
| TS13 | 3.47 | 1.65 | 1.42 | 1.94 | 2.01 | 0.71 | 3.18 |
| TS14 | 3.48 | 1.74 | 1.30 | 1.99 | 1.99 | 0.77 | 3.17 |
| CP1 (C3–C4) | CP2 (N1–C5) | |||
|---|---|---|---|---|
| ρ | ρ | |||
| TS1 | 0.056 | 0.049 | 0.043 | 0.079 |
| TS2 | 0.060 | 0.047 | 0.044 | 0.082 |
| TS3 | 0.056 | 0.048 | 0.052 | 0.085 |
| TS4 | 0.057 | 0.049 | 0.055 | 0.087 |
| TS5 | 0.057 | 0.047 | 0.053 | 0.086 |
| TS6 | 0.059 | 0.045 | 0.055 | 0.088 |
| TS7 | 0.054 | 0.049 | 0.053 | 0.085 |
| TS8 | 0.057 | 0.046 | 0.058 | 0.089 |
| TS9 | 0.058 | 0.046 | 0.052 | 0.084 |
| TS10 | 0.058 | 0.045 | 0.059 | 0.089 |
| TS11 | 0.052 | 0.048 | 0.070 | 0.087 |
| TS12 | 0.052 | 0.048 | 0.073 | 0.088 |
| TS13 | 0.052 | 0.048 | 0.069 | 0.087 |
| TS14 | 0.053 | 0.047 | 0.073 | 0.088 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Domingo, L.R.; Ríos-Gutiérrez, M.; Acharjee, N. Unveiling the Unexpected Reactivity of Electrophilic Diazoalkanes in [3+2] Cycloaddition Reactions within Molecular Electron Density Theory. Chemistry 2021, 3, 74-93. https://doi.org/10.3390/chemistry3010006
Domingo LR, Ríos-Gutiérrez M, Acharjee N. Unveiling the Unexpected Reactivity of Electrophilic Diazoalkanes in [3+2] Cycloaddition Reactions within Molecular Electron Density Theory. Chemistry. 2021; 3(1):74-93. https://doi.org/10.3390/chemistry3010006
Chicago/Turabian StyleDomingo, Luis R., Mar Ríos-Gutiérrez, and Nivedita Acharjee. 2021. "Unveiling the Unexpected Reactivity of Electrophilic Diazoalkanes in [3+2] Cycloaddition Reactions within Molecular Electron Density Theory" Chemistry 3, no. 1: 74-93. https://doi.org/10.3390/chemistry3010006
APA StyleDomingo, L. R., Ríos-Gutiérrez, M., & Acharjee, N. (2021). Unveiling the Unexpected Reactivity of Electrophilic Diazoalkanes in [3+2] Cycloaddition Reactions within Molecular Electron Density Theory. Chemistry, 3(1), 74-93. https://doi.org/10.3390/chemistry3010006