Electronically Excited States of Closed-Shell, Cyano-Functionalized Polycyclic Aromatic Hydrocarbon Anions


Abstract
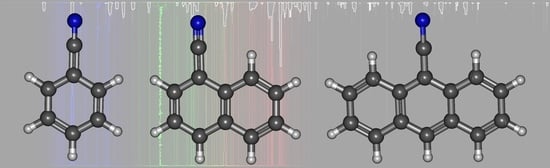
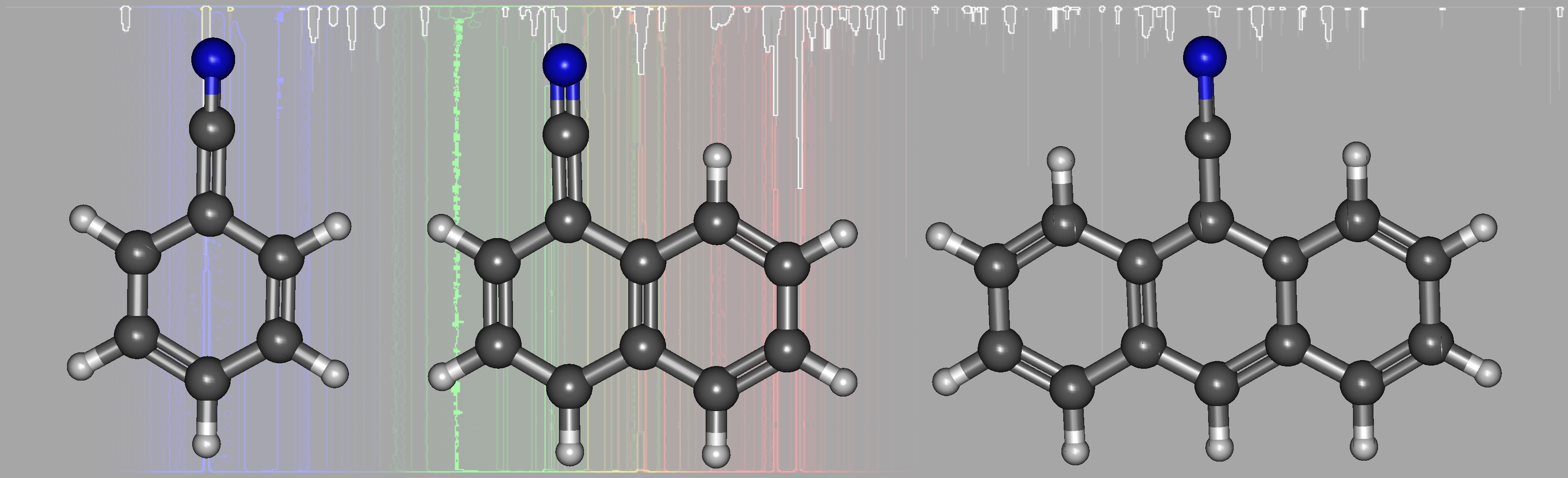
1. Introduction
2. Computational Details
3. Results
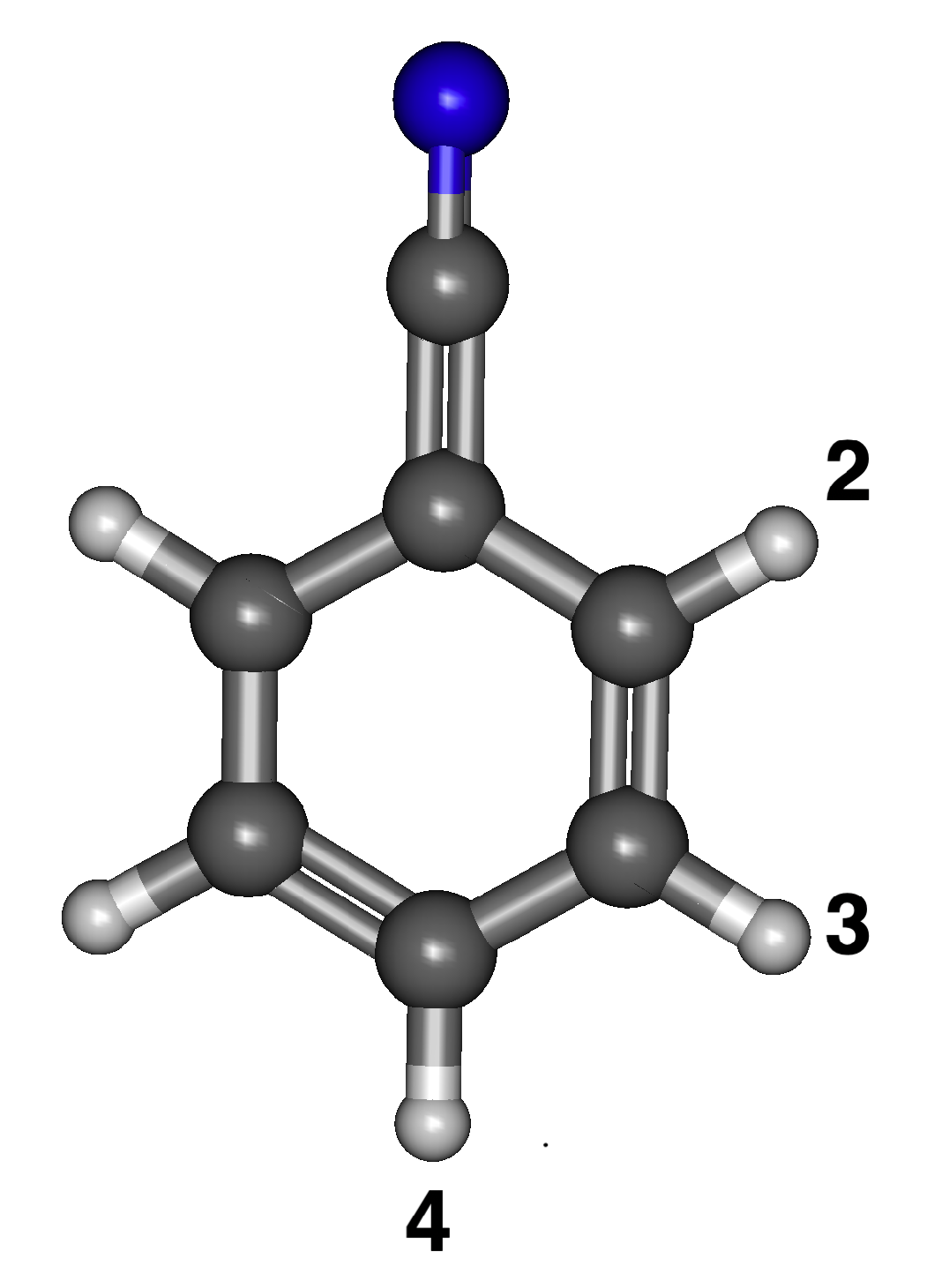
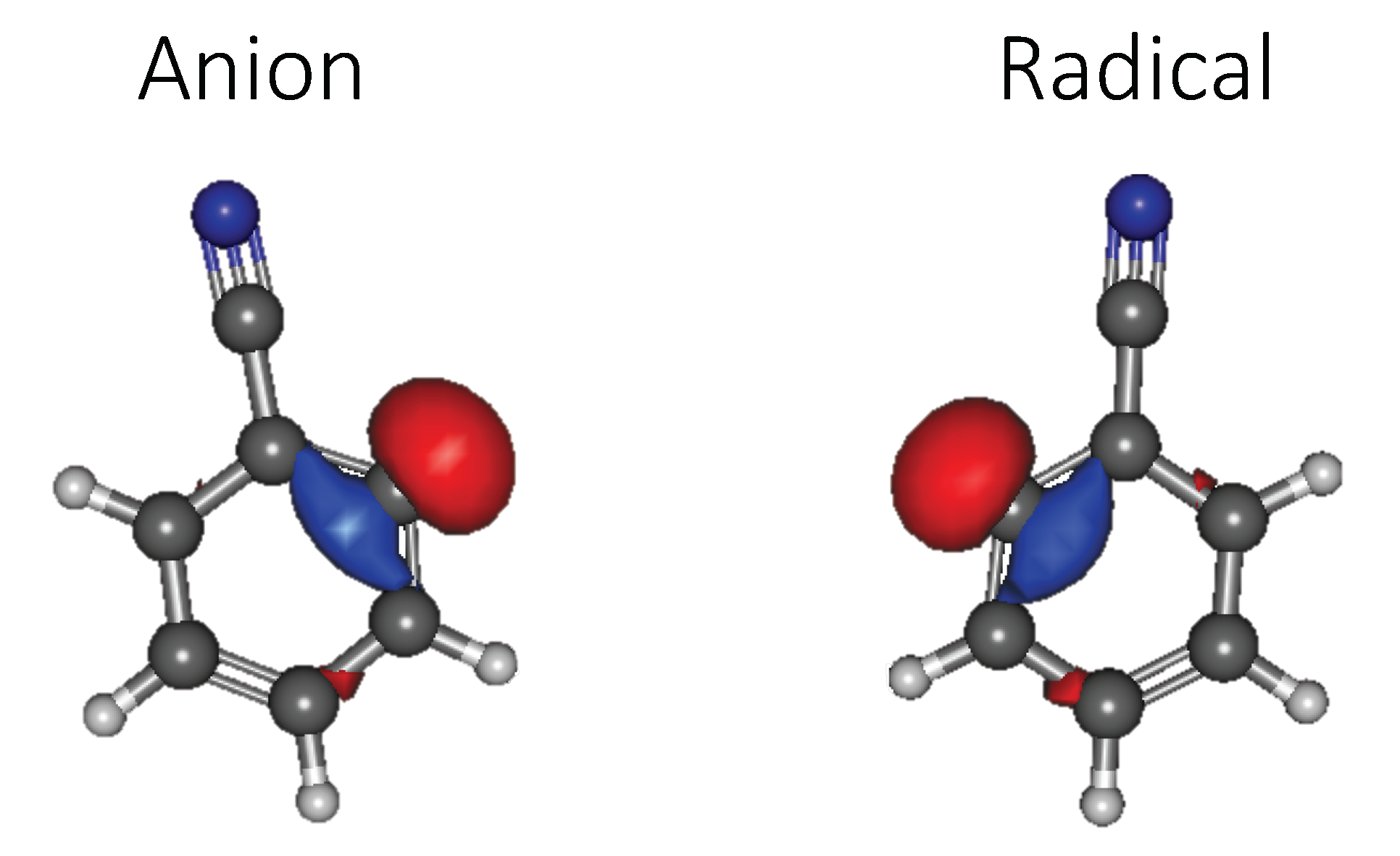
3.1. Benzenonitrile
3.1.1. Relative Energies and Dipole Moments
3.1.2. Vertical Excitation Energies
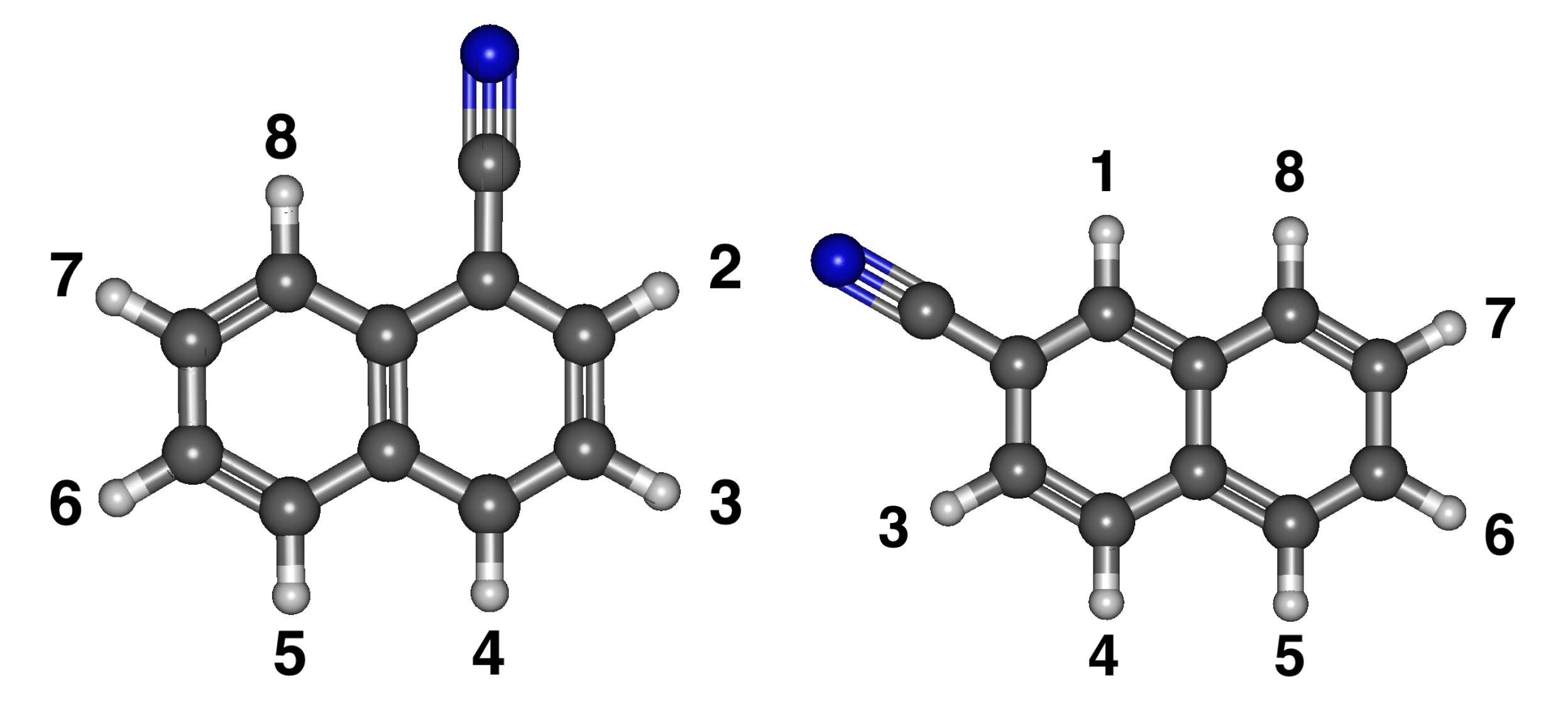
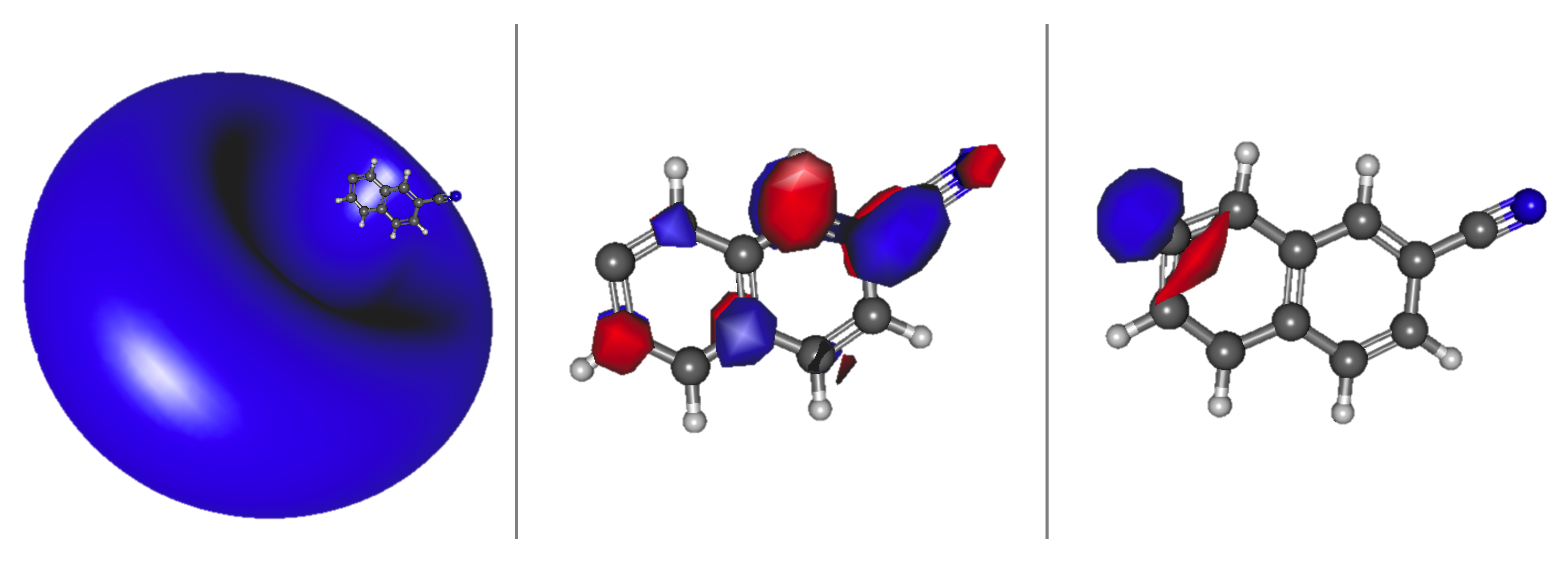
3.2. Cyanonaphthalene
Relative Energies and Dipole Moments
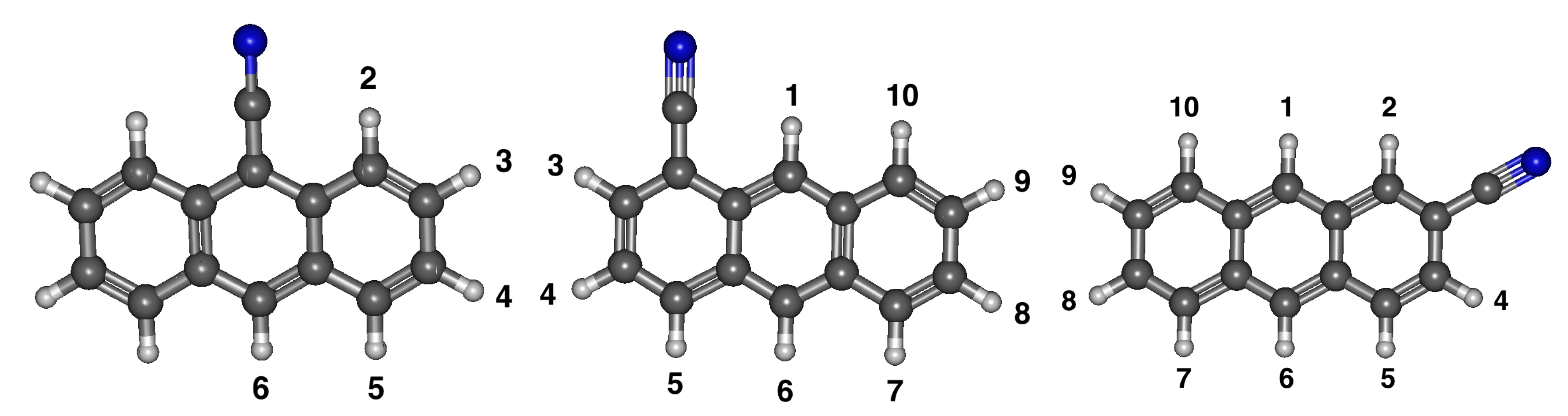
3.3. Cyanoanthracene
3.3.1. Relative Energies and Dipole Moments
3.3.2. Vertical Excitation Energies
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sidorkin, V.; Belogolova, E.F.; Doronina, E.P.; Liu, G.; Ciborowski, S.M.; Bowen, K.H. “Outlaw” Dipole Bound Anions of Intra-Molecular Complexes. J. Am. Chem. Soc. 2020, 142, 4. [Google Scholar] [CrossRef] [PubMed]
- Castellani, M.E.; Anstöter, C.S.; Verlet, J.R.R. On the Stability of a Dipole-Bound State in the Presence of a Molecule. PCCP Commun. 2019, 21, 242826. [Google Scholar] [CrossRef]
- Hendricks, J.; Lyapustina, S.; de Clercq, H.; Bowen, K. The Dipole Bound-to-Covalent Anion Transformation in Uracil. T J. Chem. Phys. 1998, 108, 8. [Google Scholar] [CrossRef]
- Skurski, P. An Excess Electron Bound to Urea. I. Canonical and Zwitterionic Tautomers. J. Chem. Phys. 2001, 115, 8373. [Google Scholar] [CrossRef][Green Version]
- Boschloo, G.; Hagfeldt, A. Characteristics of the Iodide/Triiodide Redox Mediator in Dye-Sensitized Solar Cells. Acc. Chem. Res. 2009, 42, 1819–1826. [Google Scholar] [CrossRef] [PubMed]
- Fermi, E.; Teller, E. The Capture of Negative Mesotrons in Matter. Phys. Rev. J. 1947, 72, 399–408. [Google Scholar] [CrossRef]
- Turner, J.E.; Fox, K. Minimum Dipole Moment Required to Bind an Electron to a Finite Dipole. Phys. Lett. 1966, 23, 547–549. [Google Scholar] [CrossRef]
- Mittleman, M.H.; Myerscough, V.P. Minimum Moment Required to Bind a Charged Particle to an Extended Dipole. Phys. Lett. 1966, 23, 545–546. [Google Scholar] [CrossRef]
- Gutsev, G.L.; Adamowicz, L. The Valence and Dipole-Bound States of the Cyanomethide Ion, CH2CN−. Chem. Phys. Lett. 1995, 246, 245–250. [Google Scholar] [CrossRef]
- Crawford, O.; Dalgarno, A. Bound States of an Electron in a Dipole Field. Chem. Phys. Lett. 1967, 1, 23. [Google Scholar] [CrossRef]
- Jordan, K.D. Theoretical Study of the Binding of an Electron to a Molecular Dipole:LiCl−. J. Chem. Phys. 1976, 64. [Google Scholar] [CrossRef]
- Turner, J. Minimum Dipole Moment Required to Bind an Electron-Molecular Theorist Rediscover Phenomenon Mentioned in Fermi-Teller Paper Twenty Years Earlier. Am. J. Phys. 1977, 45, 758–766. [Google Scholar] [CrossRef]
- Crawford, O.H. Electron Affinities of Polar Molecular. J. Chem. Phys. 1977, 66, 4968–4970. [Google Scholar] [CrossRef]
- Gutowski, M.; Skurski, P.; Boldrev, A.I.; Simons, J.; Jordan, K.D. Contribution of Electron Correlation to the Stability of Dipole-Bound Anionic States. Phys. Rev. A 1996, 54, 1906–1909. [Google Scholar] [CrossRef] [PubMed]
- Jordan, K.D.; Wang, F. Theory of Dipole Bound Anions. Annu. Rev. Phys. Chem. 2003, 54, 367–396. [Google Scholar] [CrossRef]
- Compton, R.; Carman, H.; Desfrancois, C.; Hendricks, J.; Lyapustina, S.A.; Bowen, K.H.; Abdoul-Carime, H.; Schermann, J.P. On the Binding of Electrons to Nitromethane: Dipole and Valence Bound Anions. J. Chem. Phys. 1996, 105, 3472–3478. [Google Scholar] [CrossRef]
- Fortenberry, R.C.; Crawford, T.D. Theoretical Prediction of New Dipole-Bound Singlet States for Anions of Interstellar Interest. J. Chem. Phys. 2011, 134, 154304. [Google Scholar] [CrossRef] [PubMed]
- Coulson, C.; Walmsley, M. The Minimum Dipole Moment Required to Bind an Electron to a Finite Electric Dipole. Proc. Phys. Soc. 1967, 91. [Google Scholar] [CrossRef]
- Lykke, K.R.; Neumark, D.M.; Andersen, T.; Trapa, V.J.; Lineberger, W.C. Autodetachment Spectroscopy and Dynamics of CH2CN− and CD2CN−. J. Chem. Phys. 1987, 87, 6842–6853. [Google Scholar] [CrossRef]
- Mullin, A.S.; Murray, K.K.; Schulz, C.P.; Lineberger, W.C. Autodetachment Dynamics of Acetaldhyde Enolate Anion, CH2CHO-. J. Phys. Chem. 1993, 97, 10281–10286. [Google Scholar] [CrossRef]
- Mullin, A.S.; Murray, K.K.; Schulz, C.; Szaflarski, D.M.; Lineberger, W. Autodetachment Spectroscopy of Vibrationally Excited Acetaldehyde Enolate Anion, CH2CHO-. Chem. Phys. 1992, 166, 207–213. [Google Scholar] [CrossRef]
- Fortenberry, R.C. Interstellar Anions: The Role of Quantum Chemistry. J. Phys. Chem. A 2015, 119, 9941–9953. [Google Scholar] [CrossRef] [PubMed]
- Sarre, P. The Diffuse Interstellar Bands: A Dipole-Bound State Hypothesis. Mon. Not. R. Astron. Soc. 2000, 313, L14–L16. [Google Scholar] [CrossRef][Green Version]
- Cordiner, M.A.; Sarre, P.J. The CH2CN− Molecule: Carrier of the λ8037 Diffuse Interstellar Band. Astron. Astrophys. 2007, 472, 537–545. [Google Scholar] [CrossRef][Green Version]
- Foing, B.; Ehrenfreund, P. Detection of Two Interstellar Absorption Bands Coincident with Spectral Features of C. Nature 1994, 396, 296–298. [Google Scholar] [CrossRef]
- Maier, J.P. Interstellar Detection of C. Nature 1994, 370, 423–424. [Google Scholar] [CrossRef]
- Campbell, E.; Holz, M.; Gerlich, D.; Maier, J. Laboratory Confirmation of C as the Carrier of Two Diffuse Interstellar Bands. Nature 2015, 523, 322–323. [Google Scholar] [CrossRef]
- Cordiner, M.A.; Linnartz, H.; Cox, N.L.J.; Cami, J.; Najarro, F.; Proffitt, C.R.; Lallement, R.; Ehrenfreund, P.; Foing, B.H.; Gull, T.R.; et al. Confirming Interstellar C Using the Hubble Space Telescope. Astrophys. J. Lett. 2019, 875, L28. [Google Scholar] [CrossRef]
- Czekner, J.; Cheung, L.F.; Johnson, E.L.; Fortenberry, R. A High Resolution Photoelectron Imaging and Theoretical Study of CP- and C2P-. J. Phys. Chem. 2018, 148, 044301. [Google Scholar] [CrossRef]
- Simons, J. Molecular Anions. J. Phys. Chem. 2008, 112, 6401–6511. [Google Scholar] [CrossRef]
- Terzieva, R.; Herbst, E. Radiative Electron Attachment to Small Linear Carbon Cluster and its Significance for the Chemistry of Diffuse Interstellar Clouds. Int. J. Mass Spectrom. 2000, 201, 135–142. [Google Scholar] [CrossRef]
- Millar, T.J.; Herbst, E.; Bettens, R.P.A. Large Molecules in the Envelope Surroundings IRC+10216. Mon. Not. R. Astron. Soc. 2000, 316, 195–200. [Google Scholar] [CrossRef]
- McCarthy, M.C.; Gottlieb, C.A.; Gupta, H.; Thaddeus, P. Laboratory and Astronomical Identification of the Negative Molecular Ion C6H−. Astrophys. J. 2006, 652, L141–L144. [Google Scholar] [CrossRef]
- Lepp, S.; Dalgarno, A. Polycyclic Aromatic Hydrocarbons in Interstellar Chemistry. Astrophys. J. 1988, 324, 533. [Google Scholar] [CrossRef]
- Schild, R.; Chaffee, F.; Frogel, J.; Persson, S. The Nature of Infrared Excesses in Extreme Be Stars. Astrophys. J. 1974, 190, 73–83. [Google Scholar] [CrossRef]
- Snow, T. A Search for H− in the Shell Surrounding Chi Ophiuchi. Astrophys. J. 1975, 198, 361–367. [Google Scholar] [CrossRef]
- Ross, T.; Baker, E.; Snow, T.; Destree, J.; Rachford, B. The Search for H− in Astrophysical Environments. Astrophys. J. 2008, 684, 358–363. [Google Scholar] [CrossRef]
- Tulej, M.; Kirkwoods, D.; Pachkov, M.; Maier, J. Gas-Phase Electronic Transitions of Carbon Chain Anions Coinciding with Diffuse Interstellar Bands. Astrophys. J. Lett. 1998, 506, L69–L73. [Google Scholar] [CrossRef]
- Kawaguchi, K.; Kasai, Y.; Ichi Ishikawa, S.; Kaifu, N. A Spectral-Line Survey Observation of IRC+10216 between 28 and 50 GHz. Publ. Astron. Soc. Jpn. 1995, 47, 853–876. [Google Scholar]
- Aoki, K. Candidates for U-Lines at 1377 and 1394 MHz in IRC+10216: Ab Intio Molecular Orbital Study. Chem. Phys. Lett. 2000, 323, 55–58. [Google Scholar] [CrossRef]
- Brünken, S.; Gupta, H.; Gottlieb, C.A.; McCarthy, M.C.; Thaddeus, P. Detection of the Carbon Chain Negative Ion C8H-in TMC-1. Astrophys. J. 2007, 664, L43–L46. [Google Scholar] [CrossRef]
- Cernicharo, J.; Guélin, M.; Agúndez, M.; Kawaguchi, K.; McCarthy, M.; Thaddeus, P. Astronomical Detection of C4H−, the Second Interstellar Anion. Astron. Astrophys. 2007, 467, L37–L40. [Google Scholar] [CrossRef]
- Thaddeus, P.; Gottlieb, C.A.; Gupta, H.; Brünken, S.; McCarthy, M.C.; Agúndez, M.; Guélin, M.; Cernicharo, J. Laboratory and Astronomical Detection of the Negative Molecular Ion C3N−. Astrophys. J. 2008, 677, 1132–1139. [Google Scholar] [CrossRef]
- Cernicharo, J.; Guélin, M.; Agúndez, M.; McCarthy, M.C.; Thaddeus, P. Detection of C5N− and Vibrationally Excited C6H in IRC +10216. Astrophys. J. 2008, 688, L83–L86. [Google Scholar] [CrossRef]
- Agúndez, M.; Cernicharo, J.; Guélin, M.; Kahane, C.; Roueff, E.; Klos, J.; Aoiz, F.J.; Lique, F.; Marcelino, N.; Goicoechea, J.R.; et al. Astronomical Identification of CN−, the Smallest Observed Molecular Anion. Astron. Astrophys. 2010, 517, L2. [Google Scholar] [CrossRef]
- Cami, J.; Bernard-Salas, J.; Peeters, E.; Malek, S.E. Detection of C60 and C70 in Young Planetary Nebulea. Science 2010, 329, 1180–1192. [Google Scholar] [CrossRef]
- McGuire, B.A.; Burkhardt, A.M.; Kalenskii, S.; Shingledecker, C.; Remijan, A.J.; Herbst, E.; McCarthy, M.C. Detection of the Aromatic Molecule Benzonitrile (c-C6H5CN) in the Interstellar Medium. Science 2018, 359, 202–205. [Google Scholar] [CrossRef] [PubMed]
- Hanel, R.; Conrath, B.; Flasar, F.M.; Kunde, V.; Maguire, W.; Pearl, J.; Pirraglia, J.; Samuelson, R.; Herath, L.; Allison, M.; et al. Infrared Observations of the Saturnian System from Voyager 1. Science 1981, 212, 192–200. [Google Scholar] [CrossRef]
- López-Puertas, M.; Dinelli, B.M.; Adriani, A.; Funke, B.; García-Comas, M.; Moriconi, M.L.; D’Aversa, E.; Boersma, C.; Allamandola, L.J. Large Abundances of Polycyclic Aromatic Hydrocarbons in Titan’s Upper Atmosphere. Astrophys. J. 2013, 770, 132. [Google Scholar] [CrossRef]
- Molina-Cuberos, G.J.; Schwingenschuh, K.; López-Moreno, J.J.; Rodrigo, R.; Lara, L.M.; Anicich, V. Nitriles Produced by ion Chemistry in the Lower Ionosphere of Titan. J. Geophys. Res. Planets 2002, 107, 9-1–9-11. [Google Scholar] [CrossRef]
- Kunde, V.; Aikin, A.C.; Hanel, R.A.; Jennings, D.E.; Maguire, W.; Samuelson, R.E. C4H2,HC3N and C2N2 in Titan’s Atmosphere. Nature 1981, 292, 686–688. [Google Scholar] [CrossRef]
- Samuelson, R.; Hanel, R.; Kunde, V.; Maguire, W. Mean Molecular Weight and Hydrogen Abundance of Titan’s Atmosphere. Nature 1981, 292, 688–693. [Google Scholar] [CrossRef]
- Maguire, W.; Hanel, R.; Jennings, D.; Samuelson, V.K.R. C3H8 and C3H4 in Titan’s Atmosphere. Nature 1981, 292, 683–686. [Google Scholar] [CrossRef]
- Yung, Y.L.; Allen, M.; Pinto, J.P. Photochemistry of the Atmosphere of Titan-Comparison between Model and Observation. Astrophys. Suppl. Ser. 1984, 55, 465–506. [Google Scholar] [CrossRef]
- Coates, A.J.; Crary, F.J.; Lewis, G.R.; Young, D.T.; Waite, J.H., Jr.; Sittler, E.C., Jr. Discovery of Heavy Negative Ions in Titan’s Ionosphere. Geophys. Res. Lett. 2007, 34, L22103. [Google Scholar] [CrossRef]
- Yung, Y. An Update of Nitrile Photochemistry on Titan. Icarus 1987, 72, 468–472. [Google Scholar] [CrossRef]
- Bradforth, S.E.; Kim, E.H.; Arnold, D.W.; Neumark, D.M. Photoelectron Spectroscopy of CN−, NCO−, and NCS−. J. Chem. Phys. 1993, 98, 800–810. [Google Scholar] [CrossRef]
- Clifford, E.P.; Wenthold, P.G.; Lineberger, W.C.; Petersson, G.A.; Ellison, G.B. Photoelectron Spectroscopy of the NCN− and HNCN− Ions. J. Phys. Chem. A 1997, 101, 4338–4345. [Google Scholar] [CrossRef]
- Aoki, K.; Ikuta, S.; Murakami, A. Equilibrium Geometries and Stabilities of the C3H Radical: Ab Initio MO Study. J. Mol. Struct. 1996, 365, 103–110. [Google Scholar] [CrossRef]
- Moustefaoui, T.; Rebrion-Rowe, C.; Le Garrec, J.-L.; Rowe, C.-B.R.; Mitchell, J.B.A. Low Temperature Electron Attachment to Polycyclic Aromatic Hydrocarbons. Faraday Discuss. 1998, 109, 71–82. [Google Scholar] [CrossRef]
- Dryza, V.; Sanelli, J.; Robertson, E.; Bieske, E. Electronic Spectra of Gas-Phase Polycyclic Aromatic Nitrogen Heterocycle Cations: Isoquinoline+ and quinoline+. J. Phys. Chem. A 2012, 17, 4323–4329. [Google Scholar] [CrossRef]
- Fortenberry, R.C.; Crawford, T.D. Singlet Excited States of Silicon-Containing Anions Relevant to Interstellar Chemistry. J. Phys. Chem. A 2011, 28, 8119–8124. [Google Scholar] [CrossRef] [PubMed]
- Theis, M.L.; Candian, A.; Tielens, A.G.G.M.; Leec, T.J.; Fortenberry, R.C. Electronically Excited States of PANH anions. Phys. Chem. Chem. Phys. 2015, 17, 14761. [Google Scholar] [CrossRef] [PubMed]
- Bassett, M.K.; Fortenberry, R.C. Symmetry Breaking and Spectral Considerations of the Surprisingly Floppy c-C3H Radical and the Related Dipole-Bound Excited State of c-C3H−. J. Chem. Phys. 2017, 146, 224303. [Google Scholar] [CrossRef] [PubMed]
- Theis, M.L.; Candian, A.; Tielens, A.G.G.M.; Lee, T.J.; Fortenberry, R.C. Electronically Excited States of Anisotropically Extended Singly-Deprotonated PAH Anions. J. Phys. Chem. A 2015, 119, 13048–13054. [Google Scholar] [CrossRef] [PubMed]
- Fortenberry, R.C.; Moore, M.M.; Lee, T.J. Excited State Trends in Bidirectionally Expanded Closed-Shell PAH and PANH Anions. J. Phys. Chem. A 2016, 120, 7327–7334. [Google Scholar] [CrossRef] [PubMed]
- Santaloci, T.J.; Fortenberry, R.C. On the Possibilities of Electronically Excited States in Stable Amine Anions: Dicyanoamine, Cyanoethynylamine, and diethynylamine. Mol. Astrophys. 2020, 19, 100070. [Google Scholar] [CrossRef]
- Becke, A.D. Density-Functional Themochemistry. III. The Excact Exchange. J. Chem. Phys. 1993, 98, 5648. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H. Gaussian˜16 Revision C.01. 2016; Gaussian Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Møller, C.; Plesset, M.S. Note on an Approximation Treatment for Many-Electron Systems. Phys. Rev. J. 1934, 46, 618–622. [Google Scholar] [CrossRef]
- Chai, J.-D.; Head-Gordon, M. Long-Ranged Corrected Hybrid Density Functionals with Damped Atom-Atom Dispersion Corrections. Phys. Chem. Chem. Phys. 2008, 10, 6615–6620. [Google Scholar] [CrossRef]
- Quynh, N.; Peters, W.; Fortenberry, R. Highly excited State Properties of Cumulenone Chlorides in the Vacuum-Ultraviolet. Phys. Chem. Chem. Phys. 2020, 22, 11838–11849. [Google Scholar]
- Stanton, J.F.; Gauss, J.; Cheng, L.; Harding, M.E.; Matthews, D.A.; Szalay, P.G. CFOUR, Coupled-Cluster Techniques for Computational Chemistry, a Quantum-Chemical Program Package. Available online: http://www.cfour.de (accessed on 3 November 2019).
- Werner, H.-J.; Knowles, P.J.; Knizia, G.; Manby, F.R.; Schütz, M. MOLPRO, Version 2015.1, a Package of ab Initio Programs; TTI—Technologie-Transfer-Initiative GmbH an der Universität Stuttgart (TTI GmbH): Stuttgart, Germany, 2015; Available online: http://www.molpro.net (accessed on 22 February 2012).
- Krylov, A.I. Equation-of-Motion Coupled-Cluster Methods for Open-Shell and Electronically Excited Species: The Hitchhiker’s Guide to Fock Space. Annu. Rev. Phys. Chem. 2008, 59, 433–462. [Google Scholar] [CrossRef] [PubMed]
- Shavitt, I.; Bartlett, R. MBPT and Coupled-Cluster Theory; Many-Body Methods in Chemistry and Physics; Cambridge University Press: Cambridge, UK, 2009; Volume 59. [Google Scholar]
- Stanton, R.; Bartlett, J.F. The Equation of Motion Coupled-Cluster Method—A Systematic Biorthogonal Approach to Molecular Excitation Energies, Transition-Probabilities, and Excited-State Properties. J. Chem. Phys. 1993, 98, 7029–7039. [Google Scholar] [CrossRef]
- Kendall, R.A.; Dunning, T.H.; Harrison, R.J. Electron Affinities of the First-Row Atoms Revisited. Systematic Basis Sets and Wave Functions. J. Chem. Phys. 1992, 96, 6796–6806. [Google Scholar] [CrossRef]
- Morgan, W.J.; Fortenberry, R.C. Additional Diffuse Functions in Basis Sets for Dipole-Bound Excited States of Anions. Theor. Chem. Acc. 2015, 134. [Google Scholar] [CrossRef]
- Gulani, S.; Jahau, T.-C.; Sanov, A.; Krylov, A.I. The Quest to Uncover the Nature of Benzonitrile Anion. Phys. Chem. Chem. Phys. 2020, 22, 5002–5010. [Google Scholar] [CrossRef] [PubMed]
- Stanton, J.F.; Gauss, J. Analytic Energy Derivatives for Ionized States Described by the Equation-of-Motion Coupled Cluster Method. J. Phys. Chem. 1994, 101, 8938–8944. [Google Scholar] [CrossRef]
- Dunning, T.H. Gaussian Basis Sets for Use in Correlated Molecular Calculations. I. The Atoms Boron Through Neon and Hydrogen. J. Chem. Phys. 1989, 90, 1007–1023. [Google Scholar] [CrossRef]
- Jenniskens, P.F. Desert A Survey of Diffuse Interstellar Bands (3800–8680 Å). Astron. Astrophys. Suppl. Ser. 1994, 106, 39–78. [Google Scholar]
- Nichols, C.M.; Wang, Z.-C.; Yang, Z.; Lineberger, W.C.; Bierbaum, V.M. Experimental and Theoretical Studies of the Reactivity and Thermochemistry of Dicyanamide: N(CN)2−. J. Phys. Chem. A 2016, 120, 992–999. [Google Scholar] [CrossRef] [PubMed]
- Dubois, D.; Sciamma-O’Brien, E.; Fortenberry, R.C. The Fundamental Vibrational Frequencies and Spectroscopic Constants of the Dicyanoamine Anion, NCNCN- (C2N3−): Quantum Chemical Analysis for Astrophysical and Planetary Environments. J. Astrophys. 2019, 883, 109. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
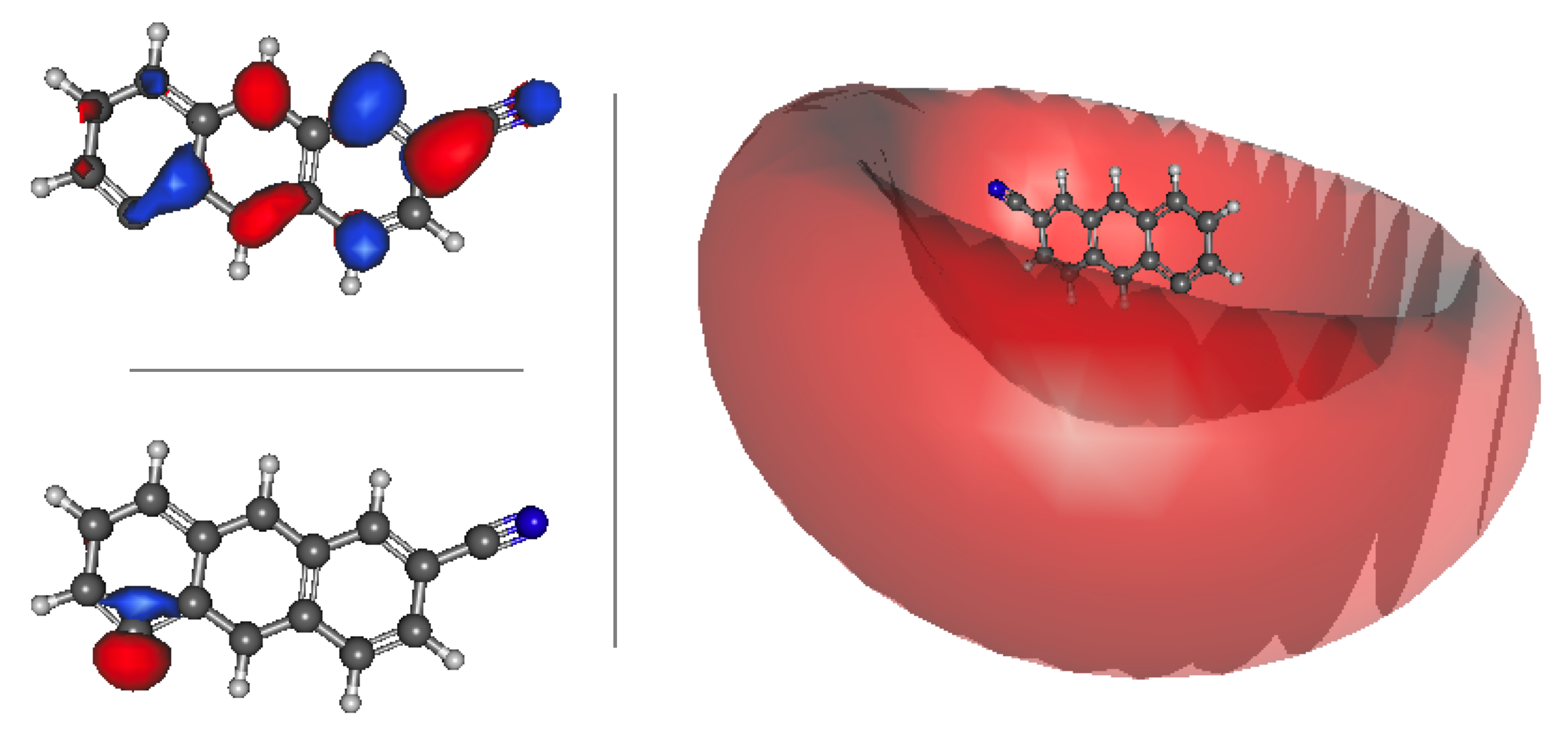{kind=link}
| B3LYP | Radical Rel.E (eV) | Anion Rel.E (eV) | Radical Dipole (D) |
|---|---|---|---|
| 2 | 0.0531 | 0.0000 | 5.18 |
| 3 | 0.0065 | 0.0771 | 4.28 |
| 4 | 0.0000 | 0.0576 | 3.82 |
| MP2 | Radical Rel.E (eV) | Anion Rel.E (eV) | |
| 2 | 1.1507 | 0.0000 | |
| 3 | 0.0000 | 0.0449 | |
| 4 | 0.0284 | 0.0863 | |
| B97XD | Radical Rel.E (eV) | Anion Rel.E (eV) | |
| 2 | 0.0634 | 0.0000 | |
| 3 | 0.0137 | 0.0986 | |
| 4 | 0.0000 | 0.0764 |
| Abs COC | 2 A/2A | 3 A/3A | 1 B/1A | 2 B/2A | 1 B/4A | 2 B/5A | 1 A/3A | 2 A/4A | eBE (eV) |
|---|---|---|---|---|---|---|---|---|---|
| 2 | 2.3003 | 2.3233 | 2.3302 | 2.3575 | 2.3219 | ||||
| 3 | 2.1393 | 2.1546 | 2.1616 | 2.1898 | 2.1519 | ||||
| 4 | 2.2196 | 2.2305 | 2.2374 | 2.2659 | 2.2371 | 2.2628 | 4.1895 | 3.1359 | 2.2230 |
| COM | |||||||||
| 2 | 1.4254 | 1.4478 | 1.4548 | 1.4821 | 1.4454 | ||||
| 3 | 1.3059 | 1.3222 | 1.3293 | 1.3584 | 1.3156 | ||||
| 4 | 1.3568 | 1.3637 | 1.3710 | 1.4040 | 1.3707 | 1.4017 | 4.2450 | 1.4017 | 1.3607 |
| Emission COC | 2 A/2 A | 3 A/3 A | 1 B/1 A | 2 B/2 A | 1 B | 2 B | 1 A | 2 A | eBE (eV) |
| 2 | 1.4254 | 1.4478 | 1.4548 | 1.4821 | 1.4454 | ||||
| 3 | 1.3059 | 1.3222 | 1.3293 | 1.3584 | 1.3156 | ||||
| 4 | 1.3568 | 1.3637 | 1.3710 | 1.4040 | 1.3707 | 1.4017 | 4.2450 | 2.1496 | 1.3607 |
| 1-Cyano | Radical Rel.E (eV) | Anion Rel.E (eV) | Radical Dipole (D) |
|---|---|---|---|
| 2 | 0.0500 | 0.0000 | 5.36 |
| 3 | 0.0176 | 0.0923 | 4.40 |
| 4 | 0.0027 | 0.0140 | 4.00 |
| 5 | 0.0072 | 0.1584 | 3.88 |
| 6 | 0.0003 | 0.2799 | 4.15 |
| 7 | 0.0000 | 0.3102 | 5.15 |
| 8 | 0.0306 | 0.2827 | 5.46 |
| 2-Cyano | Radical Rel.E (eV) | Anion Rel.E (eV) | Radical Dipole (D) |
| 1 | 0.0634 | 0.0000 | 5.84 |
| 3 | 0.0701 | 0.1391 | 5.82 |
| 4 | 0.0178 | 0.1400 | 5.14 |
| 5 | 0.0123 | 0.2201 | 4.75 |
| 6 | 0.0000 | 0.2742 | 4.30 |
| 7 | 0.0066 | 0.3002 | 4.76 |
| 8 | 0.0149 | 0.2024 | 5.48 |
| 1-Cyanonaph. | Excited States | eBE (eV) | |
|---|---|---|---|
| 2 A | 1 A | ||
| 2 | 2.5287 | 2.5544 | 2.5463 |
| 3 | 2.3528 | 2.3721 | 2.3635 |
| 4 | 2.4862 | 2.4797 | 2.4944 |
| 5 | 2.2953 | 2.3131 | 2.3017 |
| 6 | 2.1671 | 2.1880 | 2.1789 |
| 7 | 2.1402 | 2.1677 | 2.1595 |
| 8 | 2.1653 | 2.1897 | 2.1815 |
| 2-Cyanonaph. | |||
| 1 | 2.6229 | 2.6411 | 2.6332 |
| 3 | 2.4745 | 2.3465 | 2.4848 |
| 4 | 2.3751 | 2.3906 | 2.3822 |
| 5 | 2.2890 | 2.3238 | 2.3142 |
| 6 | 2.2791 | 2.2851 | 2.2931 |
| 7 | 2.2174 | 2.1070 | 2.2438 |
| 8 | 2.3301 | 2.3708 | 2.3627 |
| 1-Cyano | Radical Rel.E (eV) | Anion Rel.E (eV) | Radical Dipole (D) |
|---|---|---|---|
| 2 | 0.0323 | 0.4171 | 5.48 |
| 3 | 0.0000 | 0.4273 | 5.21 |
| 4 | 0.0063 | 0.4072 | 4.43 |
| 5 | 0.0096 | 0.2672 | 3.95 |
| 6 | 0.0095 | 0.0000 | 3.97 |
| 2-Cyano | Radical Rel.E (eV) | Anion Rel.E (eV) | Radical Dipole (D) |
| 1 | 0.0436 | 0.1499 | 5.58 |
| 3 | 0.0556 | 0.0000 | 5.45 |
| 4 | 0.0277 | 0.1064 | 4.78 |
| 5 | 0.0090 | 0.0156 | 4.06 |
| 6 | 0.0197 | 0.0145 | 4.14 |
| 7 | 0.0063 | 0.2698 | 4.01 |
| 8 | 0.0000 | 0.3651 | 4.24 |
| 9 | 0.0020 | 0.3781 | 5.10 |
| 10 | 0.0198 | 0.3545 | 5.64 |
| 3-Cyano | Radical Rel.E (eV) | Anion Rel.E (eV) | Radical Dipole (D) |
| 1 | 0.0290 | 0.0431 | 6.15 |
| 2 | 0.0621 | 0.0000 | 6.32 |
| 4 | 0.0777 | 0.1766 | 6.61 |
| 5 | 0.0211 | 0.1662 | 5.65 |
| 6 | 0.0215 | 0.1059 | 5.50 |
| 7 | 0.0111 | 0.3038 | 5.31 |
| 8 | 0.0000 | 0.3653 | 4.59 |
| 9 | 0.0074 | 0.3840 | 5.00 |
| 10 | 0.0074 | 0.2930 | 6.01 |
| 1-Cyanoanthra. Abs. | Excited States | eBE (eV) | |
|---|---|---|---|
| 2 A/2A | 1 A/1B | ||
| 2 | 2.2284 | 1.2574 | 2.3085 |
| 3 | 2.2884 | 1.6124 | 2.3406 |
| 4 | 2.3079 | 1.5382 | 2.3295 |
| 5 | 2.4781 | 1.7714 | 2.4851 |
| 6 | 2.8127 | 2.1633 | 2.8250 |
| 2-Cyanoanthra. | |||
| 1 | 2.4945 | 2.0192 | 2.5035 |
| 3 | 2.6852 | 2.0828 | 2.7056 |
| 4 | 2.4985 | 1.7281 | 2.5103 |
| 5 | 2.6489 | 2.0650 | 2.6601 |
| 6 | 2.6355 | 2.0953 | 2.6453 |
| 7 | 2.3211 | 1.8081 | 2.3344 |
| 8 | 2.2214 | 1.6596 | 2.2367 |
| 9 | 2.2096 | 1.6689 | 2.2329 |
| 10 | 2.2218 | 1.7390 | 2.2422 |
| 3-Cyanoanthra. | |||
| 1 | 2.7098 | 2.1810 | 2.7281 |
| 2 | 2.7996 | 2.2930 | 2.8030 |
| 4 | 2.6041 | 1.9315 | 2.6071 |
| 5 | 2.5120 | 1.9978 | 2.5202 |
| 6 | 2.6091 | 2.1119 | 2.6240 |
| 7 | 2.3689 | 1.8407 | 2.3862 |
| 8 | 2.3294 | 1.7633 | 2.3583 |
| 9 | 2.2789 | 1.6846 | 2.3202 |
| 10 | 2.3785 | 1.8582 | 2.4237 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Santaloci, T.J.; Fortenberry, R.C. Electronically Excited States of Closed-Shell, Cyano-Functionalized Polycyclic Aromatic Hydrocarbon Anions. Chemistry 2021, 3, 296-313. https://doi.org/10.3390/chemistry3010022
Santaloci TJ, Fortenberry RC. Electronically Excited States of Closed-Shell, Cyano-Functionalized Polycyclic Aromatic Hydrocarbon Anions. Chemistry. 2021; 3(1):296-313. https://doi.org/10.3390/chemistry3010022
Chicago/Turabian StyleSantaloci, Taylor J., and Ryan C. Fortenberry. 2021. "Electronically Excited States of Closed-Shell, Cyano-Functionalized Polycyclic Aromatic Hydrocarbon Anions" Chemistry 3, no. 1: 296-313. https://doi.org/10.3390/chemistry3010022
APA StyleSantaloci, T. J., & Fortenberry, R. C. (2021). Electronically Excited States of Closed-Shell, Cyano-Functionalized Polycyclic Aromatic Hydrocarbon Anions. Chemistry, 3(1), 296-313. https://doi.org/10.3390/chemistry3010022