Hydrocracking of Polyethylene to Jet Fuel Range Hydrocarbons over Bifunctional Catalysts Containing Pt- and Al-Modified MCM-48


Abstract
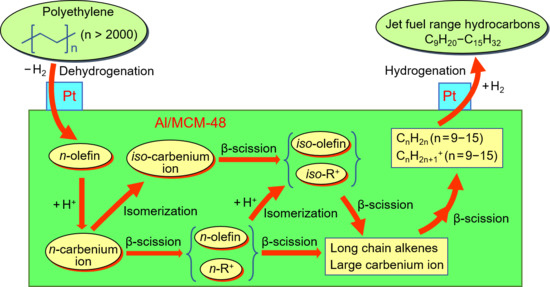
1. Introduction
2. Materials and Methods
2.1. Catalyst Synthesis
2.2. Catalyst Characterization
2.3. Catalytic Reaction
3. Results and Discussion
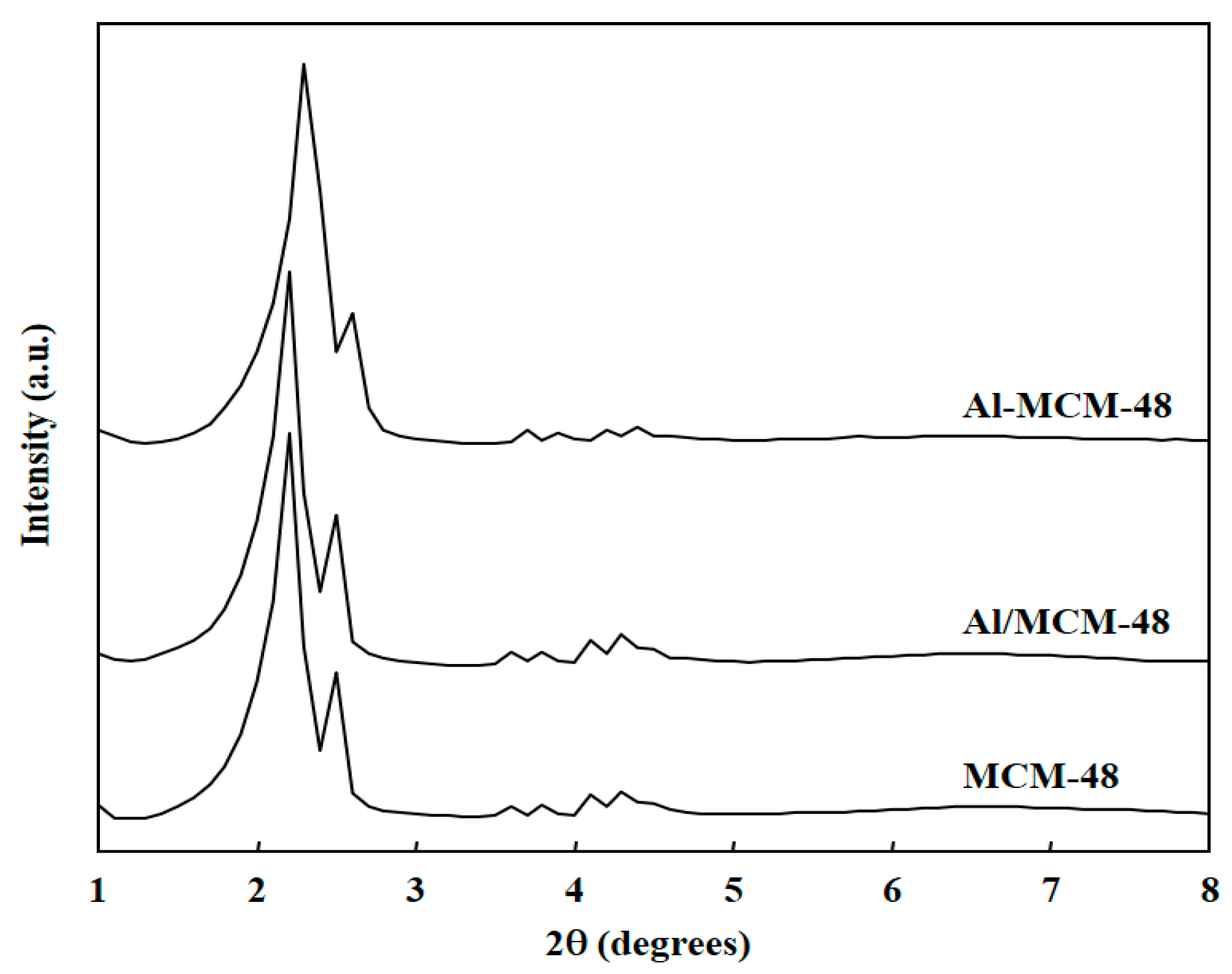
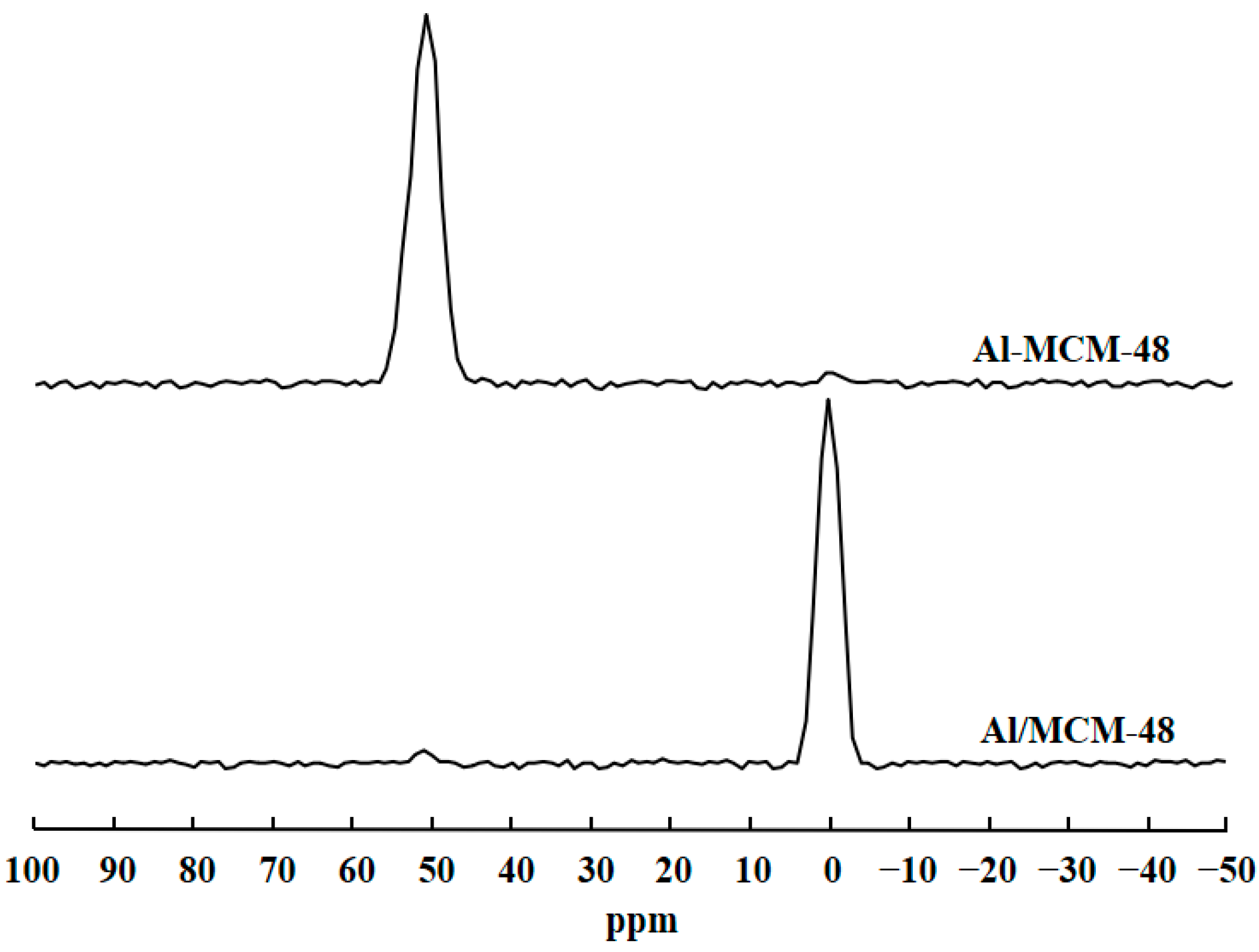
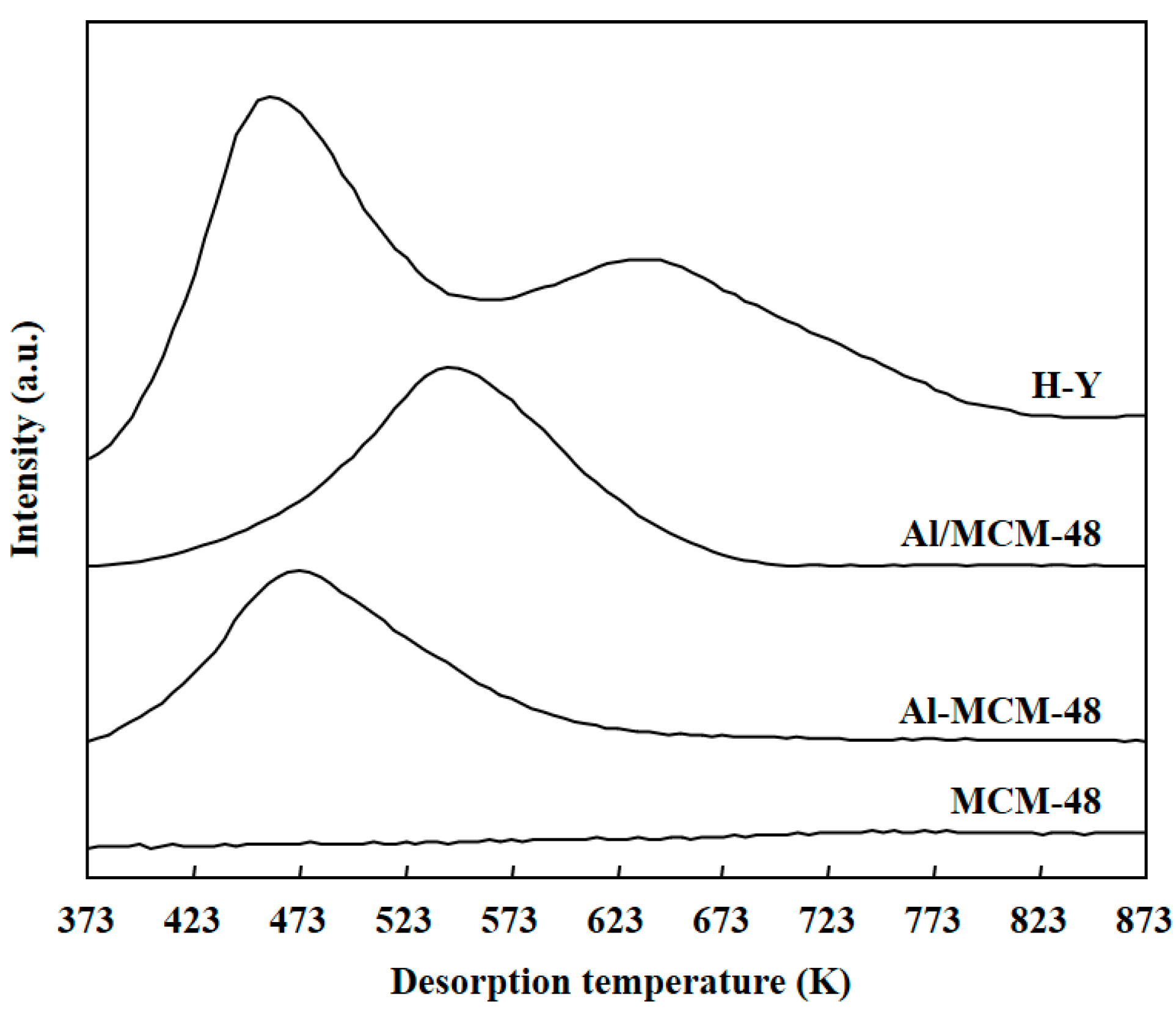
3.1. Characterization of Catalysts
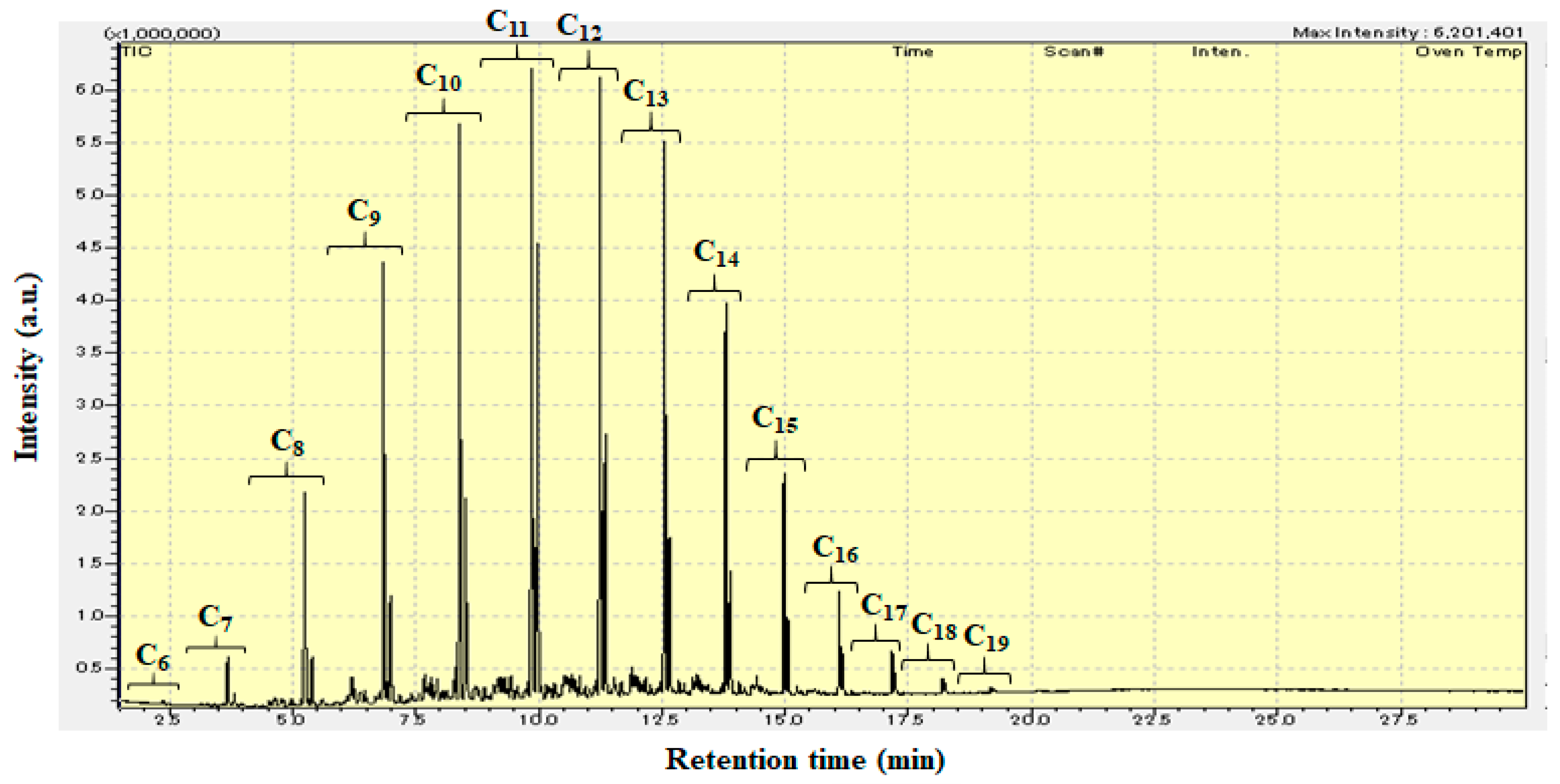
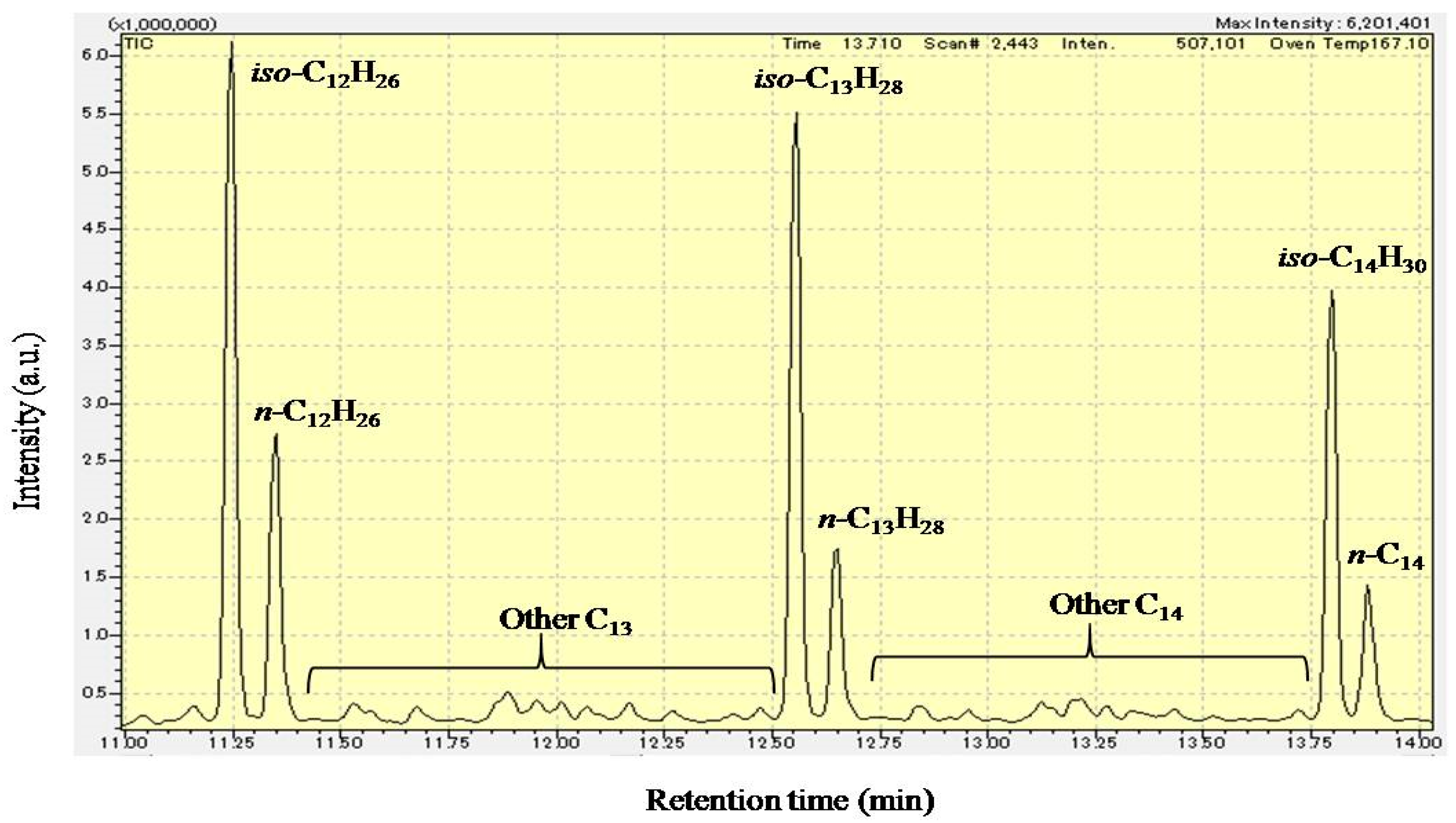
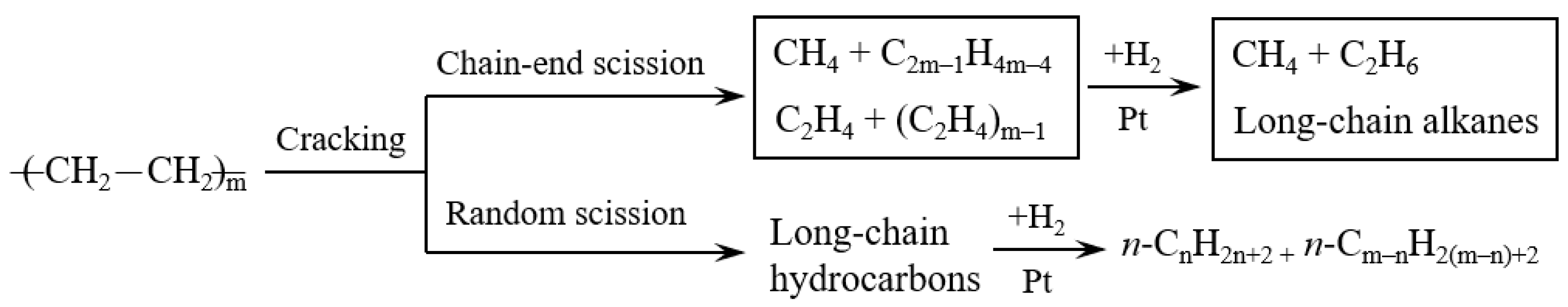
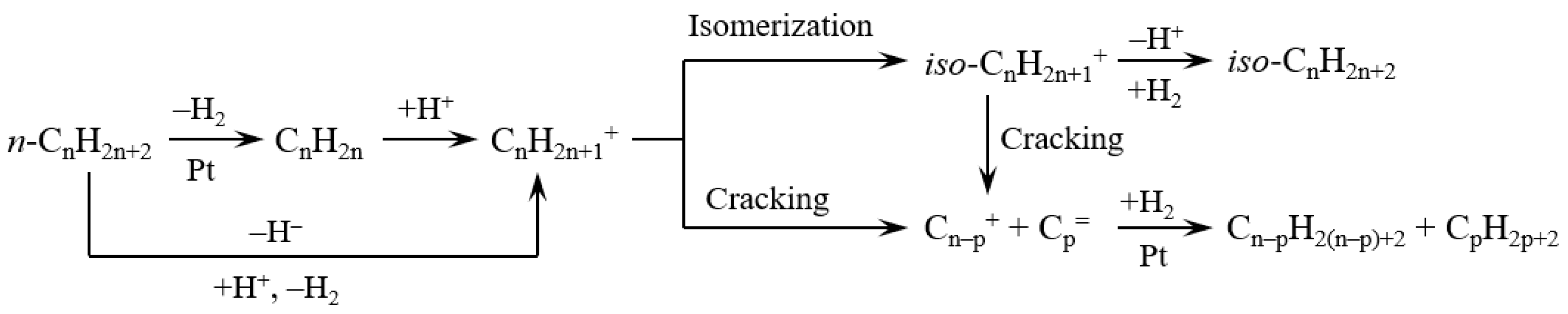
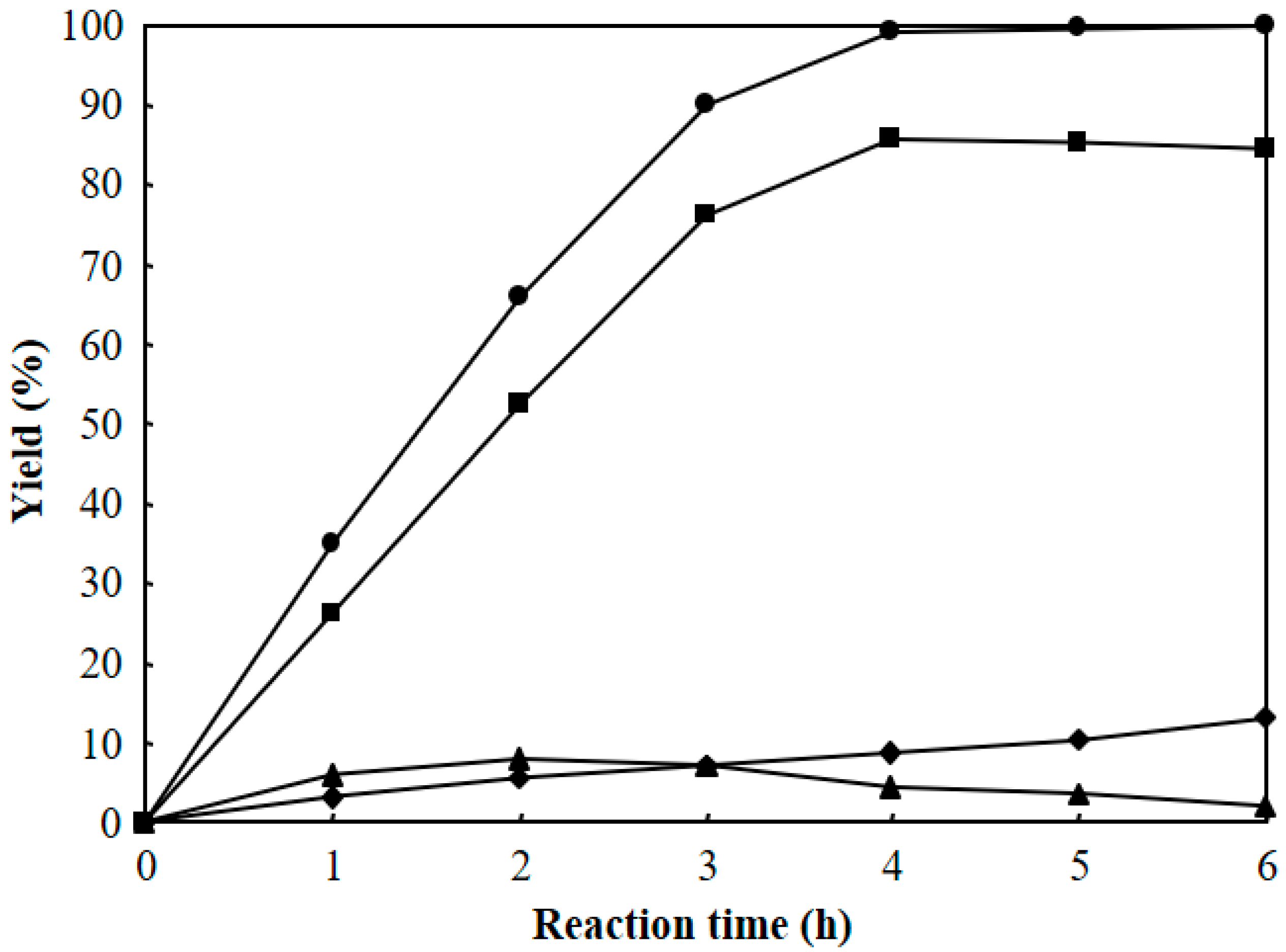
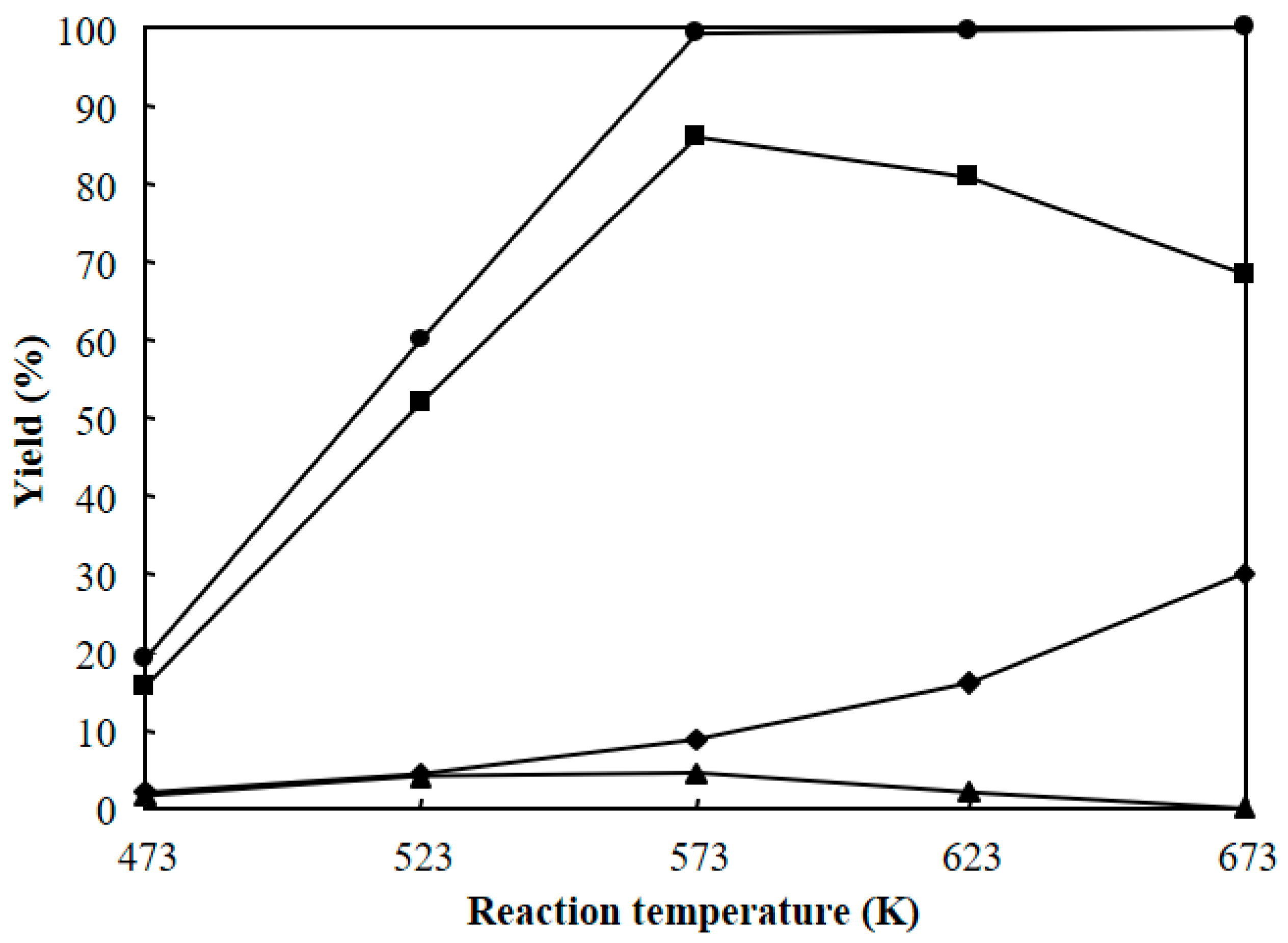
3.2. Hydrocracking of Polyethylene to Jet Fuel Range Hydrocarbons
4. Conclusions
Funding
Conflicts of Interest
References
- Huber, G.W.; Iborra, S.; Corma, A. Synthesis of transportation fuels from biomass: Chemistry, catalysts, and engineering. Chem. Rev. 2006, 106, 1044–4098. [Google Scholar] [CrossRef] [PubMed]
- Cesaro, A.; Belgiorno, V. Combined biogas and bioethanol production: Opportunities and challenge for industrial application. Energies 2015, 8, 8121–8144. [Google Scholar] [CrossRef]
- Thanh, L.T.; Okitsu, K.; Boi, L.V.; Maeda, Y. Catalytic technologies for biodiesel fuel production and utilization of glycerol: A review. Catalysts 2012, 2, 191–222. [Google Scholar] [CrossRef]
- Hanaoka, T.; Liu, Y.; Matsunaga, K.; Miyazawa, T.; Hirata, S.; Sakanishi, K. Bench-scale production of liquid fuel from woody biomass via gasification. Fuel Process. Technol. 2010, 91, 859–865. [Google Scholar] [CrossRef]
- Liu, Y.; Hanaoka, T.; Miyazawa, T.; Murata, K.; Okabe, K.; Sakanishi, K. Fischer-Tropsch synthesis in slurry-phase reactors over Mn- and Zr-modified Co/SiO2 catalysts. Fuel Process. Technol. 2009, 90, 901–908. [Google Scholar] [CrossRef]
- Liu, Y.; Sotelo-Boyas, R.; Murata, K.; Minowa, T.; Sakanishi, K. Production of bio-hydrogenated diesel by hydrotreatment of high-acid-value waste cooking oil over ruthenium catalyst supported on Al-polyoxocation-pillared pontmorillonite. Catalysts 2012, 2, 171–190. [Google Scholar] [CrossRef]
- Liu, Y.; Sotelo-Boyas, R.; Murata, K.; Minowa, T.; Sakanishi, K. Hydrotreatment of vegetable oils to produce bio-hydrogenated diesel and liquefied petroleum gas fuel over catalysts containing sulfided Ni-Mo and solid Acids. Energy Fuels 2011, 25, 4675–4685. [Google Scholar] [CrossRef]
- Liu, Y.; Murata, K.; Inaba, M. Hydrocracking of algae oil to aviation fuel-ranged hydrocarbons over NiMo-supported catalysts. Catal. Today 2019, 332, 115–121. [Google Scholar] [CrossRef]
- Liu, Y. Catalytic Deoxygenation of hexadecyl palmitate as a model compound of Euglena oil in H2 and N2 atmospheres. Catalysis 2017, 7, 333. [Google Scholar] [CrossRef]
- Yao, G.; Staples, M.D.; Malina, R.; Tyner, W.E. Stochastic teno-economic analysis of alcohol-to-jet fuel production. Biotechnol. Biofuels 2017, 10, 18. [Google Scholar] [CrossRef]
- Liu, Y. Catalytic ethylene oligomerization over Ni/Al-HMS: A key step in conversion of bio-ethanol to higher olefins. Catalysis 2018, 8, 537. [Google Scholar] [CrossRef]
- Andrei, R.D.; Popa, M.I.; Fajula, F.; Hulea, V. Hetergeneous oligomerization of ethylene over highly active and stable Ni-AlSBA-15 mesoporous catalysts. J. Catal. 2015, 323, 76–84. [Google Scholar] [CrossRef]
- Pan, Z.; Xue, X.; Zhang, C.; Wang, D.; Xie, Y.; Zhang, R. Evaluation of process parameters on high-density polyethylene hydro-liquefaction products. J. Anal. Appl. Pyrolysis 2018, 136, 146–152. [Google Scholar] [CrossRef]
- Zhang, X.; Lei, H.; Zhu, L.; Zhu, X.; Qian, M.; Yadavalli, G.; Yan, D.; Wu, J.; Chen, S. Optimizing carbon efficiency of jet fuel range alkanes from cellulose co-fed with polyethylene via catalytically combined processes. Bioresour. Technol. 2016, 214, 45–54. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Duan, D.; Lei, H.; Villota, E.; Ruan, R. Jet fuel production from waste plastics via catalytic pyrolysis with activated carbons. Appl. Energy 2019, 251, 113337. [Google Scholar] [CrossRef]
- Zhang, X.; Lei, H. Synthesis of high-density jet fuel from plastics via catalytically integral processes. RCS Adv. 2016, 6, 6154–6163. [Google Scholar] [CrossRef]
- Tao, L.; Markham, J.N.; Haq, Z.; Biddy, M.J. Techno-economic analysis for upgrading the bio-mass-derived ethanol-to-jet blendstocks. Green Chem. 2017, 19, 1082–1101. [Google Scholar] [CrossRef]
- Han, J.; Tao, L.; Wang, M. Well-to-wake analysis of ethanol-to-jet and sugar-to jet pathways. Biotechnol. Biofuels 2017, 10, 21. [Google Scholar] [CrossRef]
- Liu, Y.; Murata, K.; Inaba, M.; Mimura, N. Syntheses of Ti- and Al-containing hexagonal mesoporous silicas for gas-phase epoxidation of propylene by molecular oxygen. Appl. Catal. A Gen. 2006, 309, 91–105. [Google Scholar] [CrossRef]
- Kresge, C.T.; Leonowicz, M.E.; Roth, W.J.; Vartuli, J.C.; Beck, J.C. Ordered mesoporous molecular sieves synthesized by a liquid-crystal template mechanism. Nature 1992, 359, 710–712. [Google Scholar] [CrossRef]
- Romero, A.A.R.; Alba, M.D.; Zhou, W.; Klinoeski, J. Synthesis and characterization of the mesoporous silicate molecular sieve MCM-48. J. Phys. Chem. B 1997, 101, 5294–5300. [Google Scholar] [CrossRef]
- Kim, H.; Choi, S.J.; Kim, J.M.; Jeon, J.K.; Park, S.H.; Jung, S.C.; Kim, S.C.; Park, Y.K. Catalytic copyrolysis of particle board and polypropylene over Al-MCM-48. Mater. Res. Bull. 2016, 82, 61–66. [Google Scholar] [CrossRef]
- Park, Y.K.; Lee, H.W.; Lee, J.Y.; Kim, Y.M. The use of high density polyethylene (HDPE) as a co-feeding feedstock on the catalytic pyrolysis of yellow poplar over Al-MCM-48 and Al-MSU-F. J. Anal. Appl. Pyrolysis 2018, 135, 390–396. [Google Scholar] [CrossRef]
- Romero, A.; Nieto-Marquez, A.; Essayem, N.; Alonso, E.; Pinel, C. Improving conversion of D-glucose into short-chain alkanes over Ru/MCM-48 based catalysts. Microporous Mesoporous Mater. 2019, 286, 25–35. [Google Scholar] [CrossRef]
- Zhang, W.; Pinnavaia, T.J. Transition metal substituted derivatives of cubic MCM-48 mesoporous molecular sieves. Catal. Lett. 1996, 38, 261–265. [Google Scholar] [CrossRef]
- Chen, F.; Huang, L.; Yang, X.; Wang, Z. Synthesis of Al-substitute MCM-41 and MCM-48 solid acids with mixed cationic-anionic surfactants as templates. Mater. Lett. 2013, 109, 299–301. [Google Scholar] [CrossRef]
- Morey, M.S.; Stucky, G.D.; Schwarz, S.; Froba, M. Isomorphic substitution and postsynthesis incorporation of zirconium into MCM-48 mesoporous silica. J. Phys. Chem. B 1999, 103, 2037–2041. [Google Scholar] [CrossRef]
- Liu, Y.; Murata, K.; Okabe, K.; Hanaoka, T.; Sakanishi, K. Synthesis of Zr-grafted SBA-15 as an Effective Support for Cobalt Catalyst in Fischer-Tropsch Synthesis. Chem. Lett. 2008, 37, 984–985. [Google Scholar] [CrossRef]
- Komai, S.; Yazawa, Y.; Satsuma, A.; Hattori, T. Determination of metal dispersion of Pt/CeO2 catalyst by CO-pulse method. J. Jpn. Pet. Inst. 2005, 48, 173–177. [Google Scholar] [CrossRef][Green Version]
- Park, K.C.; Yim, D.J.; Ihm, S.K. Characteristics of Al-MCM-41 supported Pt catalysts:effect of Al distribution in Al-MCM-41 on its catalyticactivity in naphthalene hydrogenation. Catal. Today 2002, 74, 281–290. [Google Scholar] [CrossRef]
- Liu, Y.; Misono, M. Hydroisomerization of n-Butane over Platinum-Promoted Cesium Hydrogen Salt of 12-Tungstophosphoric Acid. Materials 2009, 2, 2319–2336. [Google Scholar] [CrossRef]
- Liu, Y.; Murata, K.; Sakanishi, K. Hydroisomerization-cracking of gasoline distillate from Fischer–Tropsch synthesis over bifunctional catalysts containing Pt and heteropolyacids. Fuel 2011, 90, 3056–3065. [Google Scholar] [CrossRef]
- Liu, Y.; Koyano, G.; Misono, M. Hydroisomerization of n-hexane and n-heptane over platinum-promoted Cs2.5H0.5PW12O40 (Cs2.5) studied in comparison with several other solid acids. Top. Catal. 2000, 11, 239–246. [Google Scholar] [CrossRef]
- Liu, Y.; Na, K.; Misono, M. Skeletal isomerization of n-pentane over Pt-promoted cesium hydrogen salts of 12-tungstophosphoric acid. J. Mol. Catal. A Chem. 1999, 141, 145–153. [Google Scholar] [CrossRef]
- Fernandes, F.A.N.; Teles, U.M. Modeling and optimization of Fischer-Tropsch products hydrocracking. Fuel Process. Technol. 2007, 88, 207–214. [Google Scholar] [CrossRef]
- Bouchy, C.; Hastoy, G.; Guillon, E.; Martens, J.A. Fischer-Tropsch waxes upgrading via hydrocracking and selective hydroisomerization. Oil Gas Sci. Technol. 2009, 64, 91–112. [Google Scholar]
- Liu, Y.; Murata, K.; Okabe, K.; Inaba, M.; Takahara, I.; Hanaoka, T.; Sakanishi, K. Selective hydrocracking of Fischer-Tropsch waxes to high-quality diesel fuel over Pt-promoted polyoxocation-pillared montmorillonites. Top. Catal. 2009, 52, 597–608. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalyst 1 | BET Surface Area (m2 g−1) | Pore Size (Å) | Pt particle Size (nm) |
|---|---|---|---|
| Pt/MCM-48 | 1040 | 39.2 | 2.2 |
| Pt/Al-MCM-48 | 1062 | 37.9 | 2.3 |
| Pt/Al/MCM-48 | 1023 | 38.0 | 2.1 |
| Pt/H-Y | 246 | 7.5 | 2.6 |
| Catalyst | Total Yield of C1–C21 (%) | C1–C8 Yield (%) | C9–C15 Yield (%) | C16–C21 Yield (%) |
|---|---|---|---|---|
| Blank | 1.2 | 1.2 | 0 | 0 |
| Pt/MCM-48 | 7.1 | 6.2 | 0.9 | 0 |
| Pt/Al-MCM-48 | 97.8 | 7.3 | 77.1 | 13.4 |
| Pt/Al/MCM-48 | 99.3 | 8.9 | 85.9 | 4.5 |
| Pt/H-Y | 99.6 | 80.7 | 18.9 | 0 |
| Reaction Cycle | Total Yield of C1–C21 (%) | C1–C8 Yield (%) | C9–C15 Yield (%) | C16–C21 Yield (%) |
|---|---|---|---|---|
| 1 | 99.3 | 8.9 | 85.9 | 4.5 |
| 2 | 99.2 | 8.8 | 85.8 | 4.6 |
| 3 | 99.1 | 8.8 | 85.8 | 4.5 |
| 4 | 99.2 | 8.9 | 85.9 | 4.4 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, Y. Hydrocracking of Polyethylene to Jet Fuel Range Hydrocarbons over Bifunctional Catalysts Containing Pt- and Al-Modified MCM-48. Reactions 2020, 1, 195-209. https://doi.org/10.3390/reactions1020014
Liu Y. Hydrocracking of Polyethylene to Jet Fuel Range Hydrocarbons over Bifunctional Catalysts Containing Pt- and Al-Modified MCM-48. Reactions. 2020; 1(2):195-209. https://doi.org/10.3390/reactions1020014
Chicago/Turabian StyleLiu, Yanyong. 2020. "Hydrocracking of Polyethylene to Jet Fuel Range Hydrocarbons over Bifunctional Catalysts Containing Pt- and Al-Modified MCM-48" Reactions 1, no. 2: 195-209. https://doi.org/10.3390/reactions1020014
APA StyleLiu, Y. (2020). Hydrocracking of Polyethylene to Jet Fuel Range Hydrocarbons over Bifunctional Catalysts Containing Pt- and Al-Modified MCM-48. Reactions, 1(2), 195-209. https://doi.org/10.3390/reactions1020014