Production of Fuels and Chemicals from a CO2/H2 Mixture



Abstract
1. Introduction
2. Experimental Set Up
2.1. Catalyst Preparation
2.2. Experimental Setup and Procedure
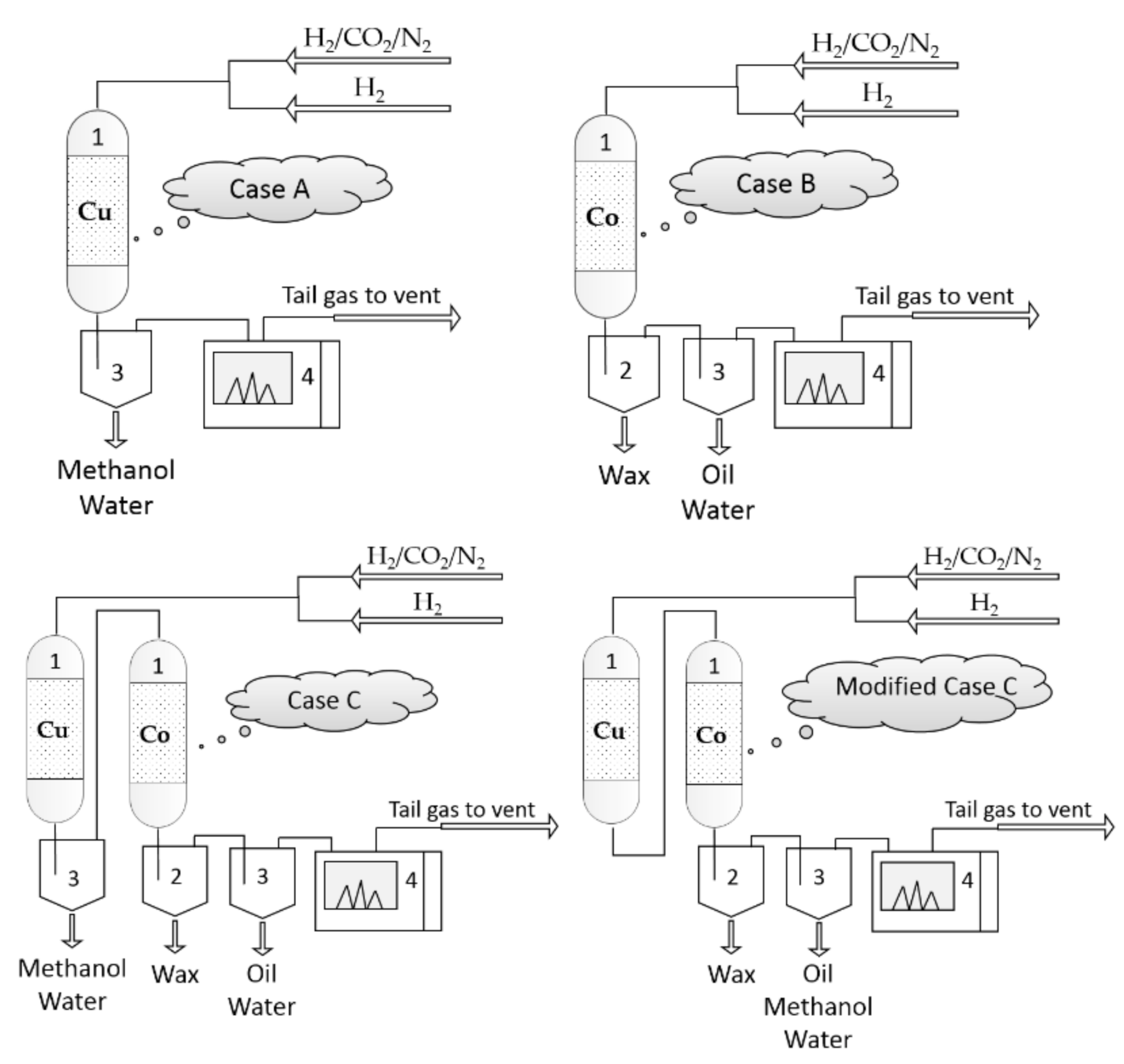
- ➢ Case A: CO2 hydrogenation over a Cu-based catalyst
- ➢ Case B: CO2 hydrogenation over a Co-based catalyst.
- ➢ Case C: CO2 hydrogenation in a reactor loaded with Cu-based catalyst in series with a reactor loaded with Co-based catalyst.
2.3. Product Analysis
3. Results
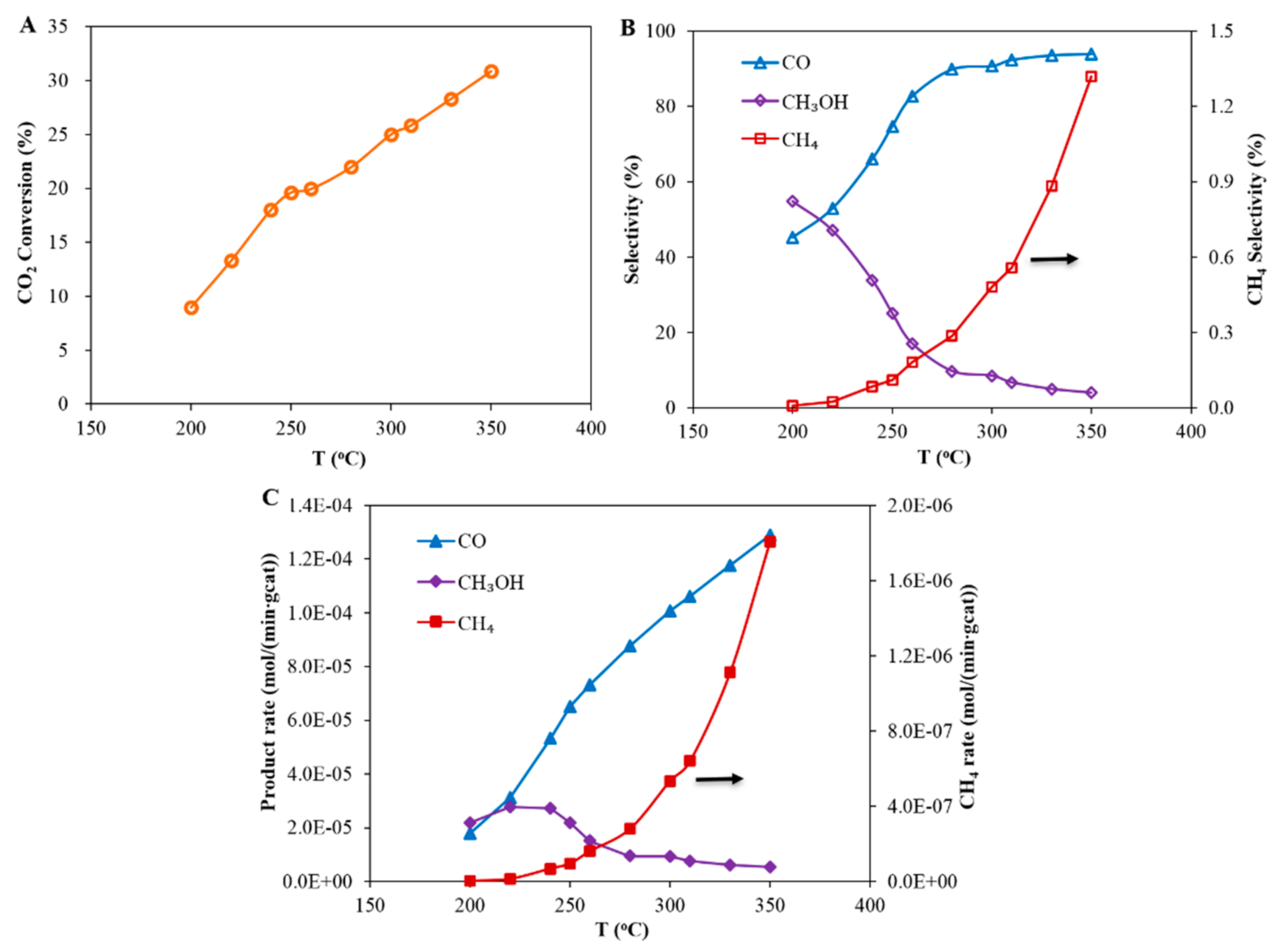
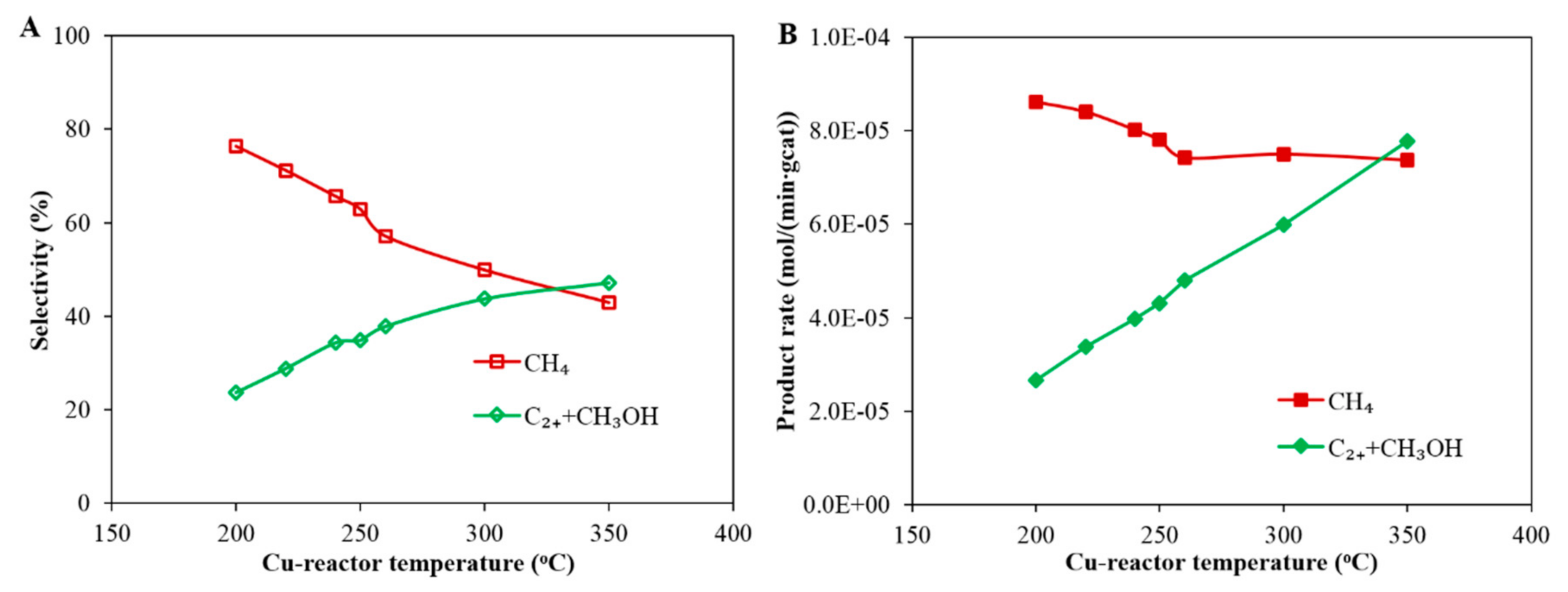
3.1. Case A: CO2 Hydrogenation Over a Cu-Based Catalyst
- The base method is the Peng-Robinson Equation of State (EOS).
- The RGibbs reactor was used to calculate equilibrium.
- The feed was: H2 with a flow rate of 3 kmol/h and CO2 with a flow rate of 1 kmol/h
- The outlet was: H2, CO, CO2, H2O and methanol
3.2. Case B: CO2 Hydrogenation Over a Co-Based Catalyst
- For CO2 hydrogenation (second column), the product is almost entirely composed of short chain paraffins, rich in CH4. The analysis results show that CH4 selectivity is more than 90% and no olefin was detected in the products.
- FT CO hydrogenation (third column) produces mainly long chain hydrocarbons (including paraffins and olefins) with CH4 selectivity less than 10%.
- For a feed which is a mixture of CO and CO2 (fourth column) with only small quantities of CO (5.9%), the product analysis shows that selectivity of CH4 is much lower compared to CO2 hydrogenation only and the products also contain longer chain hydrocarbons in a considerable amount, although still lower compared to CO hydrogenation only.
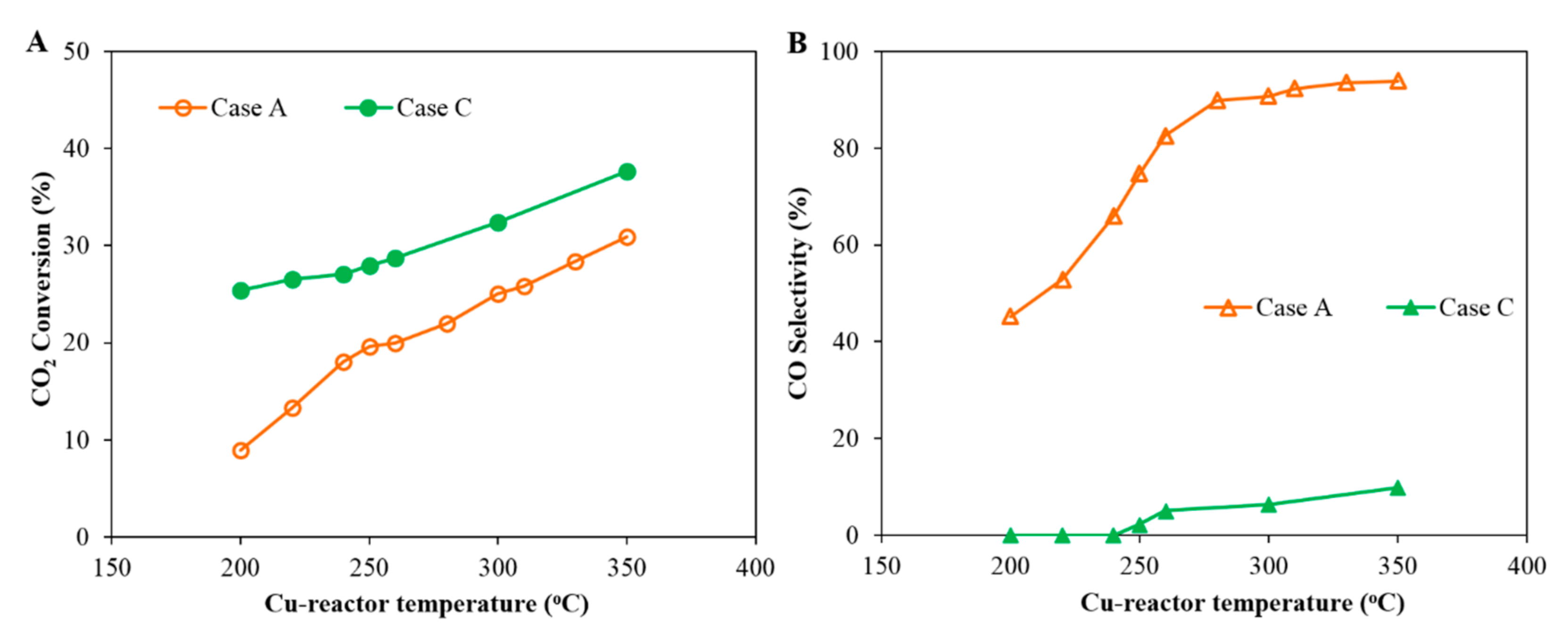
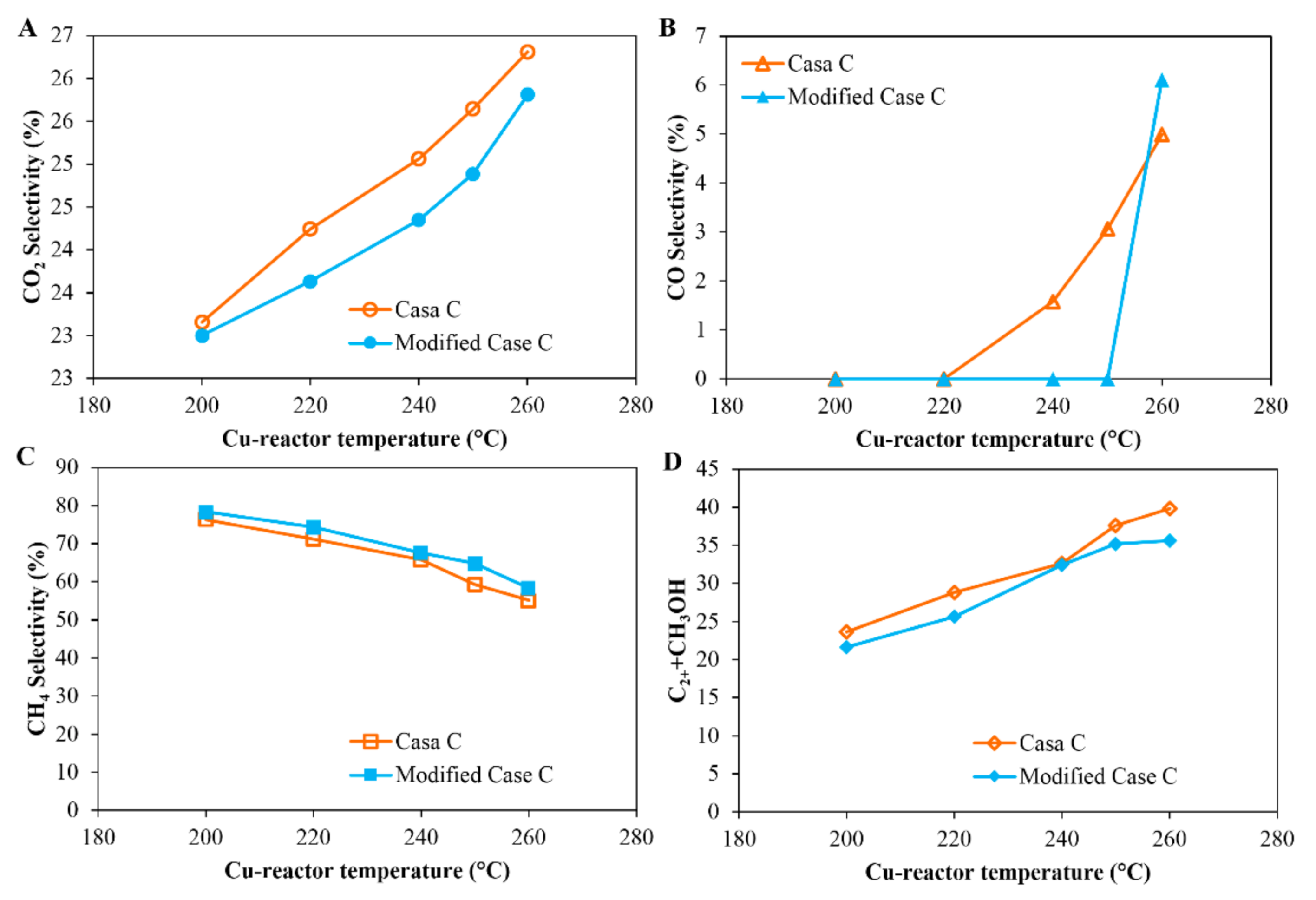
3.3. Case C: CO2 Hydrogenation Using a Cu-Based Catalyst in Series with a Co-Based Catalyst
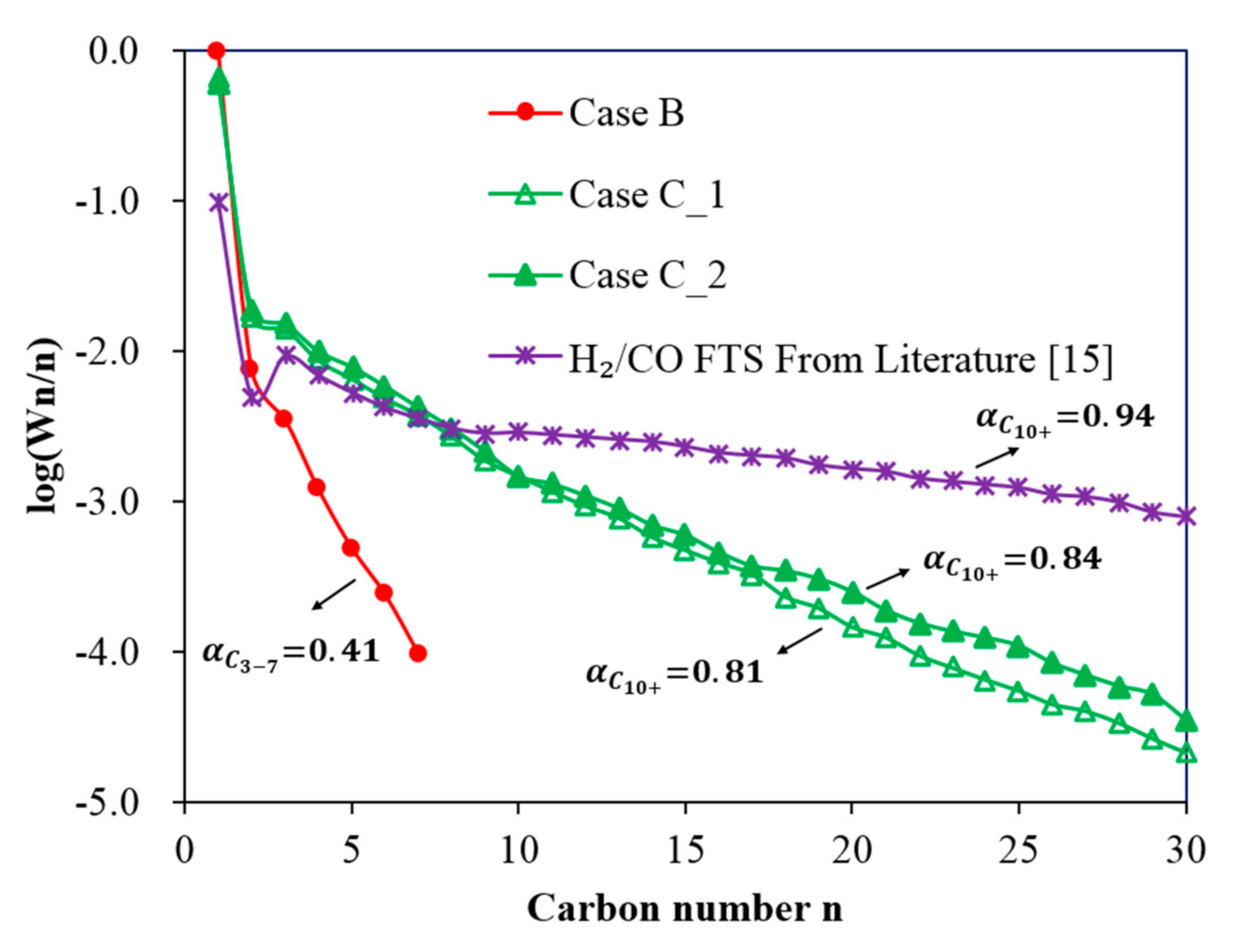
- In Case B, with a single Co-reactor (H2/CO2 feed gas, Table 1), the results show that CH4 is the dominant product with a small quantity of short chain hydrocarbons being formed. The results follow a typical ASF distribution with a low α value of 0.41.
- In Case C, with a Cu-reactor connected in series to a Co-reactor (H2/CO2 feed gas Table 1), two conditions are considered: Case C_1 and Case C_2, which consider the product distribution of the Co-reactor at two temperatures of the Cu-reactor, 300 °C and 350 °C, respectively. Figure 9 shows that the slope of the distribution of Case C_2 is slightly smaller than that of Case C_1 and thus the α value of Case C_2 is slightly higher than that of Case C_1 (0.81 < 0.84). This is due to the higher CO concentration in the product from the Cu-reactor in Case C_2, due to the higher operating temperature. The CO concentration is higher at high temperatures because, as can be seen in Figure 2B, the CO selectivity in the Cu-reactor increases with temperature. By comparing the α value in Case B with that of both cases in Case C, the results indicate that a better FT product distribution is obtained by coupling the Co-reactor to the Cu-reactor.
4. Discussion and Implications
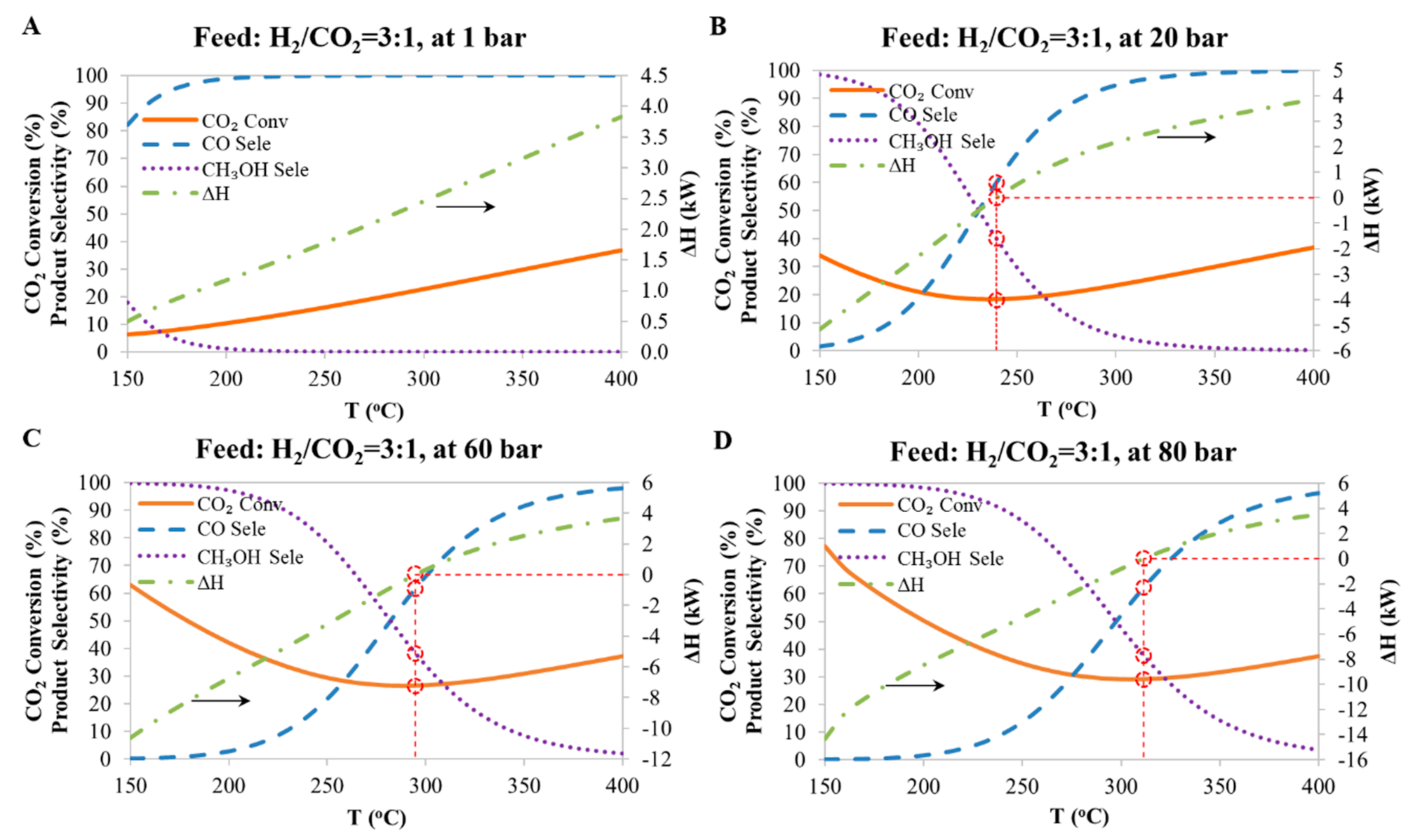
4.1. Comparason between the Current Work with the Results Reported in the Literature
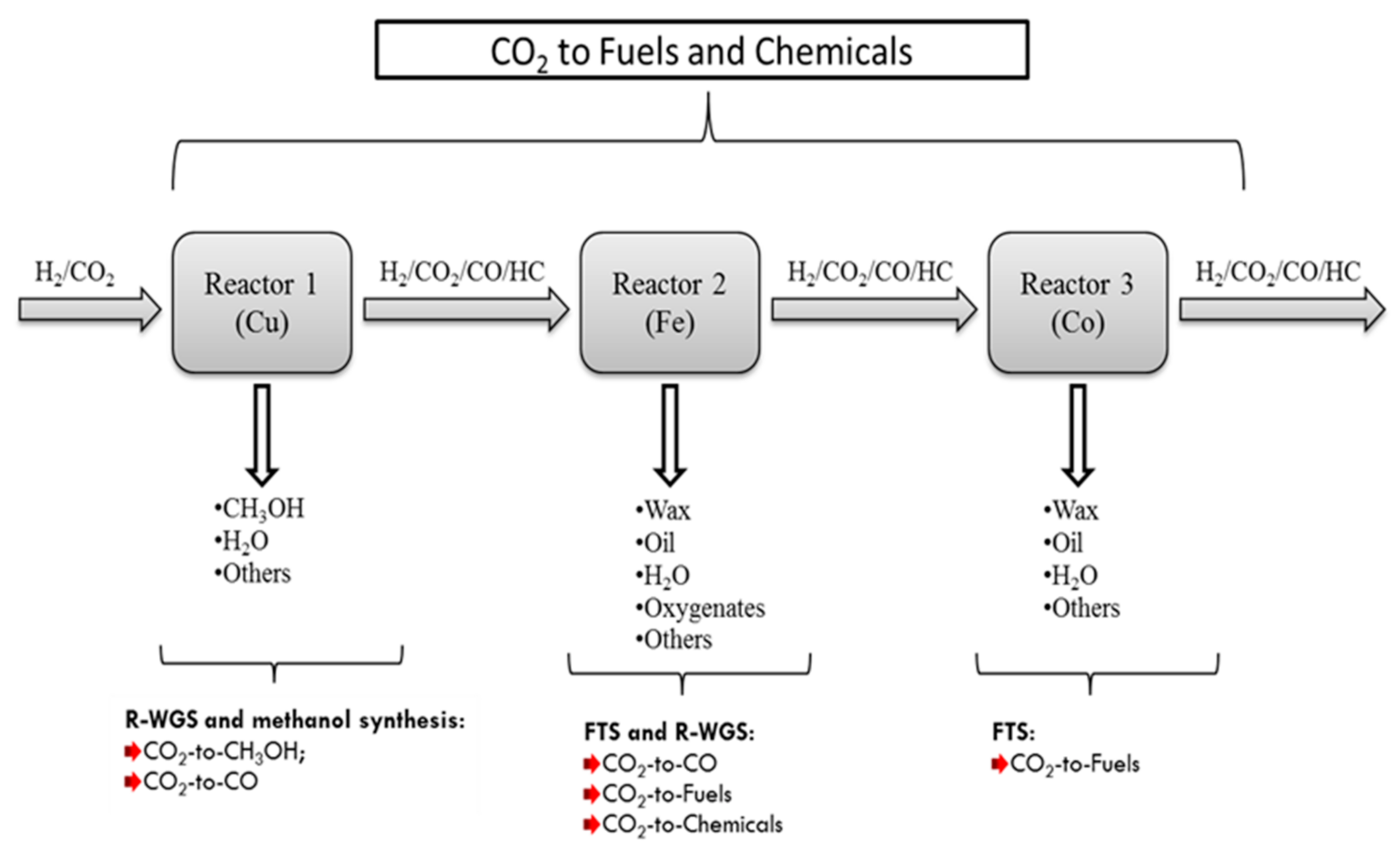
4.2. Multi-Rreactor System
- In the first reactor, as in Case C, a Cu-based catalyst is used to convert CO2 to methanol with CO as the byproduct. In this case methanol production is favored by setting reactions conditions to low temperature and high pressure. The aqueous product (a mixture of methanol and H2O) is removed from the produced stream, and the tail gas (a mixture of H2/CO/CO2 with a small amount of CH4) is introduced into the Fe-catalyst reactor.
- HTFT occurs over an Fe-based catalyst, where the tail gas from the first reactor (H2/CO/CO2 mixture) is converted to short olefins and oxygenates. Since the feed stream does not contain water, the equilibrium limitation of the R-WGS reaction occurring in the first reactor is eliminated in the second reactor and thus more CO2 is converted to CO, and because the iron catalyst is WGS active, a new equilibrium can be rapidly reached. Part of the CO formed is consumed in FT reactions within the reactor to produce light and medium olefinic hydrocarbons with small amounts of oxygenates, and the remaining CO, goes out in the tail gas (after removal of condensable FT products and water). CO2 may also react with H2 to produce FT products on the iron catalyst depending on the amount of CO undergoing FT reaction.
- The third reactor is a low temperature FT reactor over a Co-based catalyst. This reactor receives a CO rich tail gas mixture (H2/CO/CO2) from the second reactor, thus allowing a high selectivity for heavier hydrocarbons relative to lighter ones as discussed previously. Furthermore, if CO is an undesired product, this reactor will reduce the CO to low levels by converting it to FT products.
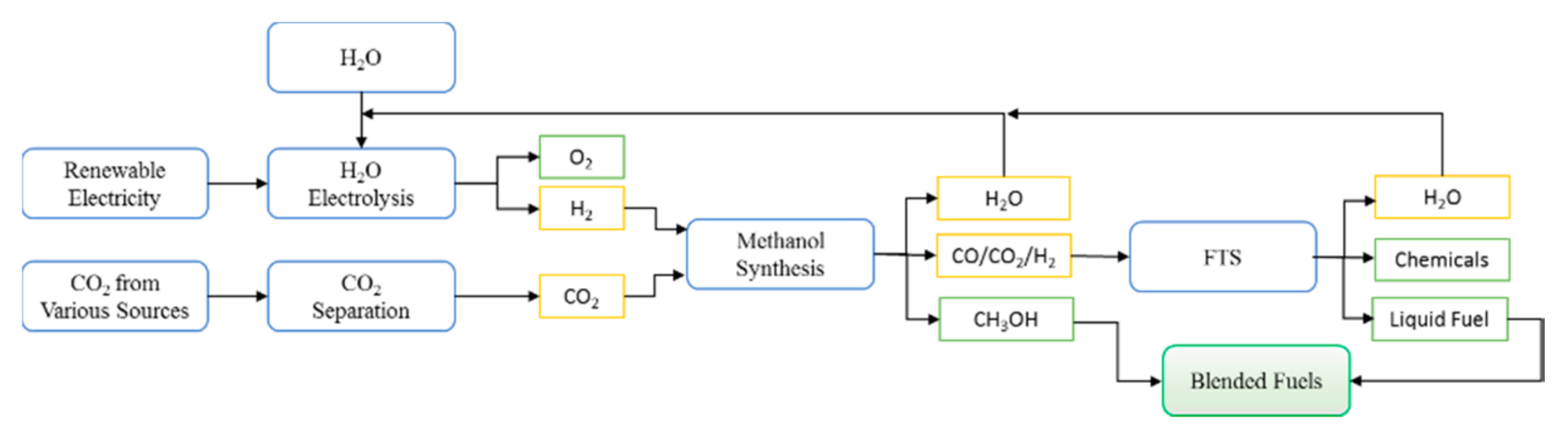
4.3. A Proposed “Greener” Process for the Conversion of CO2 to Valuable Products
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Park, S.-E.; Nam, S.S.; Choi, M.J.; Lee, K.W. Catalytic Reduction of Carbon Dioxide. The Effects of Catalysts and Reductants. Energy Convers. Manag. 1995, 36, 573–576. [Google Scholar] [CrossRef]
- Yao, L.; Shen, X.; Pan, Y.; Peng, Z. Synergy between Active Sites of Cu-In-Zr-O Catalyst in CO2 Hydrogenation to Methanol. J. Catal. 2019, 372, 74–85. [Google Scholar] [CrossRef]
- Blumberg, T.; Morosuk, T.; Tsatsaronis, G. CO2-Utilization in the Synthesis of Methanol: Potential Analysis and Exergetic Assessment. Energy 2019, 175, 730–744. [Google Scholar] [CrossRef]
- Joo, O.-S.; Jung, K.-D.; Moon, I.; Rozovskii, A.Y.; Lin, G.I.; Han, S.-H.; Uhm, S.-J. Carbon Dioxide Hydrogenation to Form Methanol via a Reverse-Water-Gas-Shift Reaction (the CAMERE Process). Ind. Eng. Chem. Res. 1999, 38, 1808–1812. [Google Scholar] [CrossRef]
- Zhang, Y.; Fei, J.; Yu, Y.; Zheng, X. Study of CO2 Hydrogenation to Methanol over Cu-V/γ-Al2O3 Catalyst. J. Nat. Gas Chem. 2007, 16, 12–15. [Google Scholar] [CrossRef]
- Bahruji, H.; Bowker, M.; Hutchings, G.; Dimitratos, N.; Wells, P.; Gibson, E.; Jones, W.; Brookes, C.; Morgan, D.; Lalev, G. Pd/ZnO Catalysts for Direct CO2 Hydrogenation to Methanol. J. Catal. 2016, 343, 133–146. [Google Scholar] [CrossRef]
- Li, S.; Guo, L.; Ishihara, T.S.C. Hydrogenation of CO2 to Methanol over Cu/AlCeO Catalyst, Catal. Today 2020, 339, 352–361. [Google Scholar] [CrossRef]
- Lonis, F.; Tola, V.; Cau, G. Renewable Methanol Production and Use through Reversible Solid Oxide Cells and Recycled CO2 Hydrogenation. Fuel 2019, 246, 500–515. [Google Scholar] [CrossRef]
- Lange, J.-P. Methanol Synthesis: A Short Review of Technology Improvements. Catal. Today 2001, 64, 3–8. [Google Scholar] [CrossRef]
- Pérez-Fortes, M.; Schöneberger, J.C.; Boulamanti, A.; Tzimas, E. Methanol Synthesis Using Captured CO2 as Raw Material: Techno-Economic and Environmental Assessment. Appl. Energy 2015, 161, 718–732. [Google Scholar] [CrossRef]
- Farsi, M.; Jahanmiri, A. Methanol Production in an Optimized Dual-Membrane Fixed-Bed Reactor. Chem. Eng. Process. Process Intensif. 2011, 50, 1177–1185. [Google Scholar] [CrossRef]
- Zhang, Y.; Jacobs, G.; Sparks, D.E.; Dry, M.E.; Davis, B.H. CO and CO2 Hydrogenation Study on Supported Cobalt Fischer-Tropsch Synthesis Catalysts. Catal. Today 2002, 71, 411–418. [Google Scholar] [CrossRef]
- Visconti, C.G.; Lietti, L.; Tronconi, E.; Forzatti, P.; Zennaro, R.; Finocchio, E. Fischer-Tropsch Synthesis on a Co/Al2O3 Catalyst with CO2 Containing Syngas. Appl. Catal. A Gen. 2009, 355, 61–68. [Google Scholar] [CrossRef]
- Riedel, T.; Claeys, M.; Schulz, H.; Schaub, G.; Nam, S.-S.; Jun, K.-W.; Choi, M.-J.; Kishan, G.; Lee, K.-W. Comparative Study of Fischer–Tropsch Synthesis with H2/CO and H2/CO2 Syngas Using Fe- and Co-Based Catalysts. Appl. Catal. A Gen. 1999, 186, 201–213. [Google Scholar] [CrossRef]
- Yao, Y.; Hildebrandt, D.; Glasser, D.; Liu, X. Fischer-Tropsch Synthesis Using H2/CO/CO2 Syngas Mixtures over a Cobalt Catalyst. Ind. Eng. Chem. Res. 2010, 49, 11061–11066. [Google Scholar] [CrossRef]
- Shafer, W.D.; Jacobs, G.; Graham, U.M.; Hamdeh, H.H.; Davis, B.H. Increased CO2 Hydrogenation to Liquid Products Using Promoted Iron Catalysts. J. Catal. 2019, 369, 239–248. [Google Scholar] [CrossRef]
- Sempuga, B.C.; Yao, Y. CO2 Hydrogenation from a Process Synthesis Perspective: Setting up Process Targets. J. CO2 Util. 2017, 20, 34–42. [Google Scholar] [CrossRef]
- Datta, A.; Dutta, S.; Mandal, B.K. Effect of methanol addition to diesel on the performance and emission characteristics of a CI engine. J. Phys. Conf. Ser. 2014, 1, 8–13. [Google Scholar] [CrossRef]
- Zhu, L.; Cheung, C.S.; Zhang, W.G.; Huang, Z.; Zhu, L.; Cheung, C.S.; Zhang, W.G.; Huang, Z. Influence of methanol—Biodiesel blends on the particulate emissions of a direct injection diesel engine influence of methanol—Biodiesel blends on the particulate emissions of a direct injection diesel engine. Aerosol Sci. Technol. 2010, 44, 362–369. [Google Scholar] [CrossRef]
- Daza, Y.A.; Kuhn, J.N. CO2 conversion by reverse water gas shift catalysis: Comparison of catalyststs, mechanisms and their consequences for CO2 conversion to liquid fuels. RSC Adv. 2011, 6, 49675–49691. [Google Scholar] [CrossRef]
- De la Osa, A.R.; De Lucas, A.; Romero, A.; Valverde, J.L.; Sánchez, P. Fischer-Tropsch Diesel Production over Calcium-Promoted Co/Alumina Catalyst: Effect of Reaction Conditions. Fuel 2011, 90, 1935–1945. [Google Scholar] [CrossRef]
- Zhou, W.; Chen, J.G.; Fang, K.G.; Sun, Y.H. The Deactivation of Co/SiO2 Catalyst for Fischer-Tropsch Synthesis at Different Ratios of H2 to CO. Fuel Process. Technol. 2006, 87, 609–616. [Google Scholar] [CrossRef]
- Tavakoli, A.; Sohrabi, M.; Kargari, A. Application of Anderson—Schulz—Flory (ASF) Equation in the Product Distribution of Slurry Phase FT Synthesis with Nanosized Iron Catalysts. Chem. Eng. J. 2008, 136, 358–363. [Google Scholar] [CrossRef]
- He, Z.; Cui, M.; Qian, Q.; Zhang, J.; Liu, H.; Han, B. Synthesis of liquid fuel via direct hydrogenation of CO2. PNAS 2019, 116, 12654–12659. [Google Scholar] [CrossRef]
- Choi, Y.H.; Jang, Y.J.; Park, H.; Kim, W.Y.; Lee, Y.H.; Choi, S.H.; Lee, J.S. Carbon dioxide Fischer-Tropsch synthesis: A new path to carbon-neutral fuels. Appl. Catal. B 2017, 202, 605–610. [Google Scholar] [CrossRef]
- Prasad, P.S.S.; Bae, J.W.; Jun, K.W.; Lee, K.W. Fischer-Tropsch synthesis by carbon dioxide hydrogenation on Fe-based catalyst. Catal. Surv. Asia 2008, 12, 170–183. [Google Scholar] [CrossRef]
- Martinelli, M.; Visconti, C.G.; Lietti, L.; Forzatti, P.; Bassano, C.; Deiana, P. CO2 Reactivity on Fe–Zn–Cu–K Fischer–Tropsch Synthesis Catalysts with Different K-Loadings. Catal. Today 2014, 228, 77–88. [Google Scholar] [CrossRef]
- Dry, M.E. The Fischer-Tropsch Process: 1950–2000. Catal. Today 2002, 71, 227–241. [Google Scholar] [CrossRef]
- Gorimbo, J.; Muleja, A.; Lu, X.; Yao, Y.; Liu, X.; Hildebrandt, D.; Glasser, D. Lu Plot and Yao Plot: Models to Analyze Product Distribution of Long-Term Gas-Phase Fischer-Tropsch Synthesis Experimental Data on an Iron Catalyst. Energy Fuels 2017, 31, 5682–5690. [Google Scholar] [CrossRef]
- Wu, Y.; Feng, J.; Li, W. System Development of Integrated High Temperature and Low Temperature Fischer-Tropsch Synthesis for High Value Chemicals. Chem. Eng. Res. Des. 2017, 1–12. [Google Scholar] [CrossRef]
- Van der Laan, G.P.; Beenackers, A.A.C.M. Intrinsic Kinetics of the Gas-solid Fischer-Tropsch and Water Gas Shift Reactions over a Precipitated Iron Catalyst. Appl. Catal. A Gen. 2000, 193, 39–53. [Google Scholar] [CrossRef]
- Steynberg, A.P.; Espinoza, R.L.; Jager, B.; Vosloo, A.C. High Temperature Fischer–Tropsch Synthesis in Commercial Practice. Appl. Catal. A Gen. 1999, 186, 41–54. [Google Scholar] [CrossRef]
- Huang, Y.; Yi, Q.; Wei, G.; Kang, J.; Li, W.; Feng, J. Energy Use, Greenhouse Gases Emission and Cost e Ff Ectiveness of an Integrated High- and Low- Temperature Fisher-Tropsch Synthesis Plant From. Appl. Energy 2018, 228, 1009–1019. [Google Scholar] [CrossRef]
- Leckel, D. Hydroprocessing Euro 4-Type Diesel from High-Temperature Fischer-Tropsch Vacuum Gas Oils. Energy Fuels 2009, 23, 38–45. [Google Scholar] [CrossRef]
- Dry, M.E. Practical and Theoretical Aspects of the Catalytic Fischer-Tropsch Process. Appl. Catal. A Gen. 1996, 138, 319–344. [Google Scholar] [CrossRef]
- Saeidi, S.; Amin, N.A.S.; Rahimpour, M.R. Hydrogenation of CO2 to Value-Added Products—A Review and Potential Future Developments. J. CO2 Util. 2014, 5, 66–81. [Google Scholar] [CrossRef]
- Satthawong, R.; Koizumi, N.; Song, C.; Prasassarakich, P. Light Olefin Synthesis from CO2 Hydrogenation over K-Promoted Fe-Co Bimetallic Catalysts. Catal. Today 2015, 251, 34–40. [Google Scholar] [CrossRef]
- Zhang, J.; Lu, S.; Su, X.; Fan, S.; Ma, Q.; Zhao, T. Selective Formation of Light Olefins from CO2 Hydrogenation over Fe–Zn–K Catalysts. J. CO2 Util. 2015, 12, 95–100. [Google Scholar] [CrossRef]
- Hildebrandt, D.; Glasser, D.; Hausberger, B.; Patel, B.; Glasser, B.J. Chemistry. Producing Transportation Fuels with Less Work. Science 2009, 323, 1680–1681. [Google Scholar] [CrossRef]
- Milani, D.; Khalilpour, R.; Zahedi, G.; Abbas, A. A Model-Based Analysis of CO2 Utilization in Methanol Synthesis Plant. J. CO2 Util. 2015, 10, 12–22. [Google Scholar] [CrossRef]
- Prieto, G. Carbon Dioxide Hydrogenation into Higher Hydrocarbons and Oxygenates: Thermodynamic and Kinetic Bounds and Progress with Heterogeneous and Homogeneous Catalysis. ChemSusChem 2017, 10, 1–16. [Google Scholar] [CrossRef] [PubMed]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Items | Case A | Case B | Case C | |
|---|---|---|---|---|
| Reactor | One fixed bed | One fixed bed | Two fixed bed reactors in series | |
| Reactor 1 | Reactor 2 | |||
| Catalyst | Cu | Co | Cu | Co |
| Catalyst weight (g) | 1 | 1 | 1 | 1 |
| Feed: H2/CO2/N2 | 67.6%/22.6%/9.8% | 67.6%/22.6%/9.8% | 67.6%/22.6%/9.8% | / |
| Temperature (°C) | 200–350 | 200 | 200–350 | 200 |
| Flow rate (ml(NTP)/(min·gcat)) a | 60 | 60 | 60 | / |
| Pressure (bar gauge) | 20 | 20 | 20 | / |
| CO2:H2 | P (bar) | T (°C) | CO2 Conversion (%) | Methanol Selectivity (%) | CO Selectivity (%) |
|---|---|---|---|---|---|
| 3:1 | 20 | 239.5 | 18.4 | 40.0 | 60.0 |
| 3:1 | 60 | 294.7 | 26.6 | 38.3 | 61.7 |
| 3:1 | 80 | 311.4 | 29.2 | 37.7 | 62.3 |
| CO2 Hydrogenation | CO Hydrogenation a | (CO+CO2) Hydrogenation a | |
|---|---|---|---|
| Feed gas | H2/CO2/N2 = 67.6%/22.6%/9.8% | H2/CO/N2 = 58.8%/30.3%/10.3% | H2/CO2/CO/N2 = 65.7%/18.3%/5.9%/9.7% |
| CO Conversion (%) | --- | 14.6 | 100.0 |
| CO2 Conversion (%) | 24.2 | --- | 13.0 |
| CH4 Selectivity (%) | 92.5 | 8.0 | 66.3 |
| C2+ Selectivity (%) | 7.5 | 92.0 | 33.7 |
| C2−4 Selectivity (%) | 6.5 | 19.8 | 24.5 |
| C5+ Selectivity (%) | 1.0 | 72.1 | 9.1 |
| O2/P2 b | 0.0 | 0.24 | 0.0 |
| O3/P3 c | 0.0 | 1.7 | 0.0 |
| O4/P4 d | 0.0 | 1.1 | 0.0 |
| Reactor | Catalyst | CO2 Conv (%) | CH4 Sel (%) | C5+ + CH3OH Sel (%) | CO Sel (%) | Reaction Conditions | Ref |
|---|---|---|---|---|---|---|---|
| One reactor | Co/TiO2 | 24.2 | 92.5 | 1.0 | 0.0 | H2/CO2 = 3, 200 °C, 60 ml(NTP)/(min·gcat), 20 bar | Current work |
| Two reactors in series | Cu-200_Co-200 | 23.2 | 76.4 | 17.6 | 0.0 | Reactor one: Cu catalyst; 200–350 °C; 60 ml(NTP)/(min·gcat), 20 bar; reactor two: Co/TiO2, 200 °C | Current work |
| Cu-250_Co-200 | 25.6 | 62.9 | 28.6 | 2.2 | |||
| Cu-300_Co-200 | 30.6 | 50.0 | 33.0 | 6.3 | |||
| Cu-350_Co-200 | 35.6 | 43.0 | 44.8 | 9.8 | |||
| One reactor | Co6/MnOx | 15.3 | 46.4 a | 53.2 | 0.4 | H2/CO2 = 1, 200 °C, 8 bar, no flow (batch mode), solvent: squalane | [24] |
| Co6/ZnOx | / | 80.7 a | 19.2 | 0.1 | |||
| Co6/CeOx | / | 89.8 a | 9.8 | 0.0 | |||
| Co6/AlOx | / | 94.2 a | 5.7 | 0.0 | |||
| One reactor | Co/MnO/SiO2/Pt | 18.0 | 95.0 | H2/CO2 = 2, 190 °C, 30(ml(NTP)/(min·gcat),10 bar | [14] | ||
| Fe/TiO2 | 11.5 | 33.3 | 4.4 | 35.7 | H2/CO2 = 3, 300 °C, 31.6 (ml(NTP)/(min·gcat),10 bar | ||
| Fe/Al2O3 | 22.8 | 38.3 | 7.8 | 11.4 | |||
| Fe/SiO2 | 6.9 | 23.4 | 0.1 | 71.0 | |||
| Fe-K/Al2O3 | 30.4 | 7.6 | 23.5 | 40.5 | |||
| One reactor | Fe/Cu/K | 10.8 | 9.1 | 26.9 | 39.3 | H2/CO2 = 3, 300 °C, 8 bar, 60ml/(min·gcat), 10–20 bar | [26] |
| Fe/Cu/Al/K | 11.3 | 8.5 | 27.1 | 45 | |||
| Fe/Cu/Si/K | 10.2 | 21.1 | 8.8 | 43.4 | |||
| Fe/Cu/Al/K (2) | 15.6 | 9.9 | 39.4 | 22.8 | |||
| One reactor | Fe2O3 | 14.3 | 40.2 | 1.5 | 33.2 | H2/CO2 = 3, 300 °C, 60ml/(min·gcat), 10 bar | [25] |
| CuFeO2-6 | 17.3 | 1.8 | 45.3 | 31.7 | |||
| Cu2O-Fe2O3 | 15.7 | 41.0 | 1.8 | 28.9 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yao, Y.; Sempuga, B.C.; Liu, X.; Hildebrandt, D. Production of Fuels and Chemicals from a CO2/H2 Mixture. Reactions 2020, 1, 130-146. https://doi.org/10.3390/reactions1020011
Yao Y, Sempuga BC, Liu X, Hildebrandt D. Production of Fuels and Chemicals from a CO2/H2 Mixture. Reactions. 2020; 1(2):130-146. https://doi.org/10.3390/reactions1020011
Chicago/Turabian StyleYao, Yali, Baraka Celestin Sempuga, Xinying Liu, and Diane Hildebrandt. 2020. "Production of Fuels and Chemicals from a CO2/H2 Mixture" Reactions 1, no. 2: 130-146. https://doi.org/10.3390/reactions1020011
APA StyleYao, Y., Sempuga, B. C., Liu, X., & Hildebrandt, D. (2020). Production of Fuels and Chemicals from a CO2/H2 Mixture. Reactions, 1(2), 130-146. https://doi.org/10.3390/reactions1020011