Pathogenesis of Inflammatory Bowel Disease: Basic Science in the Light of Real-World Epidemiology




{kind=link}
Abstract
1. Introduction
2. Overall Background
2.1. Brief History
2.2. Essential Macroscopic Structures
3. Sub-Cellular and Cellular Effectors
3.1. Epithelial Cells
3.2. The Defensins
3.3. Innate Immunity
3.4. Adaptive Immunity
- Some 70% of UC patients are pANCA (perinuclear anti-neutrophil cytoplasmic antibody) positive [25].
- Most patients with CD present antibodies to Saccharomyces cerevisiae (ASCA), anti-laminaribioside (ALCA), anti-chitobioside (ACCA), anti-mannobioside (AMCA), anti-laminarin (anti-L), and anti-chitin (anti-C) antibodies, in addition to other antibodies that target microbial antigens, such as anti-outer membrane porin C (anti-OmpC), anti-Cbir1 flagellin, and anti-12 antibody. Also, autoantibodies targeting the exocrine pancreas (PAB) were shown to be highly specific for CD [26].
- Commensal antigens may be erroneously recognized and presented as foreign to pro-inflammatory Th1 cells (wrong TLRs or wrong antigen-presenting cells may originate these errors) [27]. The issue of the antigen-presenting cells will be again dealt with below.
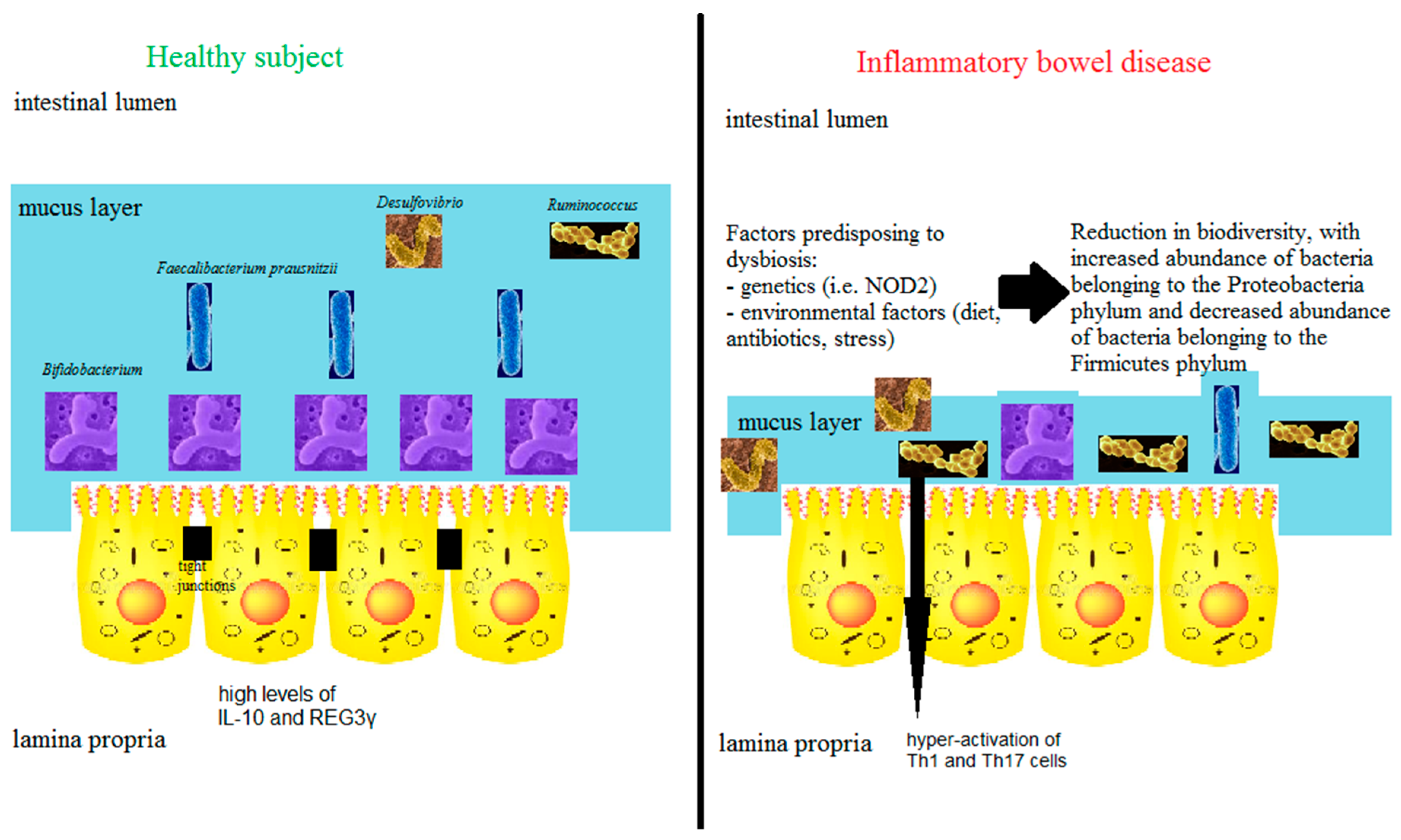
3.5. The Microbiome
4. Predominant Immune Response Bias in the Gut
- (a)
- Most of the gut antibody responses are mediated by immunoglobulin (Ig) subclasses that, incapable to fix complement, can lead to aborted responses [29].
- (b)
- (c)
- The lining epithelium itself constitutes a barrier. Its tight junctions are modulated by zonulin, a 47 KDa protein. Increased zonulin activity can enhance antigens delivery through the paracellular pathway of intestinal epithelial cells, leading to the abrogation of immune tolerance [30].
- (d)
- Many gut processes are terminated via the specific chain of apoptosis, as opposed to regular cell death. For a review of the basic effects involved in apoptosis, see reference [31]. Apoptosis is designed to effect target cell demise while maximally sparing inflammation, a mission that is pursued by targeting single cell groups to reduce to a minimum the release of chemoattractant cell debris. Briefly: (a) The intracellular apoptotic death receptors usually recognize members of the tumor necrosis factor (TNF) family as initiator signals; (b) The execution process is triggered by the caspase cascade, leading to chromatin and organelle disruption; (c) The mitochondria are the final common target: irreversible damage to membranes and permeabilization cause oozing of oxidative compounds promoting cell death.
- d.1
- Clinical relevance of apoptosis in the setting of IBDBriefly, we wish to reiterate that as many as three of the routinely used drug classes in the control of IBD can be considered apoptosis activators.
- d.1.1.
- Sulfasalazine [32] (but not 5-ASA);
- d.1.2.
- Thiopurines and their metabolites 6-thioguanines can abolish the efficacy of anti-apoptotic factors in lymphocytes via RAC-1 blocking [33];
- d.1.3.
- The anti-TNF monoclonals are caspase inducers and potentially may trigger cell lysis via complement fixation and initiation of antibody-dependent cell-cytotoxicity (ADCC) [34].
- (e)
- The Regulatory T-Lymphocytes (T-Regs)
5. The Trigger Factors of IBD: Genetic Factors, Drug Factors, Leisure Habits, Diet
5.1. Genetics (1)—The Case for Monogenic IBDs
5.1.1. X-Linked Apoptosis Inhibitor
5.1.2. Signal Transduction and Activation of Transcription (STATs)
5.1.3. A Disintegrin and Metalloprotease (ADAM) 17
5.2. Genetics (2)—The IL-23/Th17 Axis as a Significant Pathogenetic Element
5.3. Diet
Diet Could Have Effects Beyond the Bowel
5.4. Induction by Drugs
5.5. Leisure Habits: Smoking
6. The Kick-Out Factors in IBD: Antigen-Presenting (Dendritic) Cells
7. IBDs in Real Life: Clues from Epidemiology
7.1. World Epidemiology
7.2. East Meets West: A Paradigm for the Role of Environmental Factors
7.3. Similar Orders of Figures Have Long Been the Matter of Scrutiny with Regard to the Incidence of Pediatric IBD (CD) in the South Island of New Zealand
7.4. Clues from Migratory Events
8. Urbanization: IBD Epidemiology and Microbiome
8.1. Chemical Pollution
8.2. Infectious Disease
8.3. Lack of Physical Exercise and Microbiome
9. An Attempt at a Holistic Vision
10. Conclusive Thoughts
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lim, M.L.; Wallace, M.R. Infectious diarrhea in history. Infect. Dis. Clin. N. Am. 2004, 18, 261–274. [Google Scholar] [CrossRef] [PubMed]
- Francois Joseph Victor Broussais (1772–1838). System of physiological medicine. JAMA 1969, 209, 1523. [Google Scholar]
- Wilks, S. Morbid appearances in the intestines of Miss Bankes. Lond. Med. Gaz. 1859, 2, 264–265. [Google Scholar]
- Adams, E.W. Founders of modern medicine: Giovanni Battista Morgagni (1682–1771). Med. Libr. Hist. J. 1903, 1, 270–277. [Google Scholar]
- Crohn, B.B.; Ginzburg, L.; Oppenheimer, G.D. Regional ileitis: A pathologic and clinical entity. J. Am. Med. Assoc. 1932, 99, 1323–1329. [Google Scholar] [CrossRef]
- Lockhart-Mummery, H.E.; Morson, B.C. Crohn’s disease (regional enteritis) of the large intestine and its distinction from ulcerative colitis. Gut 1960, 1, 87–105. [Google Scholar] [CrossRef] [PubMed]
- Lockhart-Mummery, H.E.; Morson, B.C. Crohn’s disease of the large intestine. Gut 1964, 5, 493–509. [Google Scholar] [CrossRef] [PubMed]
- Graham, M.F.; Blomquist, P.; Zederfeldt, B. The alimentary Canal. In Wound Healing: Biochemical and Clinical Aspects; Cohen, I.K., Diegelmann, R.F., Lindblad, W., Eds.; Saunders: Philadelphia, PA, USA, 1991. [Google Scholar]
- James, S.P. Mucosal Immune Regulation. In IBD, From Bench to Bedside; Shanahan, F., Targan, S., Eds.; Williams and Wilkins: Baltimore, MD, USA, 1984; pp. 65–77. [Google Scholar]
- Strober, W.; Brown, W.R. The mucosal Immune System. In Immunologic Diseases; Samter, M., Talmage, D.W., Frank, M.M., Austen, K.F., Claman, H.N., Eds.; Little and Brown: Boston, MA, USA, 1988; Volume 1, pp. 79–139. [Google Scholar]
- Helander, H.F.; Fändriks, L. Surface area of the digestive tract—Revisited. Scand. J. Gastroenterol. 2014, 49, 681–689. [Google Scholar] [CrossRef] [PubMed]
- Van der Flier, L.G.; Clevers, H. Stem cells, self-renewal, and differentiation in the intestinal epithelium. Annu. Rev. Physiol. 2009, 71, 241–260. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, R.; Watanabe, M. Role of epithelial cells in the pathogenesis and treatment of inflammatory bowel disease. J. Gastroenterol. 2016, 51, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Surawicz, C.M.; Haggitt, R.C.; Husseman, M.; McFarland, L.V. Mucosal biopsy diagnosis of colitis: Acute self-limited colitis and idiopathic inflammatory bowel disease. Gastroenterology 1994, 107, 755–763. [Google Scholar] [CrossRef]
- Wehkamp, J.; Harder, J.; Weichenthal, M.; Schwab, M.; Schäffeler, E.; Schlee, M.; Herrlinger, K.R.; Stallmach, A.; Noack, F.; Fritz, P.; et al. NOD2 (CARD15) mutations in Crohn’s disease are associated with diminished mucosal alpha-defensin expression. Gut 2004, 53, 1658–1664. [Google Scholar] [CrossRef] [PubMed]
- Wehkamp, J.; Salzman, N.H.; Porter, E.; Nuding, S.; Weichenthal, M.; Petras, R.E.; Shen, B.; Schaeffeler, E.; Schwab, M.; Linzmeier, R.; et al. Reduced Paneth cells alpha-defensins in ileal CD. Proc. Natl. Acad. Sci. USA 2005, 102, 18129–18134. [Google Scholar] [CrossRef] [PubMed]
- Noble, C.L.; Nimmo, E.R.; Drummond, H.; Ho, G.T.; Tenesa, A.; Smith, L.; Anderson, N.; Arnott, I.D.; Satsangi, J. The contribution of OCTN1/2 variants within the IBD5 locus to disease susceptibility and severity in Crohn’s. Gastroenterology 2005, 129, 1854–1864. [Google Scholar] [CrossRef] [PubMed]
- Swidsinki, A.; Ladhoff, A.; Pernthaler, A.; Swidsinski, S.; Loening-Baucke, V.; Ortner, M.; Weber, J.; Hoffmann, U.; Schreiber, S.; Dietel, M.; et al. Mucosal flora in inflammatory bowel disease. Gastroenterology 2002, 122, 44–54. [Google Scholar] [CrossRef]
- Iwasaki, A.; Medzhitov, R. Toll-like receptor control of the adaptive immune responses. Nat. Immunol. 2004, 5, 987–995. [Google Scholar] [CrossRef] [PubMed]
- Hugot, J.P.; Chamaillard, M.; Zouali, H.; Lesage, S.; Cézard, J.P.; Belaiche, J.; Almer, S.; Tysk, C.; O’Morain, C.A.; Gassull, M.; et al. Association of NOD-2 leucine rich repeat variants with susceptibility to Crohn’s. Nature 2001, 411, 599–603. [Google Scholar] [CrossRef] [PubMed]
- Ogura, Y.; Bonen, D.K.; Inohara, N.; Nicolae, D.L.; Chen, F.F.; Ramos, R.; Britton, H.; Moran, T.; Karaliuskas, R.; Duerr, R.H.; et al. A frameshift mutation in NOD2 associated with susceptibility to Crohn’s disease. Nature 2001, 411, 603–606. [Google Scholar] [CrossRef] [PubMed]
- Strober, W.; Murray, P.J.; Kitani, A.; Watanabe, T. Signaling pathways and molecular interactions of NOD-1 and NOD-2. Nat. Rev. Immunol. 2006, 6, 9–20. [Google Scholar] [CrossRef] [PubMed]
- Gasché, C.; Nemeth, M.; Grundtner, P.; Willheim-Polli, C.; Ferenci, P.; Schwarzenbacher, R. Evolution of Crohn’s disease-associated Nod2 mutations. Immunogenetics 2008, 60, 115–120. [Google Scholar] [CrossRef] [PubMed]
- Henckaerts, L.; Vermeire, S. NOD/2 CARD 15 disease associations other than CD. Inflamm. Bowel Dis. 2007, 13, 235–241. [Google Scholar] [CrossRef] [PubMed]
- Cohavy, O.; Bruckner, D.; Gordon, L.K.; Misra, R.; Wei, B.; Eggena, M.E.; Targan, S.R.; Braun, J. Colonic bacteria express an Ulcerative Colitis p-ANCA related protein epitope. Infect. Immun. 2000, 68, 1542–1548. [Google Scholar] [CrossRef] [PubMed]
- Kuna, A.T. Serological markers of IBD. Biochem. Med. (Zagreb) 2013, 23, 28–42. [Google Scholar] [CrossRef] [PubMed]
- Niess, J.H.; Brand, S.; Gu, X.; Landsman, L.; Jung, S.; McCormick, B.A.; Vyas, J.M.; Boes, M.; Ploegh, H.L.; Fox, J.G.; et al. CX3CR1-mediated dendritic cell access to the intestinal lumen and bacterial clearance. Science 2005, 307, 254–258. [Google Scholar] [CrossRef] [PubMed]
- Actis, G.C. The Gut Microbiome. Inflamm. Allergy Drug Targets 2014, 13, 217–223. [Google Scholar] [CrossRef] [PubMed]
- Strober, W.; James, S.P. The mucosal immune system. In Basic Human Immunology; Stites, D.P., Terr, A., Norwalk, C.T., Eds.; Appleton & Lange: New York, NY, USA, 1991; pp. 175–186. [Google Scholar]
- Fasano, A. Zonulin and its regulation of intestinal barrier function: The biological door to inflammation, autoimmunity, and cancer. Physiol. Rev. 2011, 91, 151–175. [Google Scholar] [CrossRef] [PubMed]
- Müller, M.; Krammer, P.H. Integrated cell function: Apoptosis. In The liver: Biology and Pathobiology; Arias, I.M., Ed.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2001; pp. 187–205. [Google Scholar]
- Doering, J.; Begue, B.; Lentze, M.J.; Rieux-Laucat, F.; Goulet, O.; Schmitz, J.; Cerf-Bensussan, N.; Ruemmele, F.M. Induction of T-lymphocyte apoptosis by sulphasalazin in patients with Crohn’s Disease. Gut 2004, 53, 1632–1638. [Google Scholar] [CrossRef] [PubMed]
- Tiede, I.; Fritz, G.; Strand, S.; Poppe, D.; Dvorsky, R.; Strand, D.; Lehr, H.A.; Wirtz, S.; Becker, C.; Atreya, R.; et al. CD-28 dependent Rac-1 activation is the molecular target of azathioprine in primary human CD4+ T lymphocytes. J. Clin. Investig. 2003, 111, 1133–1145. [Google Scholar] [CrossRef] [PubMed]
- Levin, A.D.; Wildenberg, M.E.; van den Brink, G.R. Mechanism of action of anti-TNF therapy in IBD. J. Crohn’s Colitis 2016, 10, 989–997. [Google Scholar] [CrossRef] [PubMed]
- Lan, R.Y.; Mackay, I.R.; Gershwin, M.E. Regulatory T-cells in the prevention of mucosal inflammatory disorders: Patrolling the border. J. Autoimmun. 2007, 29, 272–280. [Google Scholar] [CrossRef] [PubMed]
- Tan, Q.K.G.; Louie, R.J.; Sleasman, J.W. IPEX Syndrome. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Stephens, K., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 2004. [Google Scholar]
- Marks, D.J.; Miyagi, K.; Rahman, F.Z.; Novelli, M.; Bloom, S.L.; Segal, A.W. Inflammatory bowel disease in CGD reproduces the clinicopathological features of Crohn’s disease. Am. J. Gastroenterol. 2009, 104, 117–124. [Google Scholar] [CrossRef] [PubMed]
- Uhlig, H.H.; Schwerd, T. From Genes to Mechanisms: The Expanding Spectrum of Monogenic Disorders Associated with Inflammatory Bowel Disease. Inflamm. Bowel Dis. 2016, 22, 202–212. [Google Scholar] [CrossRef] [PubMed]
- Actis, G.C.; Pellicano, R. The pathologic galaxy modulating the genotype and phenotype of inflammatory bowel disease: Comorbidity, contiguity, and genetic and epigenetic factors. Min. Med. 2016, 107, 401–412. [Google Scholar]
- Blank, M.; Shiloh, Y. Programs for cell death: Apoptosis is only one way to go. Cell Cycle 2007, 6, 6–95. [Google Scholar] [CrossRef] [PubMed]
- Obexer, P.; Ausserlechner, M.J. X-linked inhibitor of apoptosis protein—A critical death resistance regulator and therapeutic target for personalized cancer therapy. Front. Oncol. 2014, 4, 197. [Google Scholar] [CrossRef] [PubMed]
- Latour, S.; Aguilar, C. XIAP deficiency syndrome in humans. Semin. Cell Dev. Biol. 2015, 39, 115–123. [Google Scholar] [CrossRef] [PubMed]
- Worthey, E.A.; Mayer, A.N.; Syverson, G.D.; Helbling, D.; Bonacci, B.B.; Decker, B.; Serpe, J.M.; Dasu, T.; Tschannen, M.R.; Veith, R.L.; et al. Making a definitive diagnosis: Successful clinical application of whole exome sequencing in a child with intractable inflammatory bowel disease. Genet. Med. 2011, 13, 255–262. [Google Scholar] [CrossRef] [PubMed]
- Coelho, R.; Peixoto, A.; Amil-Dias, J.; Trindade, E.; Campos, M.; Magina, S.; Charbit-Henrion, F.; Lenoir, C.; Latour, S.; Magro, F.; et al. Refractory monogenic Crohn’s disease due to X-linked inhibitor of apoptosis deficiency. Int. J. Colorectal Dis. 2016, 31, 1235–1236. [Google Scholar] [CrossRef] [PubMed]
- Darnell, J.E., Jr.; Kerr, I.M.; Stark, G.R. Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science 1994, 264, 1415–1421. [Google Scholar] [CrossRef] [PubMed]
- Fu, X.Y. STAT3 in immune responses and inflammatory bowel diseases. Cell Res. 2006, 16, 214–219. [Google Scholar] [CrossRef] [PubMed]
- Glocker, E.O.; Kotlarz, D.; Boztug, K.; Gertz, E.M.; Schäffer, A.A.; Noyan, F.; Perro, M.; Diestelhorst, J.; Allroth, A.; Murugan, D.; et al. Inflammatory bowel disease and mutations affecting the interleukin-10 receptor. N. Engl. J. Med. 2009, 361, 2033–2045. [Google Scholar] [CrossRef] [PubMed]
- Chalaris, A.; Adam, N.; Sina, C.; Rosenstiel, P.; Lehmann-Koch, J.; Schirmacher, P.; Hartmann, D.; Cichy, J.; Gavrilova, O.; Schreiber, S.; et al. Critical role of the disintegrin metalloprotease ADAM17 for intestinal inflammation and regeneration in mice. J. Exp. Med. 2010, 207, 1617–1624. [Google Scholar] [CrossRef] [PubMed]
- Blaydon, D.C.; Biancheri, P.; Di, W.L.; Plagnol, V.; Cabral, R.M.; Brooke, M.A.; van Heel, D.A.; Ruschendorf, F.; Toynbee, M.; Walne, A.; et al. Inflammatory skin and bowel disease linked to ADAM17 deletion. N. Engl. J. Med. 2011, 365, 1502–1508. [Google Scholar] [CrossRef] [PubMed]
- Fagoonee, S.; Pellicano, R.; Actis, G.C. ADAM17 and gastrointestinal tract diseases: Clinical aspects with translational messages. Minerva Biotechnol. 2018, 30, 22–28. [Google Scholar]
- Abraham, C.; Cho, J. Interleukin-23/Th17 pathways and inflammatory bowel disease. Inflamm. Bowel Dis. 2009, 15, 1090–1100. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Bhatia, A.; Krugliak Cleveland, N.; Gupta, N.; Dalal, S.; Rubin, D.T.; Sakuraba, A. Rapid Onset of Inflammatory Bowel Disease after Receiving Secukinumab Infusion. ACG Case Rep. J. 2018, 5, e56. [Google Scholar] [CrossRef] [PubMed]
- Rozin, A.P.; Hasin, T.; Toledano, K.; Guralnik, L.; Balbir-Gurman, A. Seronegative polyarthritis as severe systemic disease. Neth. J. Med. 2010, 68, 236–241. [Google Scholar] [PubMed]
- Tilg, H.; Moschen, A.R. Food, immunity, and the microbiome. Gastroenterology 2015, 148, 1107–1119. [Google Scholar] [CrossRef] [PubMed]
- Martinez, I.; Stegen, J.C.; Maldonado-Gómez, M.X.; Eren, A.M.; Siba, P.M.; Greenhill, A.R.; Walter, J. The gut microbiota of rural papua new guineans: Composition, diversity patterns, and ecological processes. Cell Rep. 2015, 11, 527–538. [Google Scholar] [CrossRef] [PubMed]
- Hart, A.R.; Luben, R.; Olsen, A.; Tjonneland, A.; Linseisen, J.; Nagel, G.; Berglund, G.; Lindgren, S.; Grip, O.; Key, T.; et al. Diet in the aetiology of ulcerative colitis: A European prospective cohort study. Digestion 2008, 77, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Sigall-Boneh, R.; Levine, A.; Lomer, M.; Wierdsma, N.; Allan, P.; Fiorino, G.; Gatti, S.; Jonkers, D.; Kierkus, J.; Katsanos, K.H.; et al. Research Gaps in Diet and Nutrition in Inflammatory Bowel Disease. A Topical Review by D-ECCO Working Group [Dietitians of ECCO]. J. Crohns Colitis 2017, 11, 1407–1419. [Google Scholar] [CrossRef] [PubMed]
- Gerasimidis, K.; McGrogan, P.; Edwards, C.A. The aetiology and impact of malnutrition in paediatric inflammatory bowel disease. J. Hum. Nutr. Diet. 2011, 24, 313–326. [Google Scholar] [CrossRef] [PubMed]
- Brasil Lopes, M.; Rocha, R.; Castro Lyra, A.; Rosa Oliveira, V.; Gomes Coqueiro, F.; Silveira Almeida, N.; Santos Valois, S.; Oliveira Santana, G. Restriction of dairy products; A reality in inflammatory bowel disease patients. Nutr. Hosp. 2014, 29, 575–581. [Google Scholar] [PubMed]
- Carolan, E.; Hogan, A.E.; Corrigan, M.; Gaotswe, G.; O’Connell, J.; Foley, N.; O’Neill, L.A.; Cody, D.; O’Shea, D. The impact of childhood obesity on inflammation, innate immune cell frequency, and metabolic microRNA expression. J. Clin. Endocrinol. Metab. 2014, 99, E474–E478. [Google Scholar] [CrossRef] [PubMed]
- Ozawa, M.; Shipley, M.; Kivimaki, M.; Singh-Manoux, A.; Brunner, E.J. Dietary pattern, inflammation and cognitive decline: The Whitehall II prospective cohort study. Clin. Nutr. 2017, 36, 506–512. [Google Scholar] [CrossRef] [PubMed]
- Dror, E.; Dalmas, E.; Meier, D.T.; Wueest, S.; Thévenet, J.; Thienel, C.; Timper, K.; Nordmann, T.M.; Traub, S.; Schulze, F.; et al. Postprandial macrophage-derived IL-1β stimulates insulin, and both synergistically promote glucose disposal and inflammation. Nat. Immunol. 2017, 18, 283–292. [Google Scholar] [CrossRef] [PubMed]
- Maciver, N.J.; Jacobs, S.R.; Wieman, H.L.; Wofford, J.A.; Coloff, J.L.; Rathmell, J.C. Glucose metabolism in lymphocytes is a regulated process with significant effects on immune cell function and survival. J. Leukoc. Biol. 2008, 84, 949–957. [Google Scholar] [CrossRef] [PubMed]
- Fedrigo, O.; Pfefferle, A.D.; Babbitt, C.C.; Haygood, R.; Wall, C.E.; Wray, G.A. A potential role for glucose transporters in the evolution of human brain size. Brain Behav. Evol. 2011, 78, 315–326. [Google Scholar] [CrossRef] [PubMed]
- Ananthakrishnan, A.N.; Higuchi, L.M.; Huang, E.S.; Khalili, H.; Richter, J.M.; Fuchs, C.S.; Chan, A.T. Aspirin, nonsteroidal anti-inflammatory drug use, and risk for Crohn disease and ulcerative colitis: A cohort study. Ann. Int. Med. 2012, 156, 350–359. [Google Scholar] [CrossRef] [PubMed]
- Toussirot, E.; Houvenagel, É.; Goëb, V.; Fouache, D.; Martin, A.; Le Dantec, P.; Dernis, E.; Wendling, D.; Ansemant, T.; Berthelot, J.M.; et al. Development of inflammatory bowel disease during anti-TNF-α therapy for inflammatory rheumatic disease: A nationwide series. Jt. Bone Spine 2012, 79, 457–463. [Google Scholar] [CrossRef] [PubMed]
- Dubeau, M.F.; Iacucci, M.; Beck, P.L.; Moran, G.W.; Kaplan, G.G.; Ghosh, S.; Panaccione, R. Drug-induced inflammatory bowel disease and IBD-like conditions. Inflamm. Bowel Dis. 2013, 19, 445–456. [Google Scholar] [CrossRef] [PubMed]
- Kelsen, J.; Agnholt, J.; Dige, A.; Christensen, L.A.; Dahlerup, J.F. Discrete changes in circulating regulatory T cells during infliximab treatment of Crohn’s disease. Autoimmunity 2010, 43, 325–333. [Google Scholar] [CrossRef]
- Actis, G.C.; Lagget, M.; Pellicano, R.; Rosina, F. Pancolitis during etanercept treatment of rheumatoid arthritis relapsing on the administration of further two TNF-alpha inhibitors. Int. J. Colorectal Dis. 2012, 27, 547–548. [Google Scholar] [CrossRef] [PubMed]
- Parkes, G.C.; Whelan, K.; Lindsay, J.O. Smoking in inflammatory bowel disease: Impact on disease course and insights into the aetiology of its effect. J. Crohn’s Colitis 2014, 8, 717–725. [Google Scholar] [CrossRef] [PubMed]
- Meregnani, J.; Clarençon, D.; Vivier, M.; Peinnequin, A.; Mouret, C.; Sinniger, V.; Picq, C.; Job, A.; Canini, F.; Jacquier-Sarlin, M.; et al. Anti-inflammatory effect of vagus nerve stimulation in a rat model of inflammatory bowel disease. Auton. Neurosci. 2011, 160, 82–89. [Google Scholar] [CrossRef] [PubMed]
- Cuoco, J.A.; Fennie, C.N.; Cheriyan, G.K. The Cholinergic Anti-Inflammatory Pathway: A Novel Paradigm for Translational Research in Neuroimmunology. J. Neurol. Neurosci. 2016, 7, 1–7. [Google Scholar] [CrossRef]
- Bonaz, B.; Sinniger, V.; Hoffmann, D.; Clarençon, D.; Mathieu, N.; Dantzer, C.; Vercueil, L.; Picq, C.; Trocmé, C.; Faure, P.; et al. Chronic vagus nerve stimulation in Crohn’s disease: A 6-month follow-up pilot study. Neurogastroenterol. Motil. 2016, 28, 948–953. [Google Scholar] [CrossRef] [PubMed]
- Kushwah, R.; Hu, J. Dendritic cell apoptosis. J. Immunol. 2010, 185, 795–802. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Wang, Y.H.; Wang, Y.; Huang, L.; Sandoval, H.; Liu, Y.J.; Wang, J. Dendritic cell apoptosis in the maintenance of immune tolerance. Science 2006, 311, 1160–1164. [Google Scholar] [CrossRef] [PubMed]
- Roda, G.; Sartini, A.; Zambon, E.; Calafiore, A.; Marocchi, M.; Caponi, A.; Belluzzi, A.; Roda, E. Intestinal epithelial cells in inflammatory bowel diseases. World J. Gastroenterol. 2010, 16, 4264–4271. [Google Scholar] [CrossRef] [PubMed]
- Molodecky, N.A.; Soon, I.S.; Rabi, D.M.; Ghali, W.A.; Ferris, M.; Chernoff, G.; Benchimol, E.I.; Panaccione, R.; Ghosh, S.; Barkema, H.W.; et al. Increasing Incidence and Prevalence of the IBDs with time, based on systematic review. Gastroenterology 2012, 142, 46–54.e42. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Owyang, C.; Wu, G.D. East meets the West: The Increasing Incidence of Inflammatory Bowel Disease in Asia as a Paradigm for Environmental Effects on the Pathogenesis of Immune-Mediated Disease. Gastroenterology 2016, 151, e1–e5. [Google Scholar] [CrossRef] [PubMed]
- Ng, S.C. Emerging leadership lecture: Inflammatory bowel disease in Asia: Emergence of a “Western” disease. J. Gastroenterol. Hepatol. 2015, 30, 440–445. [Google Scholar] [CrossRef] [PubMed]
- Malekzadeh, M.M.; Vahedi, H.; Gohari, K.; Mehdipour, P.; Sepanlou, S.G.; Ebrahimi Daryani, N.; Zali, M.R.; Mansour-Ghanaei, F.; Safaripour, A.; Aghazadeh, R.; et al. Emerging Epidemic of Inflammatory Bowel Disease in a Middle Income Country: A Nation-wide Study from Iran. Arch. Iran. Med. 2016, 19, 2–15. [Google Scholar] [PubMed]
- Lopez, R.N.; Appleton, L.; Gearry, R.B.; Day, A.S. Rising Incidence of Paediatric Inflammatory Bowel Disease in Canterbury, New Zealand, 1996–2015. J. Pediatr. Gastroenterol. Nutr. 2018, 66, e45. [Google Scholar] [CrossRef] [PubMed]
- Benchimol, E.; Mack, D.R.; Guttmann, A.; Nguyen, G.C.; To, T.; Mojaverian, N.; Quach, P.; Manuel, D.G. Inflammatory bowel disease in immigrants to Canada and their children: A population-based cohort study. Am. J. Gastroenterol. 2015, 110, 553–563. [Google Scholar] [CrossRef] [PubMed]
- Damas, O.M.; Avalos, D.J.; Palacio, A.M.; Gomez, L.; Quintero, M.A.; Deshpande, A.R.; Sussman, D.A.; McCauley, J.L.; Lopez, J.; Schwartz, S.J.; et al. Inflammatory bowel disease is presenting sooner after immigration in more recent US immigrants from Cuba. Aliment. Pharmacol. Ther. 2017, 46, 303–309. [Google Scholar] [CrossRef] [PubMed]
- Actis, G.C.; Pellicano, R. Letter: Cuban immigrants to the US developing IBD have a progressive shortening of the lag between time of arrival and disease onset. Aliment. Pharmacol. Ther. 2017, 46, 768. [Google Scholar] [CrossRef] [PubMed]
- Isaksson, C. Urbanization, oxidative stress and inflammation: A question of evolving, acclimatizing or coping with urban environmental stress. Funct. Ecol. 2015, 29, 913–923. [Google Scholar] [CrossRef]
- Aller, M.A.; Arias, N.; Fuentes-Julian, S.; Blazquez-Martinez, A.; Argudo, S.; Miguel, M.P.; Arias, J.L.; Arias, J. Coupling inflammation with evo-devo. Med. Hypotheses 2012, 78, 721–731. [Google Scholar] [CrossRef] [PubMed]
- Actis, G.C. Gastroenterology: 2018 Forecast. J. Gastroenterol. Forecast. 2018, 1, 1007. [Google Scholar]
- Koivula, M.J.; Eeva, T. Metal-related oxidative stress in birds. Environ. Pollut. 2010, 158, 2359–2370. [Google Scholar] [CrossRef] [PubMed]
- LaDeau, S.L.; Allan, B.F.; Leisnham, P.T.; Levy, M.Z. The ecological foundations of transmission potential and vector-borne disease in urban landscapes. Funct. Ecol. 2015, 29, 889–901. [Google Scholar] [CrossRef] [PubMed]
- Kosek, M.; Bern, C.; Guerrant, R.L. The global burden of diarrhoeal disease, as estimated from studies published between 1992 and 2000. Bull. World Health Organ. 2003, 81, 197–204. [Google Scholar] [PubMed]
- Ribaldone, D.G.; Pellicano, R.; Actis, G.C. Inflammation: A highly conserved, Janus-like phenomenon—A gastroenterologist’ perspective. J. Mol. Med. 2018. [Google Scholar] [CrossRef] [PubMed]
- Oda, H.; Wakabayashi, H.; Yamauchi, K.; Abe, F. Lactoferrin and bifidobacteria. Biometals 2014, 27, 915–922. [Google Scholar] [CrossRef] [PubMed]
- LeGrand, D. Overview of Lactoferrin as a Natural Immune Modulator. J. Pediatr. 2016, 173, S10–S15. [Google Scholar] [CrossRef] [PubMed]
- Zuo, T.; Kamm, M.A.; Colombel, J.F.; Ng, S.C. Urbanization and the gut microbiota in health and inflammatory bowel disease. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 440–452. [Google Scholar] [CrossRef] [PubMed]
- Ng, S.C.; Tang, W.; Ching, J.Y.; Wong, M.; Chow, C.M.; Hui, A.J.; Wong, T.C.; Leung, V.K.; Tsang, S.W.; Yu, H.H.; et al. Incidence and phenotype of inflammatory bowel disease based on results from the Asia-pacific Crohn’s and colitis epidemiology study. Gastroenterology 2013, 145, 158–165.e2. [Google Scholar] [CrossRef] [PubMed]
- Soon, S.; Molodecky, N.A.; Rabi, D.M.; Ghali, W.A.; Barkema, H.W.; Kaplan, G.G. The relationship between urban environment and the inflammatory bowel diseases: A systematic review and meta-analysis. BMC Gastroenterol. 2012, 12, 51. [Google Scholar] [CrossRef] [PubMed]
- De Filippo, C.; Cavalieri, D.; Di Paola, M.; Ramazzotti, M.; Poullet, J.B.; Massart, S.; Collini, S.; Pieraccini, G.; Lionetti, P. Impact of diet in shaping gut microbiota revealed by a comparative study in children from Europe and rural Africa. Proc. Natl. Acad. Sci. USA 2010, 107, 14691–14696. [Google Scholar] [CrossRef] [PubMed]
- Kostic, A.D.; Xavier, R.J.; Gevers, D. The microbiome in inflammatory bowel disease: Current status and the future ahead. Gastroenterology 2014, 146, 1489–1499. [Google Scholar] [CrossRef] [PubMed]
- Ottman, N.; Smidt, H.; de Vos, W.M.; Belzer, C. The function of our microbiota: Who is out there and what do they do? Front. Cell. Infect. Microbiol. 2012, 2, 104. [Google Scholar] [CrossRef] [PubMed]
- Moustafa, A.; Li, W.; Anderson, E.L.; Wong, E.H.M.; Dulai, P.S.; Sandborn, W.J.; Biggs, W.; Yooseph, S.; Jones, M.B.; Venter, J.C.; et al. Genetic risk, dysbiosis, and treatment stratification using host genome and gut microbiome in inflammatory bowel disease. Clin. Transl. Gastroenterol. 2018, 9, e132. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Xu, Z.Z.; He, Y.; Yang, Y.; Liu, L.; Lin, Q.; Nie, Y.; Li, M.; Zhi, F.; Liu, S.; et al. Gut Microbiota Offers Universal Biomarkers across Ethnicity in Inflammatory Bowel Disease Diagnosis and Infliximab Response Prediction. mSystems 2018, 3, e00188-17. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Tian, Y.; Zhu, W.; Gong, J.; Gu, L.; Zhang, W.; Guo, Z.; Li, N.; Li, J. Cesarean delivery and risk of inflammatory bowel disease: A systematic review and meta-analysis. Scand. J. Gastroenterol. 2014, 49, 834–844. [Google Scholar] [CrossRef] [PubMed]
- Browne, H.P.; Neville, B.A.; Forster, S.C.; Lawley, T.D. Transmission of the gut microbiota: Spreading of health. Nat. Rev. Microbiol. 2017, 15, 531–543. [Google Scholar] [CrossRef] [PubMed]
- Ribaldone, D.G.; Fagoonee, S.; Colombini, J.; Saracco, G.; Astegiano, M.; Pellicano, R. Helicobacter Pylori infection and asthma: Is there a direct or an inverse association? A meta-analysis. World J. Meta-Anal. 2016, 4, 63–68. [Google Scholar] [CrossRef]
- Alonso-López, D.; Gutierrez, M.A.; Lopes, K.P.; Prieto, C.; Santamaría, R.; De Las Rivas, J. APID interactomes: Providing proteome-based interactomes with controlled quality for multiple species and derived networks. Nucleic Acid Res. 2016, 44, W529–W535. [Google Scholar] [CrossRef] [PubMed]
- Fiocchi, C. Inflammatory Bowel Disease: Complexity and Variability Need Integration. Front. Med. (Lausanne) 2018, 5, 75. [Google Scholar] [CrossRef] [PubMed]
- De Souza, H.S.P.; Fiocchi, C.; Iliopoulos, D. The IBD interactome: An integrated view of aetiology, pathogenesis and therapy. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 739–749. [Google Scholar] [CrossRef] [PubMed]

© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ribaldone, D.G.; Pellicano, R.; Actis, G.C. Pathogenesis of Inflammatory Bowel Disease: Basic Science in the Light of Real-World Epidemiology. Gastrointest. Disord. 2019, 1, 129-146. https://doi.org/10.3390/gidisord1010010
Ribaldone DG, Pellicano R, Actis GC. Pathogenesis of Inflammatory Bowel Disease: Basic Science in the Light of Real-World Epidemiology. Gastrointestinal Disorders. 2019; 1(1):129-146. https://doi.org/10.3390/gidisord1010010
Chicago/Turabian StyleRibaldone, Davide Giuseppe, Rinaldo Pellicano, and Giovanni C. Actis. 2019. "Pathogenesis of Inflammatory Bowel Disease: Basic Science in the Light of Real-World Epidemiology" Gastrointestinal Disorders 1, no. 1: 129-146. https://doi.org/10.3390/gidisord1010010
APA StyleRibaldone, D. G., Pellicano, R., & Actis, G. C. (2019). Pathogenesis of Inflammatory Bowel Disease: Basic Science in the Light of Real-World Epidemiology. Gastrointestinal Disorders, 1(1), 129-146. https://doi.org/10.3390/gidisord1010010