Fluorinated Surfactant Adsorption on Mineral Surfaces: Implications for PFAS Fate and Transport in the Environment



Abstract
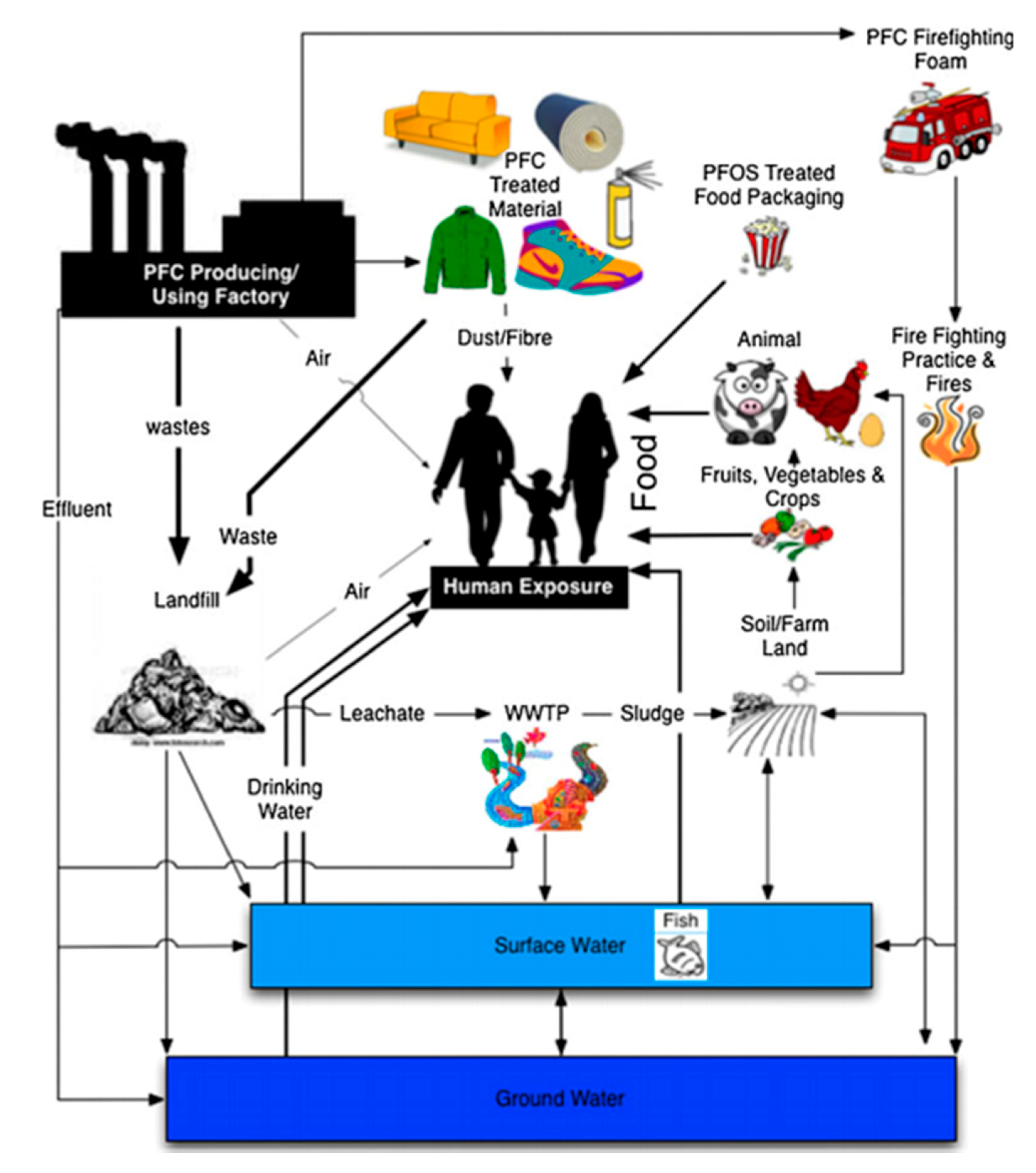
1. Introduction
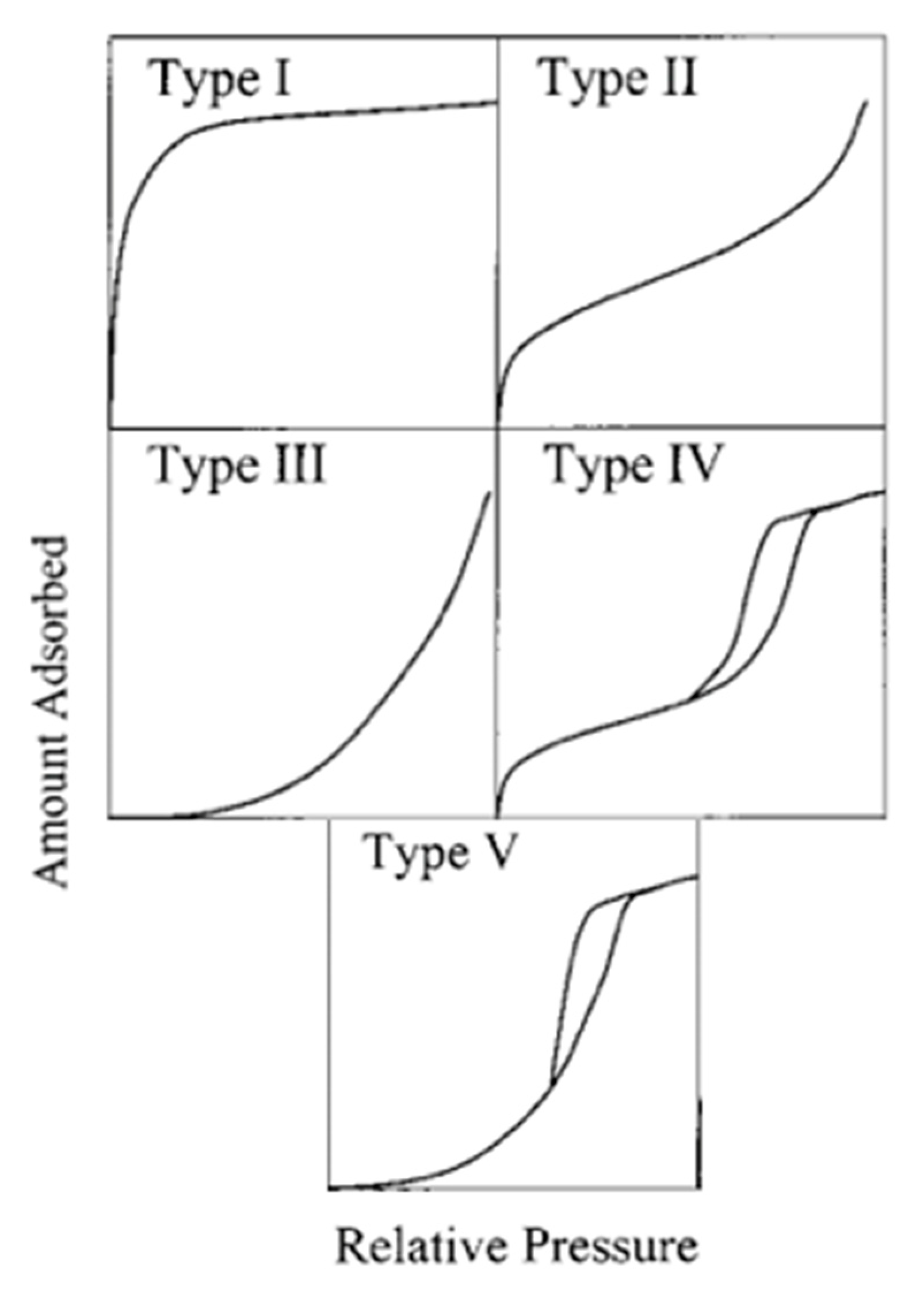
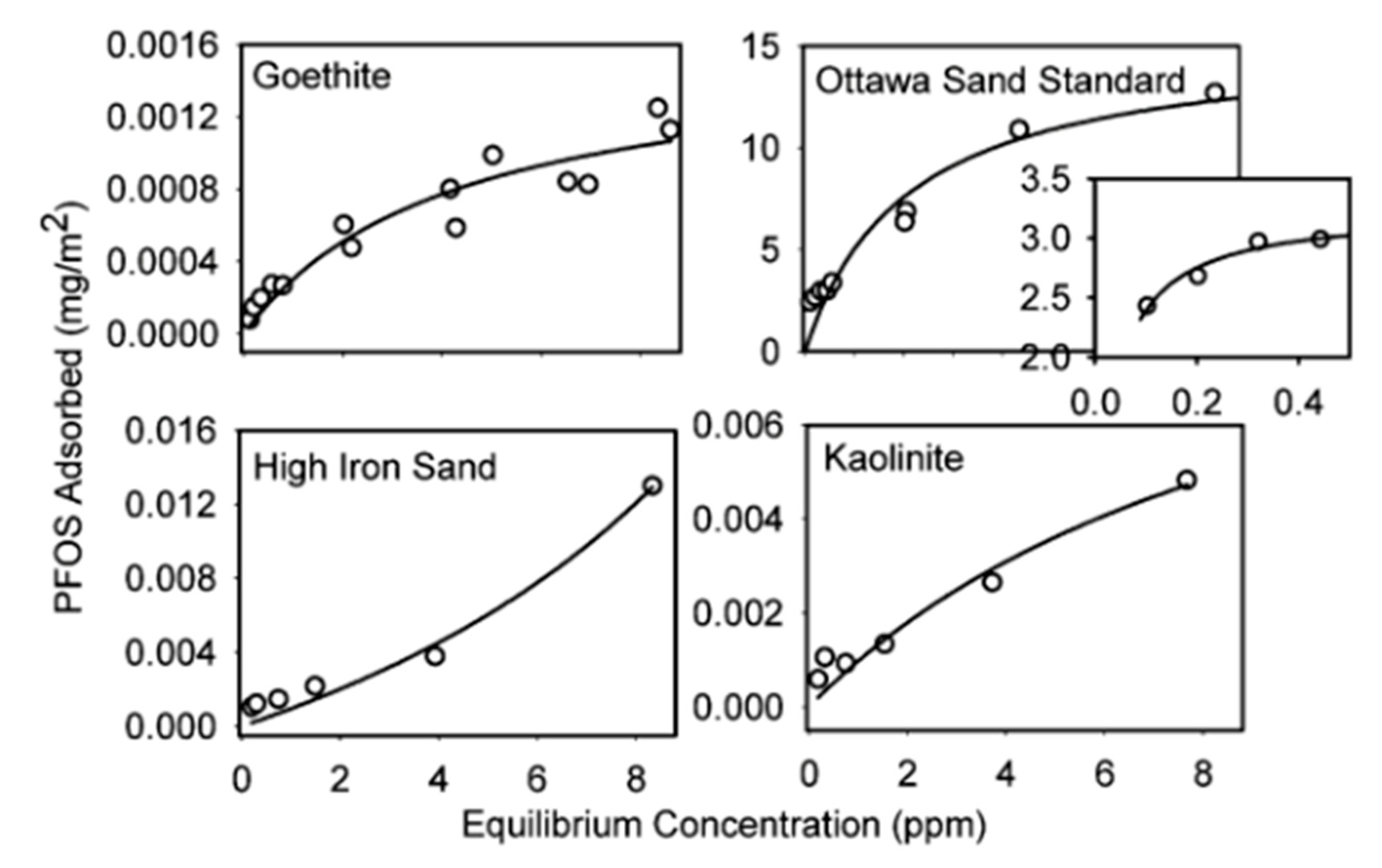
2. Adsorption Isotherm
3. Adsorption Thermodynamics
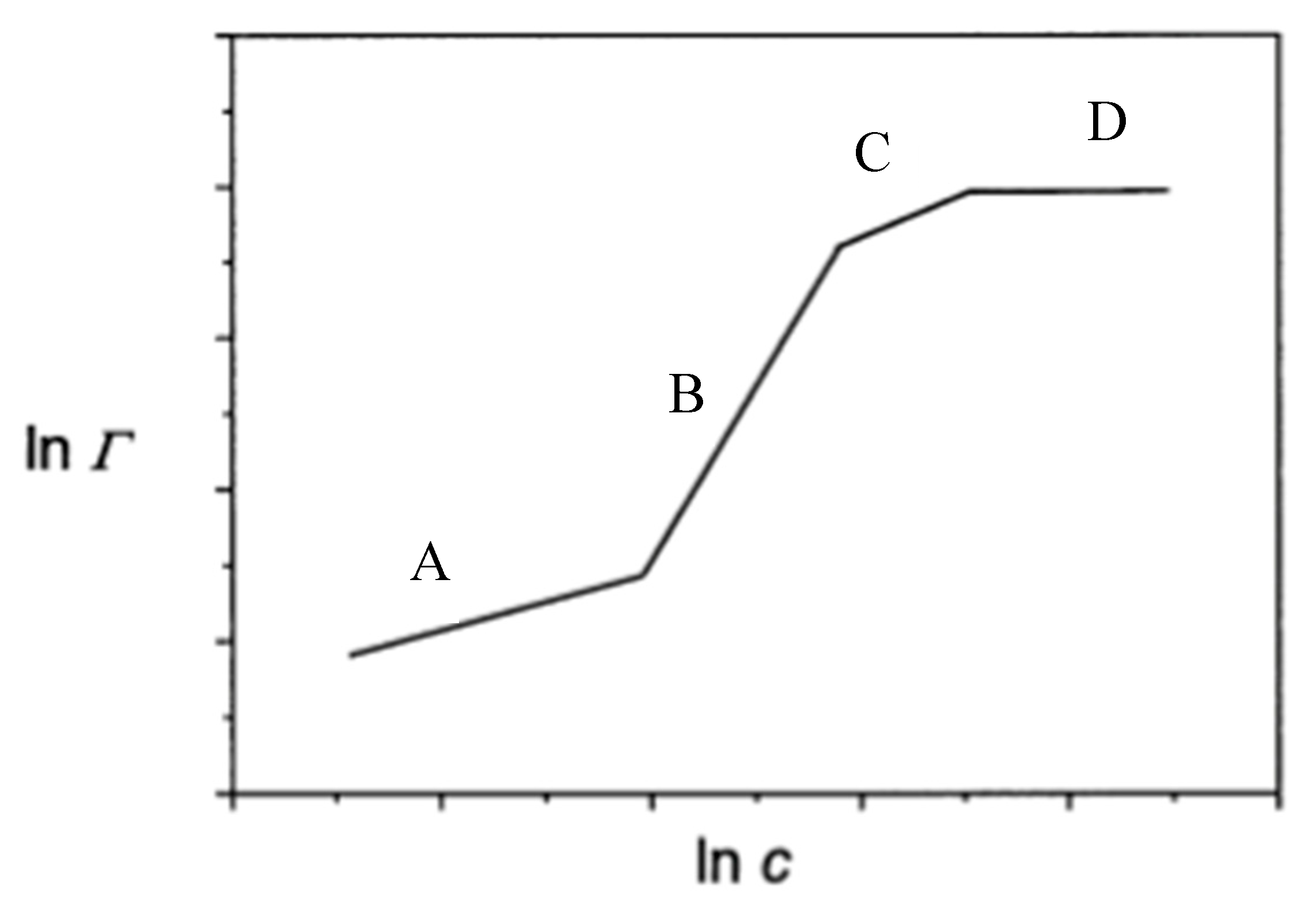
4. Adsorption Mechanism
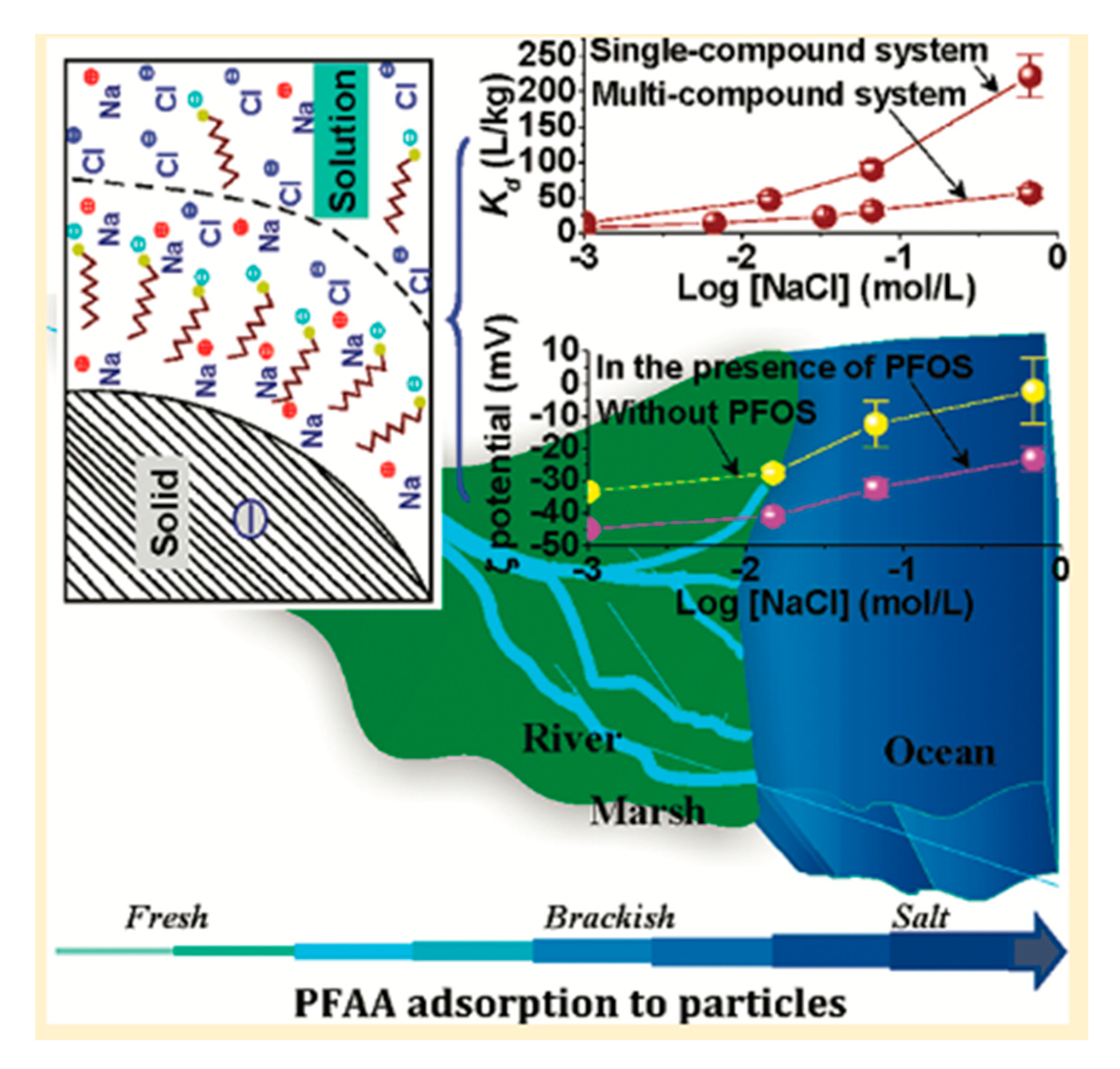
4.1. Electrostatic Interactions
4.2. Hydrophobic Interactions
4.3. Hydrogen Bonding
4.4. Ligand Ion Exchange
5. Factors That Affect Adsorption
5.1. Change in pH on Minerals and Aqueous Environment
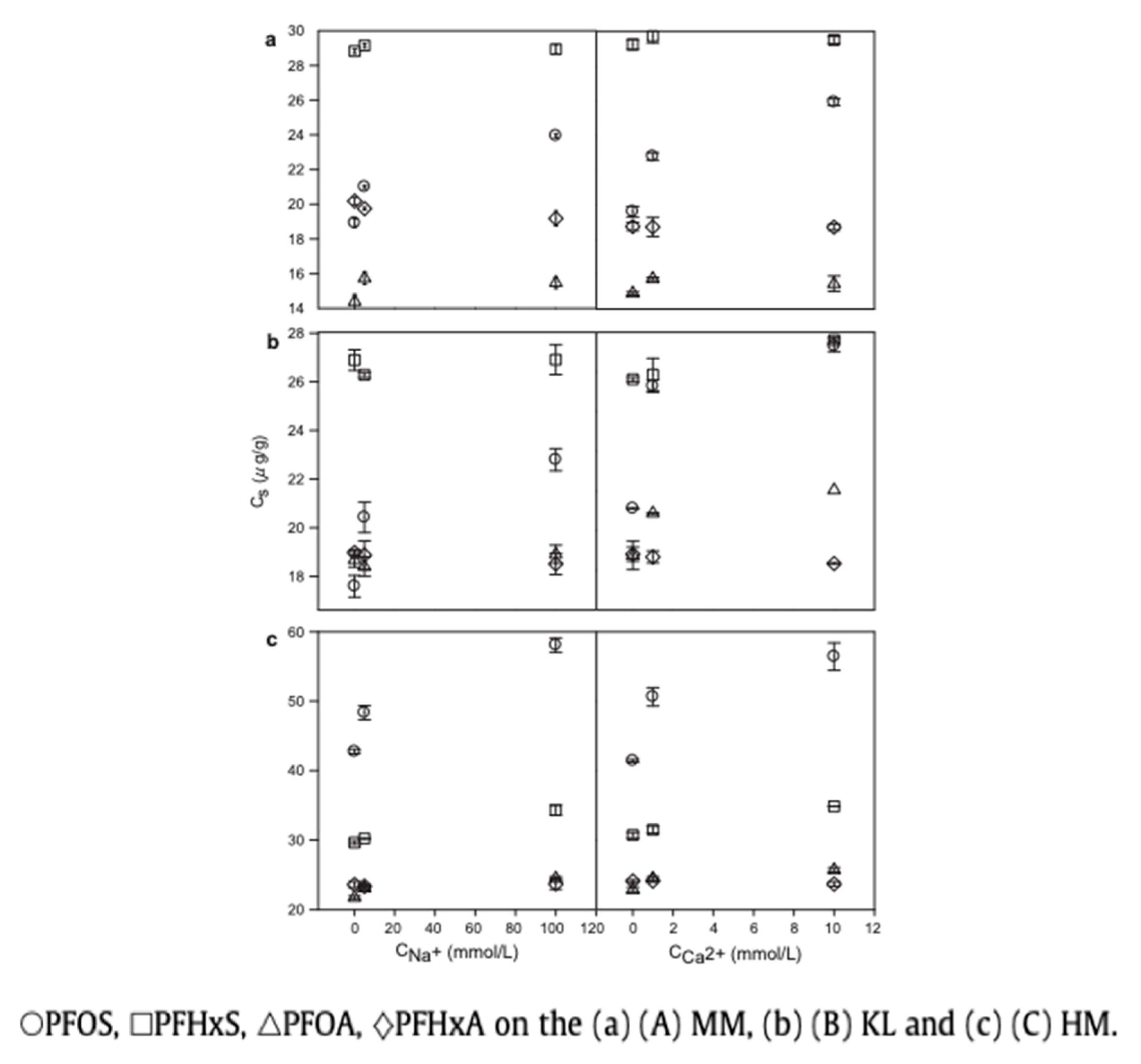
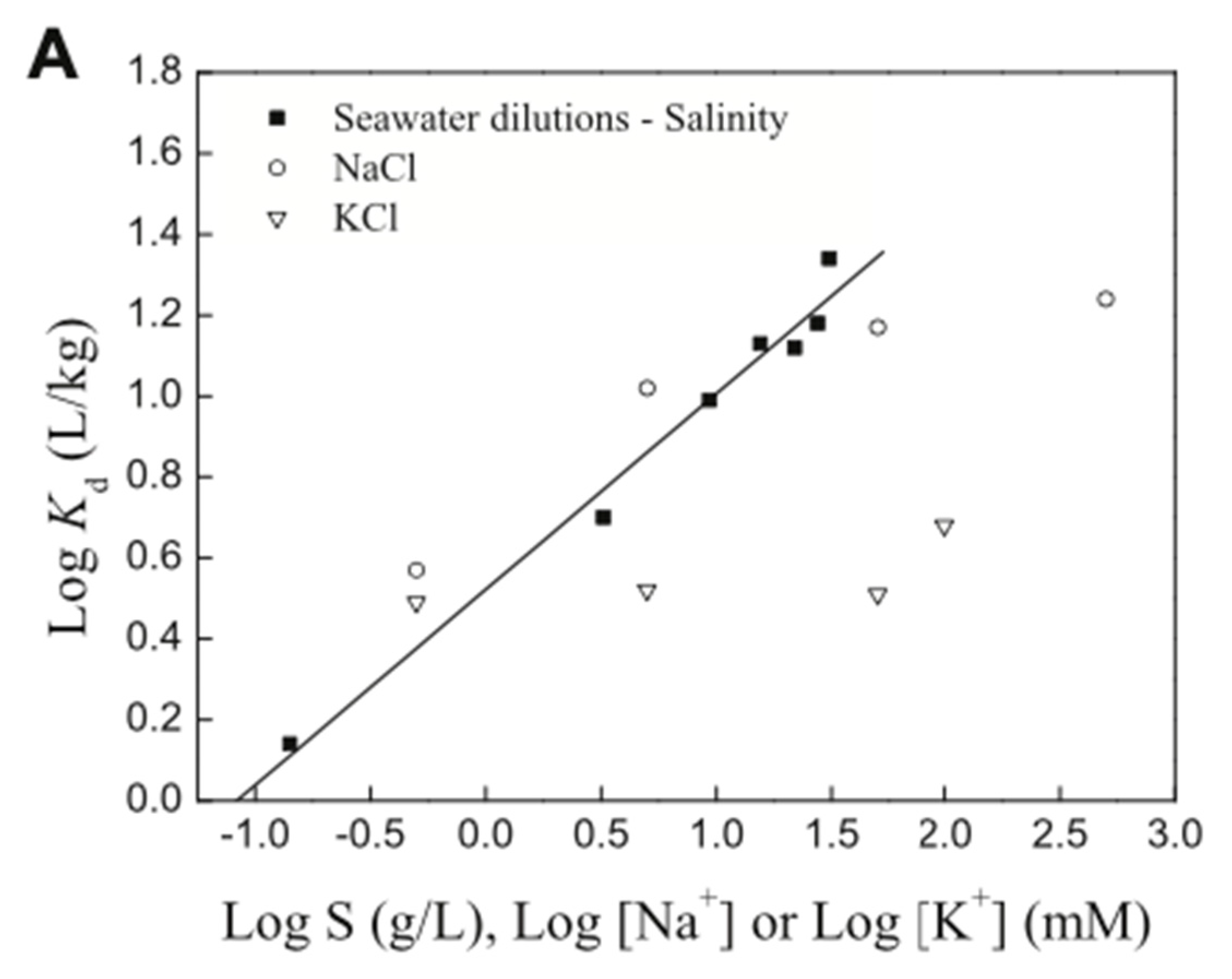
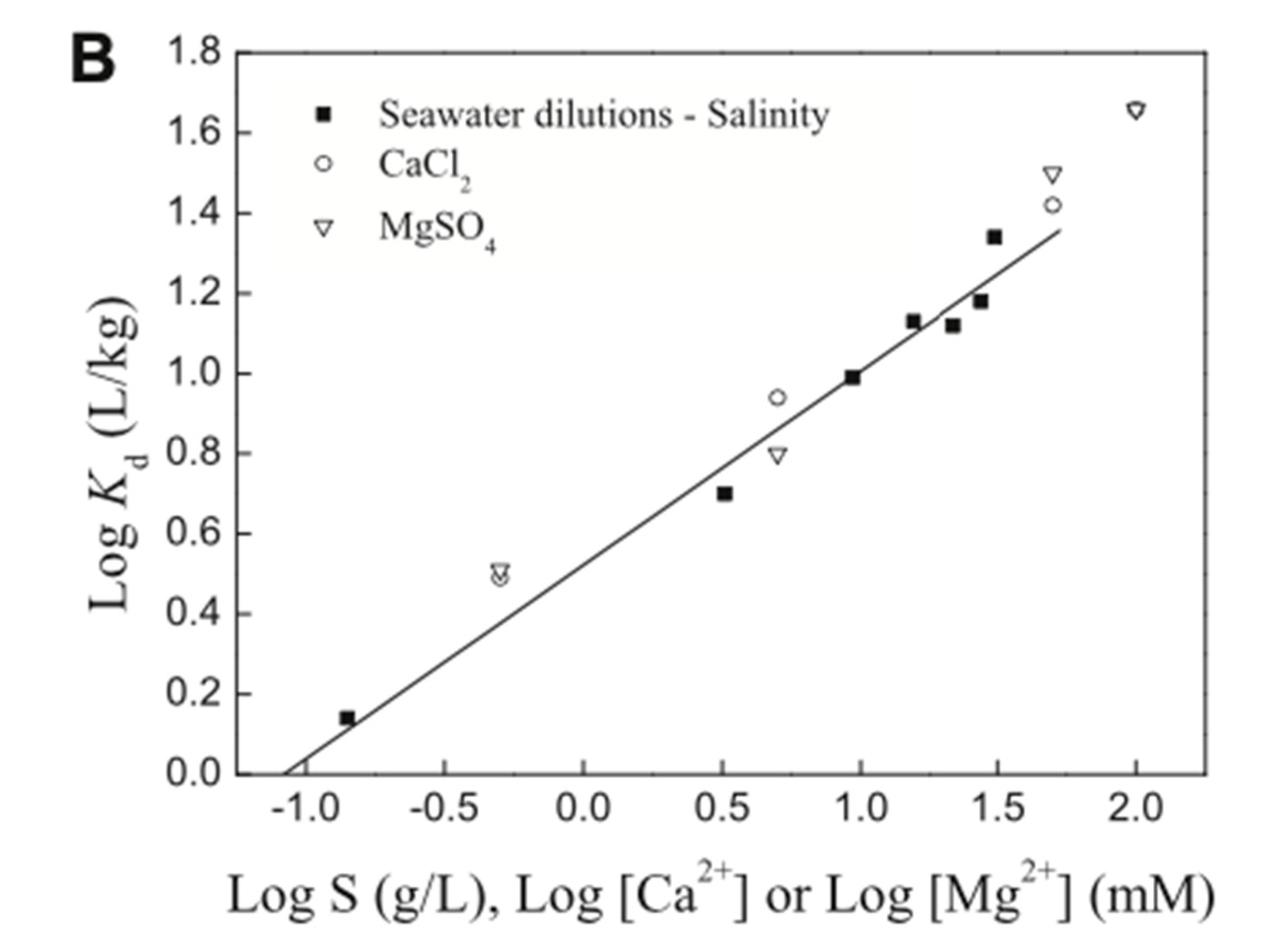
5.2. Presence of Na+, Ca2+, and Other Ions in the Aqueous Environment
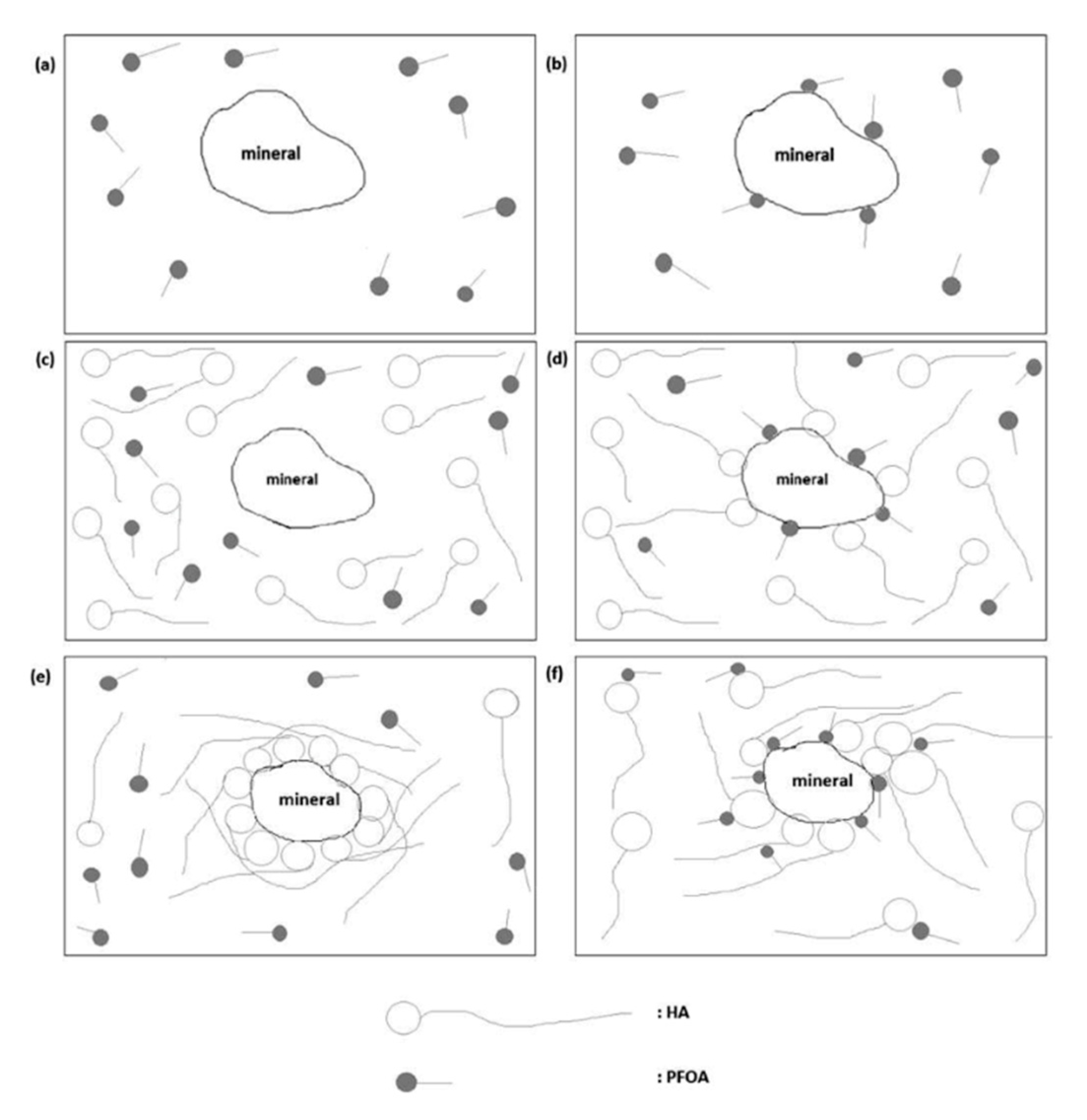
5.3. Humic Acid Presence during Adsorption
5.4. Surfactant Chemistry
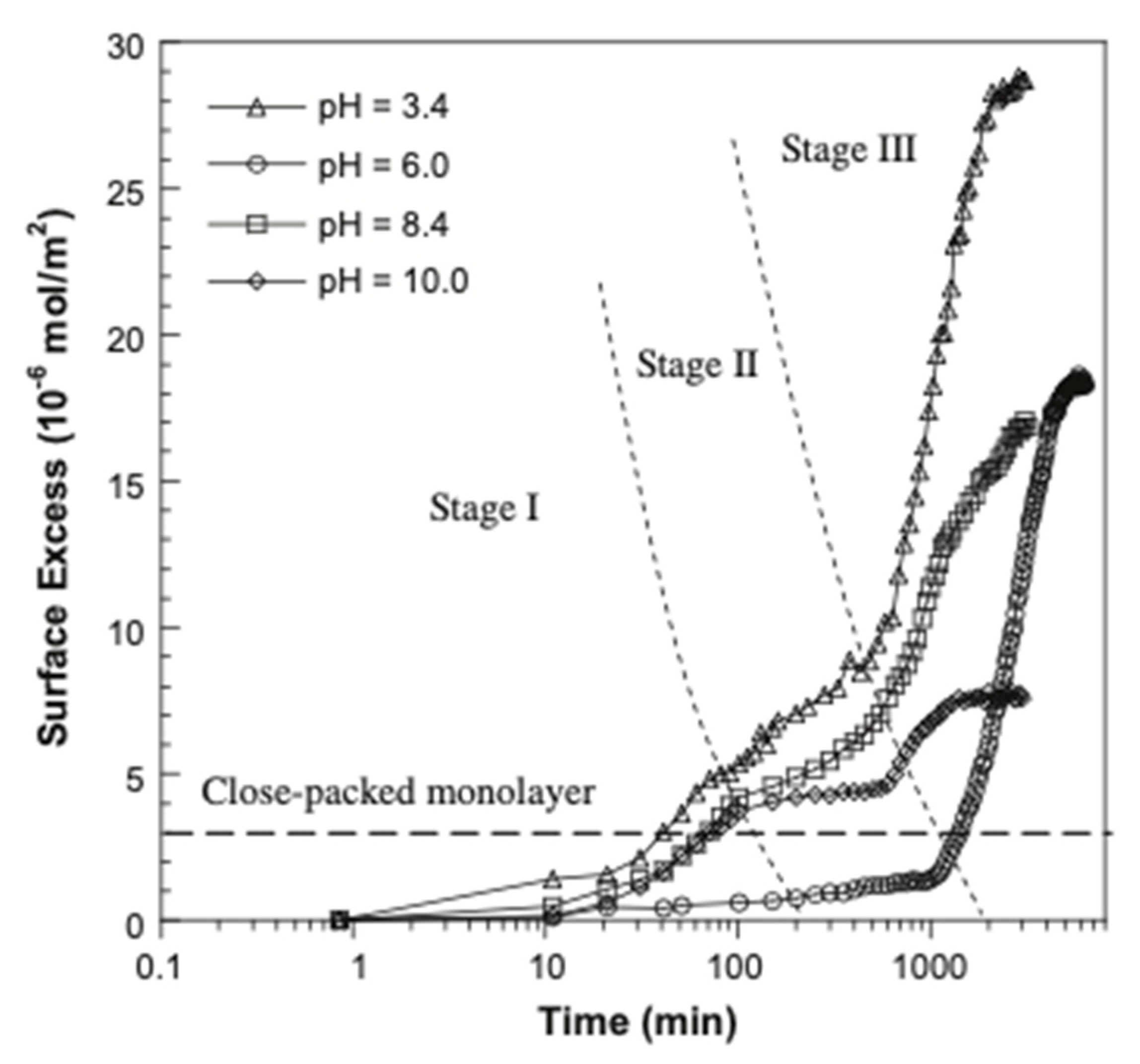
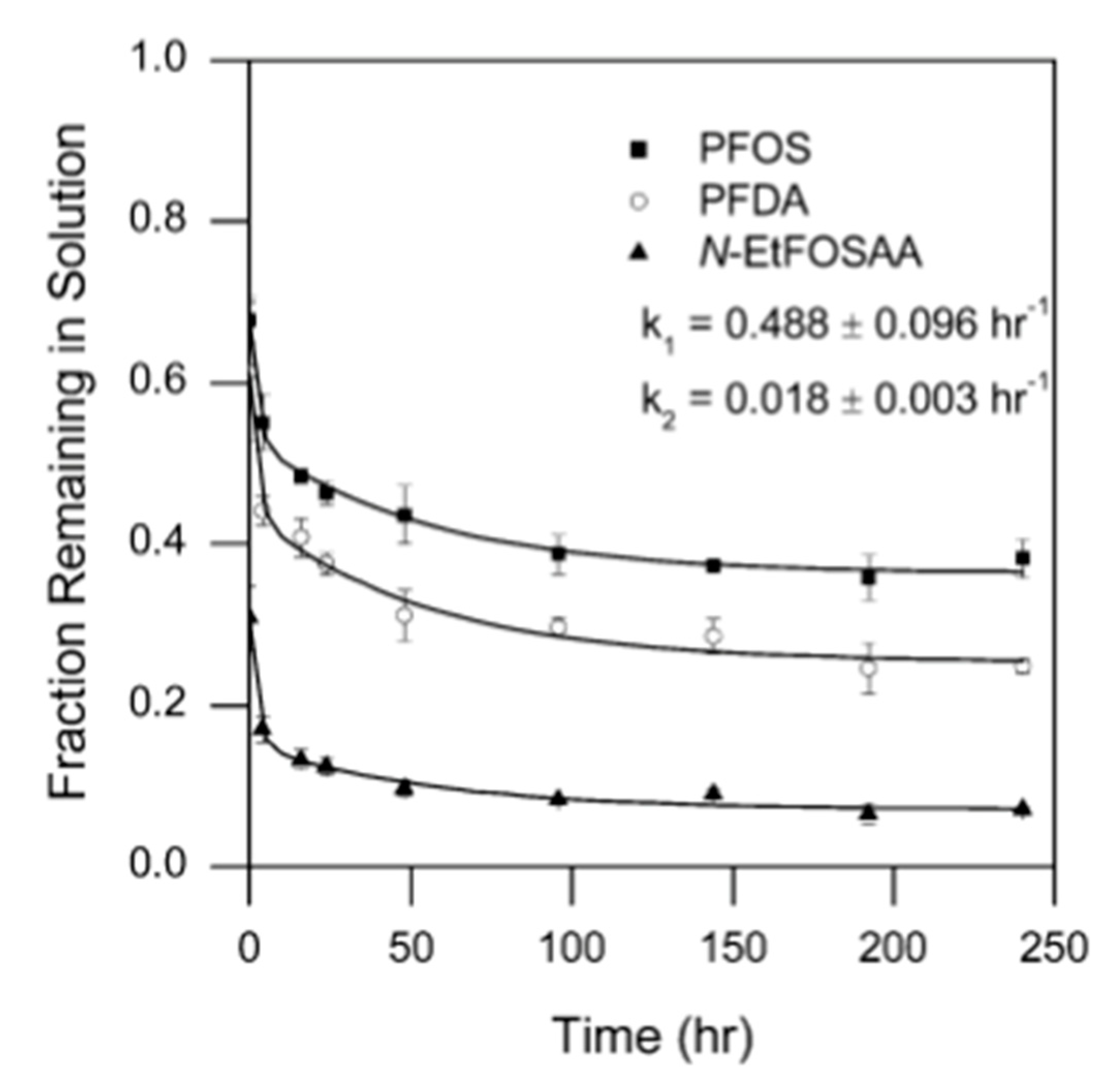
6. Adsorption Kinetics
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Villanueva, C.M.; Durand, G.; Coutte, M.B.; Chevrier, C.; Cordier, S. Atrazine in municipal drinking water and risk of low birth weight, preterm delivery, and small-for-gestational-age status. Occup. Environ. Med. 2005, 62, 400–405. [Google Scholar] [CrossRef] [PubMed]
- Agency for Toxic Substances and Disease Registry. Toxicological Profile for Arsenic; ATSDR/TP-88/02; CAS Number: 7440-38-2; U.S. Public Health Service: Atlanta, GA, USA, 1989. Available online: https://www.atsdr.cdc.gov/ToxProfiles/tp2.pdf (accessed on 22 September 2020).
- Cragin, L.A.; Kesner, J.S.; Bachand, A.M.; Barr, D.B.; Meadows, J.W.; Krieg, E.F.; Reif, J.S. Menstrual cycle characteristics and reproductive hormone levels in women exposed to atrazine in drinking water. Environ. Res. 2011, 111, 1293–1301. [Google Scholar] [CrossRef] [PubMed]
- Winchester, P.D.; Huskins, J.; Ying, J. Agrichemicals in surface water and birth defects in the United States. Acta Paediatr. 2009, 98, 664–669. [Google Scholar] [CrossRef] [PubMed]
- Vahter, M. Mechanisms of arsenic biotransformation. Toxicology 2002, 18, 211–217. [Google Scholar] [CrossRef]
- Tchounwou, P.B.; Yedjou, C.G.; Patlolla, A.K.; Sutton, D.J. Heavy metal toxicity and the environment. Exp. Suppl. 2012, 101, 133–164. [Google Scholar] [CrossRef]
- Ali, H.; Khan, E.; Ilahi, I. Environmental chemistry and ecotoxicology of hazardous heavy metals: Environmental persistence, toxicity, and bioaccumulation. J. Chem. 2019, 2019, 6730305. [Google Scholar] [CrossRef]
- Ashraf, M.A. Persistent organic pollutants (POPs): A global issue, a global challenge. Environ. Sci. Pollut. Res. 2017, 24, 4223–4227. [Google Scholar] [CrossRef]
- Guo, W.; Pan, B.; Sakkiah, S.; Yavas, G.; Ge, W.; Zou, W.; Tong, W.; Hong, H. Persistent organic pollutants in food: Contamination sources, health effects and detection methods. Int. J. Environ. Res. Public Health 2019, 16, 4361. [Google Scholar] [CrossRef]
- United States Environmental Protection Agency. DuPont Agrees to Lower Limit of PFOA in Drinking Water, Dupont Washington Works; United States Environmental Protection Agency: Washington, DC, USA, 2009.
- Lindstrom, A.B.; Strynar, M.J.; Libelo, E.L. Polyfluorinated compounds: Past, present, and future. Environ. Sci. Technol. 2011, 45, 7954–7961. [Google Scholar] [CrossRef]
- Hellsing, M.S.; Josefsson, S.; Hughes, A.V.; Ahrens, L. Sorption of perfluoroalkyl substances to two types of minerals. Chemosphere 2016, 159, 385–391. [Google Scholar] [CrossRef]
- Xing, R.; Rankin, S.E. Three stage multilayer formation kinetics during adsorption of an anionic fluorinated surfactant onto germanium. 1. Concentration Effect. J. Phys. Chem. B 2006, 110, 295–304. [Google Scholar] [CrossRef] [PubMed]
- Kothawala, N.; Köhler, S.J.; Östlund, A.; Wiberg, K.; Ahres, L. Influence of dissolved organic matter concentration and composition on the removal efficiency of perfluoroalkyl substances (PFASs) during drinking water treatment. Water Res. 2017, 121, 320–328. [Google Scholar] [CrossRef] [PubMed]
- MacManus-Spencer, L.A.; Tse, M.L.; Hebert, P.C.; Bischel, H.N.; Luthy, R.G. Binding of perfluorocarboxylates to serum albumin: A comparison of analytical methods. Anal. Chem. 2010, 82, 974–981. [Google Scholar] [CrossRef]
- Sorli, J.B.; Lag, M.; Ekeren, L.; Perez-Gil, J.; Haug, L.S.; Da Silva, E.; Matrod, M.N.; Gutzkow, K.B.; Lindeman, B. Per- and polyfluoroalkyl substances (PFASs) modify lung surfactant function and pro-inflammatory responses in human bronchial epithelial cells. Toxicol. In Vitro 2020, 62, 104656. [Google Scholar] [CrossRef]
- Kissa, E. Fluorinated Surfactants and Repellents; CRC Press: Boca Raton, FL, USA, 2001. [Google Scholar]
- Mukerjee, P.; Gumkowski, M.J.; Chan, C.C.; Sharma, R. Determination of critical micellization concentrations of perfluorocarboxylates using ultraviolet spectroscopy: Some unusual counterion effects. J. Phys. Chem. 1990, 94, 8832–8835. [Google Scholar] [CrossRef]
- Wang, Z.; MacLeod, M.; Cousins, I.T.; Scheringer, M.; Hungerbühler, K. Using COSMOtherm to predict physicochemical properties of poly- and perfluorinated alkyl substances (PFASs). Environ. Chem. 2011, 8, 389–398. [Google Scholar] [CrossRef]
- Centers for Disease Control and Prevention. An Overview of Perfluoroalkyl and Polyfluoroalkyl Substances and Interim Guidance for Clinicians Responding to Patient Exposure Concerns. 2017. Available online: https://stacks.cdc.gov/view/cdc/77114 (accessed on 22 September 2020).
- Interstate Technology and Regulatory Council. History and Use of Per- and Polyfluoroalkyl Substances (PFAS). 2017. Available online: https://pfas-1.itrcweb.org/fact_sheets_page/PFAS_Fact_Sheet_History_and_Use_April2020.pdf (accessed on 22 September 2020).
- Du, Z.W.; Deng, S.B.; Bei, Y.; Huang, Q.; Wang, B.; Huang, J.; Yu, G. Adsorption behavior and mechanism of perfluorinated compounds on various adsorbents—A review. J. Hazard. Mater. 2014, 274, 443–454. [Google Scholar] [CrossRef]
- Trudel, D.; Horowitz, L.; Wormuth, M.; Scheringer, M.; Cousins, I.T.; Hungerbuhler, K. Estimating consumer exposure to PFOS and PFOA. Risk Anal. 2008, 28, 251–269. [Google Scholar] [CrossRef]
- Hu, X.C.; Andrews, D.Q.; Lindstrom, A.B.; Bruton, T.A.; Schaider, L.A.; Grandjean, P.; Lohmann, R.; Carignan, C.C.; Blum, A.; Balan, S.A.; et al. Detection of poly- and perfluoroalkyl substances (PFASs) in U.S. drinking water linked to industrial sites, military fire training areas, and wastewater treatment plants. Environ. Sci. Technol. Lett. 2016, 3, 344–350. [Google Scholar] [CrossRef]
- Banzhaf, S.; Filipovic, M.; Lewis, J.; Sparrenbom, C.J.; Barthel, R. A review of contamination of surface-, ground-, and drinking water in Sweden by perfluoroalkyl and polyfluoroalkyl substances (PFASs). Ambio 2017, 46, 335–346. [Google Scholar] [CrossRef]
- Oliaei, F.; Kriens, D.; Weber, R.; Watson, A. PFOS and PFC releases and associated pollution from a PFC production plant in Minnesota (USA). Environ. Sci. Pollut. Res. 2013, 20, 1977–1992. [Google Scholar] [CrossRef] [PubMed]
- Steenland, K.; Fletcher, T.; Savitz, D.A. Epidemiologic evidence on the health effects of perfluorooctanoic acid (PFOA). Environ. Health Perspect. 2010, 118, 1100–1108. [Google Scholar] [CrossRef] [PubMed]
- United States Environmental Protection Agency. Technical Fact Sheet-Perfluorooctane Sulfonate (PFOS) and Perfluorooctanoic Acid (PFOA); EPA: Washington, DC, USA, 2017.
- Buck, R.C.; Franklin, J.; Berger, U.; Conder, J.M.; Cousins, I.T.; De Voogt, P.; Jensen, A.A.; Kannan, K.; Mabury, S.A.; Van Leeuwen, S.P. Perfluoroalkyl and polyfluoroalkyl substances in the environment: Terminology, classification, and origins. Integr. Environ. Assess. Manag. 2011, 7, 513–541. [Google Scholar] [CrossRef] [PubMed]
- Higgins, C.P.; Field, J.A.; Criddle, C.S.; Luthy, R.G. Quantitative determination of perfluorochemicals in sediments and domestic sludge. Environ. Sci. Technol. 2005, 39, 3946–3956. [Google Scholar] [CrossRef] [PubMed]
- Moody, C.A.; Hebert, G.N.; Strauss, S.H.; Field, J.A. Occurrence and persistence of perfluorooctanesulfonate and other perfluorinated surfactants in groundwater at a fire-training area at Wurtsmith Air Force Base, Michigan, USA. J. Environ. Monit. 2003, 5, 341–345. [Google Scholar] [CrossRef]
- Moody, C.A.; Martin, J.W.; Kwan, W.C.; Muir, D.C.; Mabury, S.A. Monitoring perfluorinated surfactants in biota and surface water samples following an accidental release of fire-fighting foam into Etobicoke Creek. Environ. Sci. Technol. 2002, 36, 545–551. [Google Scholar] [CrossRef]
- Wang, Z.Y.; DeWitt, J.C.; Higgins, C.P.; Cousins, I.T. A never-ending story of per- and polyfluoroalkyl substances (PFASs)? Environ. Sci. Technol. 2017, 51, 2508–2518. [Google Scholar] [CrossRef]
- 3M Company. Fluorochemical Use, Distribution and Release Overview; US EPA Public Docket AR226–0550; 3M Company: Saint Paul, MN, USA, 1999. [Google Scholar]
- Favreau, P.; Poncioni-Rothlisberger, C.; Place, B.J.; Bouchex-Bellomie, H.; Weber, A.; Tremp, J.; Field, J.A.; Kohler, M. Multianalyte profiling of per- and polyfluoroalkyl substances (PFASs) in liquid commercial products. Chemosphere 2017, 171, 491–501. [Google Scholar] [CrossRef]
- Bečanová, J.; Melymuk, L.; Vojta, Š.; Komprdová, K.; Klánová, J. Screening for perfluoroalkyl acids in consumer products, building materials and wastes. Chemosphere 2016, 164, 322–329. [Google Scholar] [CrossRef]
- United States Environmental Protection Agency. Lifetime Health Advisories and Health Effects Support Documents for Perfluorooctanoic Acid and Perfluorooctane Sulfonate; Document Number: 2016-12361; Environmental Protection Agency: Washington, DC, USA, 2016; pp. 33250–33251. Available online: https://www.federalregister.gov/documents/2016/05/25/2016-12361/lifetime-health-advisories-and-health-effects-support-documents-for-perfluorooctanoic-acid-and (accessed on 22 September 2020).
- Blum, A.; Balan, S.A.; Scheringer, M.; Trier, X.; Goldenman, G.; Cousins, I.T.; Diamond, M.; Fletcher, T.; Higgins, C.; Lindeman, A.E.; et al. The Madrid statement on poly- and perfluoroalkyl substances (PFASs). Environ. Health Perspect. 2015, 123, A107–A111. [Google Scholar] [CrossRef]
- Ng, C.; Hungerbuhler, K. Bioaccumulation of perfluorinated alkyl acids: Observations and models. Environ. Sci. Technol. 2014, 48, 4637–4648. [Google Scholar] [CrossRef]
- Chang, E.T.; Adami, H.-O.; Boffetta, P.; Cole, P.; Starr, T.B.; Mandel, J.S. A critical review of perfluorooctanoate and perfluorooctanesulfonate exposure and cancer risk in humans. Crit. Rev. Toxicol. 2014, 44, 1–81. [Google Scholar] [CrossRef] [PubMed]
- Chang, E.T.; Adami, H.O.; Boffetta, P.; Wedner, H.J.; Mandel, J.S. A critical review of perfluorooctanoate and perfluorooctanesulfonate exposure and immunological health conditions in humans. Crit. Rev. Toxicol. 2016, 46, 279–331. [Google Scholar] [CrossRef] [PubMed]
- Xiao, F.; Zhang, X.R.; Penn, L.; Gulliver, J.S.; Simcik, M.F. Effects of monovalent cations on the competitive adsorption of perfluoroalkyl acids by kaolinite: Experimental studies and modeling. Environ. Sci. Technol. 2011, 45, 10028–10035. [Google Scholar] [CrossRef] [PubMed]
- Mudumbi, J.B.N.; Ntwampe, S.K.O.; Matsha, T.; Mekuto, L.; Itoba-Tombo, E.F. Recent developments in polyfluoroalkyl compounds research: A focus on human/environmental health impact, suggested substitutes and removal strategies. Environ. Monit. Assess. 2017, 189, 1–29. [Google Scholar] [CrossRef]
- Li, F.; Duan, J.; Tian, S.; Ji, H.; Zhu, Y.; Wei, Z.; Zhao, D. Short-chain per- and polyfluoroalkyl substances in aquatic systems: Occurrence, impacts and treatment. Chem. Eng. J. 2020, 380, 122506. [Google Scholar] [CrossRef]
- Bowman, J.S. Fluorotechnology is critical to modern life: The FluoroCouncil counterpoint to the Madrid statement. Environ. Health Perspect. 2015, 123, A112–A113. [Google Scholar] [CrossRef]
- Cousins, I.T.; Goldenman, G.; Herzke, D.; Lohmann, R.; Miller, M.; Ng, C.A.; Patton, S.; Scheringer, M.; Trier, X.; Vierke, L.; et al. The concept of essential use for determining when uses of PFASs can be phased out. J. Environ. Monit. 2019, 21, 1803–1815. [Google Scholar] [CrossRef]
- Ritscher, A.; Wang, Z.; Scheringer, M.; Boucher, J.M.; Ahrens, L.; Berger, U.; Bintein, S.; Bopp, S.K.; Borg, D.; Buser, A.M.; et al. Zurich statement on future actions on per- and polyfluoroalkyl substances (PFASs). Environ. Health Perspect. 2018, 126, 084502. [Google Scholar] [CrossRef]
- Zhang, D.; Luo, Q.; Gao, B.; Chiang, S.-Y.D.; Woodward, D.; Huang, Q. Sorption of perfluorooctanoic acid, perfluorooctane sulfonate and perfluoroheptanoic acid on granular activated carbon. Chemosphere 2016, 144, 2336–2342. [Google Scholar] [CrossRef]
- Wang, Y.J.; Niu, J.F.; Li, Y.; Zheng, T.J.; Xu, Y.; Liu, Y. Performance and mechanisms for removal of perfluorooctanoate (PFOA) from aqueous solution by activated carbon fiber. RSC Adv. 2015, 5, 86927–86933. [Google Scholar] [CrossRef]
- Rattanaoudom, R.; Visvanathan, C.; Boontanon, S.K. Removal of concentrated PFOS and PFOA in synthetic industrial wastewater by powder activated carbon and hydrotalcite. J. Water Sustain. 2012, 2, 245–258. [Google Scholar]
- Yu, J.; Lv, L.; Lan, P.; Zhang, S.; Pan, B.; Zhang, W. Effect of effluent organic matter on the adsorption of perfluorinated compounds onto activated carbon. J. Hazard. Mater. 2012, 225–226, 99–106. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Liu, Y.; Gao, B.; Ji, R.; Li, C.; Wang, S. Removal of perfluorooctanoic acid (PFOA) and perfluorooctane sulfonate (PFOS) from water by carbonaceous nanomaterials: A review. Crit. Rev. Environ. Sci. Technol. 2019, 50, 1–36. [Google Scholar] [CrossRef]
- Ateia, M.; Alsbaiee, A.; Karanfil, T.; Dichtel, W. Efficient PFAS removal by amine-functionalized sorbents: Critical review of the current literature. Environ. Sci. Technol. Lett. 2019, 6, 688–695. [Google Scholar] [CrossRef]
- Zhi, Y.; Liu, J. Adsorption of perfluoroalkyl acids by carbonaceous adsorbents: Effect of carbon surface chemistry. Environ. Pollut. 2015, 202, 168–176. [Google Scholar] [CrossRef]
- Saeidi, N.; Kopinke, F.-D.; Georgi, A. Understanding the effect of carbon surface chemistry on adsorption of perfluorinated alkyl substances. Chem. Eng. J. 2020, 381, 122689. [Google Scholar] [CrossRef]
- Ochoa-Herrera, V.; Sierra-Alvarez, R. Removal of perfluorinated surfactants by sorption onto granular activated carbon, zeolite and sludge. Chemosphere 2008, 72, 1588–1593. [Google Scholar] [CrossRef]
- Ji, B.; Kang, P.; Wei, T.; Zhao, Y. Challenges of aqueous per- and polyfluoroalkyl substances (PFASs) and their foreseeable removal strategies. Chemosphere 2020, 250, 126316. [Google Scholar] [CrossRef]
- Sunderland, E.M.; Hu, X.C.; Dassuncao, C.; Tokranov, A.K.; Wagner, C.C.; Allen, J.G. A review of the pathways of human exposure to poly- and perfluoroalkyl substances (PFASs) and present understanding of health effects. J. Expo. Sci. Environ. Epidemiol. 2019, 29, 131–147. [Google Scholar] [CrossRef]
- Brusseau, M.L.; Khan, N.; Wang, Y.; Yan, N.; Van Glubt, S.; Carroll, K.C. Nonideal transport and extended elution tailing of PFOS in soil. Environ. Sci. Technol. 2019, 53, 10654–10664. [Google Scholar] [CrossRef] [PubMed]
- Lyu, X.; Liu, X.; Sun, Y.; Ji, R.; Gao, B.; Wu, J. Transport and retention of perfluorooctanoic acid (PFOA) in natural soils: Importance of soil organic matter and mineral contents, and solution ionic strength. J. Contam. Hydrol. 2019, 225, 103477. [Google Scholar] [CrossRef]
- Kwok, K.Y.; Yamazaki, E.; Yamashita, N.; Taniyasu, S.; Murphy, M.B.; Horii, Y.; Petrick, G.; Kallerborn, R.; Kannan, K.; Murano, K.; et al. Transport of perfluoroalkyl substances (PFAS) from an arctic glacier to downstream locations: Implications for sources. Sci. Total Environ. 2013, 447, 46–55. [Google Scholar] [CrossRef] [PubMed]
- Simon, J.A.; Abrams, S.; Bradburne, T.; Bryant, D.; Burns, M.; Cassidy, D.; Cherry, J.; Chiang, S.Y.; Cox, D.; Crimi, M.; et al. PFAS Experts Symposium: Statements on regulatory policy, chemistry and analytics, toxicology, transport/fate, and remediation for per- and polyfluoroalkyl substances (PFAS) contamination issues. Remediation 2019, 29, 31–48. [Google Scholar] [CrossRef]
- Liu, T.; Gu, Y.; Xing, D.; Dong, W.; Wu, X. Rapid and high-capacity adsorption of PFOS and PFOA by regenerable ammoniated magnetic particle. Environ. Sci. Pollut. Res. 2018, 25, 13813–13822. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.Q.; Zhang, W.L.; Liang, Y.N. Adsorption of perfluoroalkyl and polyfluoroalkyl substances (PFASs) from aqueous solution—A review. Sci. Total Environ. 2019, 694, 133606. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Zhang, C.; Yu, Y.X.; Han, J.B. Sorption of perfluorooctane sulfonate (PFOS) on marine sediments. Mar. Pollut. Bull. 2012, 64, 902–906. [Google Scholar] [CrossRef]
- Stebel, E.K.; Pike, K.A.; Huan, N.; Hartmann, H.A.; Klonowski, M.J.; Lawrence, M.G.; Collins, R.M.; Hefner, C.E.; Edmiston, P.L. Absorption of short-chain to long-chain perfluoroalkyl substances using swellable organically modified silica. Environ. Sci. Water Res. Technol. 2019, 5, 1854–1866. [Google Scholar] [CrossRef]
- Mejia-Avendano, S.; Zhi, Y.; Yan, B.; Liu, J. Sorption of polyfluoroalkyl surfactants on surface soils: Effect of molecular structures, soil properties, and solution chemistry. Environ. Sci. Technol. 2020, 54, 1513–1521. [Google Scholar] [CrossRef]
- Xiao, F.; Jin, B.; Golovko, S.A.; Golovko, M.Y.; Xing, B. Sorption and desorption mechanisms of cationic and zwitterionic per- and polyfluoroalkyl substances in natural soils: Thermodynamics and hysteresis. Environ. Sci. Technol. 2019, 53, 11818–11827. [Google Scholar] [CrossRef]
- Oliver, D.P.; Li, Y.; Orr, R.; Nelson, P.; Barnes, M.; McLaughlin, M.J.; Kookana, R.S. The role of surface charge and pH changes in tropical soils on sorption behaviour of per- and polyfluoroalkyl substances (PFASs). Sci. Total Environ. 2019, 673, 197–206. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Wang, P.; Qian, J.; Wang, C.; Xing, L.; Liu, J.; Tian, X.; Lu, B.; Tang, W. Effects of sediment components and TiO2 nanoparticles on perfluorooctane sulfonate adsorption properties. J. Soils Sediments 2019, 19, 2034–2047. [Google Scholar] [CrossRef]
- Li, F.; Fang, X.; Zhou, Z.; Liao, X.; Zou, J.; Yuan, B.; Sun, W. Adsorption of perfluorinated acids onto soils: Kinetics, isotherms, and influences of soil properties. Sci. Total Environ. 2019, 649, 504–514. [Google Scholar] [CrossRef] [PubMed]
- Wei, C.; Song, X.; Wang, Q.; Hu, Z. Sorption kinetics, isotherms and mechanisms of PFOS on soils with different physicochemical properties. Ecotoxicol. Environ. Saf. 2017, 142, 40–50. [Google Scholar] [CrossRef]
- Ahrens, L.; Yeung, L.W.Y.; Taniyasu, S.; Lam, P.K.S.; Yamashita, N. Partitioning of perfluorooctanoate (PFOA), perfluorooctane sulfonate (PFOS) and perfluorooctane sulfonamide (PFOSA) between water and sediment. Chemosphere 2011, 85, 731–737. [Google Scholar] [CrossRef]
- Xiang, L.; Xiao, T.; Yu, P.-F.; Zhao, H.-M.; Mo, C.-H.; Li, Y.-W.; Li, H.; Cai, Q.-Y.; Zhou, D.-M.; Wong, M.-H. Mechanism and implication of the sorption of perfluorooctanoic acid by varying soil size fractions. J. Agric. Food Chem. 2018, 66, 11569–11579. [Google Scholar] [CrossRef]
- Ololade, I.A.; Zhou, Q.; Pan, G. Influence of oxic/anoxic condition on sorption behavior of PFOS in sediment. Chemosphere 2016, 150, 798–803. [Google Scholar] [CrossRef]
- Li, Y.; Oliver, D.P.; Kookana, R.S. A critical analysis of published data to discern the role of soil and sediment properties in determining sorption of per and polyfluoroalkyl substances (PFASs). Sci. Total Environ. 2018, 628–629, 110–120. [Google Scholar] [CrossRef]
- Knight, E.R.; Janik, L.J.; Navarro, D.A.; Kookana, R.S.; McLaughlin, M.J. Predicting partitioning of radiolabelled 14C-PFOA in a range of soils using diffuse reflectance infrared spectroscopy. Sci. Total Environ. 2019, 686, 505–513. [Google Scholar] [CrossRef]
- Zhang, R.M.; Yan, W.; Jing, C.Y. Mechanistic study of PFOS adsorption on kaolinite and montmorillonite. Colloid Surf. A-Physicochem. Eng. Asp. 2014, 462, 252–258. [Google Scholar] [CrossRef]
- Yang, K.H.; Ruan, C.J.; Lin, Y.C.; Fang, M.D.; Wu, C.H.; Hong, P.K.A.; Lin, C.F. Role of dissolved organic matter in sorption of perfluorooctanoic acid to metal oxides. Water Environ. Res. 2016, 88, 779–784. [Google Scholar] [CrossRef] [PubMed]
- Bodratti, A.M.; Sarkar, B.; Alexandridis, P. Adsorption of poly(ethylene oxide)-containing amphiphilic polymers on solid-liquid interfaces: Fundamentals and applications. Adv. Colloid Interface Sci. 2017, 244, 132–163. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Somasundaran, P. Advances in adsorption of surfactants and their mixtures at solid/solution interfaces. Adv. Colloid Interface Sci. 2006, 123, 213–229. [Google Scholar] [CrossRef]
- Chang, Z.Y.; Chen, X.M.; Peng, Y.J. The adsorption behavior of surfactants on mineral surfaces in the presence of electrolytes—A critical review. Miner. Eng. 2018, 121, 66–76. [Google Scholar] [CrossRef]
- Evenas, L.; Furo, I.; Stilbs, P.; Valiullin, R. Adsorption isotherm and aggregate properties of fluorosurfactants on alumina measured by F-19 NMR. Langmuir 2002, 18, 8096–8101. [Google Scholar] [CrossRef]
- Zhou, Q.; Deng, S.B.; Yu, Q.; Zhang, Q.Y.; Yu, G.; Huang, J.; He, H.P. Sorption of perfluorooctane sulfonate on organo-montmorillonites. Chemosphere 2010, 78, 688–694. [Google Scholar] [CrossRef]
- Shafique, U.; Dorn, V.; Paschke, A.; Schuurmann, G. Adsorption of perfluorocarboylic acids at the silica surface. Chem. Commun. 2017, 53, 589–592. [Google Scholar] [CrossRef]
- De Gisi, S.; Lofrano, G.; Grassi, M.; Notarnicola, M. Characteristics and adsorption capacities of low-cost sorbents for wastewater treatment: A review. Sustain. Mater. Technol. 2016, 9, 10–40. [Google Scholar] [CrossRef]
- Jian, J.M.; Zhang, C.; Wang, F.; Lu, X.W.; Wang, F.; Zeng, E.Y. Effect of solution chemistry and aggregation on adsorption of perfluorooctanesulphonate (PFOS) to nano-sized alumina. Environ. Pollut. 2019, 251, 425–433. [Google Scholar] [CrossRef]
- Elmorsi, T.M. Equilibrium isotherms and kinetic studies of removal of methylene blue dye by adsorption onto miswak leaves as a natural adsorbent. J. Environ. Prot. 2011, 2, 817–827. [Google Scholar] [CrossRef]
- Kruk, M.; Jaroniec, M. Gas adsorption characterization of ordered organic-inorganic nanocomposite materials. Chem. Mater. 2001, 13, 3169–3183. [Google Scholar] [CrossRef]
- Johnson, R.L.; Anschutz, A.J.; Smolen, J.M.; Simcik, M.F.; Penn, R.L. The adsorption of perfluorooctane sulfonate onto sand, clay, and iron oxide surfaces. J. Chem. Eng. Data 2007, 52, 1165–1170. [Google Scholar] [CrossRef]
- Wang, F.; Liu, C.S.; Shih, K.M. Adsorption behavior of perfluorooctanesulfonate (PFOS) and perfluorooctanoate (PFOA) on boehmite. Chemosphere 2012, 89, 1009–1014. [Google Scholar] [CrossRef] [PubMed]
- Shih, K.M.; Wang, F. Adsorption behavior of perfluorochemicals (PFCs) on boehmite: Influence of solution chemistry. Procedia Environ. Sci. 2013, 18, 106–113. [Google Scholar] [CrossRef]
- Qian, J.; Shen, M.M.; Wang, P.F.; Wang, C.; Hu, J.; Hou, J.; Ao, Y.H.; Zheng, H.; Li, K.; Liu, J.J. Co-adsorption of perfluorooctane sulfonate and phosphate on boehmite: Influence of temperature, phosphate initial concentration and pH. Ecotoxicol. Environ. Saf. 2017, 137, 71–77. [Google Scholar] [CrossRef]
- IUPAC Commission on Colloid and Surface Chemistry Including Catalysis. Reporting physisorption data for gas/solid systems with special reference to the determination of surface area and porosity (Recommendations 1984). Pure Appl. Chem. 1985, 57, 603–619. [Google Scholar] [CrossRef]
- Lu, X.Y.; Deng, S.B.; Wang, B.; Huang, J.; Wang, Y.J.; Yu, G. Adsorption behavior and mechanism of perfluorooctane sulfonate on nanosized inorganic oxides. J. Colloid Interface Sci. 2016, 474, 199–205. [Google Scholar] [CrossRef]
- Zhao, L.X.; Bian, J.N.; Zhang, Y.H.; Zhu, L.Y.; Liu, Z.T. Comparison of the sorption behaviors and mechanisms of perfluorosulfonates and perfluorocarboxylic acids on three kinds of clay minerals. Chemosphere 2014, 114, 51–58. [Google Scholar] [CrossRef]
- Wang, F.; Shih, K.M. Adsorption of perfluorooctanesulfonate (PFOS) and perfluorooctanoate (PFOA) on alumina: Influence of solution pH and cations. Water Res. 2011, 45, 2925–2930. [Google Scholar] [CrossRef]
- Xing, R.; Rankin, S.E. Three stage multilayer formation kinetics during adsorption of an anionic fluorinated surfactant onto germanium: Solution pH and salt effects. J. Colloid Interface Sci. 2013, 401, 88–96. [Google Scholar] [CrossRef]
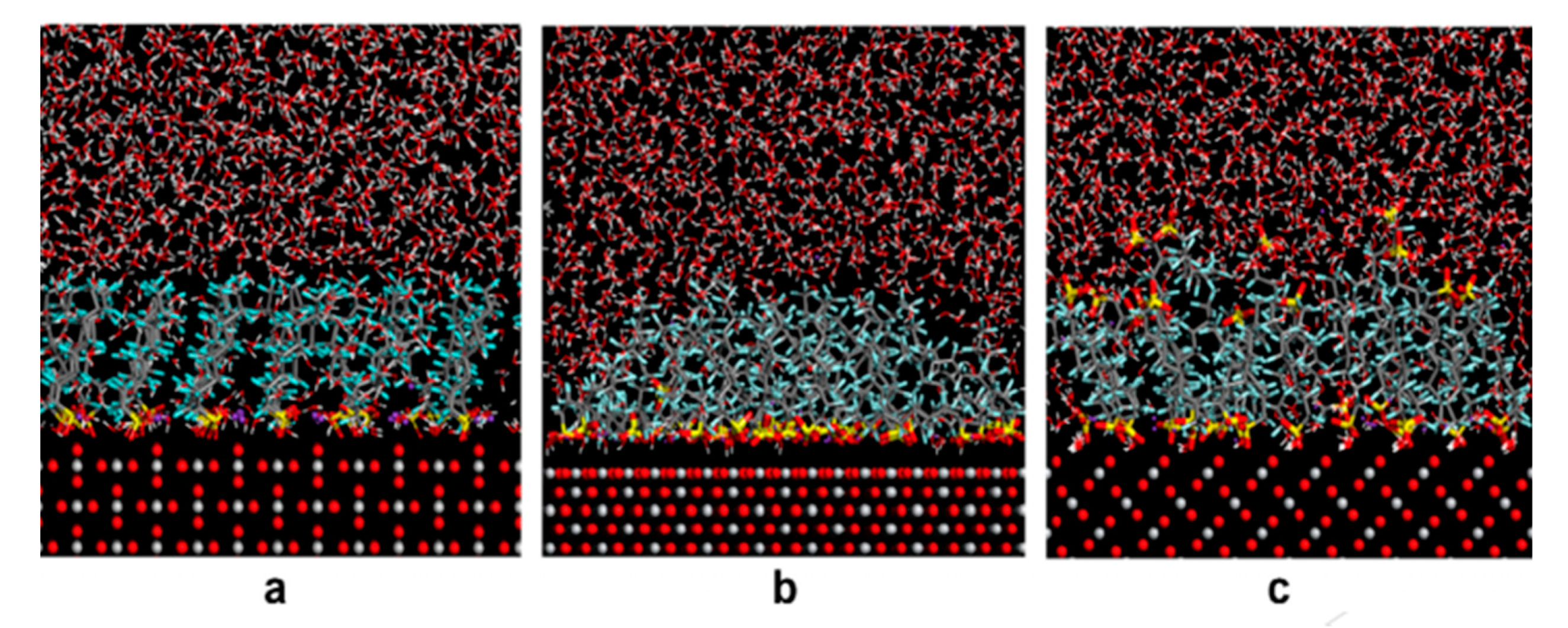
- He, G.Z.; Pan, G.; Zhang, M. Assembling structures and dynamics properties of perfluorooctane sulfonate (PFOS) at water–titanium oxide interfaces. J. Colloid Interface Sci. 2013, 405, 189–194. [Google Scholar] [CrossRef] [PubMed]
- He, G.Z.; Zhang, M.Y.; Zhou, Q.; Pan, G. Molecular dynamics simulations of structural transformation of perfluorooctane sulfonate (PFOS) at water/rutile interfaces. Chemosphere 2015, 134, 272–278. [Google Scholar] [CrossRef] [PubMed]
- He, Z.; Alexandridis, P. Micellization thermodynamics of Pluronic P123 (EO20PO70EO20) amphiphilic block copolymer in aqueous ethylammonium nitrate (EAN) solutions. Polymers 2018, 10, 32. [Google Scholar] [CrossRef] [PubMed]
- Deng, S.B.; Zhang, Q.Y.; Nie, Y.; Wei, H.R.; Wang, B.; Huang, J.; Yu, G.; Xing, B.S. Sorption mechanisms of perfluorinated compounds on carbon nanotubes. Environ. Pollut. 2012, 168, 138–144. [Google Scholar] [CrossRef]
- Gao, X.D.; Chorover, J. Adsorption of perfluorooctanoic acid and perfluorooctanesulfonic acid to iron oxide surfaces as studied by flow-through ATRFTIR spectroscopy. Environ. Chem. 2012, 9, 148–157. [Google Scholar] [CrossRef]
- Li, K.X.; Zeng, Z.X.; Xiong, J.J.; Yan, L.S.; Guo, H.Q.; Liu, S.F.; Dai, Y.H.; Chen, T. Fabrication of mesoporous Fe3O4, SiO2, CTAB-SiO2 magnetic microspheres with a core/shell structure and their efficient adsorption performance for the removal of trace PFOS from water. Colloids Surf. A Physicochem. Eng. Asp. 2015, 465, 113–123. [Google Scholar] [CrossRef]
- Trevisan, S.; Francioso, O.; Quaggiotti, S.; Nardi, S. Humic substances biological activity at the plant-soil interface: From environmental aspects to molecular factors. Plant Signal. Behav. 2010, 5, 635–643. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhi, Y.; Liu, J.; Ghoshal, S. Sorption of perfluoroalkyl acids to fresh and aged nanoscale zerovalent iron particles. Environ. Sci. Technol. 2018, 52, 6300–6308. [Google Scholar] [CrossRef]
- McNaught, A.D.; Wilkinson, A. IUPAC, Critical Micelle Concentration, Compendium of Chemical Terminology, 2nd ed.; Blackwell Scientific Publications: Oxford, UK, 1997; Available online: http://goldbook.iupac.org/terms/view/C01395 (accessed on 22 September 2020).
- Kancharla, S.; Canales, E.; Alexandridis, P. Perfluorooctanoate in aqueous urea solutions: Micelle formation, structure, and microenvironment. Int. J. Mol. Sci. 2019, 20, 5761. [Google Scholar] [CrossRef]
- Lin, Y.; Smith, T.W.; Alexandridis, P. Adsorption properties of a polymeric siloxane surfactant onto carbon black particles dispersed in mixtures of water with polar solvents. J. Colloid Interface Sci. 2002, 255, 1–9. [Google Scholar] [CrossRef]
- Sarkar, B.; Venugopal, V.; Tsianou, M.; Alexandridis, P. Adsorption of Pluronic block copolymers on silica nanoparticles. Colloids Surf. A Physicochem. Eng. Asp. 2013, 422, 155–164. [Google Scholar] [CrossRef]
- Reddy, D.H.K.; Lee, S.M. Application of magnetic chitosan composites for the removal of toxic metal and dyes from aqueous solutions. Adv. Colloid Interface Sci. 2013, 201, 68–93. [Google Scholar] [CrossRef] [PubMed]
- Atkins, R.; Craig, V.S.J.; Wanless, E.J.; Biggs, S. Mechanism of cationic surfactant adsorption at the solid-aqueous interface. Adv. Colloid Interface Sci. 2003, 103, 219–304. [Google Scholar] [CrossRef]
- Higgins, C.P.; Luthy, R.G. Sorption of perfluorinated surfactants on sediments. Environ. Sci. Technol. 2006, 40, 7251–7256. [Google Scholar] [CrossRef] [PubMed]
- Nassar, N.N. Asphaltene adsorption onto alumina nanoparticles: Kinetics and thermodynamic studies. Energy Fuels 2010, 24, 4116–4122. [Google Scholar] [CrossRef]

















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Perfluorinated Surfactant | Full Name | Chemical Formula | Chemical Structure | Molecular Weight (g/mol) | CMC (mM) | KOC |
|---|---|---|---|---|---|---|
| PFBA | Perfluorobutanoic acid | C4HF7O2 |  | 214.04 | 750 (f) | 1.12 (c) |
| PFBS | Perfluorobutanesulfonic acid | C4HF9O3S |  | 300.10 | 22 (e) | 1.14 × 10−4 (c) |
| PFPeA | Perfluoropentanoic acid | C5HF9O2 |  | 264.05 | ~250 | 0.79 (c) |
| PFHxA | Perfluorohexanoic acid | C6HF11O2 |  | 314.05 | 82 (b) | 6.92 (c) |
| PFHxS | Perfluorohexanesulfonic acid | C6HF13O3S |  | 400.12 | 12 (e) | 3.55 × 10−4 (c) |
| PFHpA | Perfluoroheptanoic acid | C7HF13O2 |  | 364.06 | 33 (g) | 6.61 (c) |
| PFOA | Perfluorooctanoic acid | C8HF15O2 |  | 414.07 | 9 (b) | 7.94 (c) |
| PFNA | Perfluorononanoic acid | C9HF17O |  | 464.08 | 3.1 (b) | 245.47 (c) |
| PFOS | Perfluorooctanesulfonic acid | C8HF17O3S |  | 500.13 | 3.1 (b) | 3.89 × 10−4 (c) |
| TEA-FOS | Tetraethylammonium Perfluorooctylsulfonate | [C8F17O3S]− [C8NH12]+ |  | 629.40 | 1.0 (a) | |
| PFDA | Perfluorodecanoic acid | C10HF19O2 |  | 514.08 | 1.5 (d) | 575.44 (c) |
| PFUnDA | Perfluoroundecanoic acid | C11HF21O2 |  | 564.09 | 0.32 (d) | 1995.26 (c) |
| PFDoDA | Perfluorododecanoic acid | C12HF23O2 |  | 614.10 | ~0.1 | 527 (h) |
| Perfluorinated Surfactant | Solvent | Mineral Surface (Adsorbent) | Mineral Surface Properties | Key Results: Adsorbed Amount/Adsorption Capacity | Adsorbed Layer Structure | Comments | Reference |
|---|---|---|---|---|---|---|---|
| PFBA | Water, Temperature: 25 °C, pH: 6.3 | Organo-montmorillonites (0.5CEC-Mt) | Particle surface | Adsorption capacity: ~0.02 mmol/g | Not reported | Surfaces not well-defined | Zhou et al. (2010) [84] |
| PFBA | Deionized water (alone) and NaCl (50 mM) (separate experiments), Temperature: not reported, pH: not reported | SOMS (organosilica adsorbent) | Particle surface, Particle size: 250–450 µm; Surface area: 650 m2/g, Pore volume: 1.03 mL/g | Adsorption capacity: 0 mg/g (in deionized water), 0 mg/g (in 50 mM NaCl) | Not reported | Stebal et al. (2019) [66] | |
| PFBA | Deionized water (alone) and NaCl (50 mM) (separate experiments), Temperature: not reported, pH: not reported | F-SOMS (fluoroalkyl modified) | Particle surface, Particle size: 250–450 µm; Surface area: 660 m2/g; Pore volume: 1.04 mL/g | Adsorption capacity: 0 mg/g (in deionized water), 0 mg/g (in 50 mM NaCl) | Not reported | Stebal et al. (2019) [66] | |
| PFBS | Water, Temperature: 25 °C, pH: 6.3 | Organo-montmorillonites (0.5CEC-Mt) | Particle surface | Adsorption capacity: ~0.15 mmol/g | Not reported | Surfaces not well-defined | Zhou et al. (2010) [84] |
| PFBS | Deionized water (alone) and NaCl (50 mM) (separate experiments), Temperature: not reported, pH: not reported | SOMS (organosilica adsorbent) | Particle surface, Particle size: 250–450 µm, Surface area: 650 m2/g; Pore volume: 1.03 mL/g, | Adsorption capacity: ~3 mg/g (in deionized water), ~1 mg/g (in 50 mM NaCl) | Not reported | Stebal et al. (2019) [66] | |
| PFBS | Deionized water (alone) and NaCl (50 mM) (separate experiments), Temperature: not reported, pH: not reported | F-SOMS (fluoroalkyl modified) | Particle surface, Particle size: 250–450 µm, Surface area: 660 m2/g, Pore volume: 1.04 mL/g | Adsorption capacity: ~1 mg/g (in deionized water), ~1 mg/g (in 50 mM NaCl) | Not reported | Stebal et al. (2019) [66] | |
| PFPeA | Deionized water (alone) and NaCl (50 mM) (separate experiments). Temperature: not reported, pH: not reported | SOMS (organosilica adsorbent) | Particle surface, Particle size: 250–450 µm, Surface area: 650 m2/g, Pore volume: 1.03 mL/g | Adsorption capacity: ~3 mg/g (in deionized water), ~0.5 mg/g (in 50 mM NaCl) | Not reported | Stebal et al. (2019) [66] | |
| PFPeA | Deionized water (alone) and NaCl (50 mM) (separate experiments), Temperature: not reported, pH: not reported | F-SOMS (fluoroalkyl modified) | Particle surface, Particle size: 250–450 µm, Surface area: 660 m2/g, Pore volume: 1.04 mL/g | Adsorption capacity: ~1 mg/g (in deionized water), ~1 mg/g (in 50 mM NaCl) | Not reported | Stebal et al. (2019) [66] | |
| PFHxA | Water and D2O, Temperature: 300 K | Silica (SiO2) | Crystal surface, Crystal dimensions: 50 × 50 × 10 nm3 | Not applicable | Not applicable | Hellsing et al. (2016) [12] | |
| PFHxA | Water and D2O (studied separately), Temperature: 300 K | Alumina (Al2O3) | Crystal surface, Crystal dimensions: 50 × 50 × 10 nm3 | 0.0033 µg/m2 | Monolayer | Hellsing et al. (2016) [12] | |
| PFHxA | Water, CaCl2, and NaCl, (0.01–10 mM), (0.1–100 mM), Temperature: 25 °C, pH: 5.5–6.5 (adsorption isotherm experiment), 2.27, 4.14, 5.99, 6.04, 9.08, and 11.16 (pH-effects experiment) | Hematite | Particle surface, Specific area: 9.9 m2/g, Purity: 70% | 34–59 µg/g (sorption isotherm), at pH 2.27 and 11.16: ~21 µg/g and ~17 µg/g, from Na+ concentration 0 and 100 mmol/L: ~22 and 22 µg/g, from Ca2+ concentration 0, 1, 10 mmol/L: ~24, 24, and 24 µg/g | Monolayer | Surfaces not well-defined | Zhao et al. (2014) [96] |
| PFHxA | Water, CaCl2, and NaCl, (0.01–10 mM), (0.1–100 mM), Temperature: 25 °C, pH: 5.5–6.5 (adsorption isotherm experiment), 2.27, 4.14, 5.99, 6.04, 9.08, and 11.16 (pH-effects experiment) | Kaolinite | Particle surface, Specific area: 23.11 m2/g, Purity: >95% | 34–59 µg/g (sorption isotherm), from pH 2.27 and 11.16: ~19 µg/g and ~12 µg/g, from Na+ concentration 0 and 100 mmol/L: ~19 and 19 µg/g, from Ca2+ concentration 0, 1, 10 mmol/L: ~19, 19, and 18 µg/g | Monolayer | Surfaces not well-defined | Zhao et al. (2014) [96] |
| PFHxA | Water, CaCl2, and NaCl, (0.01–10 mM), (0.1–100 mM), Temperature: 25 °C, pH: 5.5–6.5 (adsorption isotherm experiment), 2.27, 4.14, 5.99, 6.04, 9.08, and 11.16 (pH-effects experiment) | Montmorillonite | Particle surface, Specific area: 67.52 m2/g, Purity: 99% | 34–59 µg/g (sorption isotherm), at pH 2.27 and 11.16: ~18 µg/g ~16 µg/g, from Na+ concentration 0 and 100 mmol/L: ~20 and 19 µg/g, from Ca2+ concentration 0, 1, 10 mmol/L: ~19, 19, and 19 µg/g | Monolayer | Surfaces not well-defined | Zhao et al. (2014) [96] |
| PFHxA | Water, Temperature: 25 °C, pH: 6.3 | Organo-montmorillonites (0.5CEC-Mt) | Particle surface | Adsorption capacity: ~0.03 mmol/g | Not reported | Surfaces not well-defined | Zhou et al. (2010) [84] |
| PFHxA | Deionized water (alone) and NaCl (50 mM) (separate experiments) Temperature: not reported, pH: not reported | SOMS (organosilica adsorbent) | Particle surface, Particle size: 250–450 µm Surface area: 650 m2/g, Pore volume: 1.03 mL/g | Adsorption capacity: ~3 mg/g (in deionized water), ~1 mg/g (in 50 mM NaCl) | Not reported | Stebal et al. (2019) [66] | |
| PFHxA | Deionized water (alone) and NaCl (50 mM) (separate experiments), Temperature: not reported pH: not reported | F-SOMS (fluoroalkyl modified) | Particle surface, Particle size: 250–450 µm Surface area: 660 m2/g, Pore volume: 1.04 mL/g | Adsorption capacity: ~3.5 mg/g (in deionized water), ~3 mg/g (in 50 mM NaCl) | Not reported | Stebal et al. (2019) [66] | |
| PFHxS potassium salt | Water, CaCl2, and NaCl, (0.01–10 mM), (0.1–100 mM), Temperature: 25 °C, pH: 6.8–7.1 (adsorption isotherm experiment), 2.27, 4.14, 5.99, 6.04, 9.08, and 11.16 (pH-effects experiment) | Hematite | Particle surface, Specific area: 9.9 m2/g, Purity: 70% | 312–370 µg/g (sorption isotherm), at pH 2.27 and 11.16: ~33 µg/g and ~29 µg/g, from Na+ concentration 0 and 100 mmol/L: ~30 and 34 µg/g, from Ca2+ concentration 0, 1, 10 mmol/L: ~31, 32, and 35 µg/g | Monolayer | Surfaces not well-defined | Zhao et al. (2014) [96] |
| PFHxS potassium salt | Water, CaCl2, and NaCl, (0.01–10 mM), (0.1–100 mM), Temperature: 25 °C, pH: 6.8–7.1 (adsorption isotherm experiment), 2.27, 4.14, 5.99, 6.04, 9.08, and 11.16 (pH-effects experiment) | Kaolinite | Particle surface, Specific area: 23.11 m2/g, Purity: >95% | 312–370 µg/g (sorption isotherm), at pH 2.27 and 11.16: ~27 µg/g and ~26 µg/g, from Na+ concentration 0 and 100 mmol/L: ~27 and 27 µg/g, from Ca2+ concentration 0, 1, 10 mmol/L: ~26, 26, and 26 µg/g | Monolayer | Surfaces not well-defined | Zhao et al. (2014) [96] |
| PFHxS potassium salt | Water, CaCl2, and NaCl, (0.01–10 mM), (0.1–100 mM), Temperature: 25 °C, pH: 6.8–7.1 (adsorption isotherm experiment), 2.27, 4.14, 5.99, 6.04, 9.08, and 11.16 (pH-effects experiment) | Montmorillonite | Particle surface, Specific area: 67.52 m2/g, Purity: 99% | 312–370 µg/g (sorption isotherm), at pH 2.27 and 11.16: ~31 µg/g and ~29 µg/g, from Na+ concentration 0 and 100 mmol/L: ~29 and 29 µg/g, from Ca2+ concentration 0, 1, 10 mmol/L: ~29, 30, and 29 µg/g | Monolayer | Surfaces not well-defined | Zhao et al. (2014) [96] |
| PFHxS | Water, Temperature: 25 °C, pH: 6.3 | Organo-montmorillonites (0.5CEC-Mt) | Particle surface | Adsorption capacity: ~0.44 mmol/g | Not reported | Surfaces not well-defined | Zhou et al. (2010) |
| PFHxS | Deionized water (alone) and NaCl (50 mM) (separate experiments), Temperature: not reported, pH: not reported | SOMS (organosilica adsorbent) | Particle surface, Particle size: 250–450 µm, Surface area: 650 m2/g, Pore volume: 1.03 mL/g | Adsorption capacity: ~1 mg/g (in deionized water), ~2 mg/g (in 50 mM NaCl) | Not reported | Stebal et al. (2019) [66] | |
| PFHxS | Deionized water (alone) and NaCl (50 mM) (separate experiments), Temperature: not reported, pH: not reported | F-SOMS (fluoroalkyl modified) | Particle surface, Particle size: 250–450 µm, Surface area: 660 m2/g Pore volume: 1.04 mL/g | Adsorption capacity: ~2 mg/g (in deionized water), ~3 mg/g (in 50 mM NaCl) | Not reported | Stebal et al. (2019) [66] | |
| PFHpA | Water and NaHCO3/NaCl (1.0 mM), Temperature: ~22.2 °C, pH: 7.5 | Kaolinite | Particle surface, Mean diameter: 1.1 µm (narrow size distribution) Surface area: 10 m2/g | Solid–water distribution coefficients (logkd): Single compound system = not detectable, Multi-compound system = not applicable | Not reported | Xiao et al. (2011) [42] | |
| PFHpA | Deionized water (alone) and NaCl (50 mM) (separate experiments). Temperature: not reported, pH: not reported | SOMS (organosilica adsorbent) | Particle surface, Particle size: 250–450 µm, Surface area: 650 m2/g Pore volume: 1.03 mL/g, | Adsorption capacity: ~3 mg/g (in deionized water), ~9 mg/g (in 50 mM NaCl) | Not reported | Stebal et al. (2019) [66] | |
| PFHpA | Deionized water (alone) and NaCl (50 mM) (separate experiments), Temperature: not reported, pH: not reported | F-SOMS (fluoroalkyl modified) | Particle surface, Particle size: 250–450 µm, Surface area: 660 m2/g, Pore volume: 1.04 mL/g | Adsorption capacity: ~3 mg/g (in deionized water), ~5 mg/g (in 50 mM NaCl) | Not reported | Stebal et al. (2019) [66] | |
| PFOA | Water and D2O (studied separately), Temperature: 300 K | Silica (SiO2) | Crystal surface, Crystal dimensions: 50 × 50 × 10 nm3 | 0 µg/m2 | Not applicable | Hellsing et al. (2016) [12] | |
| PFOA | Water, pH: 6, Initial HA concentration: 10 mg/L | SiO2 | Particle surface, Particle size: 2.3 µm Specific surface area: 6.1 m2/g | 0.851 µg/m2 (no HA present), Sorption density: 0.08 mg/m2 (HA added before PFOA), ~1.1 µg/m2 (concurrent addition of PFOA and HA) (at equilibrium concentration ~700 µg/L of PFOA) | Monolayer (no HA), No structure reported (HA present) | Yang et al. (2016) [79] | |
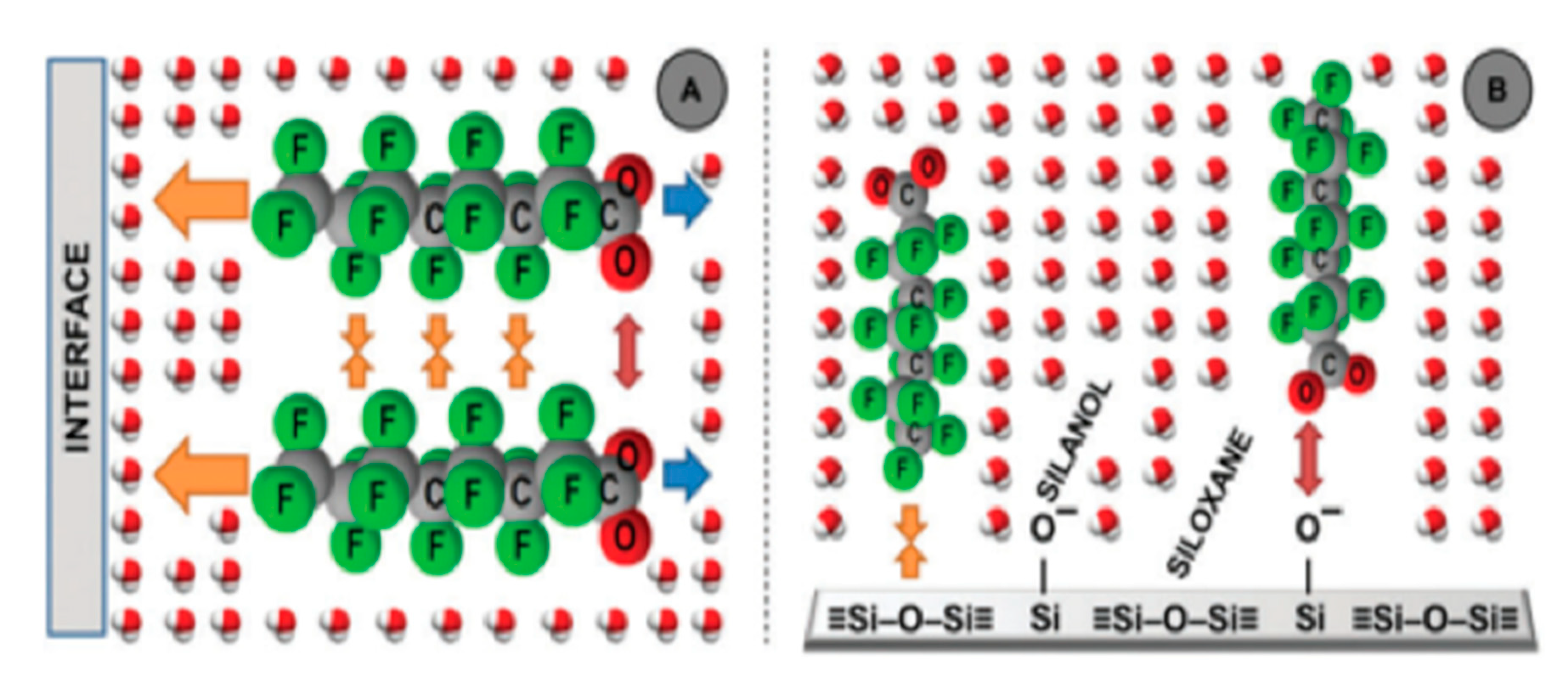
| PFOA | Water, Temperature: 20–45 °C, pH: 5, 10.2, and 1.3 | Glass silica (siloxane, silanol patches) | Flat surface; siloxane and silanol patches | Surface excess: 217 molecules/µm2, Adsorbed amount: 3.60 × 10−4 µmol/m2 (pH 5.0), 1.33 × 10−4 µmol/m2, 7.46 × 10−4 µmol/m2 (pH 10.2, 1.3) | Monolayer | Schematics are the only available information on the adsorbed structure | Shafique et al. (2017) [85] |
| PFOA | Water and D2O (studied separately), Temperature: 300 K | Alumina (Al2O3) | Crystal surface, Crystal dimensions: 50 × 50 × 10 nm3 | 0.0036 µg/m2 | Monolayer | Hellsing et al. (2016) [12] | |
| PFOA | Water, pH: 6, Initial HA concentration: 10 mg/L | Al2O3 | Particle surface, Particle size: 1.7 µm, Specific surface area: 6.0 m2/g | 1.72 µg/m2 (no HA present), Sorption density: 0.07 mg/m2 (HA added before PFOA), ~1.9 µg/m2 (concurrent addition of PFOA and HA into the aqueous system) (at equilibrium concentration ~700 µg/L of PFOA) | Monolayer (no HA), No structure reported (HA present) | Yang et al. (2016) [79] | |
| PFOA | Water, NaCl, KCl, CaCl2, and MgCl2 (0.001 M-0.1 M) (separate experiments), Temperature: 25 °C, pH: 4.3–7.2 | Alumina (Al2O3) | Particle surface, Surface area: ~88.6 m2/g, Average particle size: 87.05 µm, non-crystalline | 0.157 µg/m2, In 0.001 M and 0.1 M NaCl, KCl, CaCl2, MgCl2: ~0.065 µg/m2 and ~0.001 µg/m2, During pH experiments: ~0.078 µg/m2 (pH 4.3) and ~0.004 µg/m2 (pH 7.2) | Monolayer | Wang et al. (2011) [97] | |
| PFOA ammonium salt | Deionized water, pH: ~3.9, Temperature: 255–273 K | Porous alumina (Al2O3) | Particle surface, Particle size: 0.063–0.200 mm, Pore diameter: ~90 Å, Specific area: 120 m2/g | Adsorbed amount (Γ) = 0.46, 0.92, 1.37, 1.84, 2.58, 3.26, 4.5, 5.1, 5.1, 5.3, 5.1, 5.2 µmol/m2 | Bilayer | Evenas et al. (2002) [85] | |
| PFOA | Water, NaCl, and CaCl2 (0.0001–0.1 M), Temperature: 25 °C, pH: 4–7.5 (pH experiment) and 7 (all other experiments) | Boehmite | Particle surface, Surface area: ~299.2 m2/g, Average particle size: 37.02 µm | Adsorption capacity: 0.633 µg/m2, Adsorbed amount from kinetic experiment: 0.09 µg/m2, at pH 4 and 7.5: ~0.108 µg/m2 and ~0.09 µg/m2, In 0.0001 M NaCl and 0.1 M NaCl: ~0.0 µg/m2 and ~0.05 µg/m2, In 0.0001 M CaCl2 and 0.1 M CaCl2: ~0.0 µg/m2 and ~0.043 µg/m2 | Monolayer | Surfaces not well-defined | Wang et al. (2012) [91] |
| PFOA | Water, pH: 6, Initial HA concentration: 10 mg/L | Fe2O3 | Particle surface, Particle size: 1.2 µm, Specific surface area: 6.0 m2/g | 0.97 µg/m2 (no HA present), Sorption density: 0.07 mg/m2 (HA added before PFO, ~2.3 µg/m2 (concurrent addition of PFOA and HA) (at equilibrium concentration ~700 µg/L of PFOA) | Monolayer (no HA), No structure reported (HA present) | Yang et al. (2016) [79] | |
| PFOA | Water, CaCl2, and NaCl, (0.01–10 mM), (0.1–100 mM), Temperature: 25 °C, pH: 5.5–6.5 (adsorption isotherm experiment), 2.27, 4.14, 5.99, 6.04, 9.08, and 11.16 (pH-effects experiment) | Hematite | Particle surface, Specific area: 9.9 m2/g, Purity: 70% | 104–112 µg/g (sorption isotherm), at pH 2.27 and 11.16: ~28 µg/g and ~18 µg/g, from Na+ concentration 0 and 100 mmol/L: ~21 and 22 µg/g, from Ca2+ concentration 0, 1, 10 mmol/L: ~23, 24, and 25 µg/g | Monolayer | Surfaces not well-defined | Zhao et al. (2014) [96] |
| PFOA | Water and NaHCO3/NaCl (1.0 mM), Temperature: ~22.2 °C, pH: 7.4 | Kaolinite | Particle surface, Mean diameter: 1.1 µm (narrow size distribution), Surface area: 10 m2/g | Solid-Water Distribution Coefficients (logkd): Single compound system = ~0.36 L/kg, Multi-compound system = not detectable | Not reported | Xiao et al. (2011) [42] | |
| PFOA | Water, CaCl2, and NaCl, (0.01–10 mM), (0.1–100 mM), Temperature: 25 °C, pH: 5.5–6.5 (adsorption isotherm experiment), 2.27, 4.14, 5.99, 6.04, 9.08, and 11.16 (pH-effects experiment) | Kaolinite | Particle surface, Specific area: 23.11 m2/g, Purity: >95% | 104-112 µg/g (sorption isotherm), at pH 2.27 and 11.16: ~24 µg/g and ~16 µg/g, from Na+ concentration 0 and 100 mmol/L: ~19 and 19 µg/g from Ca2+ concentration 0, 1, 10 mmol/L: ~18, 21, and 22 µg/g | Monolayer | Surfaces not well-defined | Zhao et al. (2014) [96] |
| PFOA | Water, CaCl2, and NaCl, (0.01–10 mM), (0.1–100 mM), Temperature: 25 °C, pH: 5.5–6.5 (adsorption isotherm experiment), 2.27, 4.14, 5.99, 6.04, 9.08, and 11.16 (pH-effects experiment) | Montmorillonite | Particle surface, Specific area: 67.52 m2/g, Purity: 99% | 104–112 µg/g (sorption isotherm), at pH 2.27 and 11.16: ~23 µg/g and ~12 µg/g, from Na+ concentration 0 and 100 mmol/L: ~15 µg/g and 15 µg/g, from Ca2+ concentration 0, 1, 10 mmol/L: ~15, 16, and 15 µg/g | Monolayer | Surfaces not well-defined | Zhao et al. (2014) [96] |
| PFOA | Water, Temperature: 25 °C, pH: 6.3 | Organo-montmorillonites (0.2CEC-Mt) | Particle surface | Adsorption capacity: ~0.08 mmol/g | Not reported | Surfaces not well-defined | Zhou et al. (2010) [84] |
| PFOA | Water, Temperature: 25 °C, pH: 6.3 | Organo-montmorillonites (0.5CEC-Mt) | Particle surface | Adsorption capacity: ~0.12 mmol/g | Not reported | Surfaces not well-defined | Zhou et al. (2010) [84] |
| PFOA | Water, Temperature: 25 °C, pH: 6.3 | Organo-montmorillonites (1.0CEC-Mt) | Particle surface | Adsorption capacity: ~0.35 mmol/g | Not reported | Surfaces not well-defined | Zhou et al. (2010) [84] |
| PFOA | Deionized water (alone) and NaCl (50 mM) (separate experiments). Temperature: not reported, pH: not reported | SOMS (organosilica adsorbent) | Particle surface, Particle size: 250–450 µm, Surface area: 650 m2/g, Pore volume: 1.03 mL/g | Adsorption capacity: ~3 mg/g (in deionized water), ~23 mg/g (in 50 mM NaCl) | Not reported | Stebal et al. (2019) [66] | |
| PFOA | Deionized water (alone) and NaCl (50 mM) (separate experiments), Temperature: not reported, pH: not reported | F-SOMS (fluoroalkyl modified) | Particle surface, Particle size: 250–450 µm, Surface area: 660 m2/g, Pore volume: 1.04 mL/g | Adsorption capacity: ~3 mg/g (in deionized water), ~23 mg/g (in 50 mM NaCl) | Not reported | Stebal et al. (2019) [66] | |
| PFOS | Water and D2O (studied separately), Temperature: 300 K | Silica (SiO2) | Crystal surface, Crystal Dimensions: 50 × 50 × 10 nm3 | 0 µg/m2 | Not applicable | Hellsing et al. (2016) [12] | |
| PFOS | Water and NaNO3 (0.1 mmol/L), pH: 7, Temperature: 298K | Nanosize SiO2 | Particle surface, Average diameter: 15 nm, Surface hydroxyl density: 35.5 µmol/m2, Surface area: 64.1 m2/g | 0.1 µg/m2 (initial PFOS concentration of 0.2 µmol/L) | Bilayer | Lu et al. (2016) [95] | |
| PFOS | Water and NaCl (0.1 M), Temperature: 20–45 °C, pH: 4 | Glass silica (siloxane, silanol patches) | Flat surface; siloxane and silanol patches | Surface excess: 7241 molecules/µm2, Adsorbed amount: 1.20 × 10−2 µmol/m2 (pH 5.0) | Multilayer | Schematics are the only available information on the adsorbed surface structure | Shafique et al. (2017) [85] |
| PFOS potassium salt | Water and KNO3 (0.01–0.1 M) | Ottawa sand (SiO2) | Particle surface, Surface area: 2 × 10−3 m2/g | Adsorption isotherm experiment: 10 PFOS molecules/nm2 | Monolayer | Johnson et al. (2007) [90] | |
| PFOS | Water and KCl (concentration not reported), Temperature: 298 K | TiO2 | Flat surface; (110), (001), (100) plane, ~41 × 40 × 12 Å3 (Lx, Ly, Lz) | Not reported | Monolayer | Molecular Dynamics simulations | He et al. (2013) [99] |
| PFOS | Water and CaCl2 (concentration not reported), Temperature: 298 K | TiO2 | Flat surface; (110) plane, ~4.1 ×3.9 × 1.4 nm3 (Lx, Ly, Lz) | Not reported | Multilayer | Molecular Dynamics simulations | He et al. (2015) [100] |
| PFOS | Water and NaNO3 (0.1 mmol/L), pH: 7, Temperature: 298 K | Nanosize TiO2 | Particle surface, Average diameter: 25 nm, Surface hydroxyl density: 18.3 µmol/m2, Surface area: 278 m2/g | 0.7 µg/m2 (Initial PFOS concentration of 0.2 µmol/L) | Bilayer | Lu et al. (2016) [95] | |
| PFOS | Water and D2O (studied separately), Temperature: 300 K | Alumina (Al2O3) | Crystal surface, Crystal dimensions: 50 × 50 × 10 nm3 | 0.0035 µg/m2 | Monolayer | Hellsing et al. (2016) [12] | |
| PFOS potassium salt | Water, NaCl, KCl, CaCl2, and MgCl2 (0.001 M-0.1 M) (separate experiments), Temperature: 25 °C, pH: ~4.5–7 | Alumina (Al2O3) | Particle surface, Surface area: ~88.6 m2/g, Average particle size: 87.05 µm, non-crystalline | 0.252 µg/m2; In 0.001 M and 0.1 M NaCl, KCl, CaCl2, MgCl2: 0.085 µg/m2 and 0.025 µg/m2, during pH experiments: ~0.09 µg/m2 (pH 4.5) and ~0.02 µg/m2 (pH 7) | Monolayer | Wang et al. (2011) [97] | |
| PFOS | Water and NaNO3 (0.1 mmol/L), pH: 7, Temperature: 298 K | Nanosize Al2O3 | Particle surface, Average diameter: 50 nm, Surface hydroxyl density: 31.2 µmol/m2, Surface area: 198 m2/g | 1.1 µg/m2 (initial PFOS concentration of 0.2 µmol/L) | Bilayer | Lu et al. (2016) [95] | |
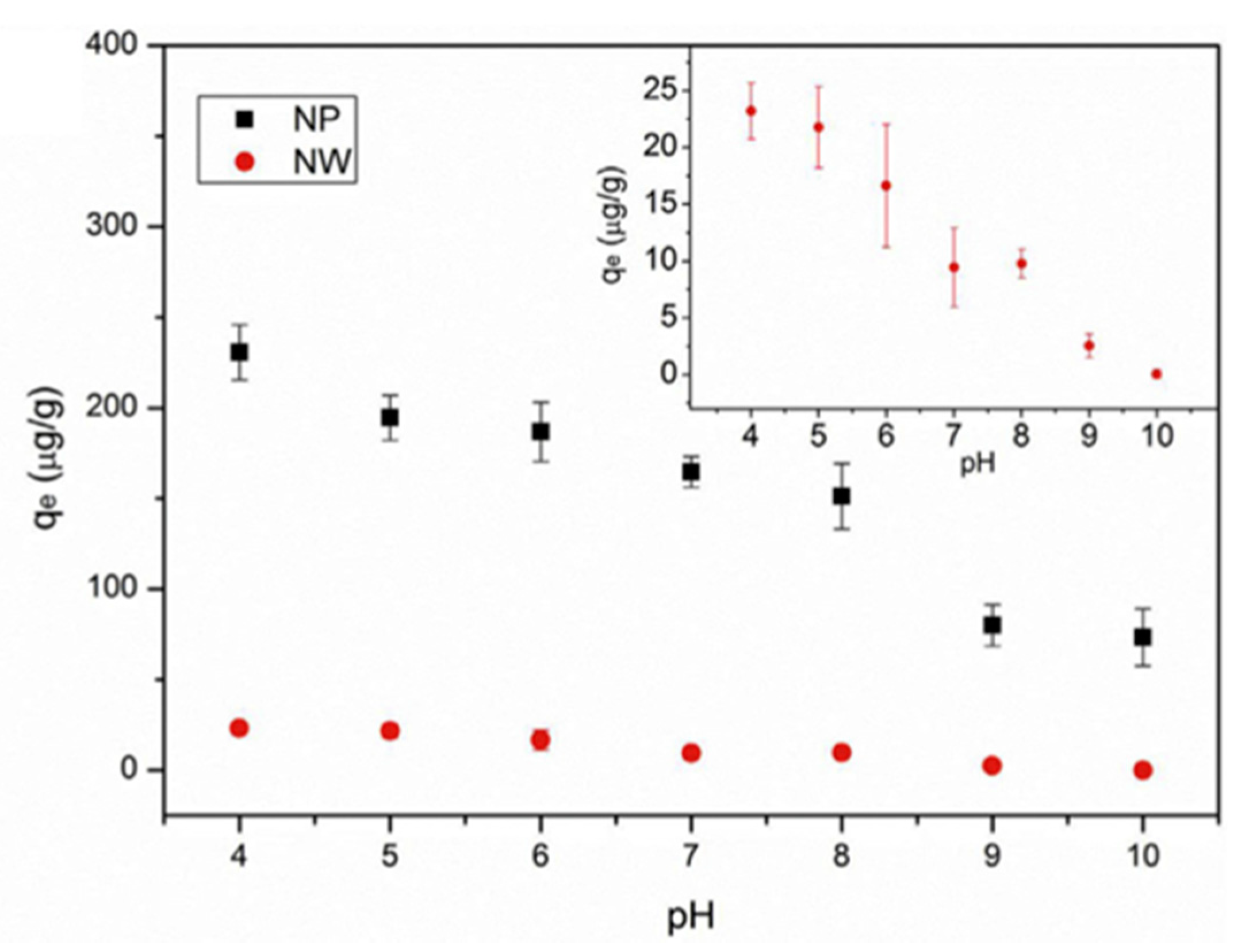
| PFOS sodium salt | Water, Temperatures: 30 °C, 40 °C, 50 °C (isotherm equilibrium experiment), and 25 °C (all other experiments), pH: 4–10 (pH-effects experiment), Humic acid concentration: 1–50 mg/L (humic acid–effects experiment), 0.001–0.1 M NaCl (salt-effects experiment) | Alumina nanoparticles | Particle surface, Particle size: 13 nm, Surface area: 83 m2 g−1 | At 30 °C: 589 mg/g, at 40 °C: 485 mg/g, at 50 °C: 447mg/g (mg/g absorbate on absorbent), At pH 4 and 10: ~240 µg/g and ~80 µg/g, In 1 mg/L and 50 mg/L HA: ~143 µg/g and 45 µg/g (pH 4), ~37 µg/g and ~27 µg/g (pH 7), ~19 µg/g and ~16 µg/g (pH 10), In 0.001 M and 0.1 M NaCl: ~180 µg/g and ~25 µg/g | Monolayer | Jian et al. (2019) [87] | |
| PFOS sodium salt | Water, Temperature: 30 °C, 40 °C, 50 °C (isotherm equilibrium experiment), and 25 °C (all other experiments), pH: 4–10 (pH change experiment) Humic acid concentration: 1–50 mg/L (humic acid effects experiment), 0.001–0.1 M NaCl (salt-effects experiment) | Alumina nanowires | Particle surface, Particle size: 2–6 nm (diameter) 13 nm (length), Surface area: 124.9 m2g−1 | At 30 °C: 589, at 40 °C: 485, at 50 °C: 447 (mg/g absorbate on absorbent), At pH 4 and 10: ~30 µg/g and ~5 µg/g, In 1 mg/L and 50 mg/L HA: ~27 µg/g and 8 µg/g (pH 4), ~9 µg/g and ~2 µg/g (pH 7), ~4 µg/g and ~1 µg/g (pH 10), In 0.001 M and 0.1 M NaCl: ~10 µg/g and ~10 µg/g | Monolayer | Sodium perfluoro-[13C8]-octanesulfonate (M8PFOS) | Jian et al. (2019) [87] |
| PFOS | Water and KH2PO4 (50 mg/L) (all experiments), Temperatures: 303 K (pH experiment), 293 K, 303 K, and 313 K (kinetic experiments), pH: 4.3 (kinetic experiment), 3–10.5 (pH experiment) | Boehmite | Particle surface, Surface area: ~299.2 m2/g, Average particle size: 37.02 µm | Adsorption capacity: 0.1529 µg/m2 (at 293K), 0.1176 µg/m2 (at 303K), 0.0980 µg/m2 (at 303 K), Adsorbed amount at pH 3 and 10.5: ~0.31 µg/m2 and ~0.07 µg/m2 | Monolayer | Surfaces not well-defined | Qian et al. (2017) [93] |
| PFOS potassium salt | Water, NaCl, and CaCl2 (0.0001 M–0.1 M), Temperature: 25 °C, pH: 4–7.5 (pH experiment) and 7 (all other experiments) | Boehmite | Particle surface, Surface area: ~299.2 ± 1.8 m2/g, Average particle size: 37.02 µm | Adsorption capacity: 0.877 µg/m2, Adsorbed amount from kinetic experiment: 0.105 µg/m2, at pH 4 and 7.5: ~0.125 µg/m2 and ~0.105 µg/m2, In 0.0001 M NaCl and 0.1 M NaCl: ~0.0 µg/m2 and ~0.07 µg/m2, In 0.0001 M CaCl2 and 0.1 M CaCl2: ~0.0 µg/m2 and ~0.055 µg/m2 | Monolayer | Surfaces not well-defined | Wang et al. (2012) [91] |
| PFOS potassium salt | Water, NaCl, and CaCl2 (0.0001 M–0.1 M), Temperature: 25 °C pH: 4–7.5, (pH experiment) and 7 (all other experiments), HA concentration: 2–50 mg/L | Boehmite | Particle surface, Surface area: ~299.2 m2/g, Average particle size: 37.02 µm | Adsorption capacity: 0.877 µg/m2, Adsorbed amount from kinetic experiment: 0.105 µg/m2, at pH 4 and 7.5: ~0.125 µg/m2 and ~0.105 µg/m2, In 0.0001 M NaCl and 0.1 M NaCl: ~0.0 µg/m2 and ~0.07 µg/m2, In 0.0001 M CaCl2 and 0.1 M CaCl2: ~0.0 µg/m2 and ~0.055 µg/m2, HA concentration 2 and 50 mg/L: ~0.09 µg/m2 and ~0.018 µg/m2 | Monolayer | Surfaces not well-defined | Shih et al. (2013) [92] |
| PFOS potassium salt | Water, CaCl2, and NaCl, (0.01–10 mM), (0.1–100 mM), Temperature: 25 °C, pH: 6.8–7.1 (adsorption isotherm experiment), 2.27, 4.14, 5.99, 6.04, 9.08, and 11.16 (pH-effects experiment) | Hematite | Particle surface, Specific area: 9.9 m2/g, Purity: 70% | 294–312 µg/g (sorption isotherm), at pH 2.27 and 11.16: ~63 µg/g and ~22 µg/g, from Na+ concentration 0 and 100 mmol/L: ~42 and 59 µg/g, from Ca2+ concentration 0, 1, 10 mmol/L: ~42, 51, and 56 µg/g | Monolayer | Surfaces not well-defined | Zhao et al. (2014) [96] |
| PFOS | Water and NaNO3 (0.1 mmol/L), pH: 7, Temperature: 298K | Nanosize Fe2O3 | Particle surface, Average diameter: 75, Surface hydroxyl density: 21 µmol/m2, Surface area: 41.7 m2/g | 4.0 µg/m2 (initial PFOS concentration of 0.2 µmol/L) | Bilayer | Lu et al. (2016) [95] | |
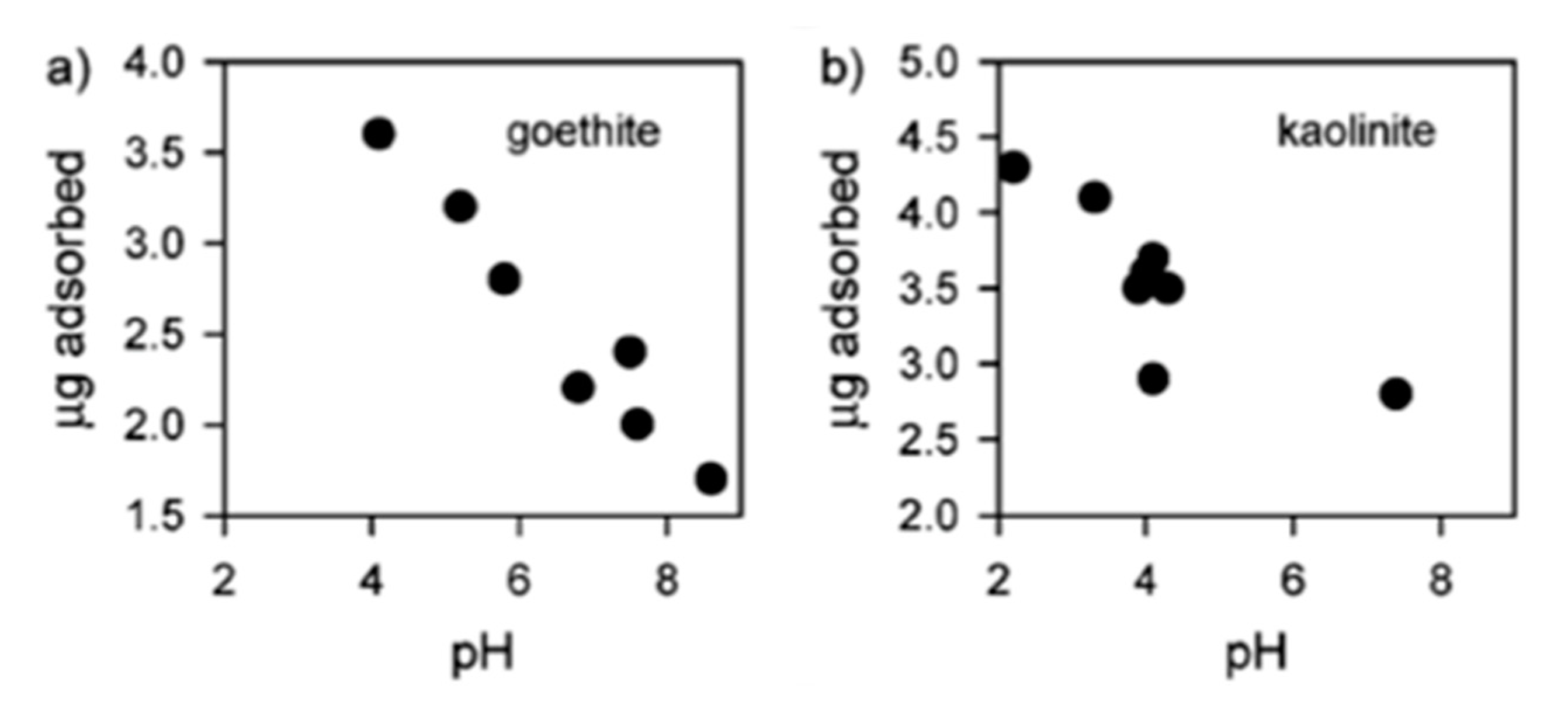
| PFOS potassium salt | Water and KNO3 (0.01–0.1 M), pH: 4.1-8.6 | Goethite (α-FeO(OH)) | Particle surface, Surface area: 58 m2/g | Adsorption isotherm experiment: 1.4 × 10−3 PFOS molecules/nm2, Mass adsorbed: (pH dependence experiments) ~3.7 µg (pH 4.1) and ~1.7 µg (pH 8.6) | Monolayer | Johnson et al. (2007) [90] | |
| PFOS potassium salt | Water and KNO3 (0.01–0.1 M) | High-iron sand (Fe3O4) | Particle surface, Surface area: 6 m2/g | Adsorption isotherm experiment: 5 × 10−3 PFOS molecules/nm2 | Multilayer | Johnson et al. (2007) [90] | |
| PFOS | Water and NaHCO3/NaCl (1.0 mM), Temperature: ~22.2 °C, pH: 7.5 | Kaolinite | Particle surface, Mean diameter: 1.1 µm (narrow size distribution), Surface area: 10 m2/g | Solid–water distribution coefficients (logkd): Single compound system = ~1.16 L/kg, Multi-compound system = ~0.88 L/kg | Not reported | Xiao et al. (2011) [42] | |
| PFOS potassium salt | Water and KNO3 (0.01–0.1 M), pH: 2.2-7.4 | Kaolinite (Al2Si2O5(OH)4) | Particle surface, Surface area: 10 m2/g | Adsorption isotherm experiment: 6.0 × 10−3 PFOS molecules/nm2, Mass adsorbed during pH dependence experiments: ~4.4 µg (pH 2.2) and ~2.8 µg (pH 7.4) | Monolayer | Johnson et al. (2007) [90] | |
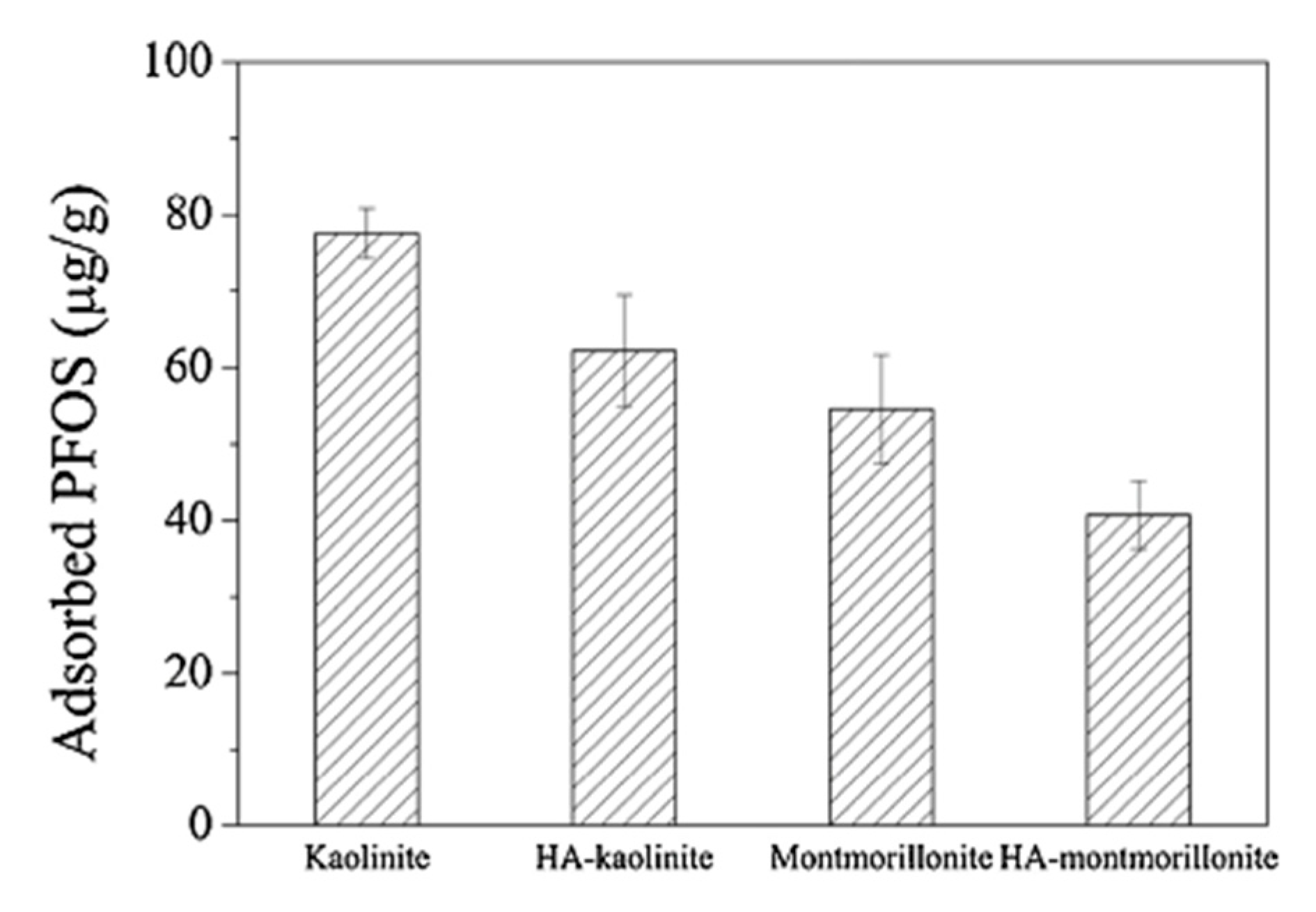
| PFOS potassium salt | Water and NaCl (10 mM), Temperature: unreported (adsorption experiments), 22 °C (adsorption experiments with HA), pH: 7 (adsorption experiments) and 3–11 (zeta potential experiments) HA concentration: 100mg/L | Kaolinite | Particle surface, particle size: 1187 ± 380 nm, Surface area: 11.9 m2/g | 77.6 ± 3.3 µg/g (without HA in solution), ~63 ± 8 µg/g (with HA in solution) | Not reported | Zhang et al. (2014) [78] | |
| PFOS potassium salt | Water, CaCl2, and NaCl, (0.01–10 mM), (0.1–100 mM), Temperature: 25 °C, pH: 6.8–7.1 (adsorption isotherm experiment), 2.27, 4.14, 5.99, 6.04, 9.08, and 11.16 (pH-effects experiment) | Kaolinite | Particle surface, Specific area: 23.11 m2/g, Purity: >95% | 294–312 µg/g (sorption isotherm), at pH 2.27 and 11.16: ~34 µg/g and ~17 µg/g, from Na+ concentration 0 and 100 mmol/L: ~19 and 23 µg/g, from Ca2+ concentration 0, 1, 10 mmol/L: ~21, 26, and 27 µg/g | Monolayer | Surfaces not well-defined | Zhao et al. (2014) [96] |
| PFOS potassium salt | Water and NaCl (10 mM), Temperature: unreported (adsorption experiments) and 22 °C (adsorption experiments with HA), pH: 7 (adsorption experiment) and 3–11 (zeta potential experiment), HA concentration: 100 mg/L | Montmorillonite | Particle surface, particle size: 842.9 ± 125.9 nm, Surface area: 82.9 m2/g | 54.5 ± 7.2 µg/g (without HA in solution), ~41 ± 5 µg/g (with HA in solution) | Not reported | Zhang et al. (2014) [78] | |
| PFOS potassium salt | Water, CaCl2, and NaCl, (0.01–10 mM), (0.1–100 mM), Temperature: 25 °C, pH: 6.8–7.1 (adsorption isotherm experiment), 2.27, 4.14, 5.99, 6.04, 9.08, and 11.16 (pH-effects experiment) | Montmorillonite | Particle surface, Specific area: 67.52 m2/g, Purity: 99% | 294–312 µg/g (sorption isotherm), at pH 2.27 and 11.16: ~37 µg/g ~17 µg/g, from Na+ concentration 0 and 100 mmol/L: ~19 and 24 µg/g, from Ca2+ concentration 0, 1, 10 mmol/L: ~19, 23, and 26 µg/g | Monolayer | Surfaces not well-defined | Zhao et al. (2014) [96] |
| PFOS potassium salt | Water, Temperature: 25 °C, pH: 6.3 | Organo-montmorillonites (Na-Mt) | Particle surface | Adsorption capacity: 0.239 mmol/g | Monolayer | Surfaces not well-defined | Zhou et al. (2010) [84] |
| PFOS potassium salt | Water, Temperature: 25° C, pH: 6.3 | Organo-montmorillonites (0.2CEC-Mt) | Particle surface | Adsorption capacity: 0.550 mmol/g | Monolayer | Surfaces not well-defined | Zhou et al. (2010) [84] |
| PFOS potassium salt | Water, Temperature: 25 °C, pH: 6.3 | Organo-montmorillonites (0.5CEC-Mt) | Particle surface | Adsorption capacity: 0.912 mmol/g | Monolayer | Surfaces not well-defined | Zhou et al. (2010) [84] |
| PFOS potassium salt | Water, Temperature: 25 °C, pH: 6.3 | Organo-montmorillonites (1.0CEC-Mt) | Particle surface | Adsorption capacity: 1.492 mmol/g | Monolayer | Surfaces not well-defined | Zhou et al. (2010) [84] |
| PFOS potassium salt | Water, Temperature: 25 °C, pH: 6.3 | Organo-montmorillonites (2.5CEC-Mt) | Particle surface | Adsorption capacity: 1.71 mmol/g | Monolayer | Surfaces not well-defined | Zhou et al. (2010) [84] |
| PFOS | Deionized water (alone) and NaCl (50 mM) (separate experiments), Temperature: not reported, pH: not reported | SOMS (organosilica adsorbent) | Particle surface, Particle size: 250–450 µm, Surface area: 650 m2/g, Pore volume: 1.03 mL/g | Adsorption capacity: ~7 mg/g (in deionized water), ~55 mg/g (in 50 mM NaCl) | Not Reported | Stebal et al. (2019) [66] | |
| PFOS | Deionized water (alone) and NaCl (50 mM) (separate experiments), Temperature: not reported, pH: not reported | F-SOMS (fluoroalkyl modified) | Particle surface, Particle size: 250–450 µm, Surface area: 660 m2/g, Pore volume: 1.04 mL/g | Adsorption capacity: ~7 mg/g (in deionized water), ~68 mg/g (in 50 mM NaCl) | Not Reported | Stebal et al. (2019) [66] | |
| PFOS potassium salt | Water and KNO3 (0.01–0.1 M) | Lake Michigan a=sediment | Particle surface, Surface area: not reported | Not reported, “unpredictable nature of the compound” | Monolayer | Johnson et al. (2007) [90] | |
| TEA-FOS | Water and NaCl (0–50 mM), Temperature: 21 °C, pH: 3.4 and 10 | Hydroxylated germanium | Flat Surface, 45-degree trapezoidal Ge, Dimensions: 80 × 10 × 4 mm, referred to as an internal reflection element (IRE). Ge surface was placed in a flow-through cell coated with Teflon. | At pH 3.4: 28.7 µg/m2 (in 0 mM NaCl), 31.9 µg/m2 (in 1 mM NaCl), 35.9 µg/m2 (in 2 mM NaCl), 42.9 µg/m2 (in 5 mM NaCl), 25.8 mM (in 10 mM Nacl), 23.0 µg/m2 (in 20 mM NaCl), 6.47 µg/m2 (in 50 mM NaCl). At pH 10: 7.58 µg/m2 (in 0 mM NaCl), 10.2 µg/m2 (in 1 mM NaCl), 12.2 µg/m2 (in 2 mM NaCl), 21.0 µg/m2 (in 5 mM NaCl), 19.6 mM (in 10 mM NaCl), 16.9 µg/m2 (in 20 mM NaCl), 9.83 µg/m2 (in 50 mM NaCl) | Multilayer | Xing et al. (2013) [98] | |
| PFNA | Water and D2O (studied separately), Temperature: 300 K | Silica (SiO2) | Crystal surface, Crystal dimensions: 50 × 50 × 10 nm3 | Not applicable | Not applicable | Hellsing et al. (2016) [12] | |
| PFNA | Water, Temperature: 20–45 °C, pH: 5 | Glass silica (siloxane, silanol patches) | Flat surface; siloxane and silanol patches | Surface excess: 397 molecules/µm2, Adsorbed amount: 6.60 × 10−4 µmol/m2 (pH 5.0) | Monolayer | Schematics are the only available information on the adsorbed surface structure | Shafique et al. (2017) [85] |
| PFNA | Water and D2O (studied separately), Temperature: 300 K | Alumina (Al2O3) | Crystal surface, Crystal dimensions: 50 × 50 × 10 nm3 | 0.0058 µg/m2 | Monolayer | Hellsing et al. (2016) [12] | |
| PFNA | Water and NaHCO3/NaCl (1.0 mM), Temperature: ~22.2 °C, pH: 7.5 | Kaolinite | Particle surface, Mean diameter: 1.1 µm (narrow size distribution), Surface area: 10 m2/g | Solid–water distribution coefficients (logkd): Single compound system = ~0.74 L/kg, Multi-compound system = ~0.30 L/kg | Not reported | Xiao et al. (2011) [42] | |
| PFNA | Deionized water (alone) and NaCl (50 mM) (separate experiments), Temperature: not reported, pH: not reported | SOMS (organosilica adsorbent) | Particle surface, Particle size: 250–450 µm, Surface area: 650 m2/g, Pore volume: 1.03 mL/g | Adsorption capacity: ~4 mg/g (in deionized water), ~13 mg/g (in 50 mM NaCl) | Not Reported | Stebal et al. (2019) [66] | |
| PFNA | Deionized water (alone) and NaCl (50 mM) (separate experiments), Temperature: not reported, pH: not reported | F-SOMS (fluoroalkyl modified) | Particle surface, Particle size: 250–450 µm, Surface area: 660 m2/g, Pore volume: 1.04 mL/g | Adsorption capacity: ~0.2 mg/g (in deionized water), ~15 mg/g (in 50 mM NaCl) | Not Reported | Stebal et al. (2019) [66] | |
| PFDA | Water, Temperature: 20–45 °C, pH: 5 | Glass silica (siloxane, silanol patches) | Flat surface; siloxane and silanol patches | Surface excess: 499 molecules/µm2, Adsorbed amount: 8.28 × 10−4 µg/m2 (pH 5.0) | Monolayer | Schematics are the only available information on the adsorbed surface structure | Shafique et al. (2017) [85] |
| PFDA | Water and NaHCO3/NaCl (1.0 mM), Temperature: ~22.2 °C, pH: 7.5 | Kaolinite | Particle surface, Mean diameter: 1.1 µm (narrow size distribution), Surface area: 10 m2/g | Solid–water distribution coefficients (logkd): Single compound system = ~1.30 L/kg, Multi-compound system = 1.05 L/kg | Not reported | Xiao et al. (2011) [42] | |
| PFDA | Deionized water (alone) and NaCl (50 mM) (separate experiments), Temperature: not reported, pH: not reported | SOMS (organosilica adsorbent) | Particle surface, Particle size: 250–450 µm, Surface area: 650 m2/g, Pore volume: 1.03 mL/g | Adsorption capacity: ~7 mg/g (in deionized water), ~17 mg/g (in 50 mM NaCl) | Not Reported | Stebal et al. (2019) [66] | |
| PFDA | Deionized water (alone) and NaCl (50 mM) (separate experiments), Temperature: not reported, pH: not reported | F-SOMS (fluoroalkyl modified) | Particle surface, Particle size: 250–450 µm, Surface area: 660 m2/g, Pore volume: 1.04 mL/g | Adsorption capacity: ~0.2 mg/g (in deionized water), ~21 mg/g (in 50 mM NaCl) | Not Reported | Stebal et al. (2019) [66] | |
| PFUnDA | Water, Temperature: 20–45 °C, pH: 5 | Glass silica (siloxane, silanol patches) | Flat surface; siloxane and silanol patches | Surface excess: 2845 molecules/µm2, Adsorbed amount: 4.73 × 10−3 µmol/m2 (pH 5.0) | Multilayer | Schematics are the only available information on the adsorbed surface structure | Shafique et al. (2017) [85] |
| PFUnDA | Water and NaHCO3/NaCl (1.0 mM), Temperature: ~22.2 °C, pH: 7.5 | Kaolinite | Particle surface, Mean diameter: 1.1 µm (narrow size distribution), Surface area: 10 m2/g | Solid–water distribution coefficients (logkd): Single compound system = ~1.70 L/kg, Multi-compound system = ~1.72 L/kg | Not reported | Xiao et al. (2011) [42] | |
| PFDoDA | Water, Temperature: 20–45 °C, pH: 5 | Glass silica (siloxane, silanol patches) | Flat surface; siloxane and silanol patches | Surface Excess: 3337 molecules/µm2, Adsorbed Amount: 5.54 × 10−3 µmol/m2 (pH 5.0), | Multilayer | Schematics are the only available information on the adsorbed surface structure | Shafique et al. (2017) [85] |
| PFDoDA | Water, Temperature: 25 °C, pH: 6.3 | Organo-montmorillonites (0.5CEC-Mt) | Particle surface | Adsorption capacity: ~0.16 mmol/g | Not reported | Surfaces not well-defined | Zhou et al. (2010) [84] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alves, A.V.; Tsianou, M.; Alexandridis, P. Fluorinated Surfactant Adsorption on Mineral Surfaces: Implications for PFAS Fate and Transport in the Environment. Surfaces 2020, 3, 516-566. https://doi.org/10.3390/surfaces3040037
Alves AV, Tsianou M, Alexandridis P. Fluorinated Surfactant Adsorption on Mineral Surfaces: Implications for PFAS Fate and Transport in the Environment. Surfaces. 2020; 3(4):516-566. https://doi.org/10.3390/surfaces3040037
Chicago/Turabian StyleAlves, Anthony V., Marina Tsianou, and Paschalis Alexandridis. 2020. "Fluorinated Surfactant Adsorption on Mineral Surfaces: Implications for PFAS Fate and Transport in the Environment" Surfaces 3, no. 4: 516-566. https://doi.org/10.3390/surfaces3040037
APA StyleAlves, A. V., Tsianou, M., & Alexandridis, P. (2020). Fluorinated Surfactant Adsorption on Mineral Surfaces: Implications for PFAS Fate and Transport in the Environment. Surfaces, 3(4), 516-566. https://doi.org/10.3390/surfaces3040037