Abstract
There have been several reports concerning the performance improving properties of additives, such as polyvinylidene difluoride (PVDF), to the membrane or electrocatalyst layer of proton exchange membrane fuel cells (PEMFC). However, it is not clear if the observed performance enhancement is due to kinetic, mass transport, or anion blocking effects of the PVDF. In a previous investigation using a thin-film rotating disk electrode (RDE) approach (of decreased complexity as compared to membrane electrode assembly (MEA) tests), a performance increase for the oxygen reduction reaction (ORR) could be confirmed. However, even in RDE measurements, reactant mass transport in the catalyst layer cannot be neglected. Therefore, in the present study, the influence of PVDF is re-examined by coating polycrystalline bulk Pt electrodes by PVDF and measuring ORR activity. The results on polycrystalline bulk Pt indicate that the effects of PVDF on the reaction kinetics and anion adsorption are limited, and that the observed performance increase on high surface area Pt/C most likely is due to an erroneous estimation of the electrochemical active surface area (ECSA) from CO stripping and Hupd.
1. Introduction
Efficient energy converters, such as the proton exchange membrane fuel cell (PEMFC), are integral parts in an environmentally friendly hydrogen economy. Although the PEMFC technology has been around for several decades, it still suffers from relatively high costs [1]. With the aim of making the fuel cell technology more economical, significant effort has been invested in finding cheaper catalysts and fabrication techniques, but also in optimizing properties of the solid membrane electrolyte [2,3,4,5,6]. There are several reports stating that the properties of the solid membrane electrolyte can be improved by including different additives. For example, a performance enhancement of PEMFCs by including additives such as polyvinylidene difluoride (PVDF) into the membrane has been reported [7].
If such studies are performed in single cell membrane electrode assembly (MEA) tests, it is very difficult to analyze the origin of observed performance increases. This is due to the complexity of the MEA system and the influence of multiple structural aspects on its performance. Therefore, the effect of PVDF additions to the membrane is not yet fully clarified. In PEMFC research, a thin-film rotating disk electrode (RDE) approach is often applied to reduce the system complexity and to study electrocatalyst layers in a more fundamental fashion [8]. The previous work of our research group used such an approach to mix PVDF into the ink of a high surface area catalyst. The electrochemical properties of catalyst layers prepared by casting the ink on a glassy carbon disk were compared with respect to PVDF free films [9]. The specific oxygen reduction reaction (ORR) activity established by RDE in phosphoric acid electrolytes was found to improve when the catalyst layer contained PVDF. Further work, however, showed that even in a RDE setup, the apparent (measured) specific ORR activity is extremely sensitive to the film quality, which in turn depends on ink properties, such as pH [10]. Thus, the observed activity increase can be due to intrinsic changes, such as oxygen solubility and suppression of anion adsorption, or it might be due to improved mesoscopic film properties enhancing the local reactant mass transport. To investigate the possible catalytic enhancement due to PVDF further, in the presented work we therefore investigated thin PVDF films on a flat surface of polycrystalline bulk Pt. This system excludes any influence of catalyst film properties, and therefore focusses on the interface between catalyst and electrolyte.
2. Experiment
2.1. Sample Preparation
To prepare PVDF films onto a Pt surface of an RDE electrode, two different amounts of PVDF (Polysciences Inc. Warrington, PA, USA) were dissolved in Tetrahydrofuran (THF) to obtain two solutions containing 0.1 mM and 0.1 M of PVDF, respectively. The electrode tip for the PVDF coating was a polycrystalline Pt disk inserted in a cylinder of polytetrafluoroethylene (PTFE), in such a manner that only a circular area of the platinum is exposed. The Pt tip was sonicated in acetone, ethanol, isopropanol, and MilliQ water, repeating each step three times. Following the sonication, the Pt tip was dip coated into one of the two PVDF solutions for 30 seconds and pulled out of the electrolyte with the surface facing down. The PDVF coated Pt electrode tip was left to dry in air in a closed glass beaker. After drying, the mirror-like shine of the bare Pt surface was lost and the surface exhibited a pale milky color. In the following, PVDF 1 will refer to the polycrystalline Pt surface dip coated in the solution containing 0.1 mM PVDF, and PVDF 2 will refer to the Pt surface dip coated in the PVDF solution containing 0.1 M PVDF. After the drying process, the coated Pt electrode was introduced in an Ar-saturated 0.05 M H2SO4 electrolyte solution for electrochemical characterization.
2.2. Electrochemical Characterization
The electrochemical characterization was performed by RDE according to the procedure described in [11,12]. In brief, the electrochemical measurements were conducted in a three- compartment Teflon cell, using an RDE system (Radiometer Analytical, Europe Hach Lange GmbH Headquarter Willstätterstr. 11, D-40549 Düsseldorf, Germany). A saturated calomel electrode (SCE) and a Pt mesh have been used as reference and counter electrodes, respectively. The working electrode (WE) was a polycrystalline Pt disk (Ø = 5 mm, A = 0.196 cm2). All the potentials in the paper are expressed with respect to the reversible hydrogen electrode (RHE) potential experimentally determined for each measurement series. The 0.05 M H2SO4 aqueous electrolyte solution was prepared using Millipore Milli Q water (> 18.2 MΩcm, TOC < 5ppb) and 96% H2SO4 Suprapur (Sigma-Aldrich). The oxygen reduction rate and specific activity (SA) are typically calculated from the positive-going polarization curves recorded in oxygen saturated electrolytes at a scan rate of 50 mVs−1 and 1600 rpm rotation rate. In all cases, a background subtraction was performed to remove any contributions from capacitive and surface oxidation processes according to [13]. The iR-drop was measured and compensated according to the methodology used in [13,14]. In brief, the solution resistance was recorded online with the potentiostat (ECi-200 Nordic Electrochemistry Aps c/o Skelvangsvej 43, DK-8920 Randers NV, Denmark) by superimposing a 5 kHz, 5 mV AC signal, and compensated for by an analogue positive feedback scheme. The effective solution resistance was around 2Ω [15]. The electrochemical active surface area (ECSA) was determined by Hupd integration and CO stripping experiments described in [12,13]. In all the measurements the potential was cycled in the potential window between 0.05 and 1.05 VRHE.
3. Results and Discussion
3.1. Surface Characterization in Ar Saturated Electrolyte
In Figure 1, the cyclic voltammograms (CVs) of the bare Pt surface and the two different PVDF films on polycrystalline Pt recorded in Ar saturated electrolyte solution are shown. The two different dip coatings were made from solutions containing 0.1 M and 0.1 mM of PVDF solution, respectively. The voltammetric profiles of polycrystalline Pt are generally divided into three electrode potential regions: (i) the hydrogen under potential deposition (Hupd) region (0.05–0.3 VRHE), (ii) the double layer region (0.3–0.7 VRHE), and (iii) the potential region of OHad and Oad formation and reduction (>0.7 VRHE) [16]. The effect of PVDF can be scrutinized by comparing the different CVs in the respective potential regions. Starting with the Hupd region, two characteristic peaks of polycrystalline Pt in contact with a H2SO4 electrolyte solution are present for all samples. However, the Hupd currents decrease according to the amount of deposited PVDF (concentration of PVDF in the dipping solution). The formation and reduction oxygenated species exhibit the same trend, i.e., the intensity of the peaks decreases as more PVDF is deposited. In contrast, the double layer potential region remains similar for all the samples. Only a small increase in the pseudo-capacity is discernible for the Pt surfaces covered by PVDF, which might be interpreted by a minor surface roughening or by a small increase in the concentration of water dipoles close to the electrode surface. However, at the same time, the presence of PVDF clearly reduces the amount of Hupd and OHad species adsorbed onto the Pt surface. Further, the analysis of the Hupd peaks is a common method to determine the ECSA used for normalizing the measured activity of a Pt electrode. Integration of the Hupd peaks and the resulting charge, , can be related to the charge of a freshly flame annealed polycrystalline Pt surface, which has a per area unit 210 [17]. From this analysis, we can determine the loss in ECSA due to the PVDF coverage to 27 % and 43 % for PVDF 1 and PVDF 2, respectively.
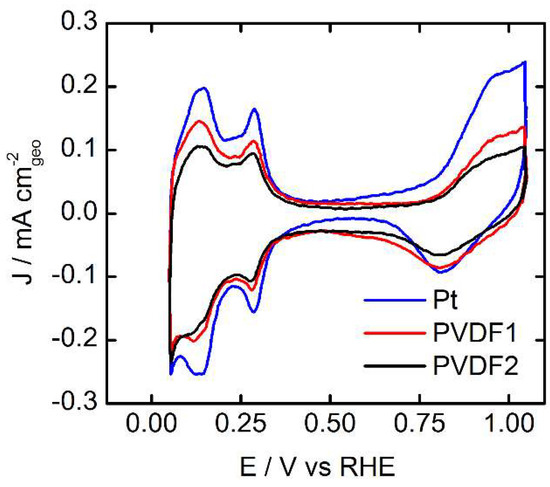
Figure 1.
Cyclic voltammograms recorded in Ar saturated 0.05 M H2SO4 electrolyte solution for bare and polyvinylidene difluoride (PVDF) modified Pt. The scan rate was 100 mVs−1 and the measurements were recorded at room temperature. PVDF 1 stands for the film prepared by dip coating the Pt electrode into 0.1 mM PVDF solution, PVDF 2 for the film prepared by dip coating into 0.1 M PVDF solution. The roughness factors (Rf), calculated by Hupd for bare Pt, PVDF1, and PVDF2, are: 1.65, 1.20, 0.94. This corresponds to an ECSA loss with respect to the bare polycrystalline Pt electrode of 27 % and 43 % for PVDF 1 and PVDF 2, respectively.
3.2. CO-Stripping Measurements
CO-stripping is an alternative technique to determine the ECSA [11]. A monolayer of CO is formed on Pt from a CO saturated electrolyte solution, keeping the electrode at a low fixed potential (typically around 50 mVRHE). Thereafter, the dissolved CO is displaced from the electrolyte solution by saturating it with Ar, and the CO adlayer is oxidized in a positive-going voltage scan. As CO oxidation is a two electron process, the surface area ACO, relating to the CO oxidation charge (QCO), is calculated by [17]:
In Figure 2, we show the CO stripping profiles for the bare surface and the two PVDF coated polycrystalline Pt surfaces. At the start of the stripping sweep starting from 0.05 VRHE, the CO covered Pt electrodes show no features, such as the Hupd, until the characteristic CO oxidation peak at around 0.8VRHE is observed [18]. The presence of the PVDF coating clearly reduces the amount of stripping charge and at the same time the oxidation onset, as well as the peak position of the maximum being shifted towards higher potentials. Calculating the ECSA loss from QCO, we determine losses of 17 % and 64%, respectively (with respect to the roughness factor of the bare Pt surface). This means that the ECSA loss calculated by Hupd and CO-oxidation led to comparable values, as seen above.
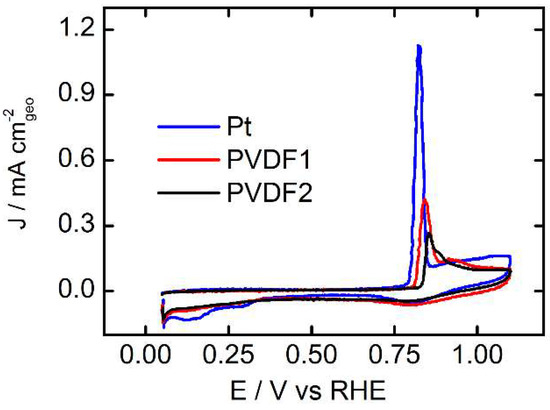
Figure 2.
CO stripping curves recorded for polycrystalline Pt and the two PVDF modified surfaces at room temperature in Ar saturated 0.05 M H2SO4 electrolyte solution. The scan rate was 50 mVs−1. PVDF 1 stands for the film prepared by dip coating the Pt electrode into 0.1 mM PVDF solution, PVDF 2 for the film prepared by dip coating into 0.1 M PVDF solution. The roughness factors (Rf) calculated by CO stripping for the bare Pt surface, PVDF1 and PVDF2 are: 1.40, 1.17, 0.51. This corresponds to an ECSA loss with respect to the bare polycrystalline Pt electrode of 17 % and 64 % for PVDF 1 and PVDF 2, respectively.
3.3. Oxygen Reduction
In order to determine the ORR activity, polarization curves of the bare and PVDF coated Pt surfaces were recorded in an oxygen saturated 0.05 M H2SO4 electrolyte solution, applying different rotations rates. The resulting background corrected data and their analysis are summarized in Figure 3.
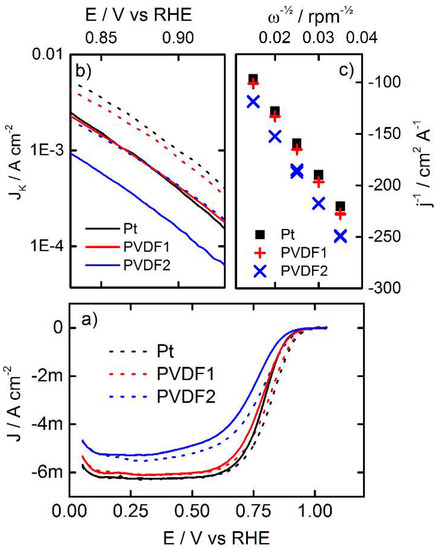
Figure 3.
(a) Oxygen reduction reaction (ORR) polarization curves recorded for bare polycrystalline Pt and PVDF modified Pt surfaces. Dotted and solid lines indicate the positive- and negative-going potential sweeps, respectively. The scan rate was 50 mVs−1 and the rotation rate 1600 rpm. The electrolyte was 0.05 M H2SO4 aqueous solution. (b) Tafel plot of the kinetic current. (c) Koutecky-Levich plot of the currents at 0.35 VRHE.
In Figure 3a, the ORR polarization curves of the three samples are separated into positive- and negative-going curves and compared with each other. Based on the current profiles, two distinct potential regions can be observed: the kinetic potential region at an electrode potential E > 0.8 VRHE, and the hydro dynamic mass transport limited potential region at E < 0.6 VRHE. The latter is easily identified by its strong rotation rate dependency (not shown). The influence of the thick PVDF coating (PVDF 2) can clearly be seen in the mass transport limited potential region, as well as in the kinetic region by a significant decrease in current. Furthermore, all curves exhibit a hysteresis, i.e., a difference in currents between the positive- and negative-going potential scan is observed. This hysteresis is a typical feature in RDE measurements and can be interpreted due to the fact that the steady state coverage of oxygenated species is not instantaneously achieved on Pt during potential sweeps when coming from the reduced or oxidized surface (lower or higher potentials). Thus, the observed ORR rate is artificially enhanced [19,20] in the positive-going polarization curves, whereas it may be too low in the negative-going polarization curves. This is explicitly shown in Figure 3b in a logarithmic Tafel plot of the kinetic current densities normalized to the geometric electrode area, i.e., JK. Note that the curves have been corrected for diffusion effects by the use of the Koutecký-Levich equation. In the positive-going potential sweeps, significantly higher kinetic ORR rates are determined than in the negative going potential sweeps. However, in the Tafel plot, the curves of the different kinetic currents form almost parallel lines, indicating that the same ORR reaction pathway is valid for all investigated surfaces. Furthermore, while larger amounts of PVDF inhibit the observed kinetic ORR current, the kinetic ORR currents on the bare Pt surface and PVDF 1 almost overlap. This means that although the ECSA on PVDF 1 is reduced, and the kinetic current is not affected.
As we consider the electrode during the negative-going potential sweep to be closer to the steady state condition, we chose the negative going potential sweeps for further analysis. In this analysis, we studied the hydrodynamic response of the system by changing of the convection (rotation rate of the RDE). In Figure 3c, the inverse of the measured current density is plotted versus ω½, in a so-called Koutecký-Levich plot based on:
where Jm is the measured current density, Jk the kinetic current density, JDL the diffusion limited current density, BL the Levich B factor, and ω the rotation rate.
It can be seen that the data points in the Koutecký-Levich plot form parallel lines, which is a clear indication that the hydrodynamic properties and the number of electrons transferred per oxygen molecule are not affected by the PVDF [21]. Thus, we conclude that the PVDF coating does not favor the formation of H2O2. The negative shift of the polarization curves of PVDF 2 can instead be related to a reduced interfacial current. The reduced interfacial current can be the result of a 3D PVDF film that reduces the diffusion to the electrode, or that a large part of the surface is blocked for the reaction by the PVDF, or a combination of both [22].
So far, we have only analyzed the kinetic current densities normalized to the geometric area, as is often done when working with bulk metal electrodes. To obtain catalytic turnover rates, or the equivalent in electrocatalysis, the (surface area) specific activity (SA), the kinetic current needs to be normalized to the ECSA of the catalyst. The determination of the ECSA of a catalyst is an essential task that can be challenging, in particular when working with catalysts for which no defined procedure is yet established [23]. Even though this is not the case for Pt based catalysts, where Hupd and CO stripping are standard procedures for the ECSA determination, the ECSA determination is not trivial. The procedure implies that the catalytic reactions with H+ and CO and the ORR probe the same type of active site on the catalyst surface. In addition, their accuracy is limited. In particular, the charge related to the Hupd process is sensitive to the lower potential limit of the CV, where Hupd overlaps with the hydrogen evolution reaction. Furthermore, it has to be mentioned that PVDF retains a dipole moment that could interact with polar molecules or protons, and thus alter the adsorption process on a PVDF coated surface. On the contrary, O2 does not retain any dipole moment, and low amounts of PVDF are not expected to affect the ORR process. Deviations in the ECSA between determination by Hupd and CO stripping, as shown here, are therefore not surprising, and can be considered within the accuracy of the method (usually around 10–15 %).
In Figure 4, comparing the JK with the normalized SA (normalized with the CO stripping charge) for Pt, PVDF 1, and PVDF 2, a complex behavior becomes apparent. In the positive-going potential scan (which is in general analyzed in RDE measurements), JK decreases with the PVDF content. However, this decrease is “overcompensated” by the loss in ECSA. This leads to a somewhat higher SA, especially for PVDF 2. However, in the negative-going potential scan—which resembles most steady state conditions—an increase in SA is observed for PVDF 1, but almost no increase in SA is observed for PVDF 2. Taking all results into account, these findings indicate that the ORR kinetics are neither significantly inhibited nor enhanced by the PVDF films. Improvement in SA seems mostly related to the fact that the surface area determination is “more affected” by the PVDF than the ORR itself, which might be related to the fact that for the ECSA determination, different probe molecules are used.
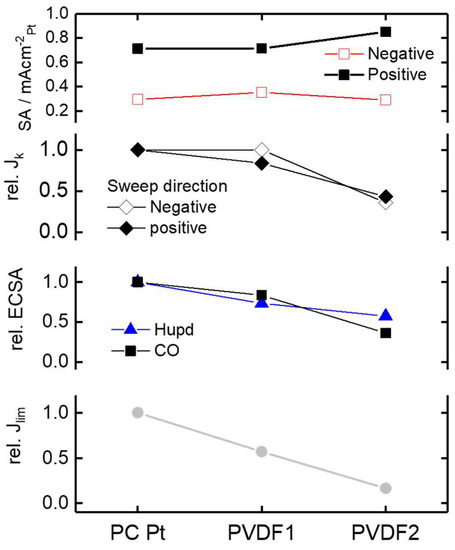
Figure 4.
Summary of results. Specific activity (SA) values at 0.9 VRHE for bare and PVDF modified Pt in the positive and negative potential scan, respectively. Relative (normalized to bare Pt) current density JK; relative ECSA based on Hupd and CO stripping charge; relative limiting current density Jlim for bare and PVDF modified Pt.
It was not an arbitrary choice to use H2SO4 as a supporting electrolyte. H2SO4 is known to be a “strongly adsorbing” electrolyte, and decreased ORR kinetics in H2SO4 as compared to a “non-absorbing” electrolyte, i.e. HClO4, are observed. This is attributed to the site blocking nature of the HSO4− and SO42− anions. In literature reporting a positive influence of PVDF on the ORR activity, it was speculated that PVDF might positively influence (inhibit) anion absorption. However, in the presented work we never observed an increase in Jk for a PVDF coated Pt surface, and thus conclude that there is no positive influence of PVDF on anion absorption, as a diminished anion absorption would result in a decreased overpotential of the ORR. The activity enhancing effect of PVDF that has been previously observed for high surface area catalysts (i.e. Pt nanoparticles supported on a carbon support (Pt/C)), therefore, is most likely due to different reasons—a combination of reduced active surface area and improved film properties that has been seen to strongly influence the apparent ORR activity of Pt/C in RDE measurements [10].
Lastly, it is seen that the diffusion limited current Jlim is affected by the amount of PVDF used to cast the Pt surface. This could be the result of two different processes: a change in the reaction pathway of the ORR, or the formation of a 3D diffusion barrier. As the Levich analysis shows a straight line with constant B values (Fig. 3) for all the samples, a change in reaction pathway has to be discarded. We therefore conclude that the main reason for the reduction of Jk is the formation of a permeable 3D layer, which partially blocks the active catalyst surface.
4. Conclusions
We have examined ORR on polycrystalline Pt with different amounts of PVDF deposited. The experimental results indicate that PVDF forms a 3D film which disturbs the mass transport of oxygen to the Pt surface. The PVDF coating also exhibits an active site-blocking effect, as both the ECSA and the apparent kinetic activity are reduced for thick PVDF layers. However, for a thin PVDF layer, the kinetic region overlaps with the one measured for bare Pt, even though the ECSA is decreased. Thus, the SA exhibits a slight increase. However, this increase in SA is associated with the loss in ECSA rather than a real kinetic enhancement effect. Depending on the scan direction, the same can be observed for thick PVDF films. Here, the kinetic current is reduced, but this reduction is overcompensated by the ECSA loss. Thus, we conclude that PVDF does not affect the ORR kinetics in 0.05 M H2SO4. The reduction in ECSA with even low PVDF coverage might be related to a difference between the interaction of the probe molecules (H+ and CO) with the PVDF covered surface and the reactant. Thus, PVDF used in small amounts inhibits the adsorption of CO and H+ without strongly blocking the ORR kinetics. This leads to the “artificial” increase in SA. Therefore, we can conclude that the addition of PVDF to Pt bulk and Pt-based catalysts does not affect the kinetics of ORR. Previously reported performance enhancing properties might be due the reduced ECSA, as well as other factors, like improved film quality.
Author Contributions
Conceptualization, A.Z. and G.K.H.W.; Data curation, A.Z.; Formal analysis, A.Z. and G.K.H.W.; Supervision, M.A.; Writing—original draft, A.Z. and G.K.H.W.; Writing—review & editing, M.A.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Debe, M.K. Electrocatalyst approaches and challenges for automotive fuel cells. Nature 2012, 486, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Gasteiger, H.A.; Kocha, S.S.; Sompalli, B.; Wagner, F.T. Activity benchmarks and requirements for Pt, Pt-alloy, and non-Pt oxygen reduction catalysts for PEMFCs. Appl. Catal. B-Environ. 2005, 56, 9–35. [Google Scholar] [CrossRef]
- Frey, T.; Linardi, M. Effects of membrane electrode assembly preparation on the polymer electrolyte membrane fuel cell performance. Electrochim. Acta 2004, 50, 99–105. [Google Scholar] [CrossRef]
- Shahgaldi, S.; Ghasemi, M.; Wan Daud, W.R.; Yaakob, Z.; Sedighi, M.; Alam, J.; Ismail, A.F. Performance enhancement of microbial fuel cell by PVDF/Nafion nanofibre composite proton exchange membrane. Fuel Process. Technol. 2014, 124, 290–295. [Google Scholar] [CrossRef]
- Li, H.-B.; Shi, W.-Y.; Zhang, Y.-F.; Liu, D.-Q.; Liu, X.-F. Effects of Additives on the Morphology and Performance of PPTA/PVDF in Situ Blend UF Membrane. Polymers 2014, 6, 1846–1861. [Google Scholar] [CrossRef]
- Mu, S.; Tian, M. Optimization of perfluorosulfonic acid ionomer loadings in catalyst layers of proton exchange membrane fuel cells. Electrochim. Acta 2012, 60, 437–442. [Google Scholar] [CrossRef]
- Brodt, M.; Wycisk, R.; Dale, N.; Pintauro, P. Power Output and Durability of Electrospun Fuel Cell Fiber Cathodes with PVDF and Nafion/PVDF Binders. J. Electrochem. Soc. 2016, 163, F401–F410. [Google Scholar] [CrossRef]
- Schmidt, T.; Gasteiger, H.; Stäb, G.; Urban, P.; Kolb, D.; Behm, R. Characterization of high-surface-area electrocatalysts using a rotating disk electrode configuration. J. Electrochem. Soc. 1998, 145, 2354–2358. [Google Scholar] [CrossRef]
- Holst-Olesen, K.; Nesselberger, M.; Perchthaler, M.; Hacker, V.; Arenz, M. Activity inhibition and its mitigation in high temperature proton exchange membrane fuel cells: The role of phosphoric acid, ammonium trifluoromethanesulfonate, and polyvinylidene difluoride. J. Power Sources 2014, 272, 1072–1077. [Google Scholar] [CrossRef]
- Inaba, M.; Quinson, J.; Arenz, M. pH matters: The influence of the catalyst ink on the oxygen reduction activity determined in thin film rotating disk electrode measurements. J. Power Sources 2017, 353, 19–27. [Google Scholar] [CrossRef]
- Mayrhofer, K.J.J.; Strmcnik, D.; Blizanac, B.B.; Stamenkovic, V.; Arenz, M.; Markovic, N.M. Measurement of oxygen reduction activities via the rotating disc electrode method: From Pt model surfaces to carbon-supported high surface area catalysts. Electrochim. Acta 2008, 53, 3181–3188. [Google Scholar] [CrossRef]
- Zana, A.; Speder, J.; Roefzaad, M.; Altmann, L.; Bäumer, M.; Arenz, M. Probing Degradation by IL-TEM: The Influence of Stress Test Conditions on the Degradation Mechanism. J. Electrochem. Soc. 2013, 160, F608–F615. [Google Scholar] [CrossRef]
- Nesselberger, M.; Ashton, S.; Meier, J.C.; Katsounaros, I.; Mayrhofer, K.J.J.; Arenz, M. The Particle Size Effect on the Oxygen Reduction Reaction Activity of Pt Catalysts: Influence of Electrolyte and Relation to Single Crystal Models. J. Am. Chem. Soc. 2011, 133, 17428–17433. [Google Scholar] [CrossRef] [PubMed]
- Wiberg, G.K.H. The Development of a State-of-the-Art Experimental Setup Demonstrated by the Investigation of Fuel Cell Reactions in Alkaline Electrolyte. Ph.D. Thesis, Technical Universtity of Munich, Munich, Germany, 2010. [Google Scholar]
- Deng, Y.-J.; Wiberg, G.K.H.; Zana, A.; Arenz, M. On the oxygen reduction reaction in phosphoric acid electrolyte: Evidence of significantly increased inhibition at steady state conditions. Electrochim. Acta 2016, 204, 78–83. [Google Scholar] [CrossRef]
- Deng, Y.-J.; Arenz, M.; Wiberg, G.K. Equilibrium coverage of OH ad in correlation with platinum catalyzed fuel cell reactions in HClO 4. Electrochem. Commun. 2015, 53, 41–44. [Google Scholar] [CrossRef]
- Trasatti, S.; Petrii, O. Real surface area measurements in electrochemistry. Pure Appl. Chem. 1991, 63, 711–734. [Google Scholar] [CrossRef]
- Arenz, M.; Mayrhofer, K.J.J.; Stamenkovic, V.; Blizanac, B.B.; Tomoyuki, T.; Ross, P.N.; Markovic, N.M. The effect of the particle size on the kinetics of CO electrooxidation on high surface area Pt catalysts. J. Am. Chem. Soc. 2005, 127, 6819–6829. [Google Scholar] [CrossRef]
- Wiberg, G.K.H.; Arenz, M. Establishing the potential dependent equilibrium oxide coverage on platinum in alkaline solution and its influence on the oxygen reduction. J. Power Sources 2012, 217, 262–267. [Google Scholar] [CrossRef]
- Hodnik, N.; Baldizzone, C.; Cherevko, S.; Zeradjanin, A.; Mayrhofer, K.J. The Effect of the Voltage Scan Rate on the Determination of the Oxygen Reduction Activity of Pt/C Fuel Cell Catalyst. Electrocatalysis 2015, 6, 1–5. [Google Scholar] [CrossRef]
- Bard, A.J.; Faulkner, L.R. Electrochemical Methods Fundamentals and Applications, 2nd ed.; Wiley: New York, NY, USA, 2001; p. 833. [Google Scholar]
- Jinnouchi, R.; Kudo, K.; Kitano, N.; Morimoto, Y. Molecular Dynamics Simulations on O2 Permeation through Nafion Ionomer on Platinum Surface. Electrochim. Acta 2016, 188, 767–776. [Google Scholar] [CrossRef]
- Wiberg, G.K.; Mayrhofer, K.J.; Arenz, M. Investigation of the oxygen reduction activity on silver—A rotating disc electrode study. Fuel Cells 2010, 10, 575–581. [Google Scholar] [CrossRef]




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).