Effects of the Interfacial Structure on the Methanol Oxidation on Platinum Single Crystal Electrodes


Abstract
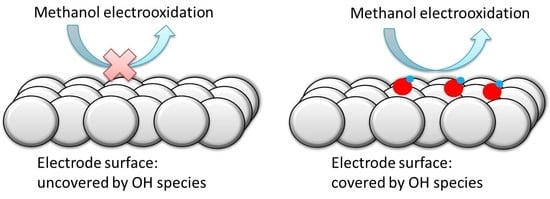
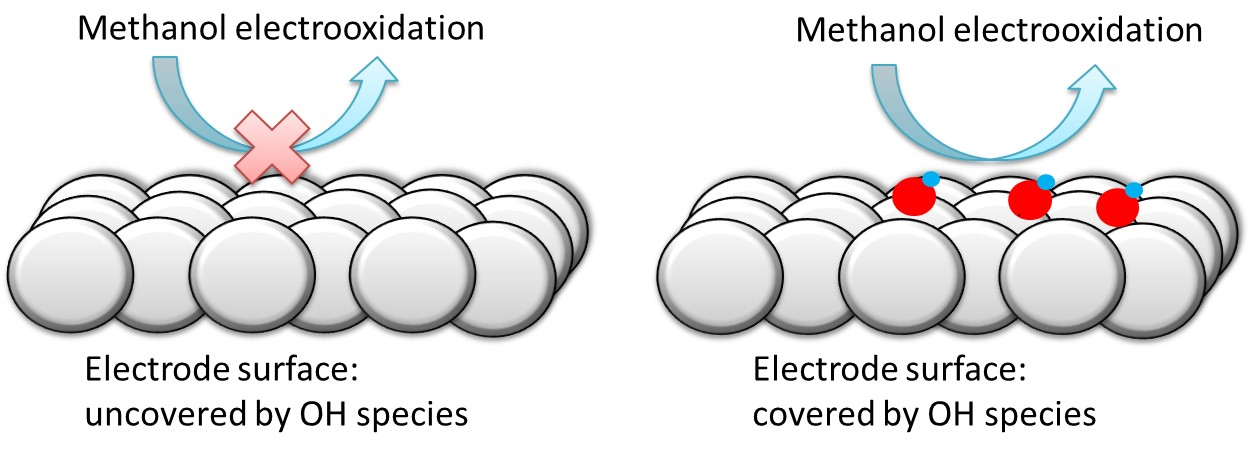{kind=link}
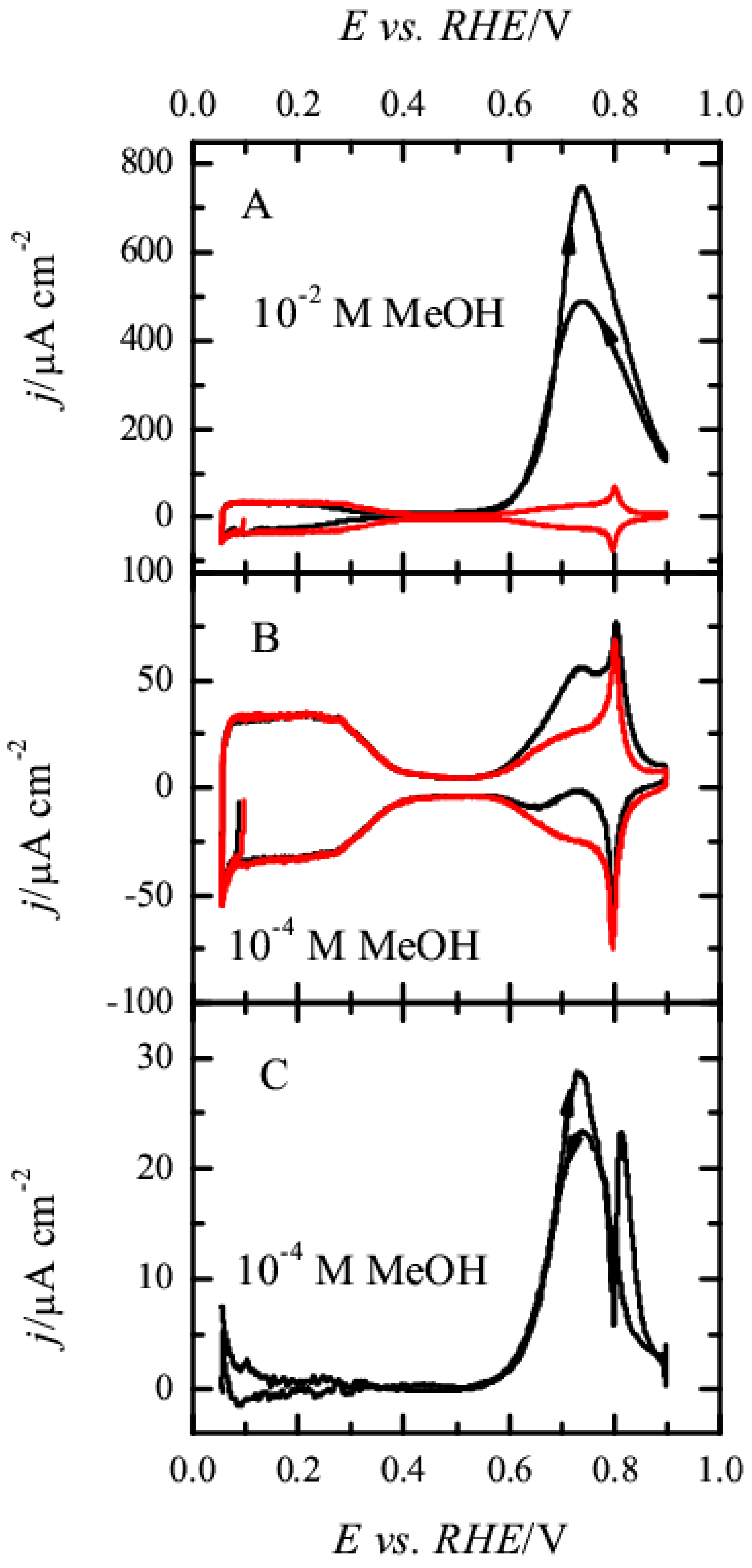{kind=link}
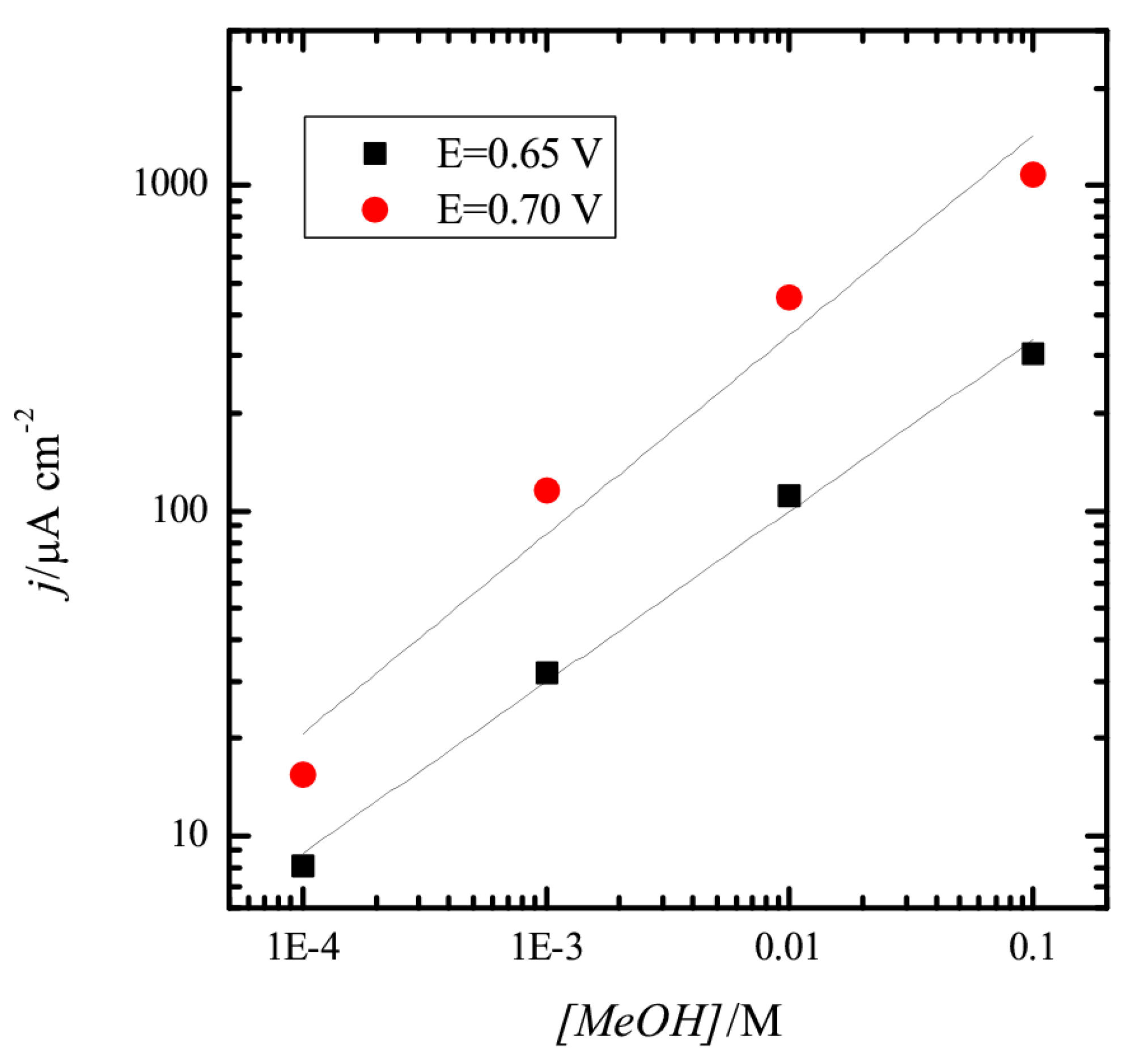{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
3. Results
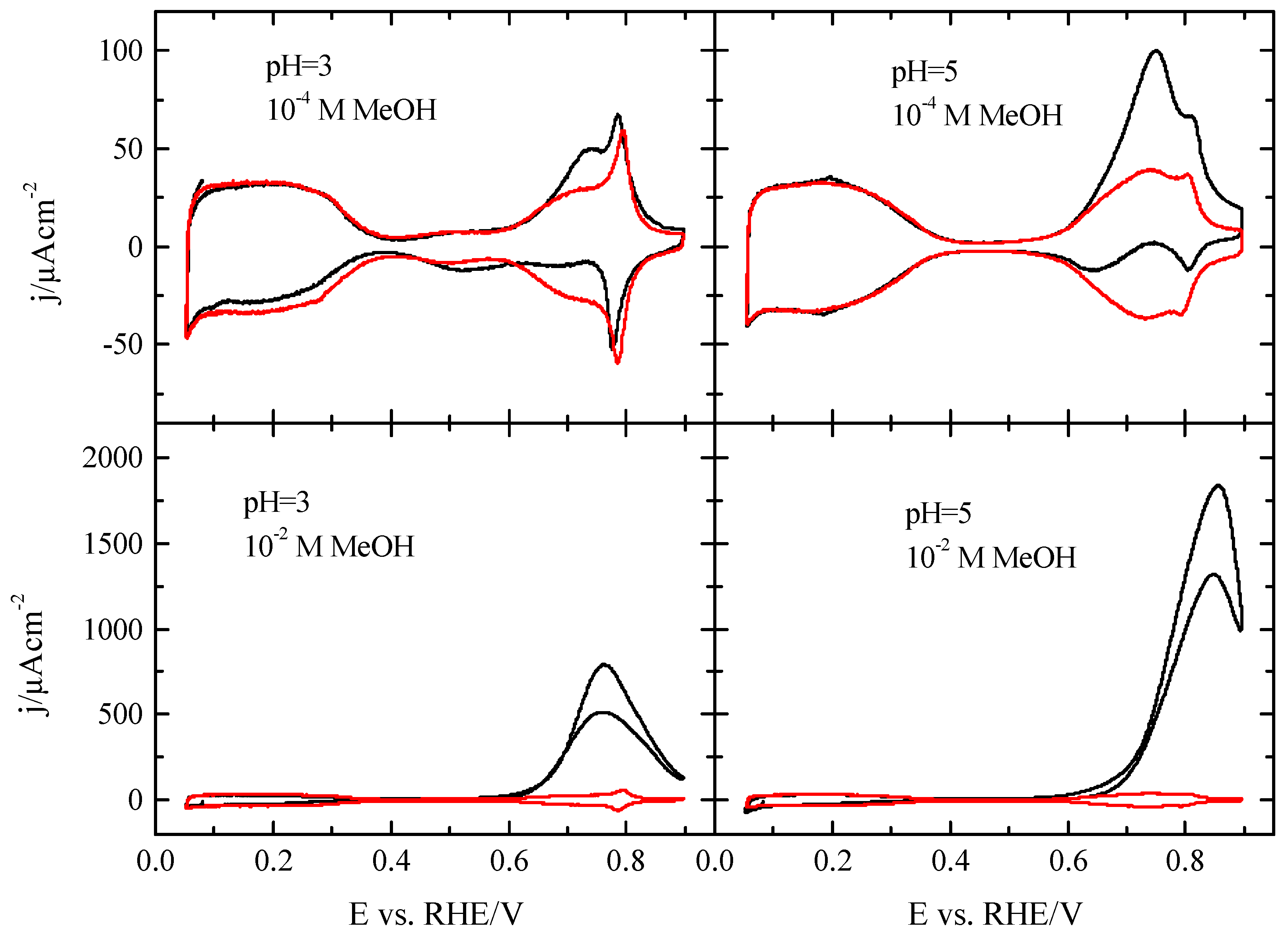
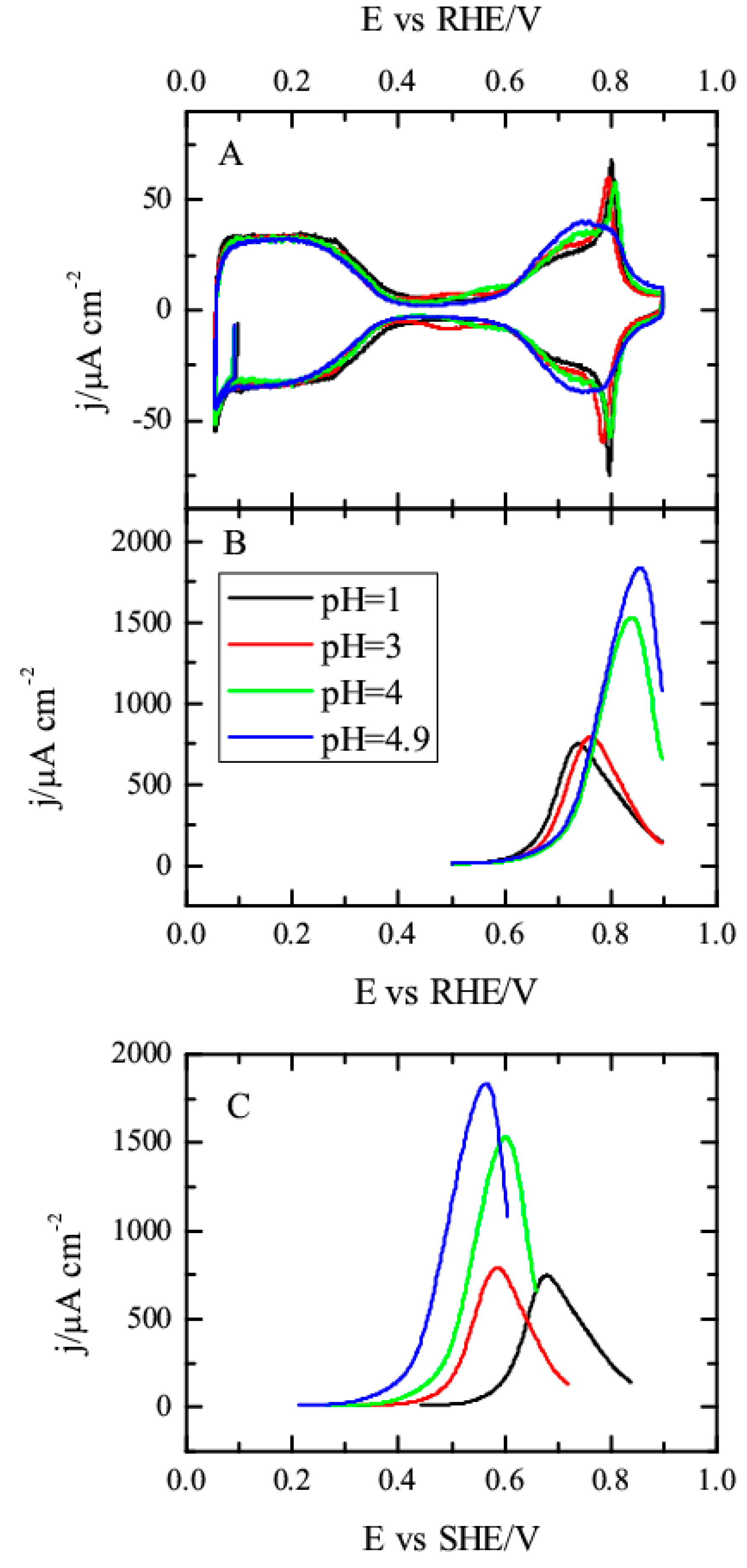
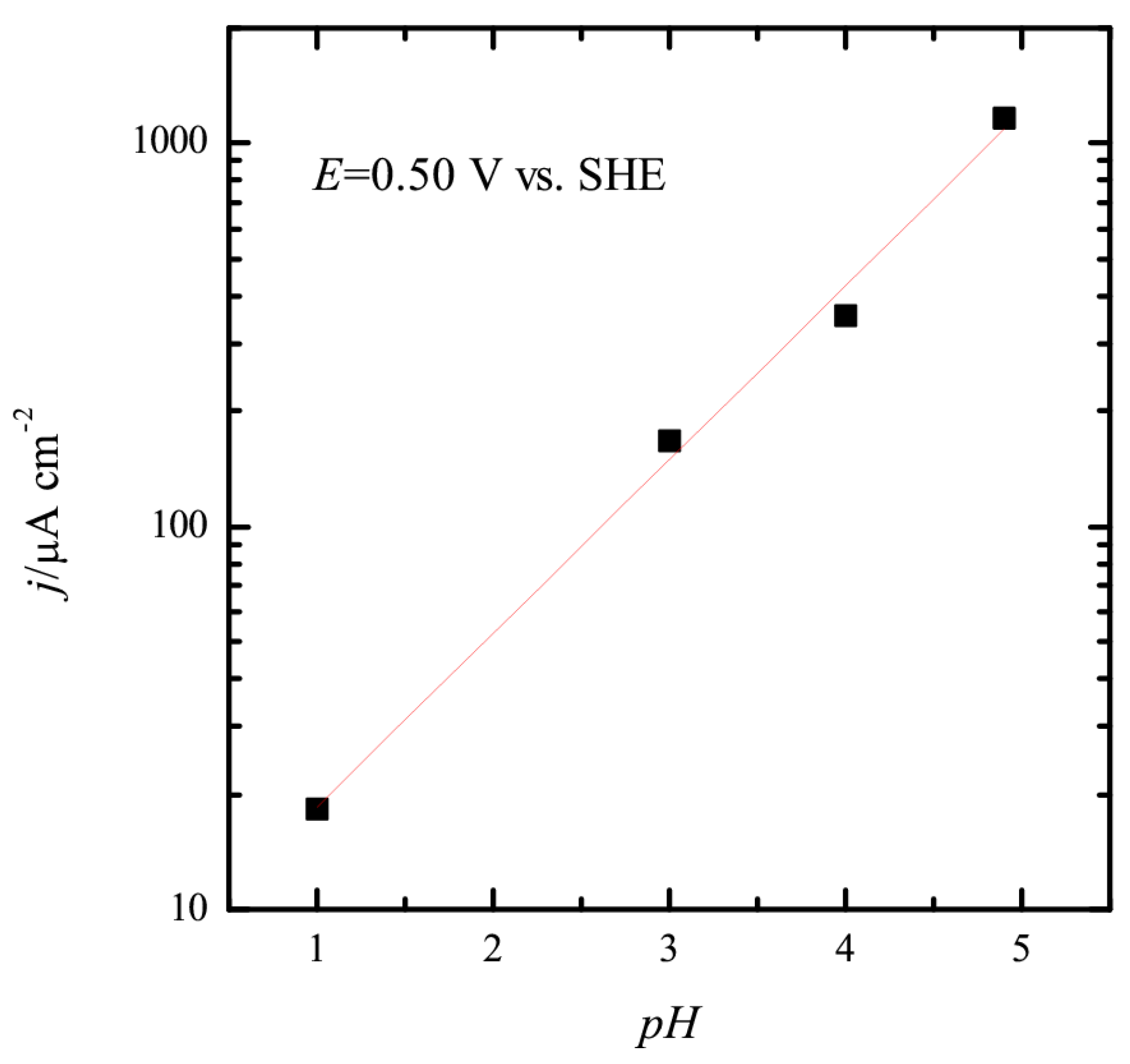
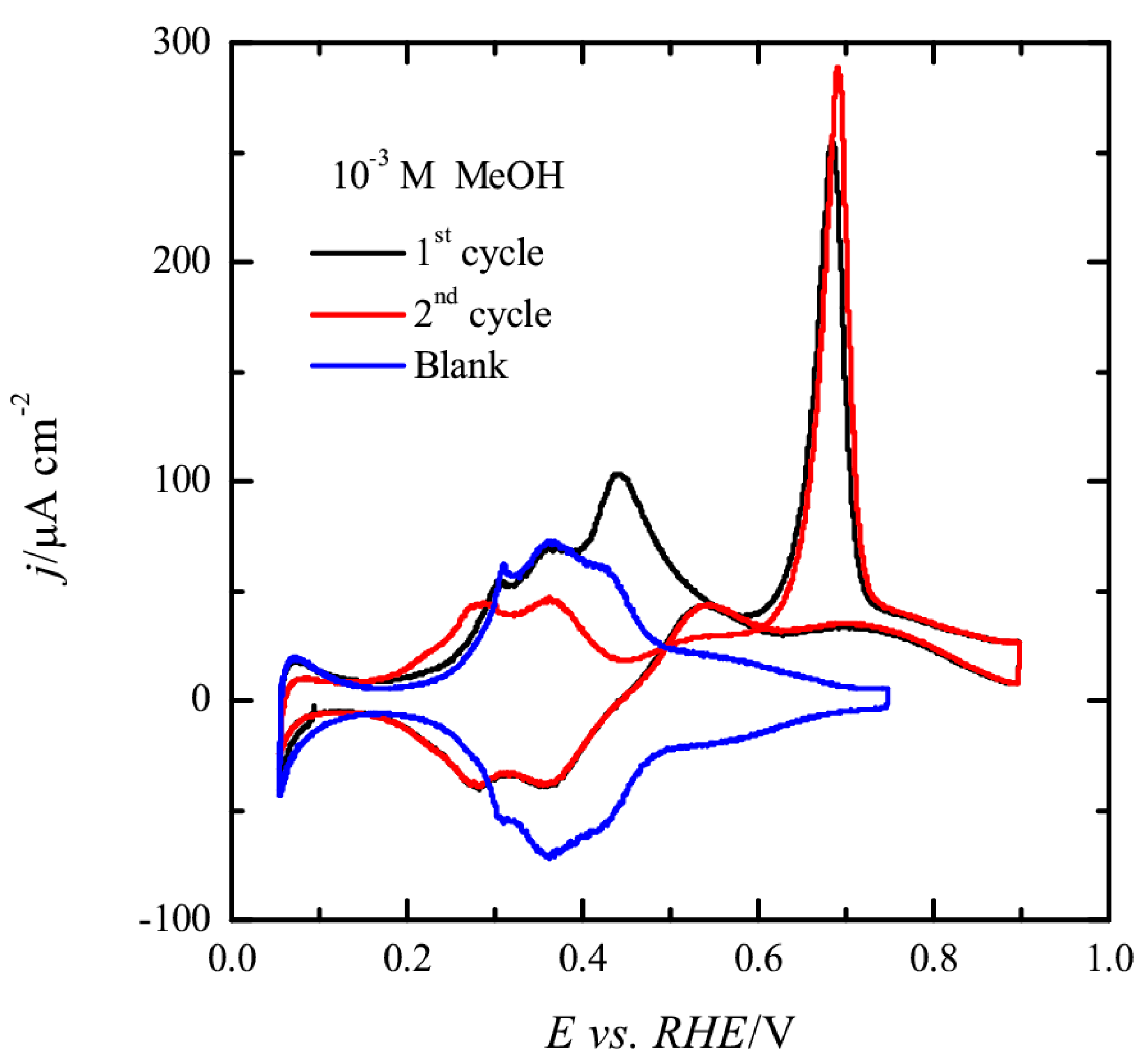
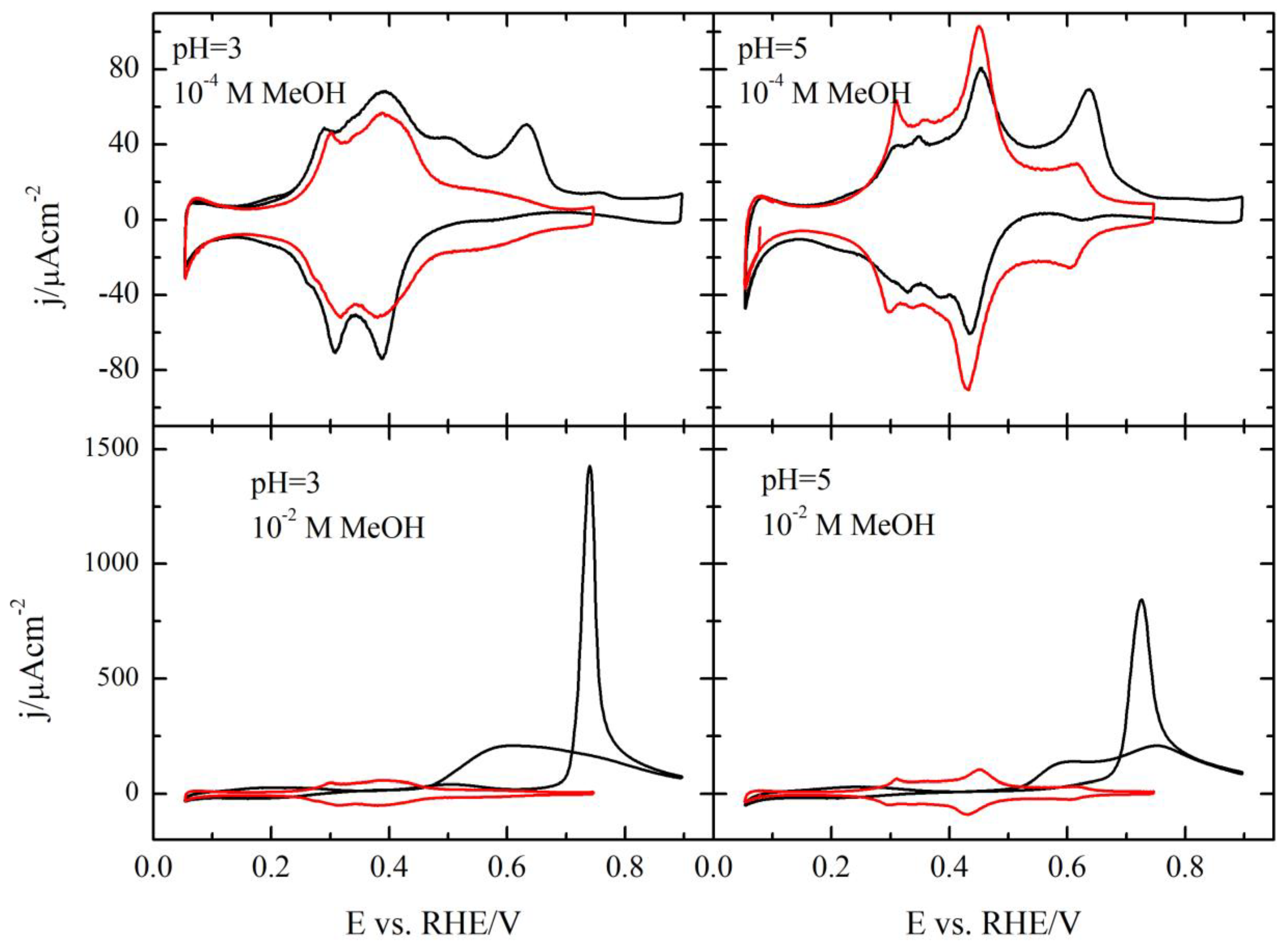
3.1. Pt(111) Electrode
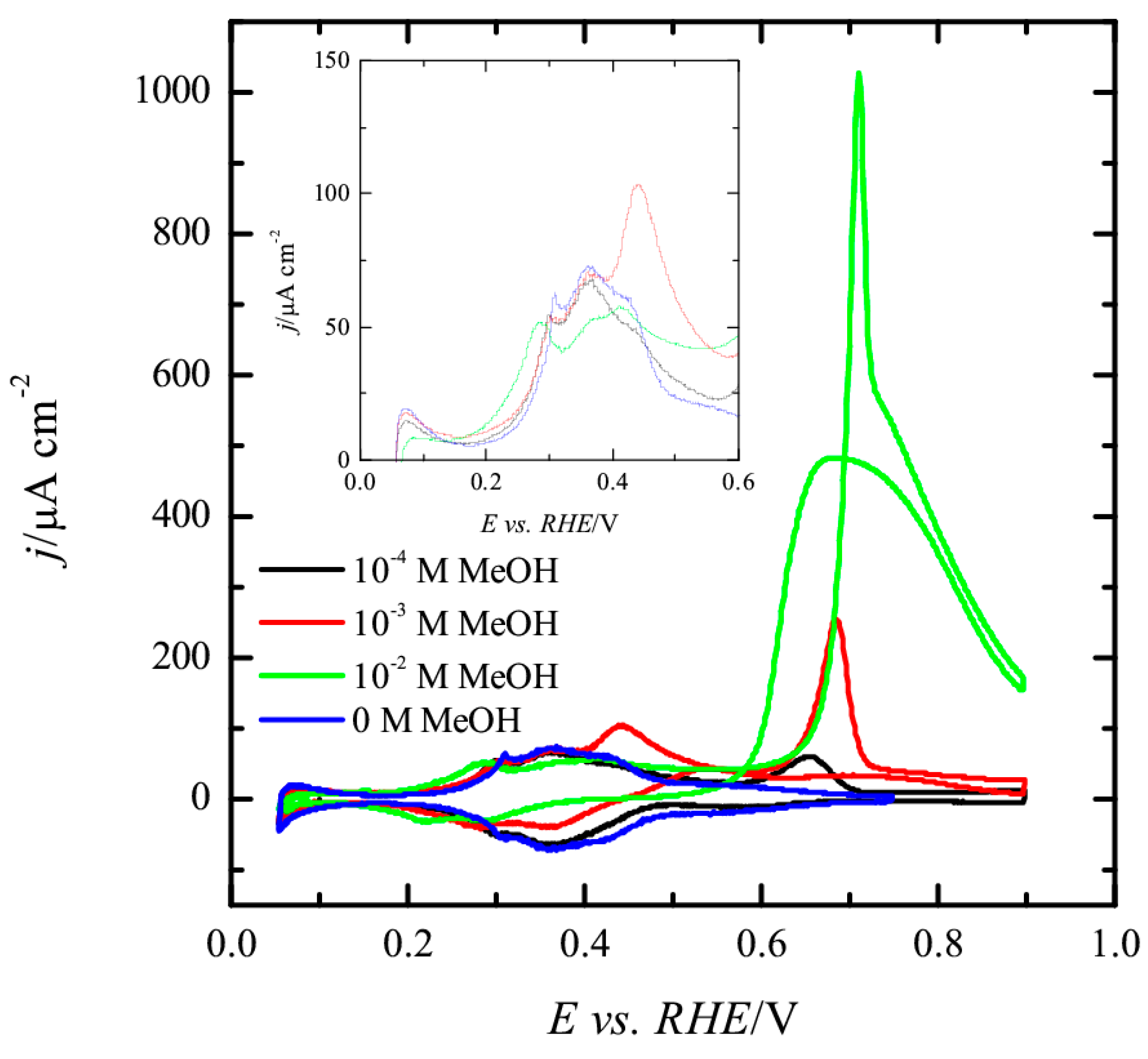
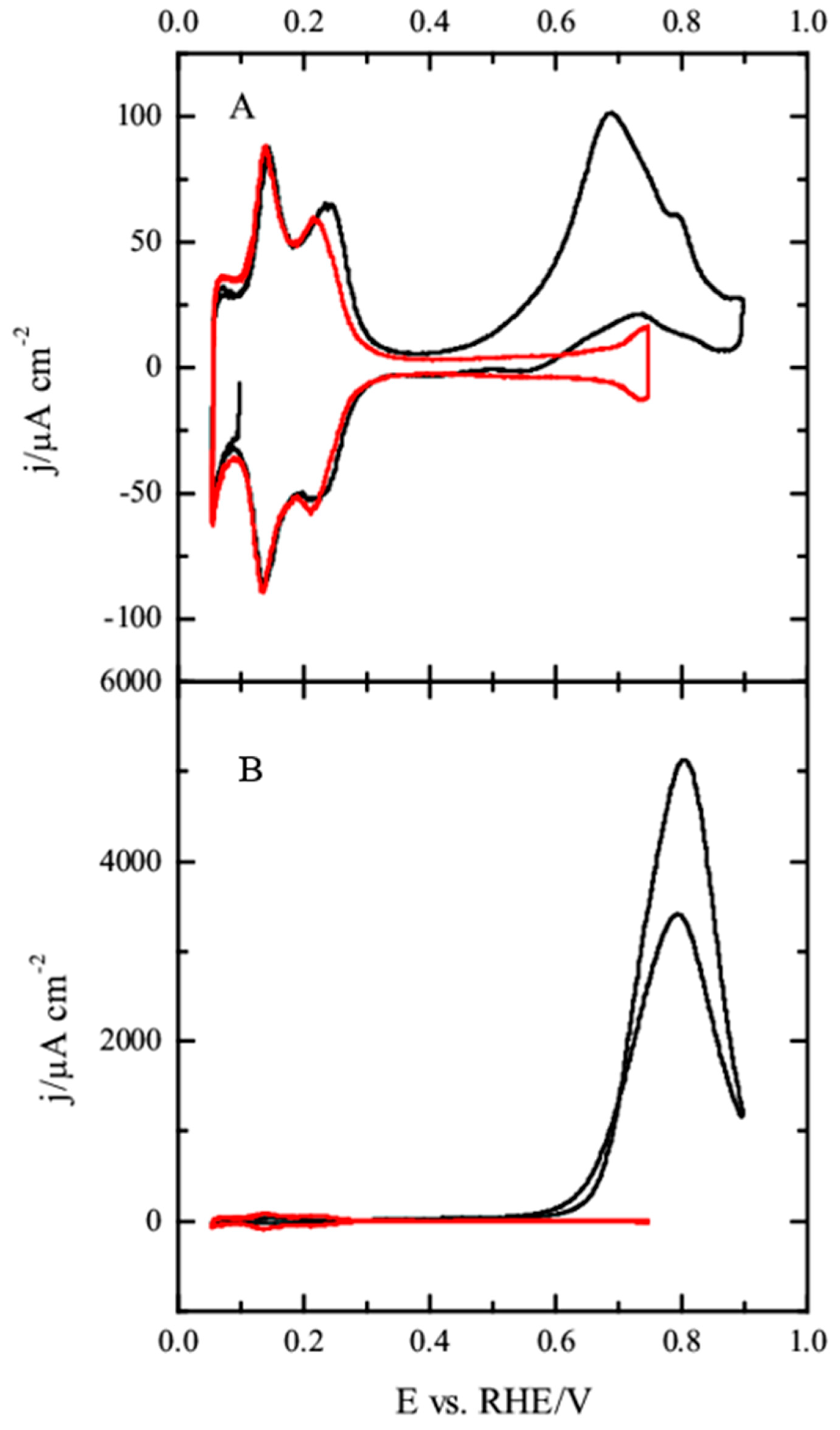
3.2. Pt(100) and Pt(110) Electrodes
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Parsons, R.; Vandernoot, T. The oxidation of small organic molecules: A survey of recent fuel cell related research. J. Electroanal. Chem. 1988, 257, 9–45. [Google Scholar] [CrossRef]
- Sriramulu, S.; Javi, T.D.; Stuve, E.M. Kinetic Modeling of Electrocatalytic Reactions: Methanol Oxidation on Platinum Electrodes. In Interfacial Electrochemistry, Theory, Experiments and Applications; Wieckowski, A., Ed.; Marcel Dekker: New York, NY, USA, 1998; p. 793. [Google Scholar]
- Iwasita, T. Electrocatalysis of methanol oxidation (vol 47, pg 3663, 2001). Electrochim. Acta 2002, 48, 289. [Google Scholar] [CrossRef]
- Markovic, N.M.; Ross, P.N. Surface science studies of model fuel cell electrocatalysts. Surf. Sci. Rep. 2002, 45, 117–229. [Google Scholar] [CrossRef]
- Koper, M.T.M.T.M.; Lai, S.C.C.S.; Herrero, E. Mechanisms of the Oxidation of Carbon Monoxide and Small Organic Molecules at Metal Electrodes. In Fuel Cell Catalysis: A Surface Science Approach; Koper, M.T.M., Ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2009; pp. 166–171. ISBN 978-0-470-13116-9. [Google Scholar]
- Cohen, J.L.; Volpe, D.J.; Abruna, H.D. Electrochemical determination of activation energies for methanol oxidation on polycrystalline platinum in acidic and alkaline electrolytes. Phys. Chem. Chem. Phys. 2007, 9, 49–77. [Google Scholar] [CrossRef] [PubMed]
- Neurock, M.; Janik, M.; Wieckowski, A. A first principles comparison of the mechanism and site requirements for the electrocatalytic oxidation of methanol and formic acid over Pt. Faraday Discuss. 2009, 140, 363–378. [Google Scholar] [CrossRef]
- Clavilier, J.; Lamy, C.; Leger, J.M. Electrocatalytic Oxidation of Methanol on Single-Crystal Platinum-Electrodes—Comparison with Polycrystalline Platinum. J. Electroanal. Chem. 1981, 125, 249–254. [Google Scholar] [CrossRef]
- Lamy, C.; Leger, J.M.; Clavilier, J.; Parsons, R. Structural effects in electrocatalysis: A comparative study of the oxidation of CO, HCOOH and CH3OH on single crystal Pt electrodes. J. Electroanal. Chem. 1983, 150, 71–77. [Google Scholar] [CrossRef]
- Franaszczuk, K.; Herrero, E.; Zelenay, P.; Wieckowski, A.; Wang, J.; Masel, R.I.I. A comparison of electrochemical and gas-phase decomposition of methanol on platinum surfaces. J. Phys. Chem. 1992, 96, 8509–8516. [Google Scholar] [CrossRef]
- Herrero, E.; Franaszczuk, K.; Wieckowski, A. Electrochemistry of methanol at low index crystal planes of platinum. An integrated voltammetric and chronoamperometric study. J. Phys. Chem. 1994, 98, 5074–5083. [Google Scholar] [CrossRef]
- Housmans, T.H.M.; Koper, M.T.M. Methanol oxidation on stepped Pt n(111) x (110) electrodes: A chronoamperometric study. J. Phys. Chem. B 2003, 107, 8557–8567. [Google Scholar] [CrossRef]
- Lai, S.C.S.; Lebedeva, N.P.; Housmans, T.H.M.; Koper, M.T.M. Mechanisms of carbon monoxide and methanol oxidation at single-crystal electrodes. Top. Catal. 2007, 46, 320–333. [Google Scholar] [CrossRef]
- Grozovski, V.; Climent, V.; Herrero, E.; Feliu, J.M. The role of the surface structure in the oxidation mechanism of methanol. J. Electroanal. Chem. 2011, 662, 43–51. [Google Scholar] [CrossRef]
- Martínez-Hincapié, R.; Sebastián-Pascual, P.; Climent, V.; Feliu, J.M. Exploring the interfacial neutral pH region of Pt(111) electrodes. Electrochem. Commun. 2015, 58, 62–64. [Google Scholar] [CrossRef]
- Sebastián, P.; Martínez-Hincapié, R.; Climent, V.; Feliu, J.M. Study of the Pt(111) | electrolyte interface in the region close to neutral pH solutions by the laser induced temperature jump technique. Electrochim. Acta 2017, 228, 667–676. [Google Scholar] [CrossRef]
- Clavilier, J.; Faure, R.; Guinet, G.; Durand, R. Preparation of monocrystalline Pt microelectrodes and electrochemical study of the plane surfaces cut in the direction of the {111} and {110} planes. J. Electroanal. Chem. 1980, 107, 205–209. [Google Scholar] [CrossRef]
- Korzeniewski, C.; Climent, V.; Feliu, J. Electrochemistry at Platinum Single Crystal Electrodes. In Electroanalytical Chemistry A Series of Advances: Volume 24; CRC Press: Boca Raton, FL, USA, 2011; pp. 75–170. ISBN 978-1-4398-3750-4. [Google Scholar]
- Clavilier, J.; Elachi, K.; Petit, M.; Rodes, A.; Zamakhchari, M.A. Electrochemical Monitoring of the Thermal Reordering of Platinum Single-Crystal Surfaces after Metallographic Polishing from the Early Stage to the Equilibrium Surfaces. J. Electroanal. Chem. 1990, 295, 333–356. [Google Scholar] [CrossRef]
- Herrero, E.; Orts, J.M.; Aldaz, A.; Feliu, J.M. Scanning tunneling microscopy and electrochemical study of the surface structure of Pt(10,10,9) and Pt(11,10,10) electrodes prepared under different cooling conditions. Surf. Sci. 1999, 440, 259–270. [Google Scholar] [CrossRef]
- García-Aráez, N.; Climent, V.; Herrero, E.; Feliu, J.M. On the electrochemical behavior of the Pt(1 0 0) vicinal surfaces in bromide solutions. Surf. Sci. 2004, 560, 269–284. [Google Scholar] [CrossRef]
- Markovic, N.M.; Grgur, B.N.; Lucas, C.A.; Ross, P.N. Surface electrochemistry of CO on Pt(110)-(1x2) and Pt(110)-(1x1) surfaces. Surf. Sci. 1997, 384, L805–L814. [Google Scholar] [CrossRef]
- Attard, G.A.; Hunter, K.; Wright, E.; Sharman, J.; Martínez-Hincapié, R.; Feliu, J.M. The voltammetry of surfaces vicinal to Pt{110}: Structural complexity simplified by CO cooling. J. Electroanal. Chem. 2017, 793, 137–146. [Google Scholar] [CrossRef]
- Herrero, E.; Wieckowski, A. Electrochemistry of Methanol at Low Index Crystal Planes. J. Phys. Chem. 1994, 98, 5074–5083. [Google Scholar] [CrossRef]
- Berna, A.; Climent, V.; Feliu, J.M. New understanding of the nature of OH adsorption on Pt(111) electrodes. Electrochem. Commun. 2007, 9, 2789–2794. [Google Scholar] [CrossRef]
- Angelucci, C.A.C.A.; Herrero, E.; Feliu, J.M.J.M. Modeling CO oxidation on Pt(111) electrodes. J. Phys. Chem. C 2010, 114, 14154–14163. [Google Scholar] [CrossRef]
- Housmans, T.H.M.; Wonders, A.H.; Koper, M.T.M. Structure Sensitivity of Methanol Electrooxidation Pathways on Platinum: An On-Line Electrochemical Mass Spectrometry Study. J. Phys. Chem. B 2006, 110, 10021–10031. [Google Scholar] [CrossRef] [PubMed]
- Herrero, E.; Fernández-Vega, A.; Feliu, J.M.; Aldaz, A. Poison formation reaction from formic acid and methanol on Pt(111) electrodes modified by irreversibly adsorbed Bi and As. J. Electroanal. Chem. 1993, 350, 73–88. [Google Scholar] [CrossRef]
- Grozovski, V.; Vidal-Iglesias, F.J.; Herrero, E.; Feliu, J.M. Adsorption of formate and its role as intermediate in formic acid oxidation on platinum electrodes. ChemPhysChem 2011, 12, 1641–1644. [Google Scholar] [CrossRef] [PubMed]
- Grozovski, V.; Climent, V.; Herrero, E.; Feliu, J.M. Intrinsic activity and poisoning rate for HCOOH oxidation on platinum stepped surfaces. Phys. Chem. Chem. Phys. 2010, 12, 8822. [Google Scholar] [CrossRef] [PubMed]
- Perales-Rondón, J.V.; Herrero, E.; Feliu, J.M. Effects of the anion adsorption and pH on the formic acid oxidation reaction on Pt(111) electrodes. Electrochim. Acta 2014, 140, 511–517. [Google Scholar] [CrossRef]
- Batista, E.A.; Malpass, G.R.P.; Motheo, A.J.; Iwasita, T. New mechanistic aspects of methanol oxidation. J. Electroanal. Chem. 2004, 571, 273–282. [Google Scholar] [CrossRef]
- Abd-El-Latif, A.A.; Baltruschat, H. Formation of methylformate during methanol oxidation revisited: The mechanism. J. Electroanal. Chem. 2011, 662, 204–212. [Google Scholar] [CrossRef]
- Mostafa, E.; Abd-El-Latif, A.E.A.A.; Baltruschat, H. Electrocatalytic oxidation and adsorption rate of methanol at Pt stepped single-crystal electrodes and effect of Ru step decoration: A DEMS study. ChemPhysChem 2014, 15, 2029–2043. [Google Scholar] [CrossRef] [PubMed]
- Ferre-Vilaplana, A.; Perales-Rondón, J.V.V.; Buso-Rogero, C.; Feliu, J.M.M.; Herrero, E. Formic acid oxidation on platinum electrodes: A detailed mechanism supported by experiments and calculations on well-defined surfaces. J. Mater. Chem. A 2017, 5, 21773–21784. [Google Scholar] [CrossRef]
- Ferre-Vilaplana, A.; Perales-Rondón, J.V.; Feliu, J.M.J.M.; Herrero, E.; Perales-Rondón, J.V.; Feliu, J.M.J.M.; Herrero, E. Understanding the effect of the adatoms in the formic acid oxidation mechanism on Pt(111) electrodes. ACS Catal. 2015, 5, 645–654. [Google Scholar] [CrossRef]
- Bergelin, M.; Herrero, E.; Feliu, J.M.M.; Wasberg, M. Oxidation of CO adlayers on Pt(111) at low potentials: An impinging jet study in H2SO4 electrolyte with mathematical modeling of the current transients. J. Electroanal. Chem. 1999, 467, 74–84. [Google Scholar] [CrossRef]
- Lebedeva, N.P.; Rodes, A.; Feliu, J.M.; Koper, M.T.M.; van Santen, R.A. Role of crystalline defects in electrocatalysis: CO adsorption and oxidation on stepped platinum electrodes as studied by in situ infrared spectroscopy. J. Phys. Chem. B 2002, 106, 9863–9872. [Google Scholar] [CrossRef]
- Rizo, R.; Sitta, E.; Herrero, E.; Climent, V.; Feliu, J.M.J.M. Towards the understanding of the interfacial pH scale at Pt(111) electrodes. Electrochim. Acta 2015, 162, 138–145. [Google Scholar] [CrossRef]
- Vidal-Iglesias, F.J.; Arán-Ais, R.M.; Solla-Gullón, J.; Herrero, E.; Feliu, J.M. Electrochemical characterization of shape-controlled Pt nanoparticles in different supporting electrolytes. ACS Catal. 2012, 2, 901–910. [Google Scholar] [CrossRef]
- Briega-Martos, V.; Herrero, E.; Feliu, J.M.J.M. Effect of pH and Water Structure on the Oxygen Reduction Reaction on platinum electrodes. Electrochim. Acta 2017, 241, 497–509. [Google Scholar] [CrossRef]
- Gamboa-Aldeco, M.E.; Herrero, E.; Zelenay, P.S.; Wieckowski, A. Adsorption of bisulfate anion on a Pt(100) electrode: A comparison with Pt(111) and Pt(poly). J. Electroanal. Chem. 1993, 348, 451–457. [Google Scholar] [CrossRef]
- Climent, V.; Gómez, R.; Orts, J.M.; Feliu, J.M. Thermodynamic analysis of the temperature dependence of OH adsorption on Pt(111) and Pt(100) electrodes in acidic media in the absence of specific anion adsorption. J. Phys. Chem. B 2006, 110, 11344–11351. [Google Scholar] [CrossRef]
- Souza-Garcia, J.; Climent, V.; Feliu, J.M. Voltammetric characterization of stepped platinum single crystal surfaces vicinal to the (110) pole. Electrochem. Commun. 2009, 11, 1515–1518. [Google Scholar] [CrossRef]
- Gómez, R.; Orts, J.M.; Alvarez-Ruiz, B.; Feliu, J.M. Effect of temperature on hydrogen adsorption on Pt(111), Pt(110), and Pt(100) electrodes in 0.1 M HClO4. J. Phys. Chem. B 2004, 108, 228–238. [Google Scholar] [CrossRef]
- Herrero, E.; Feliu, J.M.; Aldaz, A. Poison formation reaction from formic acid on Pt(100) electrodes modified by irreversibly adsorbed bismuth and antimony. J. Electroanal. Chem. 1994, 368, 101–108. [Google Scholar] [CrossRef]
- Arán-Ais, R.M.; Figueiredo, M.C.; Vidal-Iglesias, F.J.; Climent, V.; Herrero, E.; Feliu, J.M. On the behavior of the Pt(1 0 0) and vicinal surfaces in alkaline media. Electrochim. Acta 2011, 58, 184–192. [Google Scholar] [CrossRef]









© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kamyabi, M.A.; Martínez-Hincapié, R.; Feliu, J.M.; Herrero, E. Effects of the Interfacial Structure on the Methanol Oxidation on Platinum Single Crystal Electrodes. Surfaces 2019, 2, 177-192. https://doi.org/10.3390/surfaces2010014
Kamyabi MA, Martínez-Hincapié R, Feliu JM, Herrero E. Effects of the Interfacial Structure on the Methanol Oxidation on Platinum Single Crystal Electrodes. Surfaces. 2019; 2(1):177-192. https://doi.org/10.3390/surfaces2010014
Chicago/Turabian StyleKamyabi, Mohammad Ali, Ricardo Martínez-Hincapié, Juan M. Feliu, and Enrique Herrero. 2019. "Effects of the Interfacial Structure on the Methanol Oxidation on Platinum Single Crystal Electrodes" Surfaces 2, no. 1: 177-192. https://doi.org/10.3390/surfaces2010014
APA StyleKamyabi, M. A., Martínez-Hincapié, R., Feliu, J. M., & Herrero, E. (2019). Effects of the Interfacial Structure on the Methanol Oxidation on Platinum Single Crystal Electrodes. Surfaces, 2(1), 177-192. https://doi.org/10.3390/surfaces2010014