Ultrafast Vibrational Dynamics of CO Ligands on RuTPP/Cu(110) under Photodesorption Conditions


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
3. Results
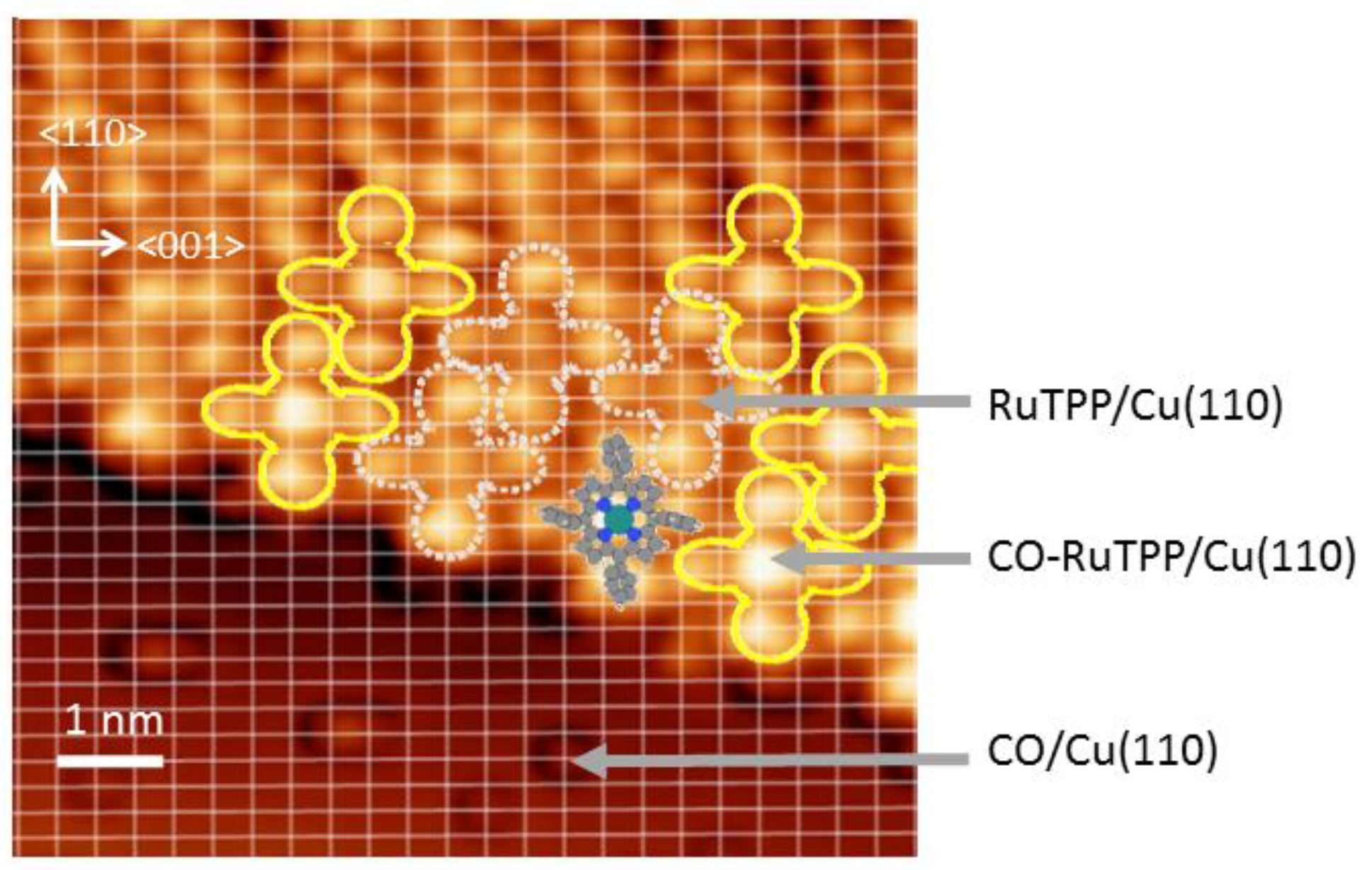
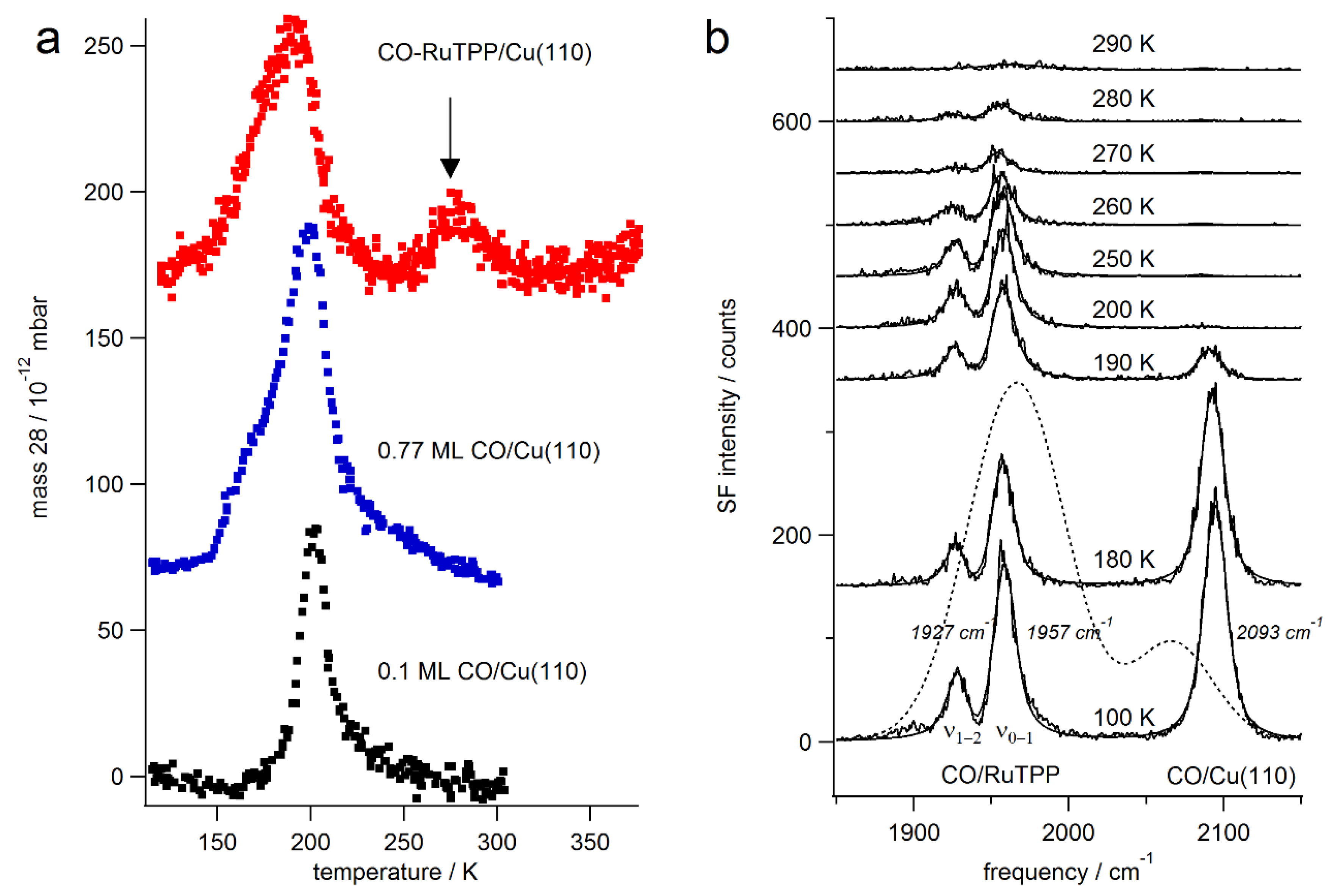
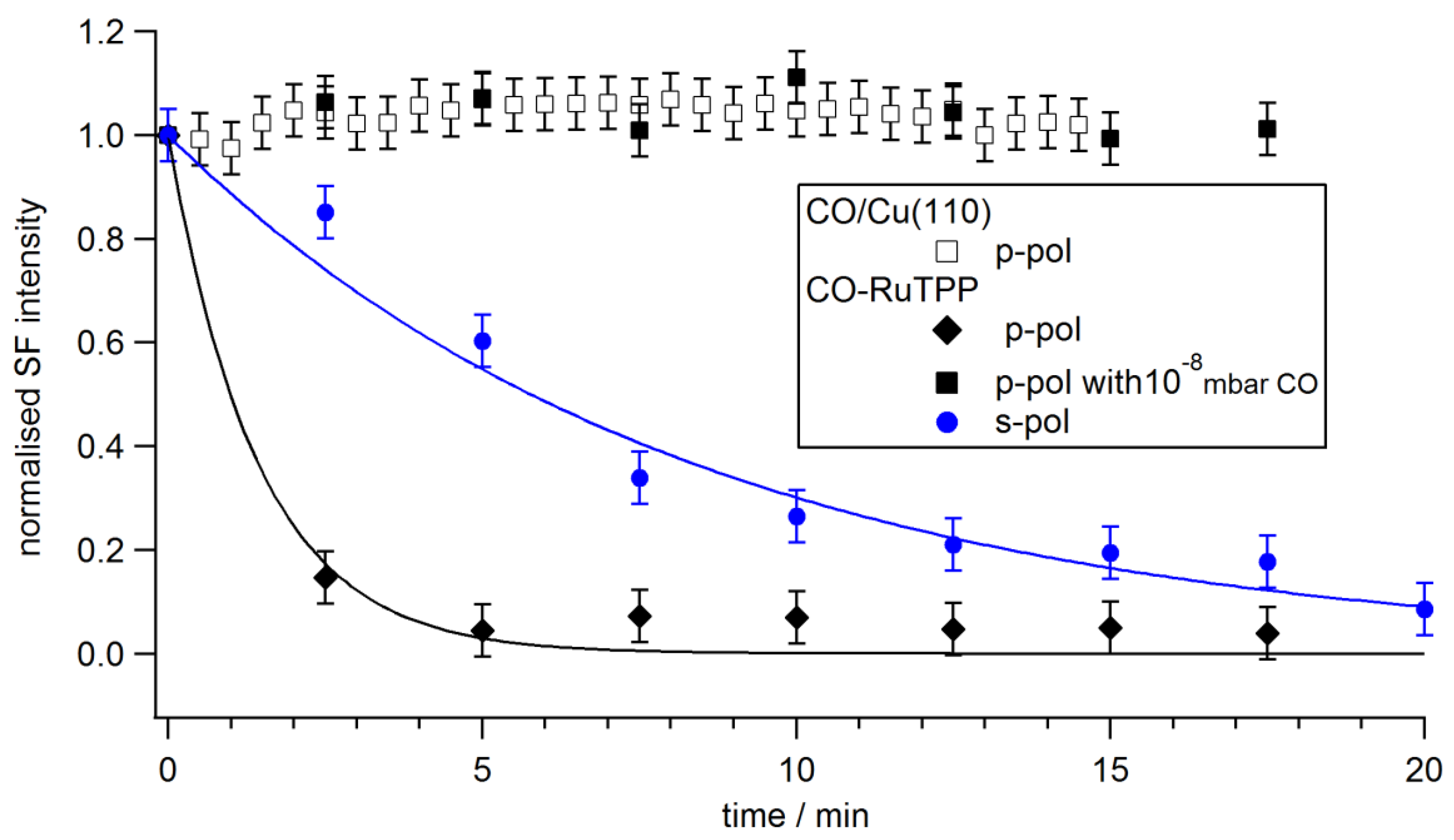
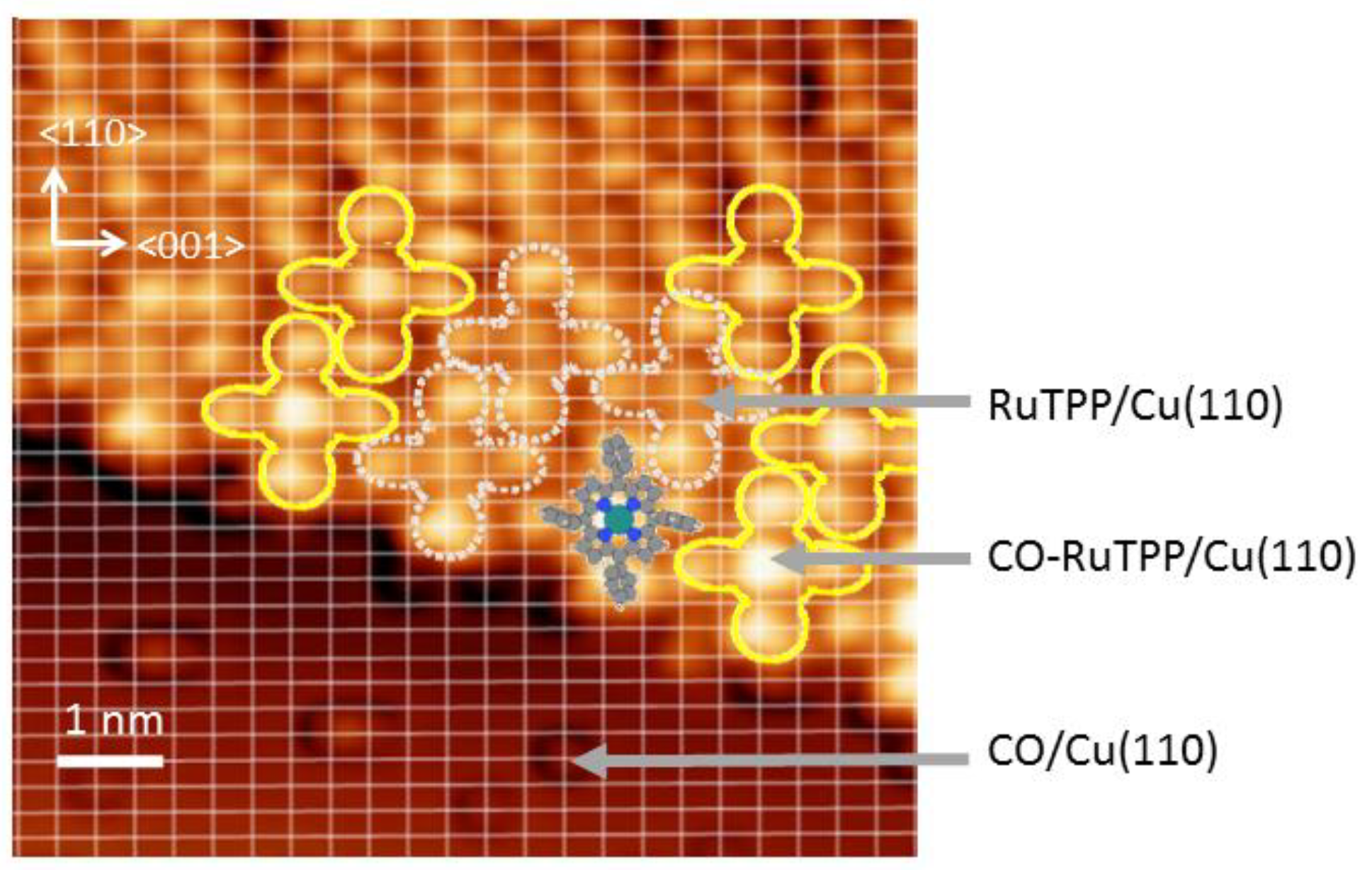
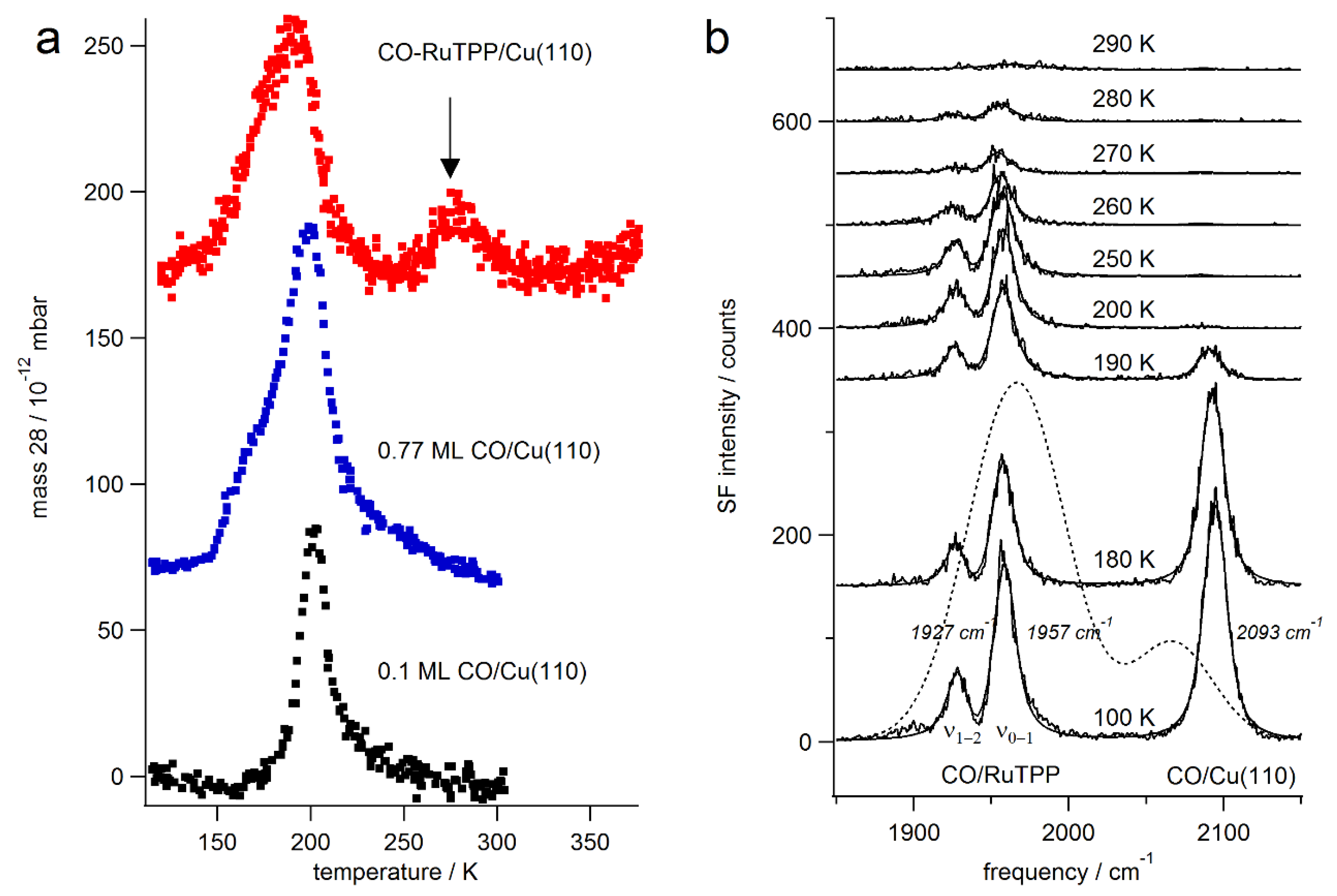
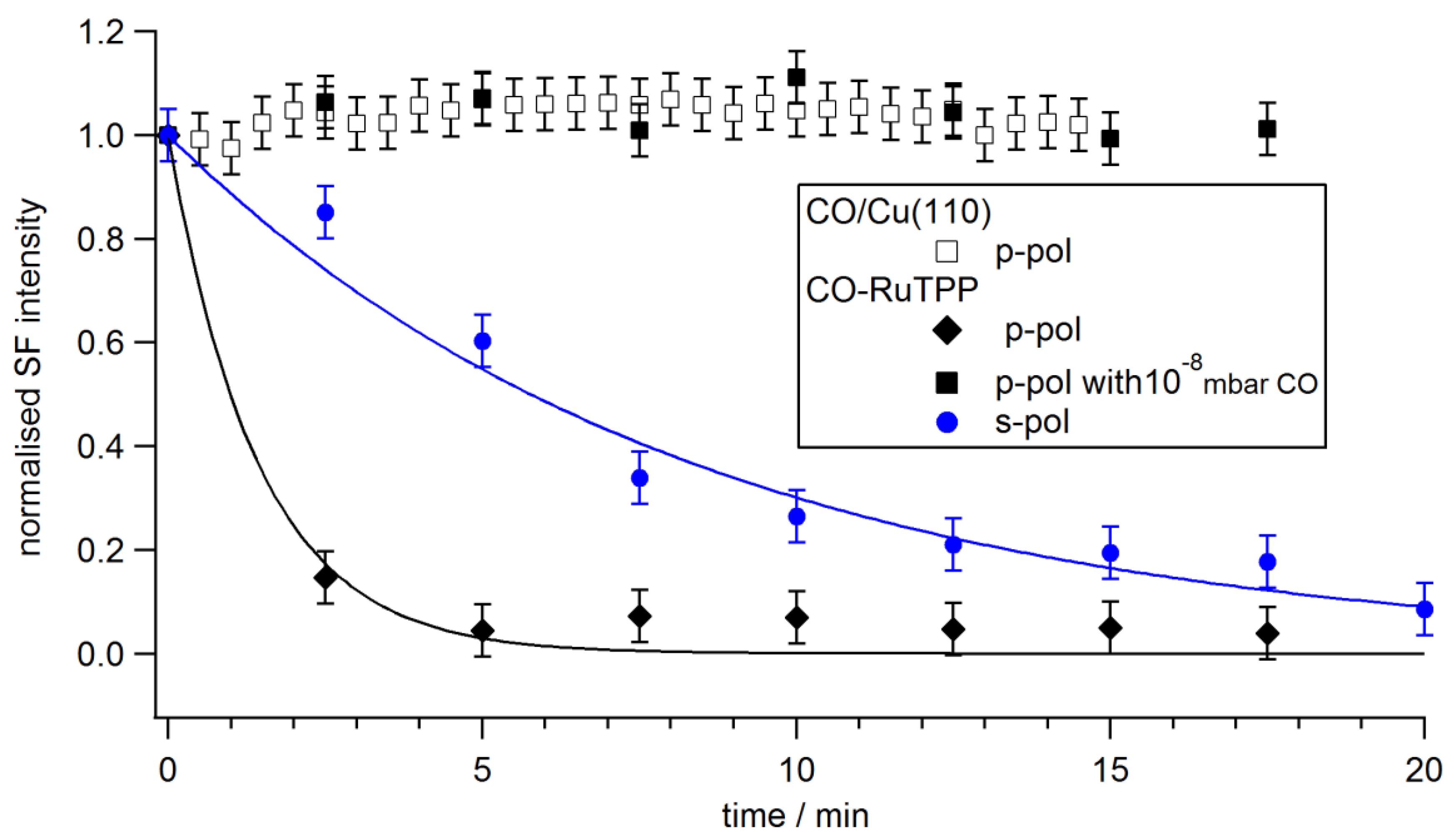
3.1. Thermal Versus Laser Induced Desorption
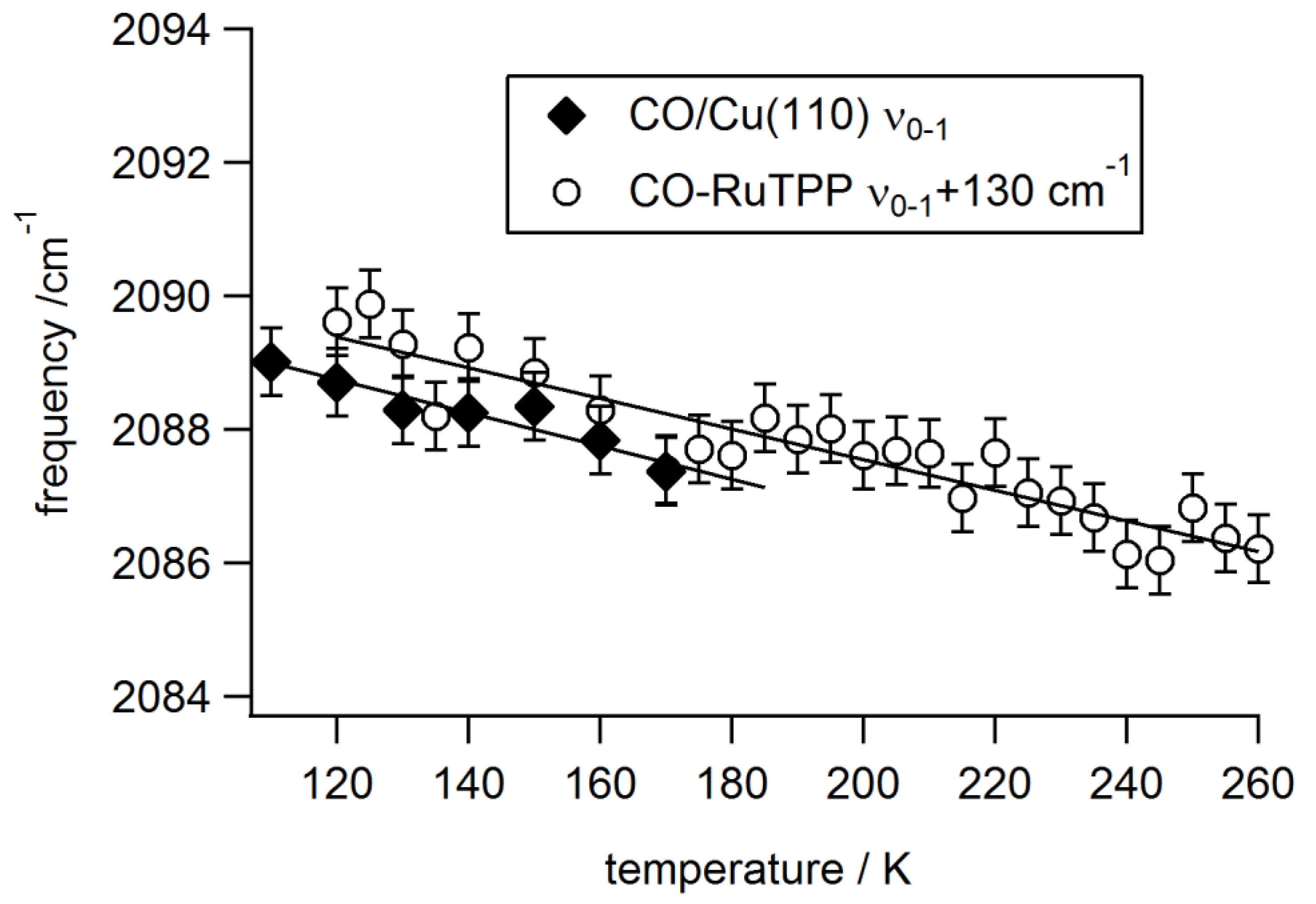
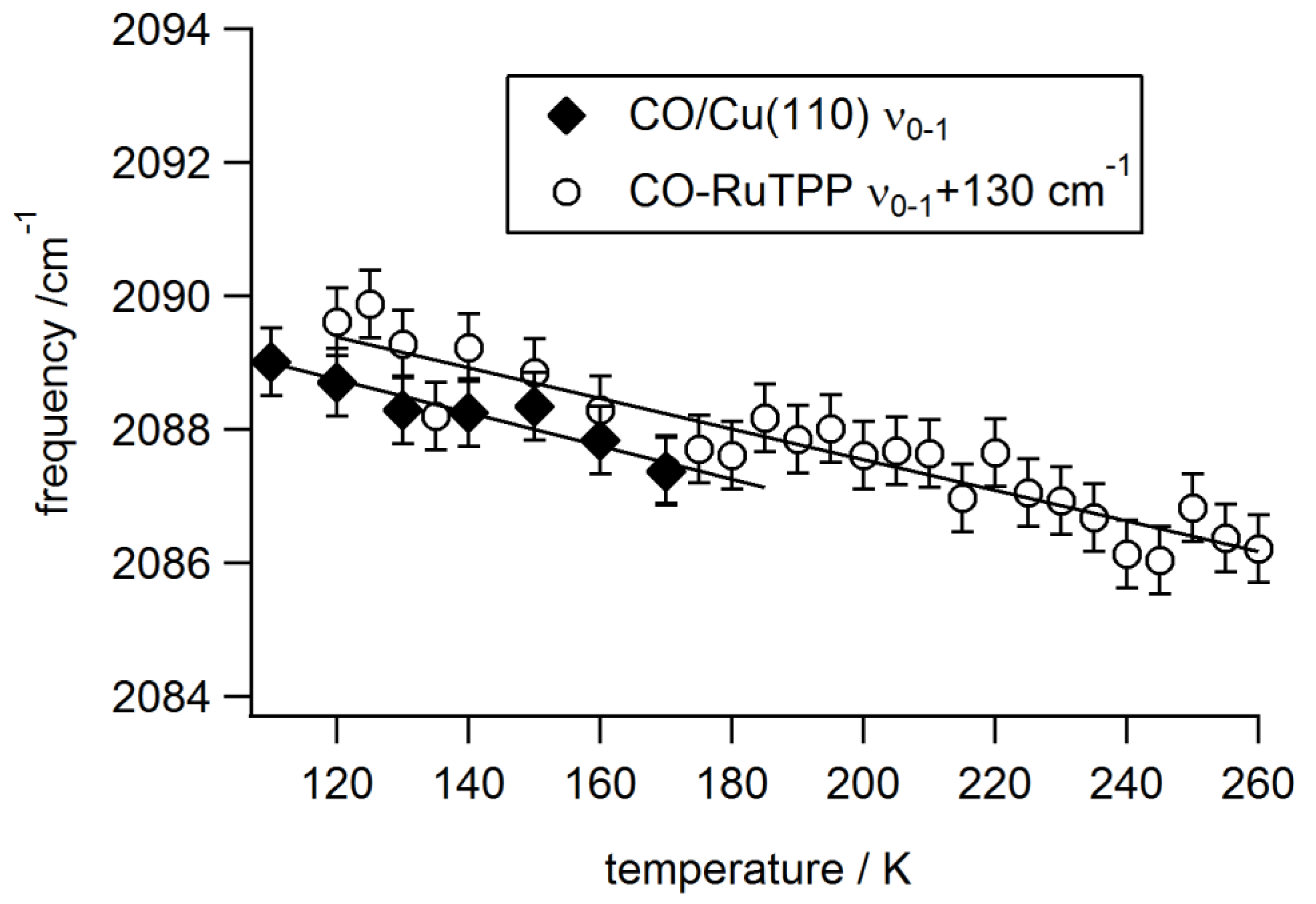
3.2. Static Temperature Dependence
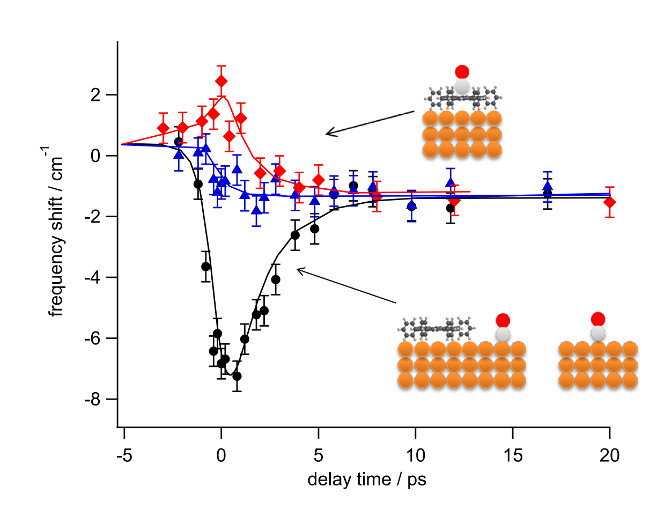
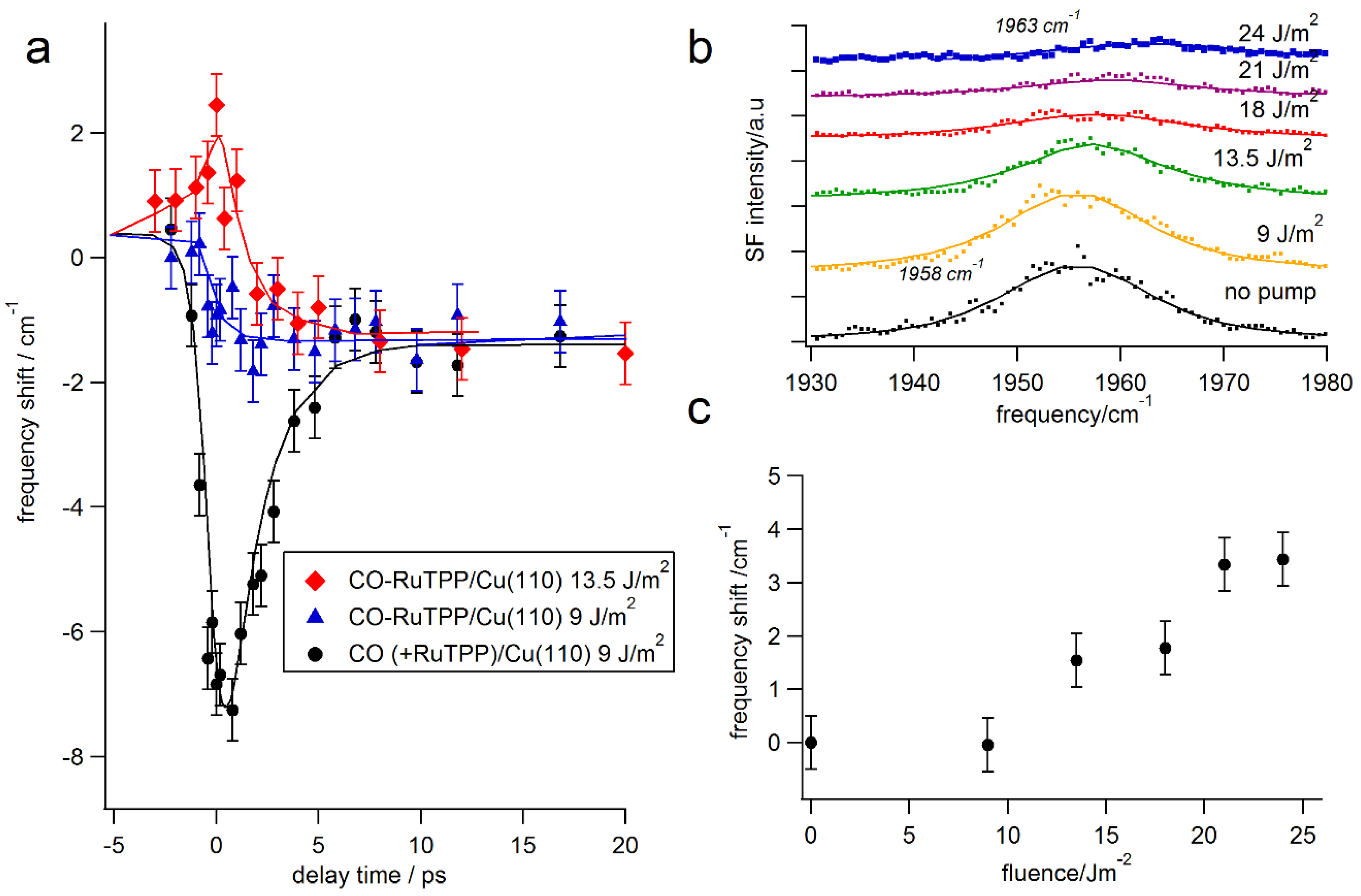
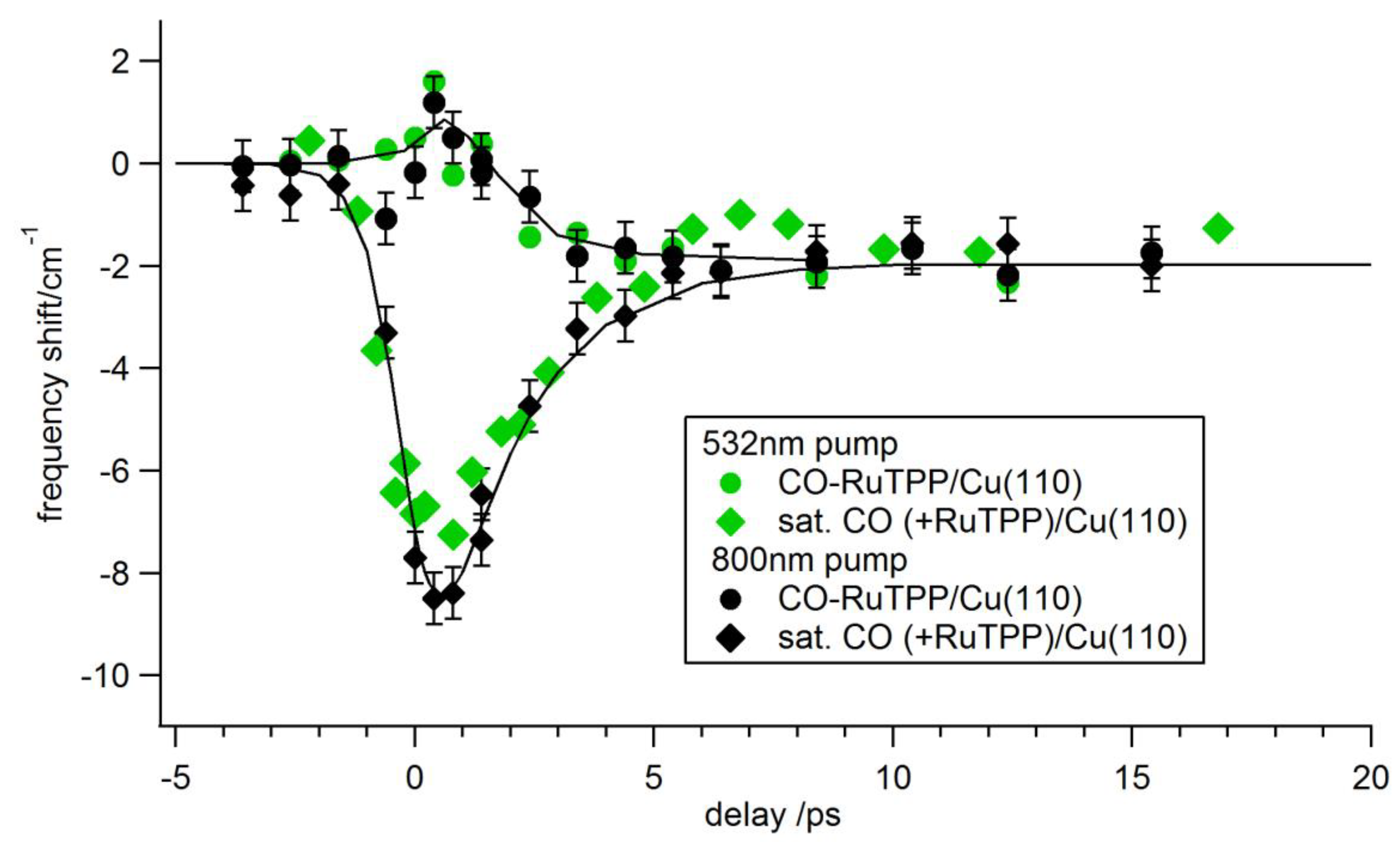
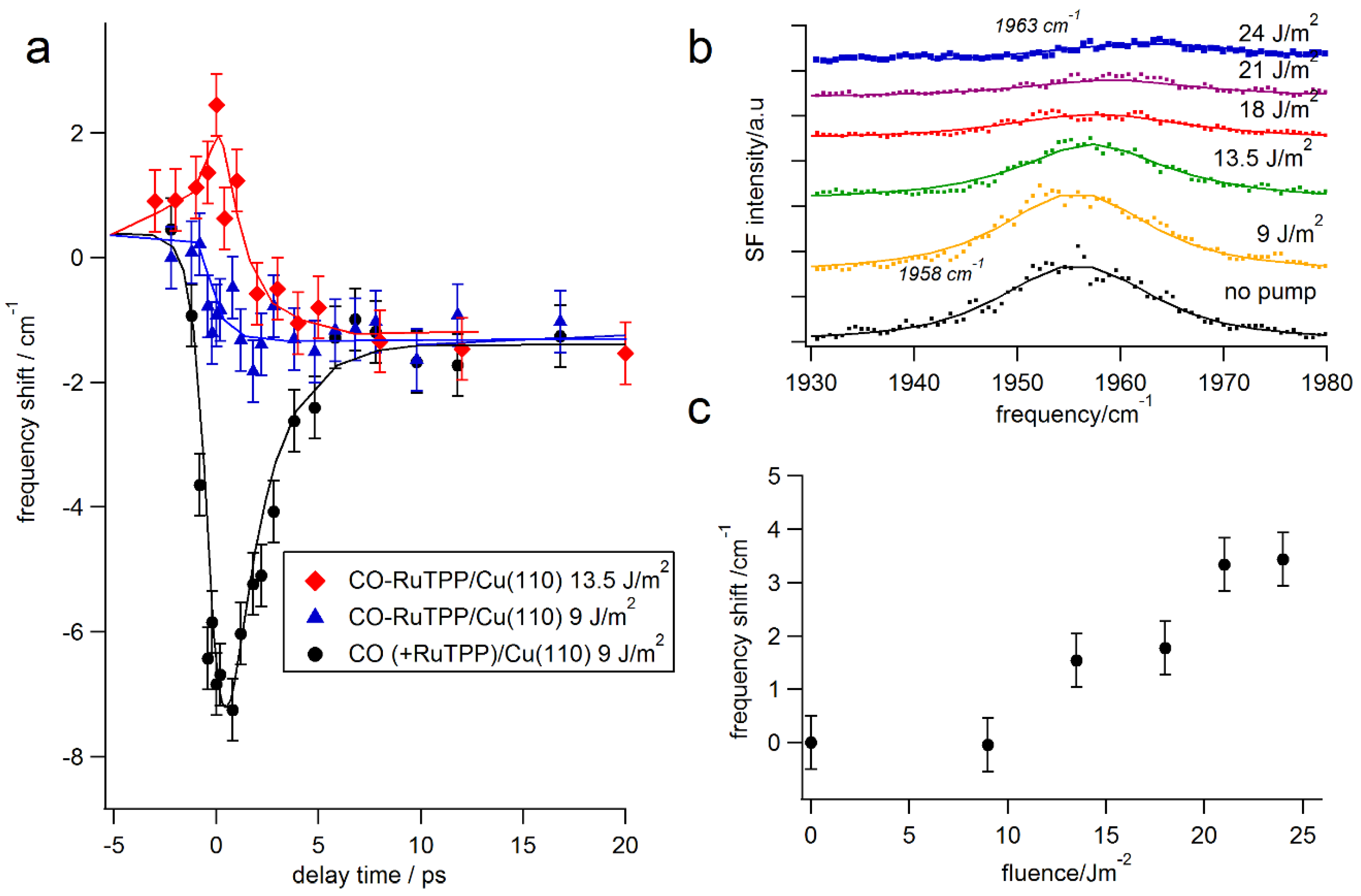
3.3. Ultrafast Dynamics
4. Discussion
5. Conclusions
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Arnolds, H. Vibrational dynamics of adsorbates—Quo vadis? Prog. Surf. Sci. 2011, 86, 1–40. [Google Scholar] [CrossRef]
- Arnolds, H.; Bonn, M. Ultrafast surface vibrational dynamics. Surf. Sci. Rep. 2010, 65, 45–66. [Google Scholar] [CrossRef]
- Kraack, J.P.; Hamm, P. Surface-Sensitive and Surface-Specific Ultrafast Two-Dimensional Vibrational Spectroscopy. Chem. Rev. 2017, 117, 10623–10664. [Google Scholar] [CrossRef] [PubMed]
- Bonn, M.; Hess, C.; Funk, S.; Miners, J.H.; Persson, B.N.J.; Wolf, M.; Ertl, G. Femtosecond surface vibrational spectroscopy of CO adsorbed on Ru(001) during desorption. Phys. Rev. Lett. 2000, 84, 4653–4656. [Google Scholar] [CrossRef] [PubMed]
- Fournier, F.; Zheng, W.; Carrez, S.; Dubost, H.; Bourguignon, B. Vibrational dynamics of adsorbed molecules under conditions of photodesorption: Pump-probe SFG spectra of CO/Pt(111). J. Chem. Phys. 2004, 121, 4839–4847. [Google Scholar] [CrossRef] [PubMed]
- Ueba, H.; Persson, B. Heating of adsorbate by vibrational-mode coupling. Phys. Rev. B 2008, 77, 035413. [Google Scholar] [CrossRef]
- Ueba, H.; Persson, B.N.J. Heat transfer between adsorbate and laser-heated hot electrons. J. Phys. Condens. Matter 2008, 20, 224016. [Google Scholar] [CrossRef]
- Backus, E.H.; Eichler, A.; Kleyn, A.W.; Bonn, M. Real-time observation of molecular motion on a surface. Science 2005, 310, 1790–1793. [Google Scholar] [CrossRef] [PubMed]
- Dell’Angela, M.; Anniyev, T.; Beye, M.; Coffee, R.; Föhlisch, A.; Gladh, J.; Katayama, T.; Kaya, S.; Krupin, O.; LaRue, J.; et al. Real-time observation of surface bond breaking with an X-ray laser. Science 2013, 339, 1302–1305. [Google Scholar] [CrossRef] [PubMed]
- Beye, M.; Anniyev, T.; Coffee, R.; Dell’Angela, M.; Föhlisch, A.; Gladh, J.; Katayama, T.; Kaya, S.; Krupin, O.; Møgelhøj, A.; et al. Selective Ultrafast Probing of Transient Hot Chemisorbed and Precursor States of CO on Ru(0001). Phys. Rev. Lett. 2013, 110, 186101. [Google Scholar] [CrossRef] [PubMed]
- Inoue, K.; Watanabe, K.; Matsumoto, Y. Instantaneous vibrational frequencies of diffusing and desorbing adsorbates: CO/Pt(111). J. Chem. Phys. 2012, 137, 024704. [Google Scholar] [CrossRef] [PubMed]
- Inoue, K.; Watanabe, K.; Sugimoto, T.; Matsumoto, Y.; Yasuike, T. Disentangling Multidimensional Nonequilibrium Dynamics of Adsorbates: CO Desorption from Cu(100). Phys. Rev. Lett. 2016, 117, 186101. [Google Scholar] [CrossRef] [PubMed]
- Alducin, M.; Díez Muiño, R.; Juaristi, J.I. Non-adiabatic effects in elementary reaction processes at metal surfaces. Prog. Surf. Sci. 2017, 92, 317–340. [Google Scholar] [CrossRef]
- Novko, D.; Alducin, M.; Blanco-Rey, M.; Juaristi, J.I. Effects of electronic relaxation processes on vibrational linewidths of adsorbates on surfaces: The case of CO/Cu(100). Phys. Rev. B 2016, 94, 224306. [Google Scholar] [CrossRef]
- Novko, D.; Tremblay, J.C.; Alducin, M.; Juaristi, J.I. Ultrafast Transient Dynamics of Adsorbates on Surfaces Deciphered: The Case of CO on Cu(100). Phys. Rev. Lett. 2019, 122, 016806. [Google Scholar] [CrossRef]
- Scholz, R.; Floß, G.; Saalfrank, P.; Füchsel, G.; Lončarić, I.; Juaristi, J.I. Femtosecond-laser induced dynamics of CO on Ru(0001): Deep insights from a hot-electron friction model including surface motion. Phys. Rev. B 2016, 94, 165447. [Google Scholar] [CrossRef]
- Ghalgaoui, A.; Horchani, R.; Wang, J.J.; Ouvrard, A.; Carrez, S.; Bourguignon, B. Identification of Active Sites in Oxidation Reaction from Real-Time Probing of Adsorbate Motion over Pd Nanoparticles. J. Phys. Chem. Lett. 2018, 9, 5202–5206. [Google Scholar] [CrossRef] [PubMed]
- Ghalgaoui, A.; Ouvrard, A.; Wang, J.J.; Carrez, S.; Zheng, W.Q.; Bourguignon, B. Electron to Adsorbate Energy Transfer in Nanoparticles: Adsorption Site, Size, and Support Matter. J. Phys. Chem. Lett. 2017, 8, 2666–2671. [Google Scholar] [CrossRef] [PubMed]
- Omiya, T.; Arnolds, H. Coverage dependent non-adiabaticity of CO on a copper surface. J. Chem. Phys. 2014, 141, 214705. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.-Y.; Xu, P.; Camillone, N.R.; White, M.G.; Camillone, N., III. Adlayer structure dependent ultrafast desorption dynamics in carbon monoxide adsorbed on Pd (111). J. Chem. Phys. 2016, 145, 014704. [Google Scholar] [CrossRef] [PubMed]
- Szymanski, P.; Harris, A.L.; Camillone, N., III. Temperature-Dependent Femtosecond Photoinduced Desorption in CO/Pd(111). J. Phys. Chem. A 2007, 111, 12524–12533. [Google Scholar] [CrossRef] [PubMed]
- Levine, L.M.A.; Holten, D. Axial-ligand control of the photophysical behavior of ruthenium(II) tetraphenyl-and octaethylporphyrin: Contrasting properties of metalloporphyrin (.pi.,.pi.*) and (d,.pi.*) excited states. J. Phys. Chem. 1988, 92, 714–720. [Google Scholar] [CrossRef]
- Hieringer, W.; Flechtner, K.; Kretschmann, A.; Seufert, K.; Auwarter, W.; Barth, J.V.; Gorling, A.; Steinruck, H.P.; Gottfried, J.M. The surface trans effect: Influence of axial ligands on the surface chemical bonds of adsorbed metalloporphyrins. J. Am. Chem. Soc. 2011, 133, 6206–6222. [Google Scholar] [CrossRef] [PubMed]
- Omiya, T.; Poli, P.; Arnolds, H.; Raval, R.; Persson, M.; Kim, Y. Desorption of CO from individual ruthenium porphyrin molecules on a copper surface via an inelastic tunnelling process. Chem. Commun. 2017, 53, 6148–6151. [Google Scholar] [CrossRef] [PubMed]
- Nuernberger, P.; Lee, K.F.; Bonvalet, A.; Bouzhir-Sima, L.; Lambry, J.C.; Liebl, U.; Joffre, M.; Vos, M.H. Strong Ligand-Protein Interactions Revealed by Ultrafast Infrared Spectroscopy of CO in the Heme Pocket of the Oxygen Sensor FixL. J. Am. Chem. Soc. 2011, 133, 17110–17113. [Google Scholar] [CrossRef] [PubMed]
- Nuernberger, P.; Lee, K.F.; Bonvalet, A.; Vos, M.H.; Joffre, M. Multiply Excited Vibration of Carbon Monoxide in the Primary Docking Site of Hemoglobin Following Photolysis from the Heme. J. Phys. Chem. Lett. 2010, 1, 2077–2081. [Google Scholar] [CrossRef]
- Van Wilderen, L.J.G.W.; Key, J.M.; Van Stokkum, I.H.M.; van Grondelle, R.; Groot, M.L. Dynamics of carbon monoxide photodissociation in bradyrhizobium japonicum FixL probed by picosecond midinfrared spectroscopy. J. Phys. Chem. B 2009, 113, 3292–3297. [Google Scholar] [CrossRef] [PubMed]
- Lim, M.H.; Jackson, T.A.; Anfinrud, P.A. Ultrafast rotation and trapping of carbon monoxide dissociated from myoglobin. Nat. Struct. Biol. 1997, 4, 209–214. [Google Scholar] [CrossRef] [PubMed]
- Lagutchev, A.; Hambir, S.A.; Dlott, D.D. Nonresonant background suppression in broadband vibrational sum-frequency generation spectroscopy. J. Phys. Chem. C 2007, 111, 13645–13647. [Google Scholar] [CrossRef]
- Donovan, P.; Robin, A.; Dyer, M.S.; Persson, M.; Raval, R. Unexpected Deformations Induced by Surface Interaction and Chiral Self-Assembly of CoII-Tetraphenylporphyrin (Co-TPP) Adsorbed on Cu(110): A Combined STM and Periodic DFT Study. Chem. Eur. J. 2010, 16, 11641–11652. [Google Scholar] [CrossRef] [PubMed]
- Woodruff, D.P.; Hayden, B.E.; Prince, K.; Bradshaw, A.M. Dipole coupling and chemical shifts in IRAS of CO adsorbed on Cu(110). Surf. Sci. 1982, 123, 397–412. [Google Scholar] [CrossRef]
- Shafizadeh, N.; Boye-Peronne, S.; Soorkia, S.; de Miranda, B.K.C.; Garcia, G.A.; Nahon, L.; Chen, S.F.; de la Lande, A.; Poisson, L.; Soep, B. The surprisingly high ligation energy of CO to ruthenium porphyrins. Phys. Chem. Chem. Phys. 2018, 20, 11730–11739. [Google Scholar] [CrossRef] [PubMed]
- Krim, L.; Sorgues, S.; Soep, B.; Shafizadeh, N. Infrared spectra of RuTPP, RuCOTPP, and Ru(CO)2TPP isolated in solid argon. J. Phys. Chem. A 2005, 109, 8268–8274. [Google Scholar] [CrossRef] [PubMed]
- Azizyan, A.S.; Kurtikyan, T.S.; Martirosyan, G.G.; Ford, P.C. Tracking reactive intermediates by FTIR monitoring of reactions in low-temperature sublimed solids: Nitric oxide disproportionation mediated by ruthenium(II) carbonyl porphyrin Ru(TPP)(CO). Inorg. Chem. 2013, 52, 5201–5205. [Google Scholar] [CrossRef] [PubMed]
- Baerends, E.J.; Ros, P. Evaluation of the LCAO Hartree—Fock—Slater method: Applications to transition-metal complexes. Int. J. Quantum Chem. 1978, 14, 169–190. [Google Scholar] [CrossRef]
- Hoffmann, F.M. Infrared reflection-absorption spectroscopy of adsorbed molecules. Surf. Sci. Rep. 1983, 3, 107–192. [Google Scholar] [CrossRef]
- Isvoranu, C.; Wang, B.; Ataman, E.; Knudsen, J.; Schulte, K.; Andersen, J.N.; Bocquet, M.-L.; Schnadt, J. Comparison of the Carbonyl and Nitrosyl Complexes Formed by Adsorption of CO and NO on Monolayers of Iron Phthalocyanine on Au(111). J. Phys. Chem. C 2011, 115, 24718–24727. [Google Scholar] [CrossRef]
- Zhou, X.L.; Zhu, X.Y.; White, J.M. Photochemistry at adsorbate/metal interfaces. Surf. Sci. Rep. 1991, 13, 73–220. [Google Scholar] [CrossRef]
- Richter, L.J.; Buntin, S.A.; King, D.S.; Cavanagh, R.R. Constraints on the use of polarization and angle-of-incidence to characterize surface photoreactions. Chem. Phys. Lett. 1991, 186, 423–426. [Google Scholar] [CrossRef]
- García Rey, N.; Arnolds, H. Hot hole-induced dissociation of NO dimers on a copper surface. J Chem Phys 2011, 135, 224708. [Google Scholar] [CrossRef] [PubMed]
- Germer, T.A.; Stephenson, J.C.; Heilweil, E.J.; Cavanagh, R.R. Picosecond time-resolved adsorbate response to substrate heating: Spectroscopy and dynamics of CO/Cu(100). J. Chem. Phys. 1994, 101, 1704–1716. [Google Scholar] [CrossRef]
- Braun, J.; Weckesser, J.; Ahner, J.; Mocuta, D.; Yates, J.T., Jr.; Wöll, C. The frustrated translational mode of CO on Cu(110): Azimuthal anisotropy studied by helium atom scattering—A comparison with time-of-flight electron stimulated desorption of ion angular distribution measurements. J. Chem. Phys. 1998, 108, 5161–5164. [Google Scholar] [CrossRef]
- Braun, J.; Kostov, K.L.; Witte, G.; Wöll, C. CO overlayers on Ru(0001) studied by helium atom scattering: Structure, dynamics, and the influence of coadsorbed H and O. J. Chem. Phys. 1997, 106, 8262–8273. [Google Scholar] [CrossRef]
- Burema, S.R.; Seufert, K.; Auwärter, W.; Barth, J.V.; Bocquet, M.-L. Probing nitrosyl ligation of surface-confined metalloporphyrins by inelastic electron tunneling spectroscopy. Acs Nano 2013, 7, 5273–5281. [Google Scholar] [CrossRef] [PubMed]
- Seufert, K.; Bocquet, M.-L.; Auwärter, W.; Weber-Bargioni, A.; Reichert, J.; Lorente, N.; Barth, J.V. Cis-dicarbonyl binding at cobalt and iron porphyrins with saddle-shape conformation. Nat. Chem. 2011, 3, 114–119. [Google Scholar] [CrossRef] [PubMed]
- Carpene, E. Ultrafast laser irradiation of metals: Beyond the two-temperature model. Phys. Rev. B 2006, 74, 024301. [Google Scholar] [CrossRef]
- Vogler, A.; Kunkely, H. Photochemistry of biologically important transition metal complexes. II. carbonylpiperidinetetraphenylporphine complexes of iron(II) and ruthenium(II). Ber. Bunsenges. Phys. Chem. 1976, 80, 425–429. [Google Scholar] [CrossRef]
- Lane, I.M.; Liu, Z.P.; King, D.A.; Arnolds, H. Ultrafast vibrational dynamics of NO and CO adsorbed on an iridium surface. J. Phys. Chem. C 2007, 111, 14198–14206. [Google Scholar] [CrossRef]
- Persson, B.N.J.; Ryberg, R. Brownian motion and vibrational phase relaxation at surfaces: CO on Ni(111). Phys. Rev. B 1985, 32, 3586–3596. [Google Scholar] [CrossRef]
- Persson, B.N.J.; Ryberg, R. Vibrational phase relaxation at surfaces: CO on Ni(111). Phys. Rev. Lett. 1985, 54, 2119–2122. [Google Scholar] [CrossRef] [PubMed]
- Cook, J.C.; Clowes, S.K.; McCash, E.M. Reflection absorption IR studies of vibrational energy transfer processes and adsorption energetics. J. Chem. Soc. Faraday Trans. 1997, 93, 2315–2322. [Google Scholar] [CrossRef]
- Li, X.Y.; Spiro, T.G. Is bound carbonyl linear or bent in heme proteins? Evidence from resonance Raman and infrared spectroscopic data. J. Am. Chem. Soc. 1988, 110, 6024–6033. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.; Bocian, D.F. Carbonyl tilting and bending potential energy surface of carbon monoxyhemes. J. Phys. Chem. 1996, 100, 6363–6367. [Google Scholar] [CrossRef]
- Graham, A.P.; Hofmann, F.; Toennies, J.P.; Williams, G.P.; Hirschmugl, C.J.; Ellis, J. A high resolution helium atom scattering and far infrared study of the dynamics and the lateral potential energy surface of CO molecules chemisorbed on Cu(001). J. Chem. Phys. 1998, 108, 7825–7834. [Google Scholar] [CrossRef]
- Hirschmugl, C.; Williams, G.; Hoffmann, F.; Chabal, Y. Adsorbate-substrate resonant interactions observed for CO on Cu(100) in the far infrared. Phys. Rev. Lett. 1990, 65, 480–483. [Google Scholar] [CrossRef] [PubMed]
- Lane, I.M.; King, D.A.; Liu, Z.-P.; Arnolds, H. Real-time observation of nonadiabatic surface dynamics: The first picosecond in the dissociation of NO on iridium. Phys. Rev. Lett. 2006, 97, 186105. [Google Scholar] [CrossRef] [PubMed]
- Benndorf, C.; Bertel, E.; Dose, V.; Jacob, W.; Memmel, N.; Rogozik, J. An inverse photoemission study of CO adsorption on clean and potassium promoted Ru(001). Surf. Sci. 1987, 191, 455–465. [Google Scholar] [CrossRef]
- Rogozik, J.; Scheidt, H.; Dose, V.; Prince, K.C.; Bradshaw, A.M. The 2π-derived level in the adsorption system CO/Cu(110). Surf. Sci. Lett. 1984, 145, L481–L487. [Google Scholar]
- Backus, E.H.G.; Forsblom, M.; Persson, M.; Bonn, M. Highly efficient ultrafast energy transfer into molecules at surface step sites. J. Phys. Chem. C 2007, 111, 6149–6153. [Google Scholar] [CrossRef]
- Avanesian, T.; Christopher, P. Adsorbate specificity in hot electron driven photochemistry on catalytic metal surfaces. J. Phys. Chem. C 2014, 118, 28017–28031. [Google Scholar] [CrossRef]
- Garduño-Mejía, J.; Higlett, M.P.; Meech, S.R. Modelling the influence of nonthermal electron dynamics in thin and ultrathin gold films. Chem. Phys. 2007, 341, 276–284. [Google Scholar] [CrossRef]
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Omiya, T.; Kim, Y.; Raval, R.; Arnolds, H. Ultrafast Vibrational Dynamics of CO Ligands on RuTPP/Cu(110) under Photodesorption Conditions. Surfaces 2019, 2, 117-130. https://doi.org/10.3390/surfaces2010010
Omiya T, Kim Y, Raval R, Arnolds H. Ultrafast Vibrational Dynamics of CO Ligands on RuTPP/Cu(110) under Photodesorption Conditions. Surfaces. 2019; 2(1):117-130. https://doi.org/10.3390/surfaces2010010
Chicago/Turabian StyleOmiya, Takuma, Yousoo Kim, Rasmita Raval, and Heike Arnolds. 2019. "Ultrafast Vibrational Dynamics of CO Ligands on RuTPP/Cu(110) under Photodesorption Conditions" Surfaces 2, no. 1: 117-130. https://doi.org/10.3390/surfaces2010010
APA StyleOmiya, T., Kim, Y., Raval, R., & Arnolds, H. (2019). Ultrafast Vibrational Dynamics of CO Ligands on RuTPP/Cu(110) under Photodesorption Conditions. Surfaces, 2(1), 117-130. https://doi.org/10.3390/surfaces2010010