
Structural Stability Analysis of Proteins Using End-to-End Distance: A 3D-RISM Approach


Abstract
:1. Introduction
2. Computational Details

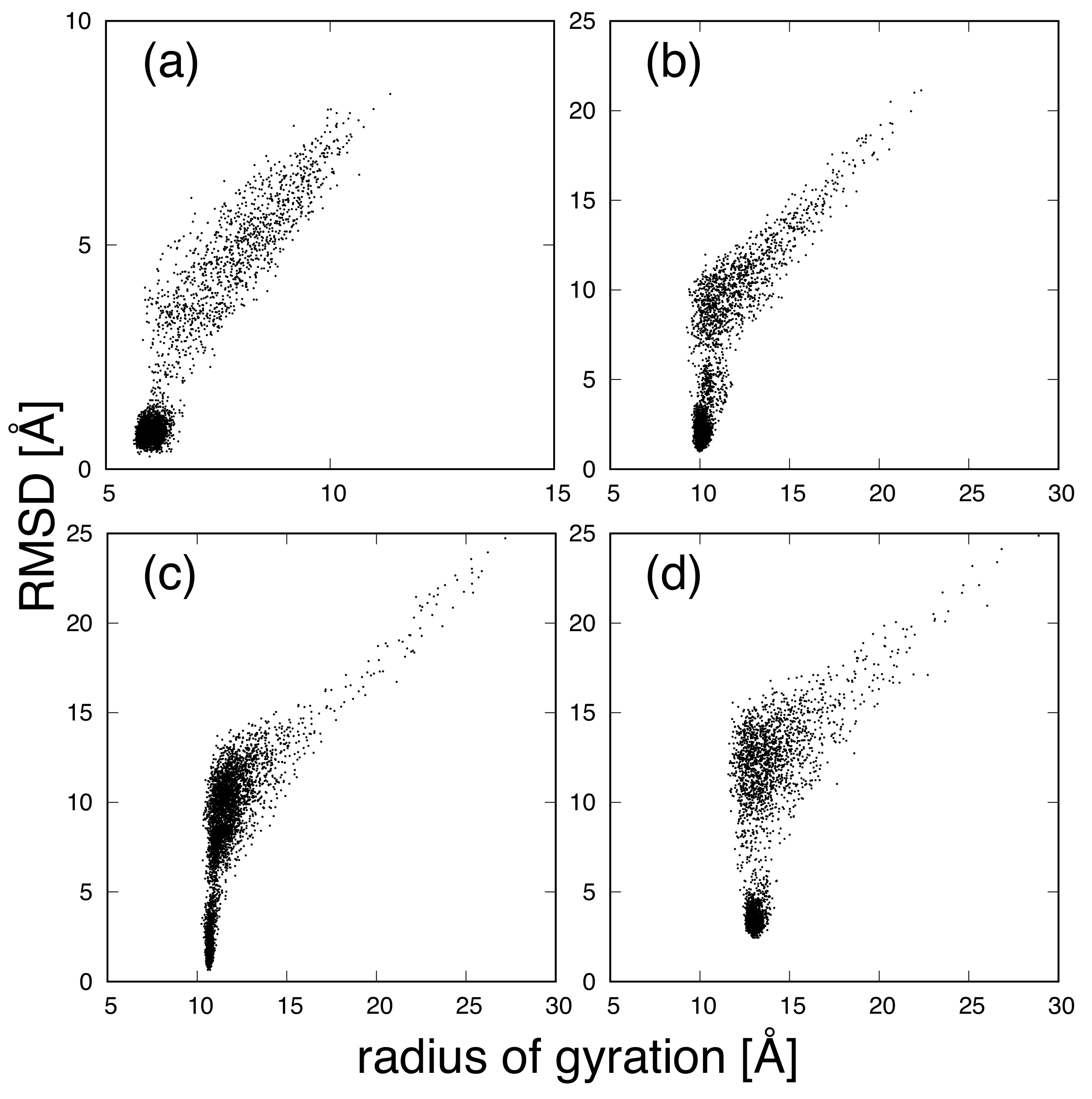{kind=link}
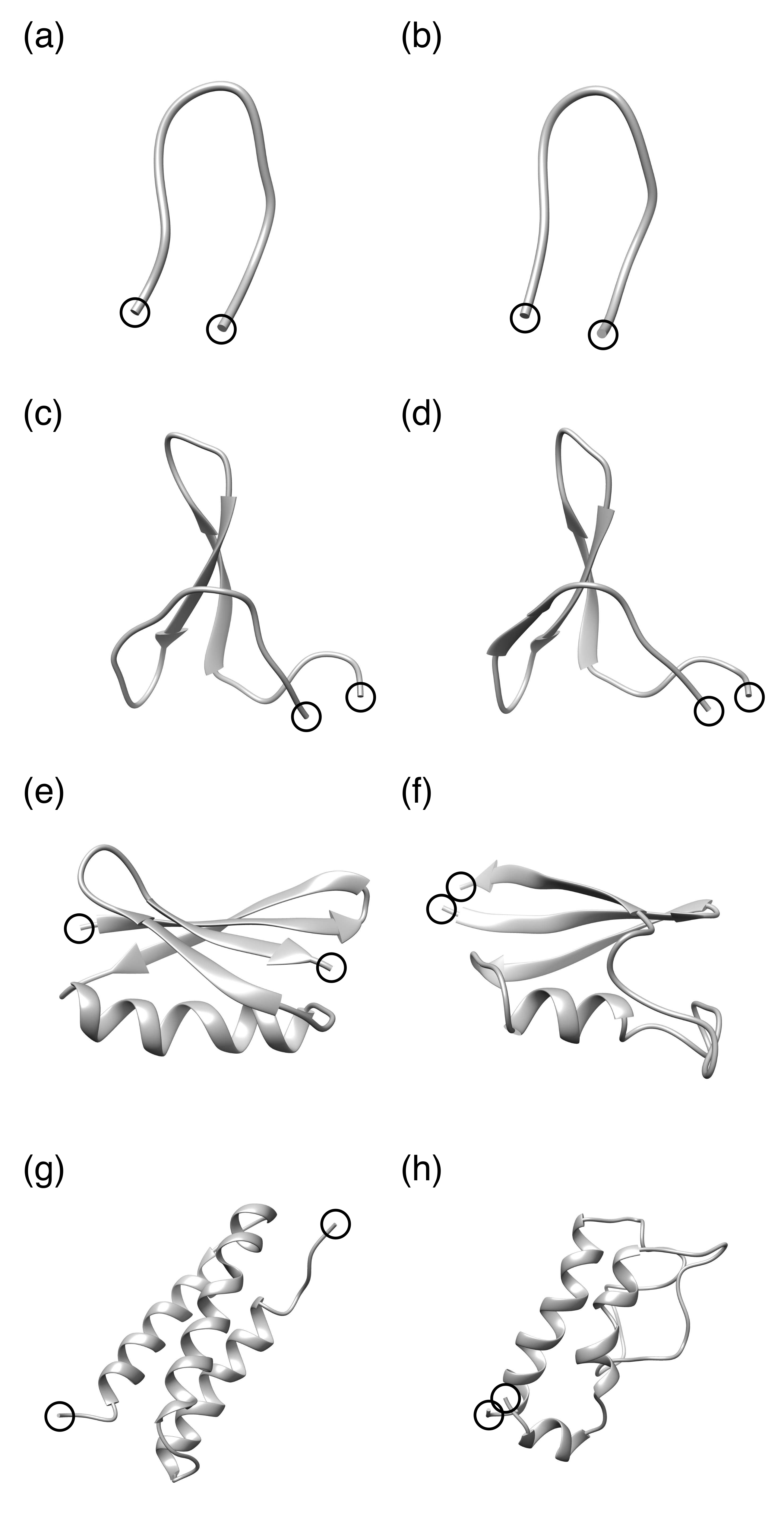{kind=link}
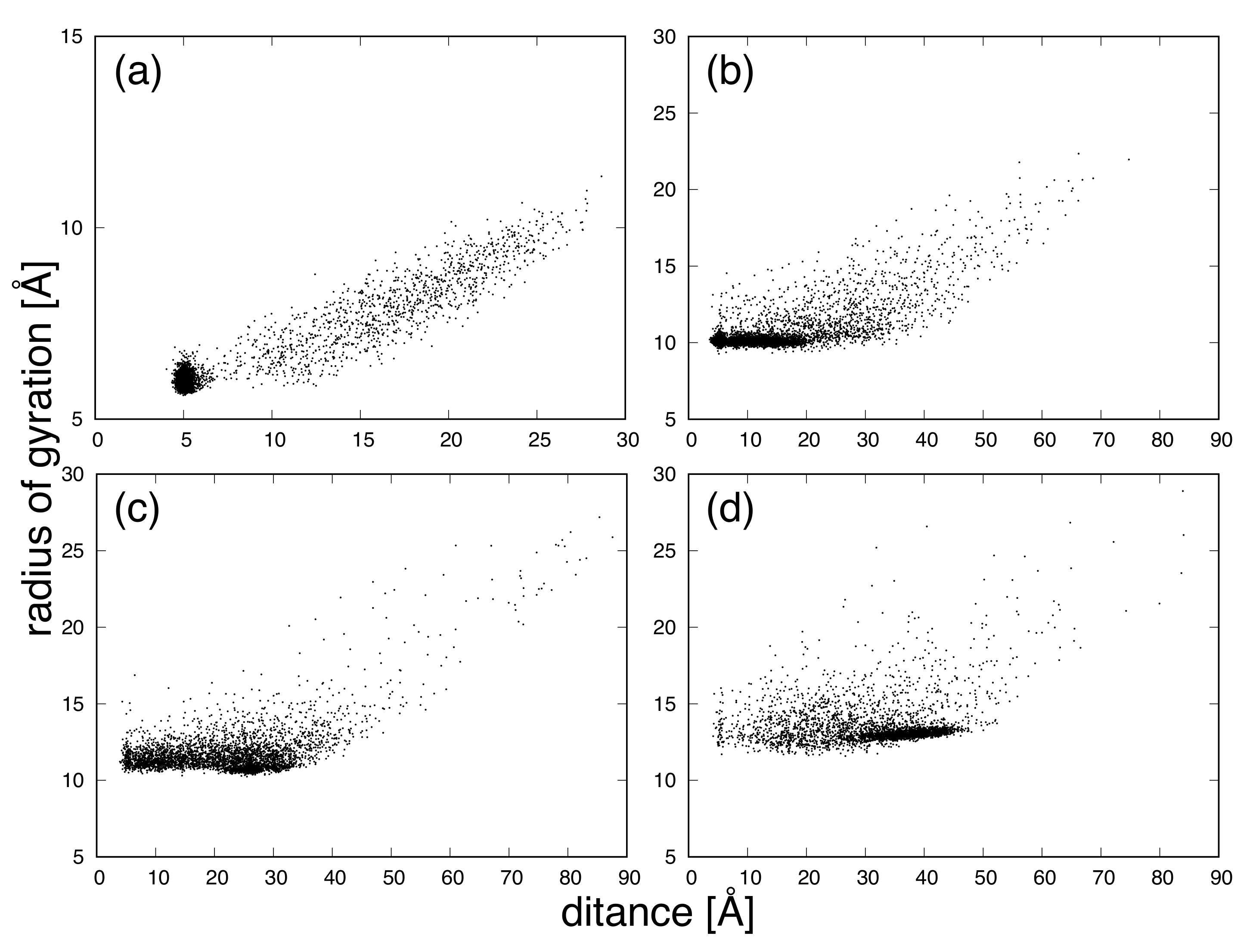{kind=link}
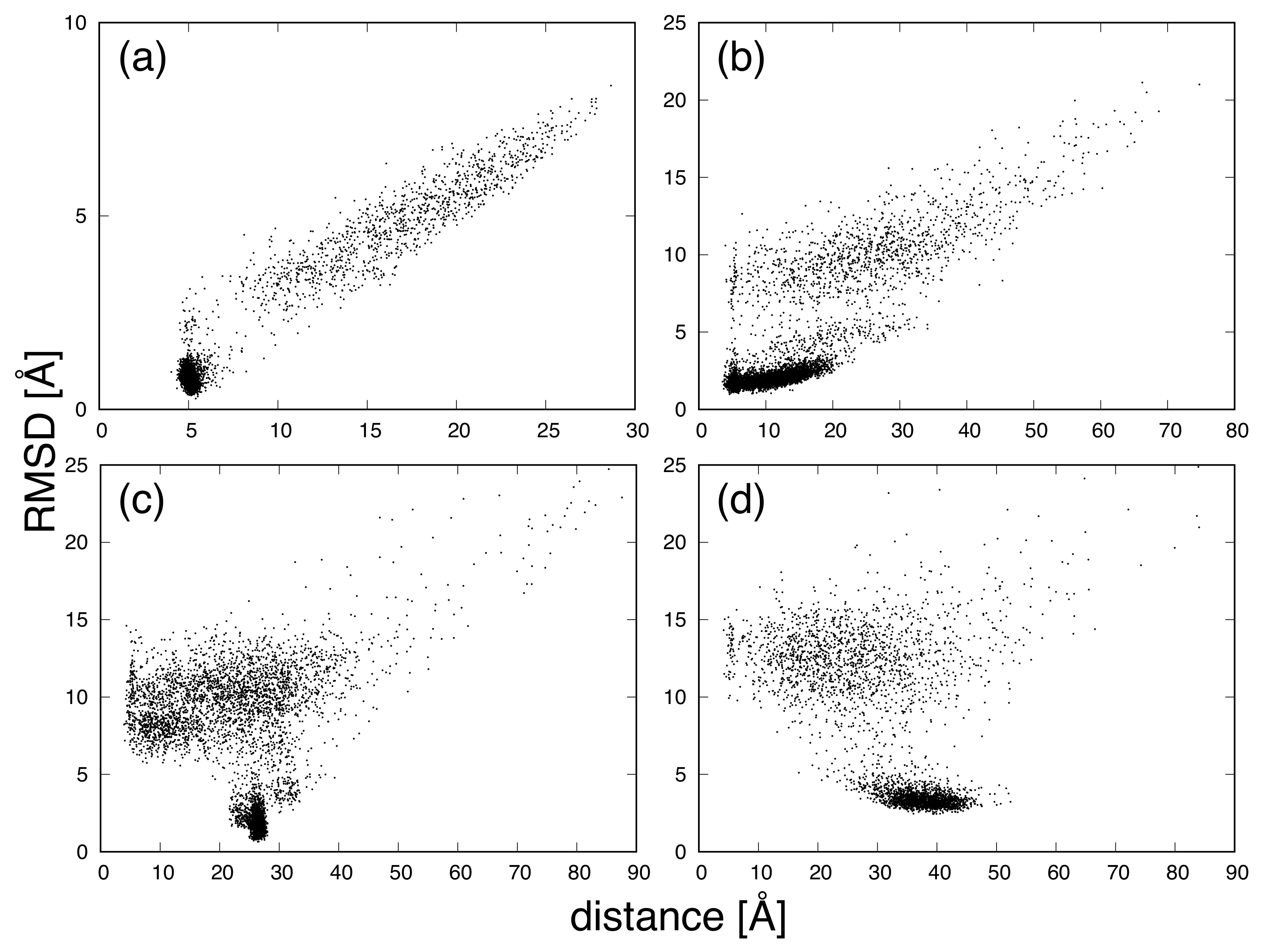{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
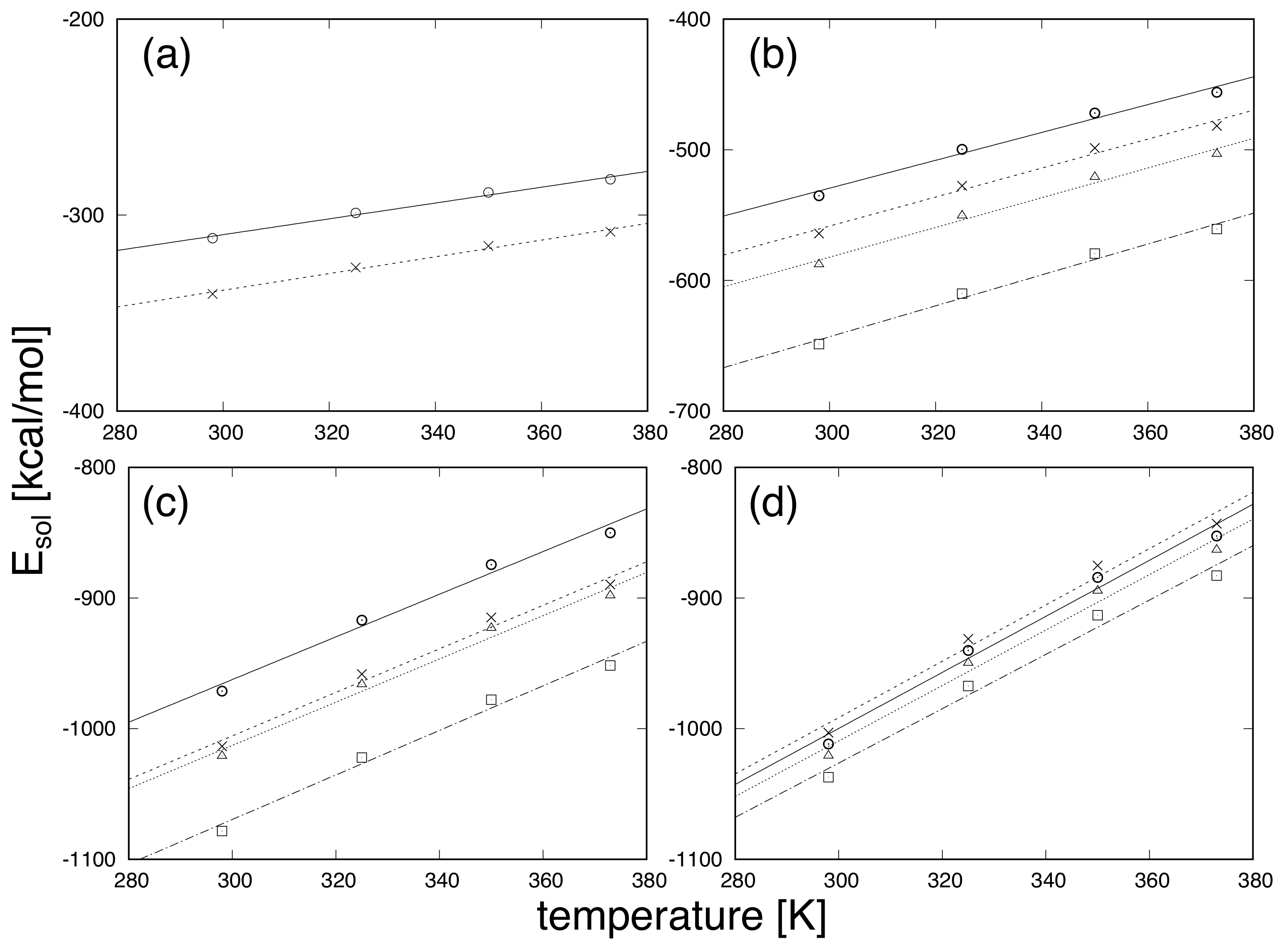
| Temperature [K] | Number Density [Å] | Diameter [Å] |
|---|---|---|
| 298 | 0.033329 | 2.87500 |
| 325 | 0.032994 | 2.87125 |
| 350 | 0.032526 | 2.87000 |
| 375 | 0.031924 | 2.87250 |
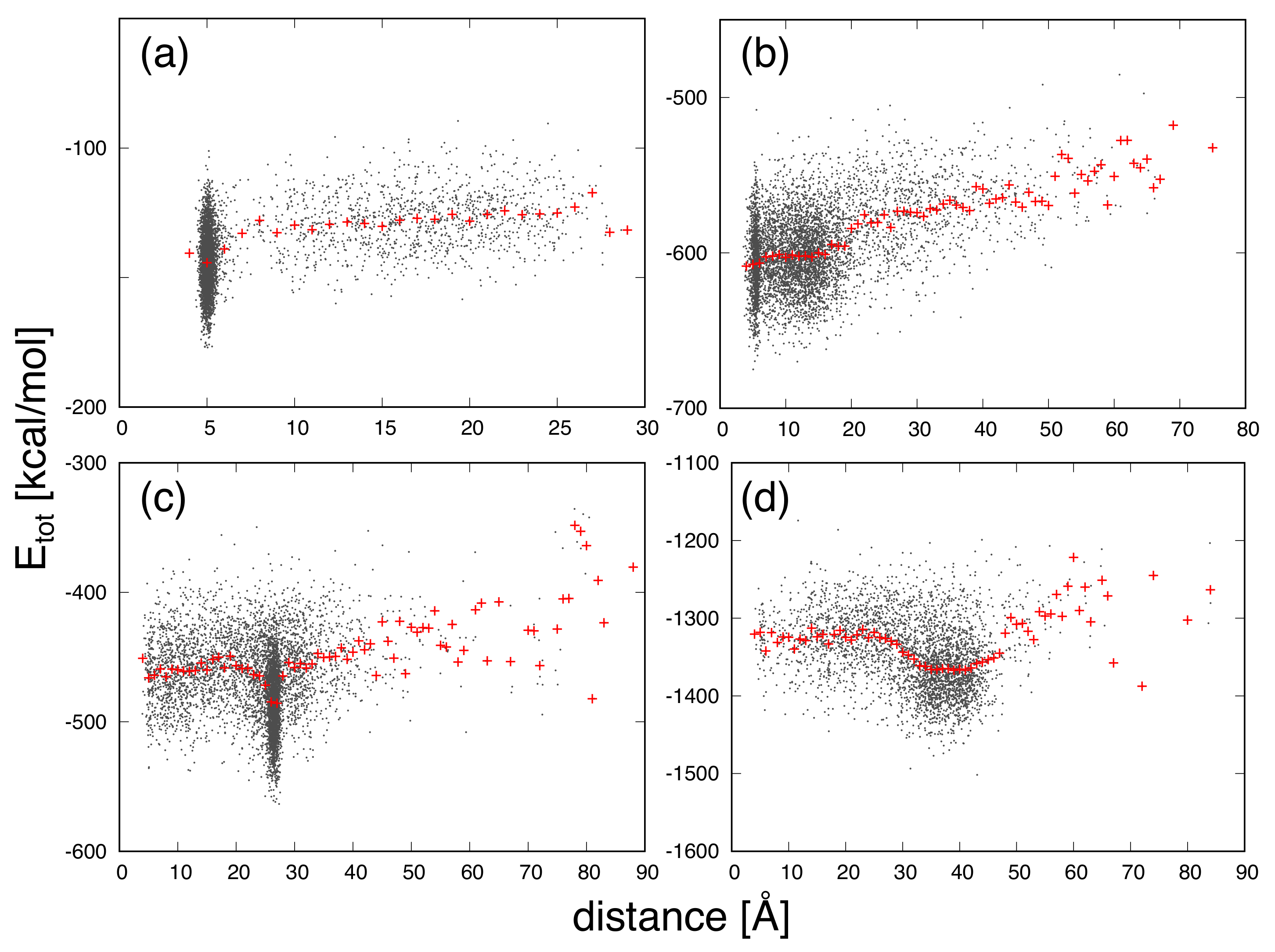
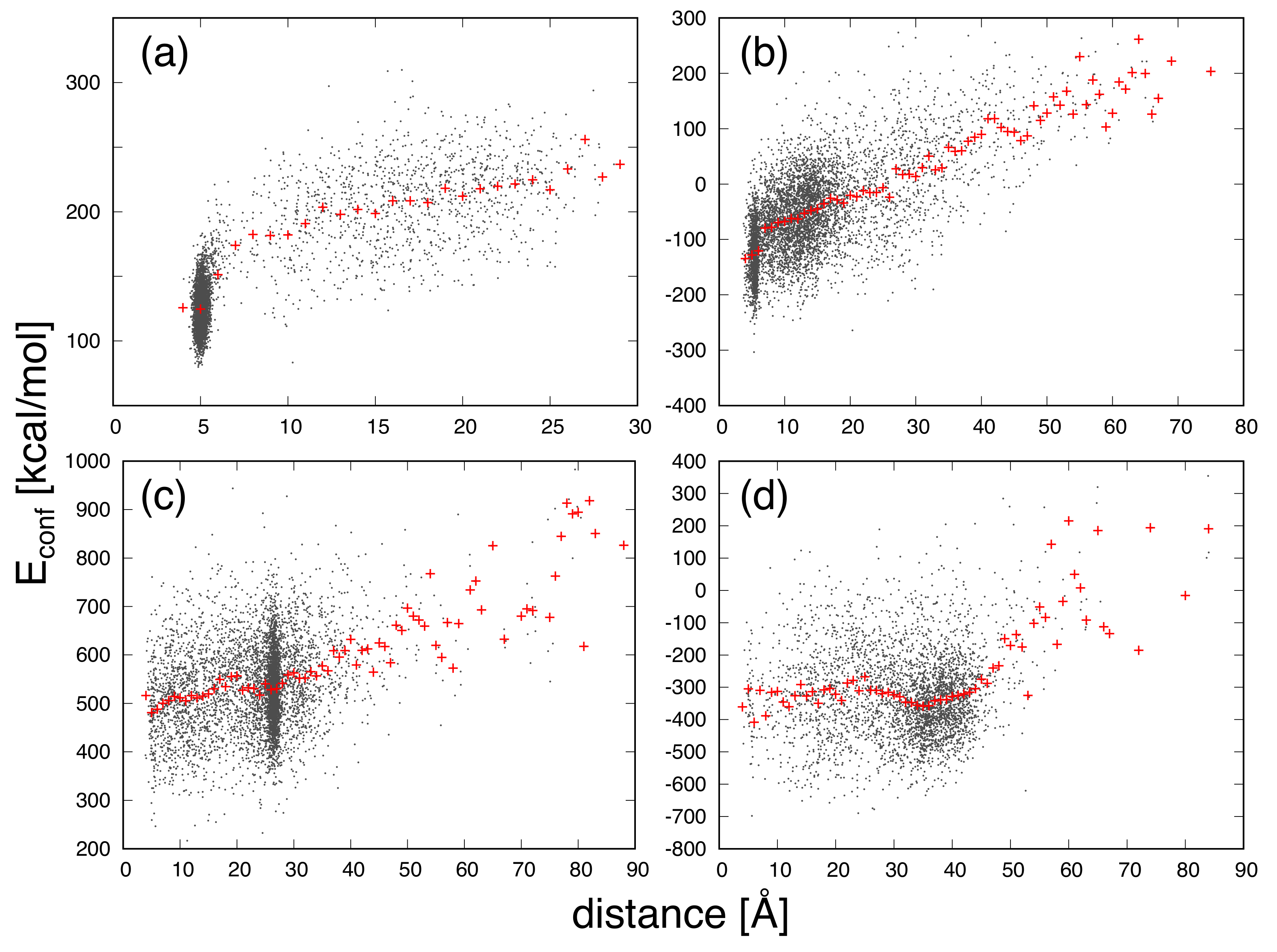
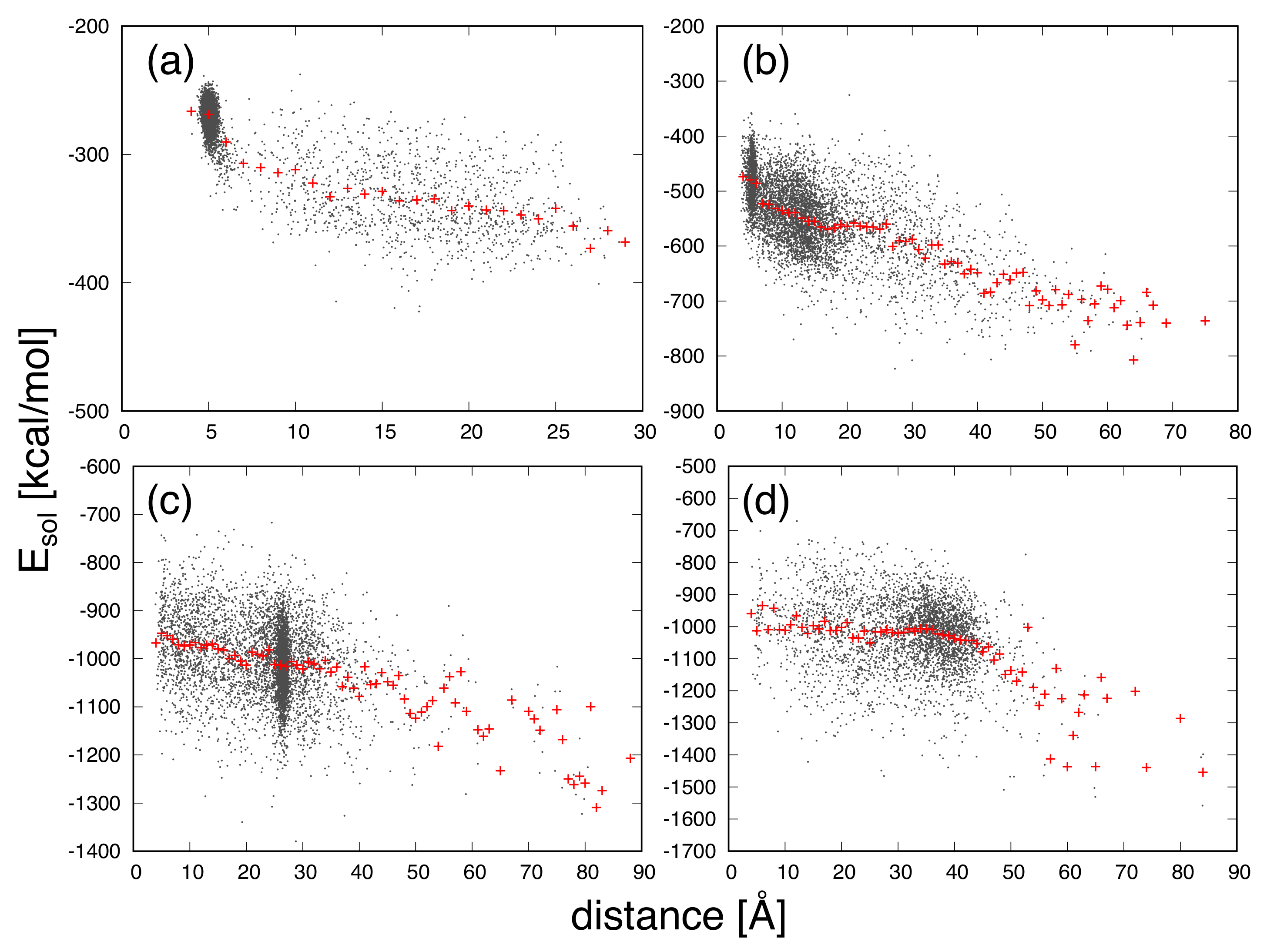
3. Results and Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| 3D-RISM | Three-dimensional reference interaction site model |
| MD | Molecular dynamics |
| RMSD | Root-mean-square deviation |
| FRET | Fluorescent resonance energy transfer |
References
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Baek, M.; DiMaio, F.; Anishchenko, I.; Dauparas, J.; Ovchinnikov, S.; Lee, G.R.; Wang, J.; Cong, Q.; Kinch, L.N.; Schaeffer, R.D.; et al. Accurate prediction of protein structures and interactions using a three-track neural network. Science 2021, 373, 871–876. [Google Scholar] [CrossRef] [PubMed]
- Varadi, M.; Anyango, S.; Deshpande, M.; Nair, S.; Natassia, C.; Yordanova, G.; Yuan, D.; Stroe, O.; Wood, G.; Laydon, A.; et al. AlphaFold Protein Structure Database: Massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res. 2021, 50, D439–D444. [Google Scholar] [CrossRef] [PubMed]
- Beglov, D.; Roux, B. An integral equation to describe the solvation of polar molecules in liquid water. J. Phys. Chem. B 1997, 101, 7821–7826. [Google Scholar] [CrossRef]
- Kovalenko, A.; Hirata, F. Three-dimensional density profiles of water in contact with a solute of arbitrary shape: A RISM approach. Chem. Phys. Lett. 1998, 290, 237–244. [Google Scholar] [CrossRef]
- Imai, T.; Hiraoka, R.; Kovalenko, A.; Hirata, F. Water molecules in a protein cavity detected by a statistical-mechanical theory. J. Am. Chem. Soc. 2005, 127, 15334–15335. [Google Scholar] [CrossRef]
- Sindhikara, D.J.; Yoshida, N.; Hirata, F. Placevent: An algorithm for prediction of explicit solvent atom distribution-Application to HIV-1 protease and F-ATP synthase. J. Comput. Chem. 2012, 33, 1536–1543. [Google Scholar] [CrossRef]
- Fusani, L.; Wall, I.; Palmer, D.; Cortes, A. Optimal water networks in protein cavities with GAsol and 3D-RISM. Bioinformatics 2018, 34, 1947–1948. [Google Scholar] [CrossRef]
- Yoshida, N.; Phongphanphanee, S.; Maruyama, Y.; Imai, T.; Hirata, F. Selective ion-binding by protein probed with the 3D-RISM theory. J. Am. Chem. Soc. 2006, 128, 12042–12043. [Google Scholar] [CrossRef]
- Yoshida, N.; Phongphanphanee, S.; Hirata, F. Selective ion binding by protein probed with the statistical mechanical integral equation theory. J. Phys. Chem. B 2007, 111, 4588–4595. [Google Scholar] [CrossRef]
- Maruyama, Y.; Matsushita, T.; Ueoka, R.; Hirata, F. Solvent and Salt Effects on Structural Stability of Human Telomere. J. Phys. Chem. B 2011, 115, 2408–2416. [Google Scholar] [CrossRef]
- Maruyama, Y.; Mitsutake, A. Stability of Unfolded and Folded Protein Structures Using a 3D-RISM with the RMDFT. J. Phys. Chem. B 2017, 121, 9881–9885. [Google Scholar] [CrossRef]
- Lindorff-Larsen, K.; Piana, S.; Dror, R.O.; Shaw, D.E. How fast-folding proteins fold. Science 2011, 334, 517–520. [Google Scholar] [CrossRef]
- Harano, Y.; Roth, R.; Sugita, Y.; Ikeguchi, M.; Kinoshita, M. Physical basis for characterizing native structures of proteins. Chem. Phys. Lett. 2007, 437, 112–116. [Google Scholar] [CrossRef]
- Kinoshita, M. Importance of translational entropy of water in biological self-assembly processes like protein folding. Int. J. Mol. Sci. 2009, 10, 1064–1080. [Google Scholar] [CrossRef]
- Oshima, H.; Kinoshita, M. A highly efficient hybrid method for calculating the hydration free energy of a protein. J. Comput. Chem. 2016, 37, 712–723. [Google Scholar] [CrossRef]
- Kajiwara, Y.; Yasuda, S.; Takamuku, Y.; Murata, T.; Kinoshita, M. Identification of thermostabilizing mutations for a membrane protein whose three-dimensional structure is unknown. J. Comput. Chem. 2017, 38, 211–223. [Google Scholar] [CrossRef]
- Maruyama, Y.; Mitsutake, A. Analysis of Structural Stability of Chignolin. J. Phys. Chem. B 2018, 122, 3801–3814. [Google Scholar] [CrossRef]
- Harano, Y.; Roth, R.; Kinoshita, M. On the energetics of protein folding in aqueous solution. Chem. Phys. Lett. 2006, 432, 275–280. [Google Scholar] [CrossRef]
- Yasuda, S.; Yoshidome, T.; Oshima, H.; Kodama, R.; Harano, Y.; Kinoshita, M. Effects of side-chain packing on the formation of secondary structures in protein folding. J. Chem. Phys. 2010, 132, 065105. [Google Scholar] [CrossRef] [Green Version]
- Maruyama, Y.; Harano, Y. Does water drive protein folding? Chem. Phys. Lett. 2013, 581, 85–90. [Google Scholar] [CrossRef]
- Lee, J.; Im, H.; Chong, S.H.; Ham, S. Role of electrostatic interactions in determining the G-quadruplex structures. Chem. Phys. Lett. 2018, 693, 216–221. [Google Scholar] [CrossRef]
- Ha, T. Single-Molecule Fluorescence Resonance Energy Transfer. Methods 2001, 25, 78–86. [Google Scholar] [CrossRef] [Green Version]
- Schuler, B.; Eaton, W.A. Protein folding studied by single-molecule FRET. Curr. Opin. Struct. Biol. 2008, 18, 16–26. [Google Scholar] [CrossRef] [Green Version]
- Schuler, B. Single-molecule FRET of protein structure and dynamics—A primer. J. Nanobiotechnol. 2013, 11 (Suppl. S1), 1–17. [Google Scholar] [CrossRef] [Green Version]
- Matsunaga, Y.; Kidera, A.; Sugita, Y. Sequential data assimilation for single-molecule FRET photon-counting data. J. Chem. Phys. 2015, 142, 214115. [Google Scholar] [CrossRef]
- Matsunaga, Y.; Sugita, Y. Refining Markov state models for conformational dynamics using ensemble-averaged data and time-series trajectories. J. Chem. Phys. 2018, 148, 241731. [Google Scholar] [CrossRef]
- Matsunaga, Y.; Sugita, Y. Linking time-series of single-molecule experiments with molecular dynamics simulations by machine learning. eLife 2018, 7, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Matsunaga, Y.; Sugita, Y. Use of single-molecule time-series data for refining conformational dynamics in molecular simulations. Curr. Opin. Struct. Biol. 2020, 61, 153–159. [Google Scholar] [CrossRef]
- Pronk, S.; Páll, S.; Schulz, R.; Larsson, P.; Bjelkmar, P.; Apostolov, R.; Shirts, M.R.; Smith, J.C.; Kasson, P.M.; Van Der Spoel, D.; et al. GROMACS 4.5: A high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics 2013, 29, 845–854. [Google Scholar] [CrossRef]
- MacKerell, A.D.; Bashford, D.; Bellott, M.; Dunbrack, R.L.; Evanseck, J.D.; Field, M.J.; Fischer, S.; Gao, J.; Guo, H.; Ha, S.; et al. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B 1998, 102, 3586–3616. [Google Scholar] [CrossRef] [PubMed]
- Mackerell, A.D.; Feig, M.; Brooks, C.L. Extending the treatment of backbone energetics in protein force fields: Limitations of gas-phase quantum mechanics in reproducing protein conformational distributions in molecular dynamics simulation. J. Comput. Chem. 2004, 25, 1400–1415. [Google Scholar] [CrossRef] [PubMed]
- Piana, S.; Lindorff-Larsen, K.; Shaw, D.E. How robust are protein folding simulations with respect to force field parameterization? Biophys. J. 2011, 100, L47–L49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sumi, T.; Mitsutake, A.; Maruyama, Y. A solvation free-energy functional: A reference-modified density functional formulation. J. Comput. Chem. 2015, 36, 1359–1369, Erratum in J. Comput. Chem. 2015, 36, 2009–2011. [Google Scholar] [CrossRef] [Green Version]
- Sumi, T.; Maruyama, Y.; Mitsutake, A.; Mochizuki, K.; Koga, K. Application of reference-modified density functional theory: Temperature and pressure dependences of solvation free energy. J. Comput. Chem. 2018, 39, 202–217. [Google Scholar] [CrossRef] [Green Version]
- Maruyama, Y.; Hirata, F. Modified anderson method for accelerating 3D-RISM calculations using graphics processing unit. J. Chem. Theory Comput. 2012, 8, 3015–3021. [Google Scholar] [CrossRef]
- Honda, S.; Akiba, T.; Kato, Y.S.; Sawada, Y.; Sekijima, M.; Ishimura, M.; Ooishi, A.; Watanabe, H.; Odahara, T.; Harata, K. Crystal structure of a ten-amino acid protein. J. Am. Chem. Soc. 2008, 130, 15327–15331. [Google Scholar] [CrossRef] [Green Version]
- Ashbaugh, H.S.; Collett, N.J.; Hatch, H.W.; Staton, J.A. Assessing the thermodynamic signatures of hydrophobic hydration for several common water models. J. Chem. Phys. 2010, 132, 124504. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [Green Version]
- Flory, P.J. Principles of Polymer Chemistry; Cornellxford University Press: Ithaca, NY, USA, 1953. [Google Scholar]
- De Gennes, P.G. Scaling Concepts in Polymer Physics; Cornellxford University Press: Ithaca, NY, USA, 1979. [Google Scholar]
- Rubinstein, M.; Colby, R.H. Polymer Physics; Oxford University Press: Oxford, UK, 2003. [Google Scholar]
- Yoshikawa, Y.; Sakumichi, N.; Chung, U.I.; Sakai, T. Negative energy elasticity in a rubber-like gel. Phys. Rev. X 2021, 11, 11045. [Google Scholar]
- Sakumichi, N.; Yoshikawa, Y.; Sakai, T. Linear elasticity of polymer gels in terms of negative energy elasticity. Polym. J. 2021, 53, 1293–1303. [Google Scholar] [CrossRef]
- Fujiyabu, T.; Sakai, T.; Kudo, R.; Yoshikawa, Y.; Katashima, T.; Chung, U.i.; Sakumichi, N. Temperature Dependence of Polymer Network Diffusion. Phys. Rev. Lett. 2021, 127, 237801. [Google Scholar] [CrossRef]
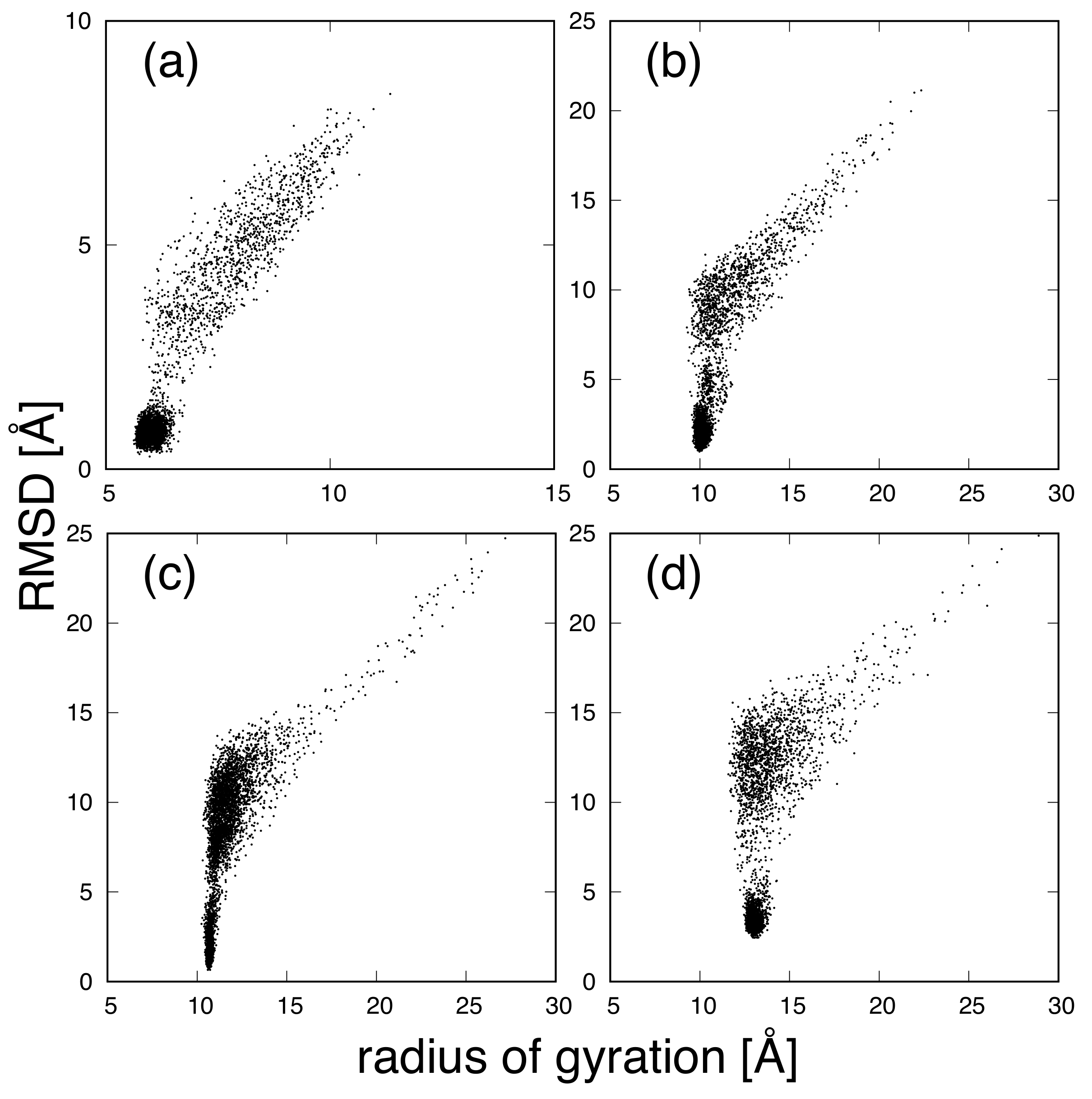
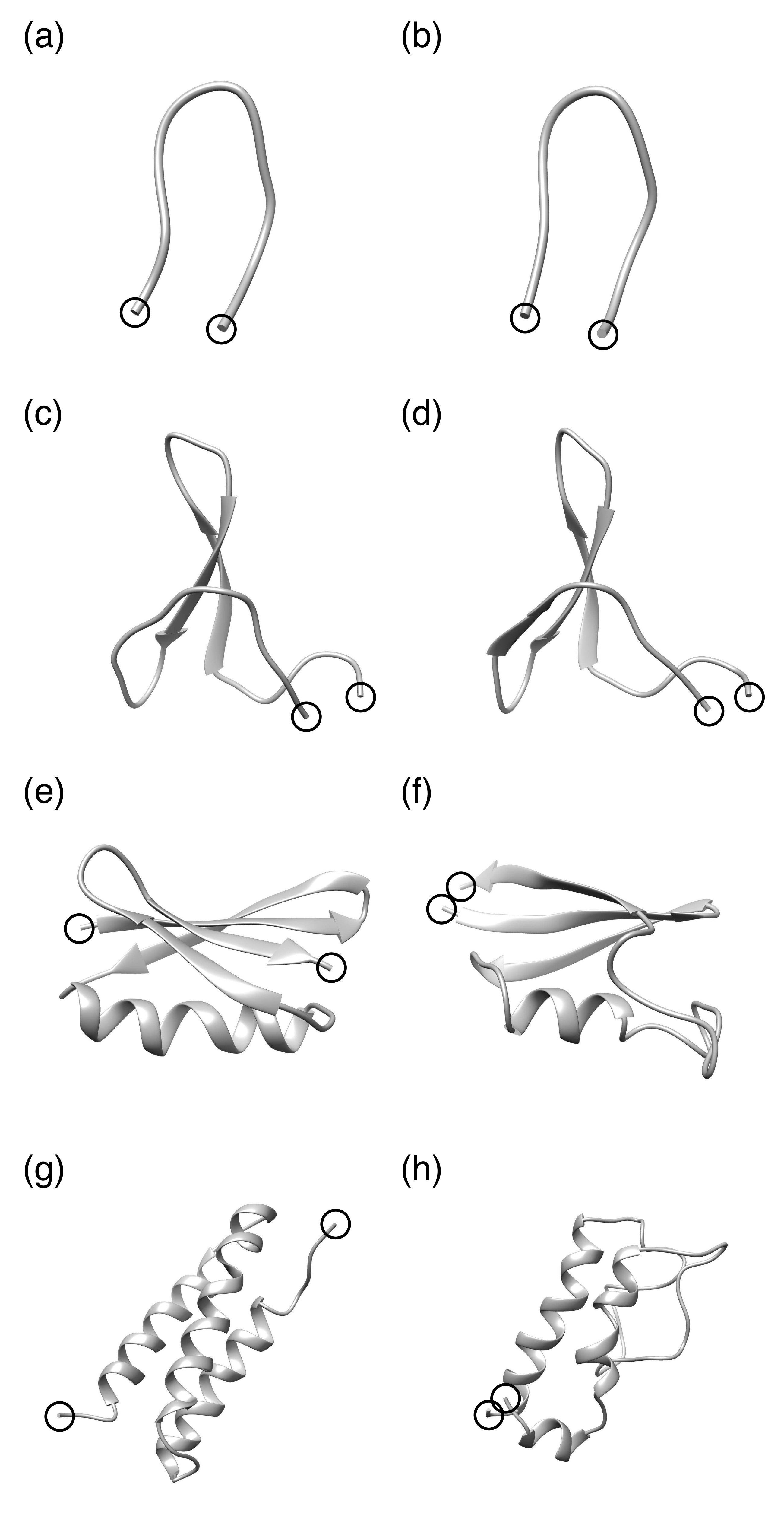
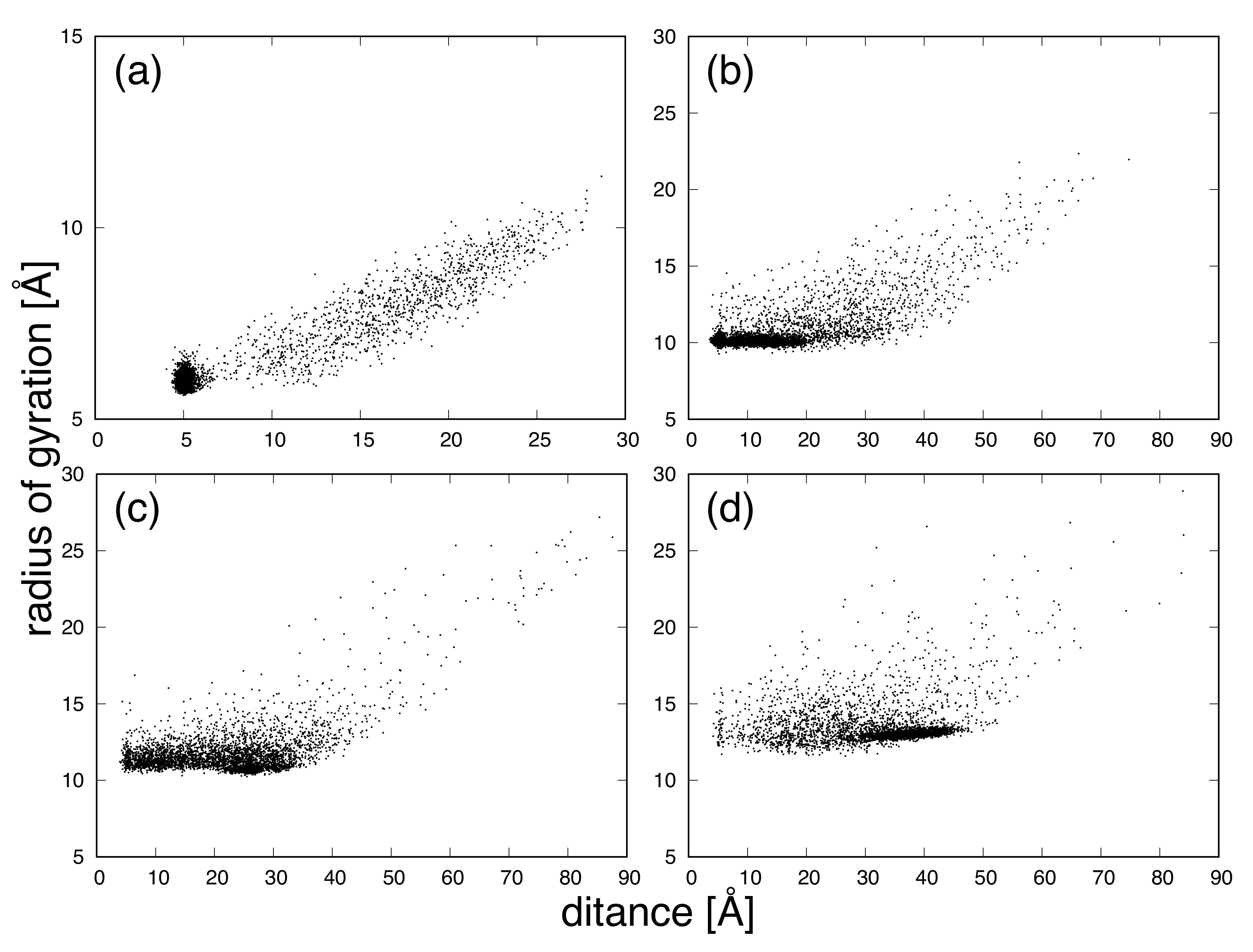
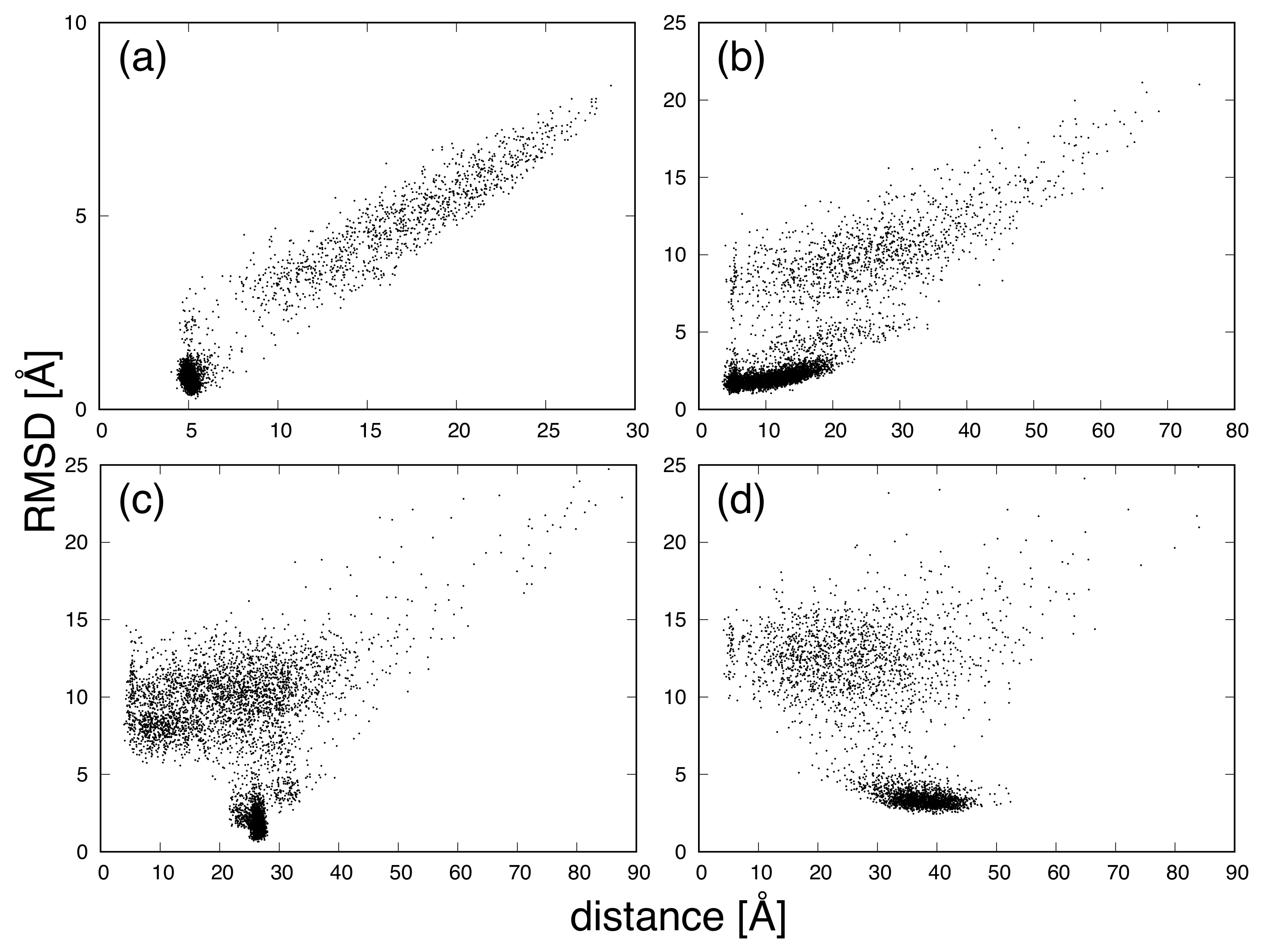
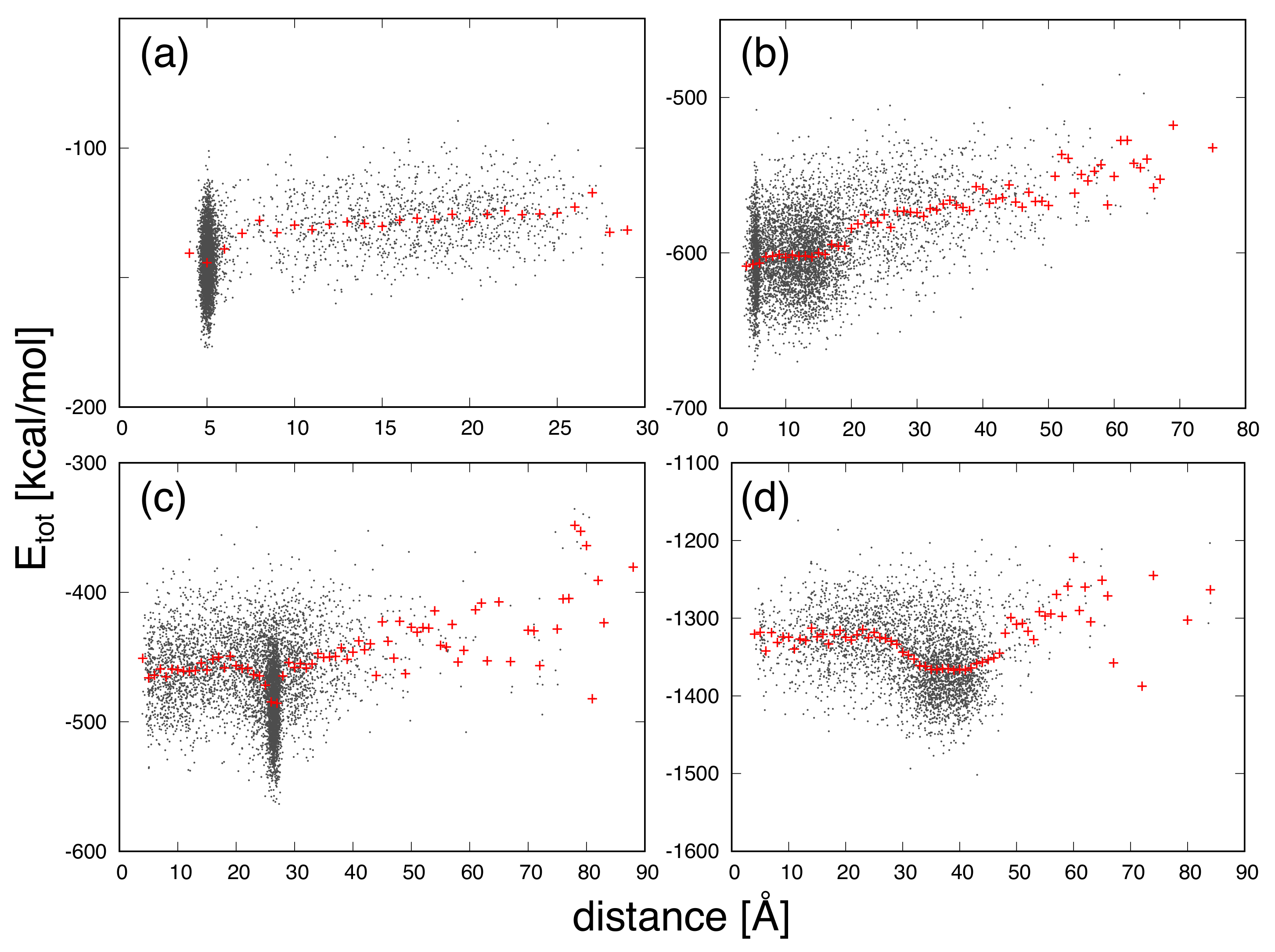
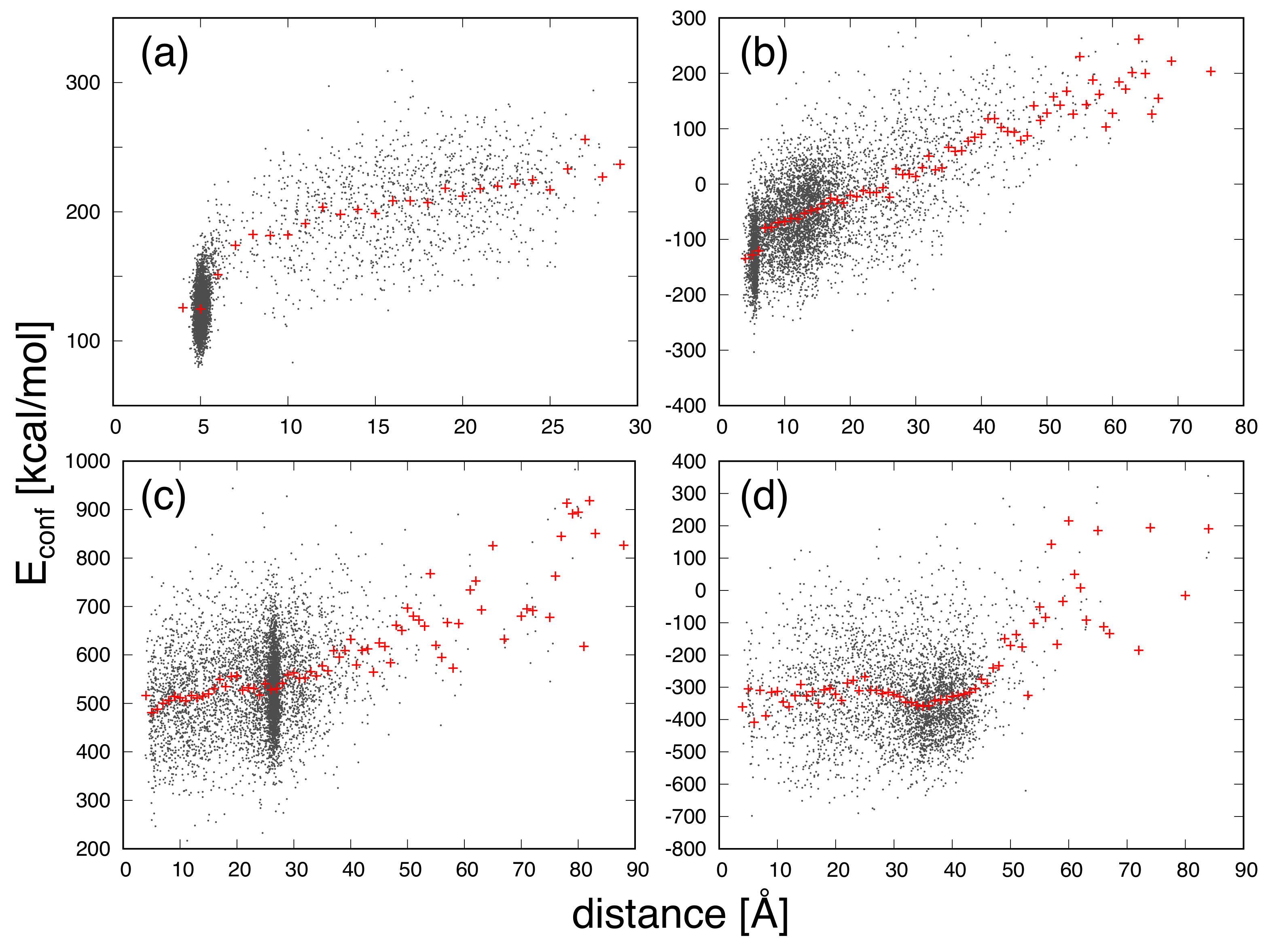
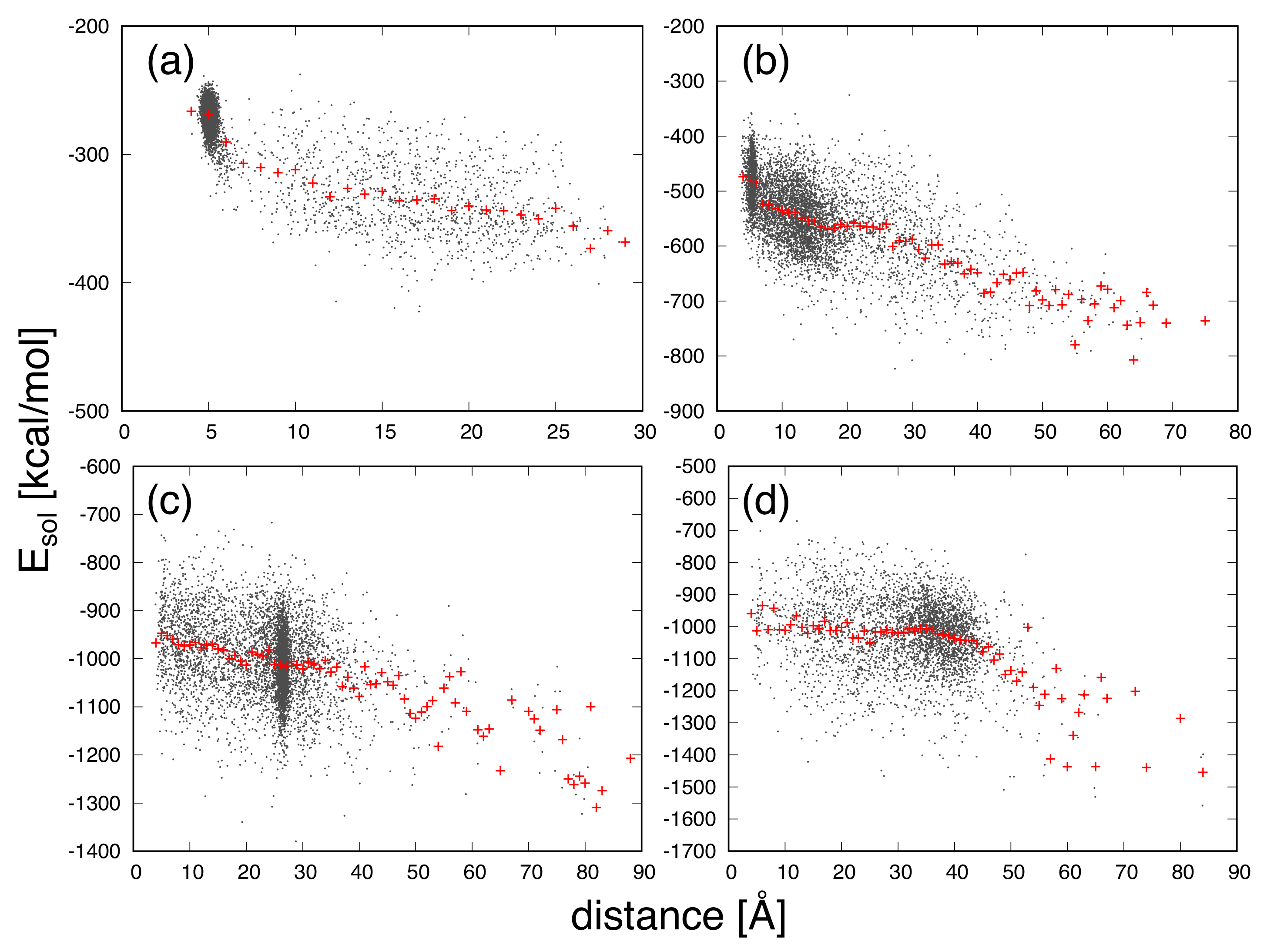
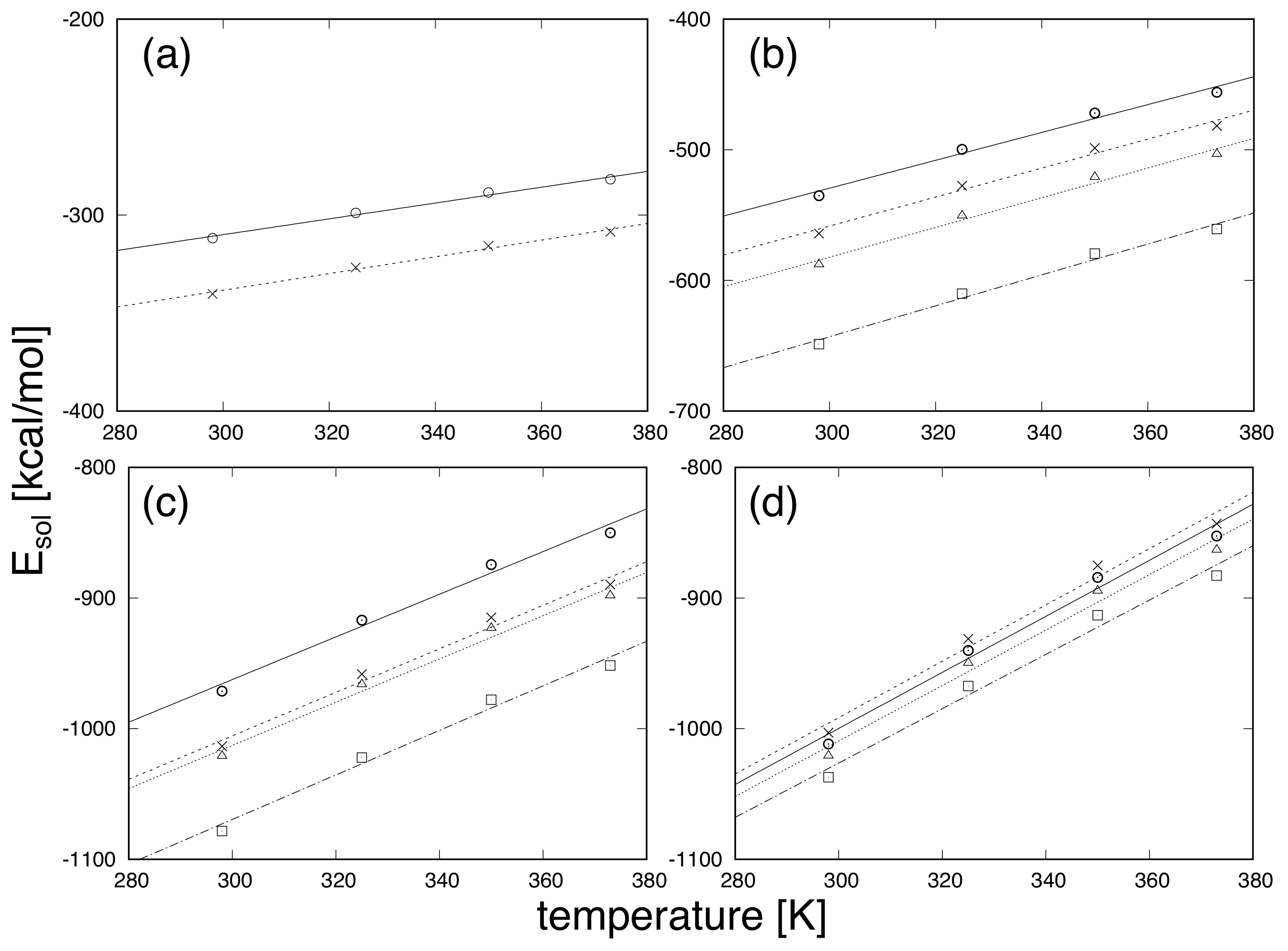
| Protein | Minimum | C-RMSD [Å] | End-to-End Distance [Å] | Rg [Å] | [kcal/mol] |
|---|---|---|---|---|---|
| CLN025 | 0.7 | 5.0 | 6.2 | −177.0 | |
| distance | 1.0 | 4.0 | 6.3 | −136.1 | |
| mGTT | 1.6 | 5.0 | 10.1 | −675.0 | |
| distance | 1.8 | 3.6 | 10.2 | −596.0 | |
| mNuG2 | 1.9 | 27.4 | 10.7 | −563.4 | |
| distance | 8.2 | 3.9 | 11.2 | −493.9 | |
| m3D | 3.6 | 43.1 | 13.2 | −1501.7 | |
| distance | 14.3 | 4.2 | 12.9 | −1287.9 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maruyama, Y.; Mitsutake, A. Structural Stability Analysis of Proteins Using End-to-End Distance: A 3D-RISM Approach. J 2022, 5, 114-125. https://doi.org/10.3390/j5010009
Maruyama Y, Mitsutake A. Structural Stability Analysis of Proteins Using End-to-End Distance: A 3D-RISM Approach. J. 2022; 5(1):114-125. https://doi.org/10.3390/j5010009
Chicago/Turabian StyleMaruyama, Yutaka, and Ayori Mitsutake. 2022. "Structural Stability Analysis of Proteins Using End-to-End Distance: A 3D-RISM Approach" J 5, no. 1: 114-125. https://doi.org/10.3390/j5010009
APA StyleMaruyama, Y., & Mitsutake, A. (2022). Structural Stability Analysis of Proteins Using End-to-End Distance: A 3D-RISM Approach. J, 5(1), 114-125. https://doi.org/10.3390/j5010009