
Applications of Time-Resolved Thermodynamics for Studies on Protein Reactions

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
−kBT (∂ln Keq/∂P)T = ΔV
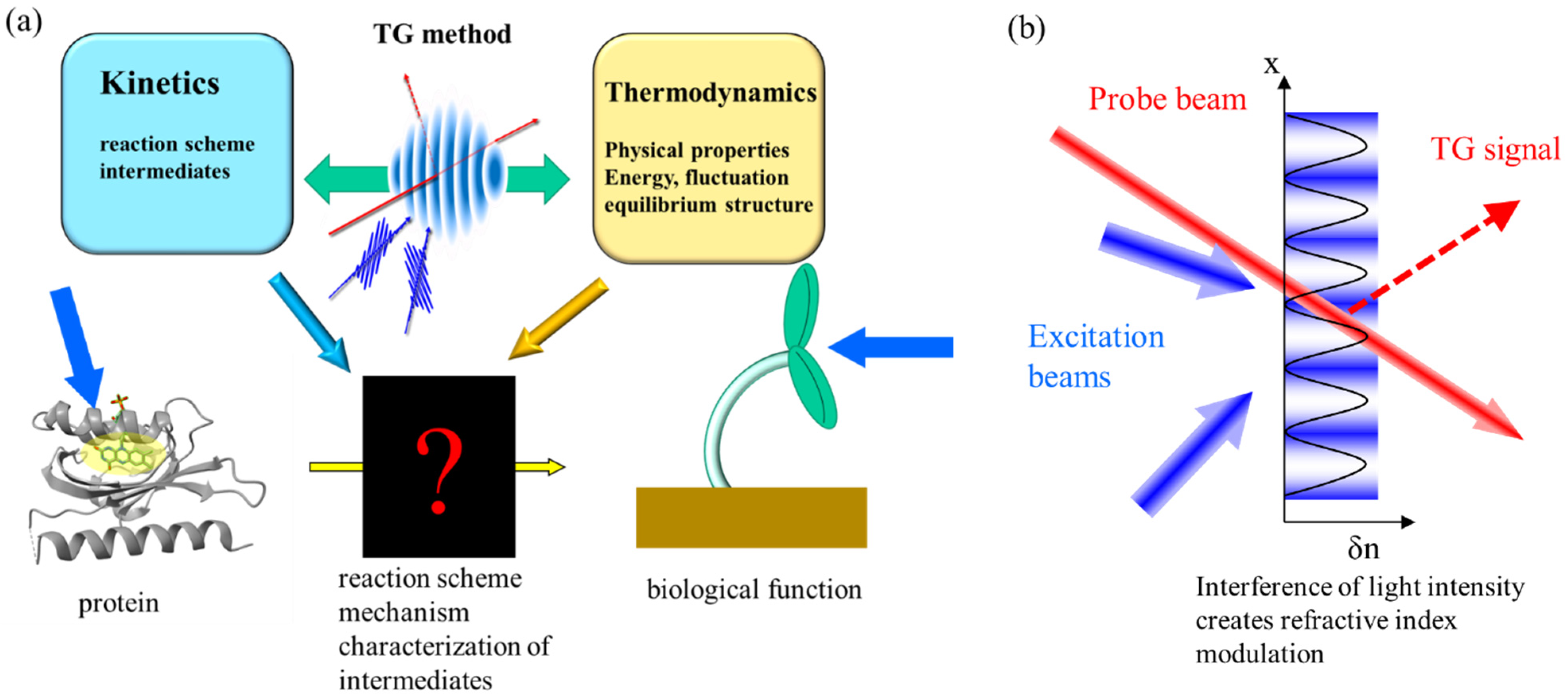
2. Principle
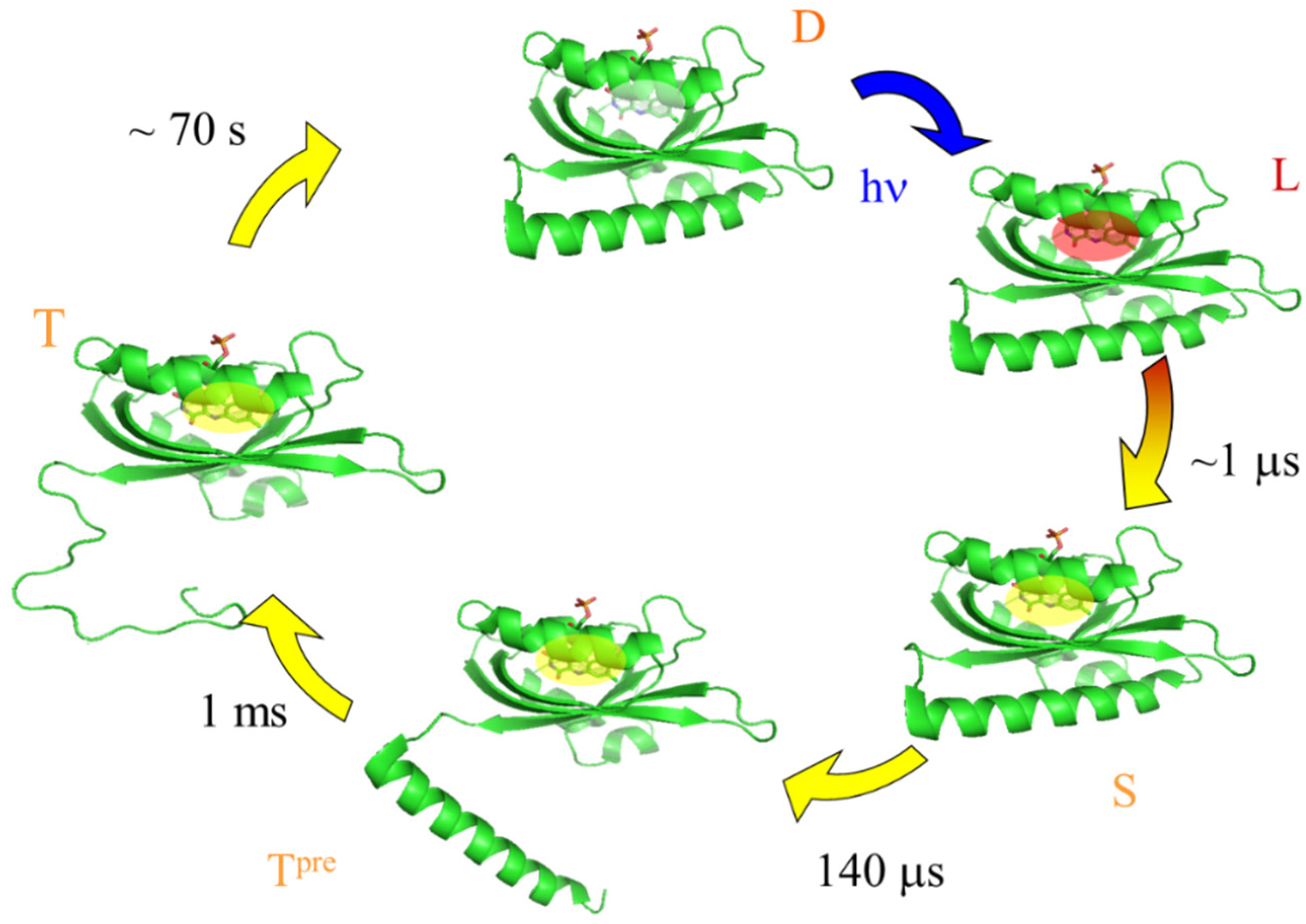
3. Results and Discussion
3.1. Diffusion Changes
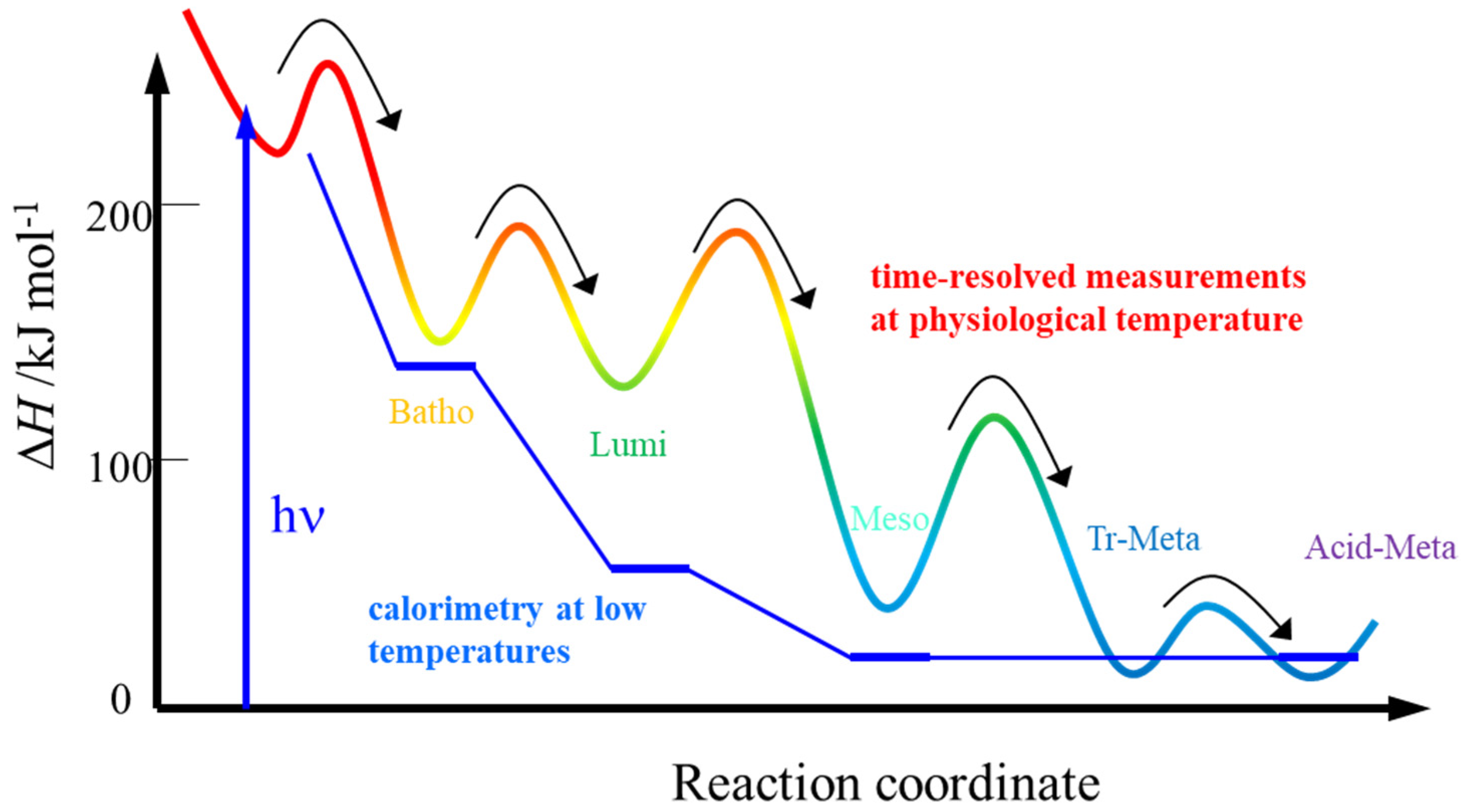
3.2. Enthalpy and Volume Change
3.3. Change in Heat Capacity
3.4. Thermal Expansion Coefficient Change
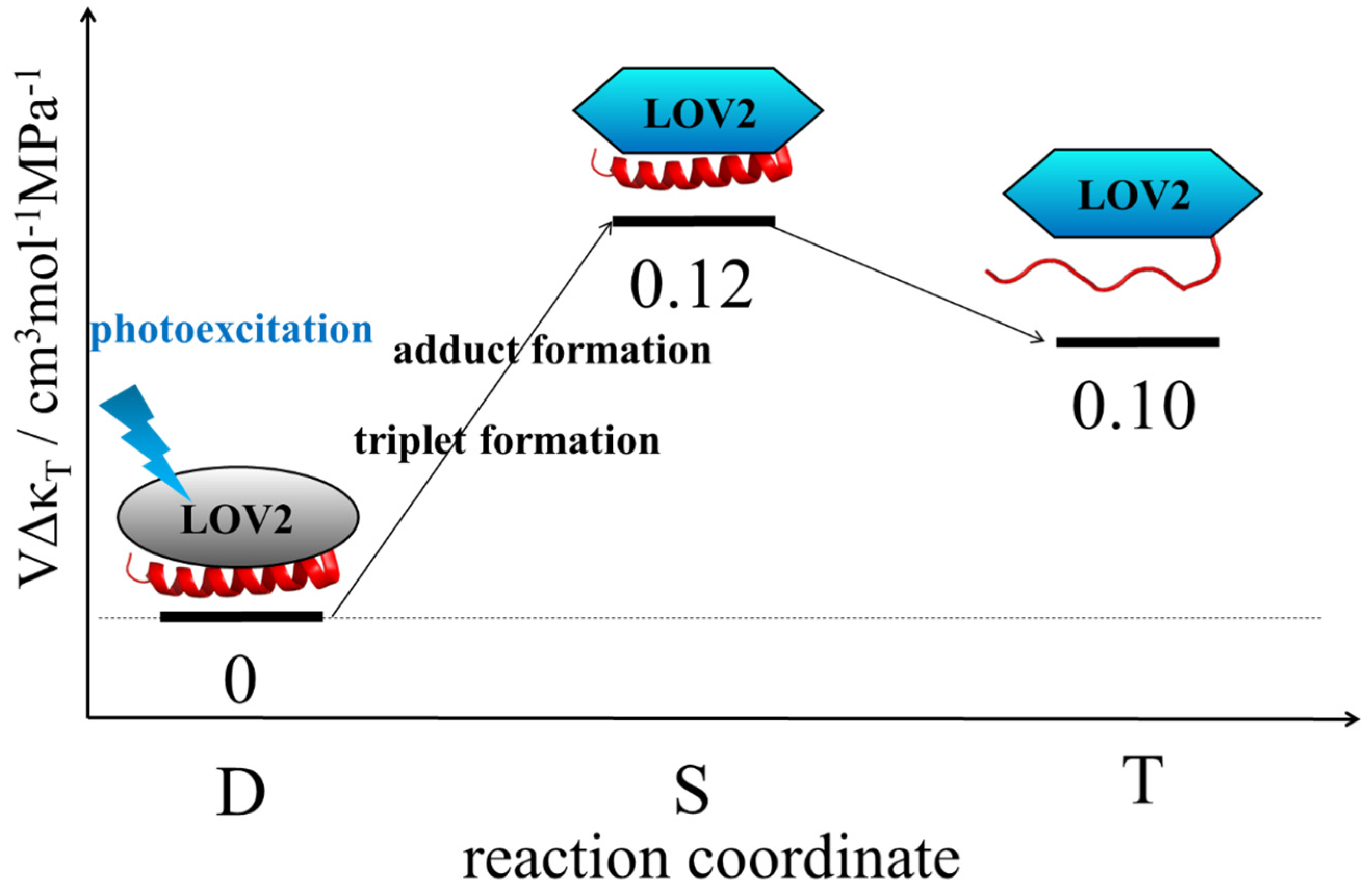
3.5. Change in Compressibility
4. Conclusions and Perspective
- This method can be used for irreversible reactions, and is also capable of measuring the thermodynamic properties and D in a time resolved manner. Using this method, the D values of various transient, short lived radicals were measured, and it was found that D was very different between stable molecules and transient radicals [15,16].
- 2.
- This TG technique has a high sensitivity because the homodyne detection of the TG method is background free. Proteins generally and easily form oligomers at high concentrations, and the reaction is altered by the formation of oligomers. High sensitivity is important for measurements at dilute concentrations. Furthermore, ΔV of a few cm3/mol was detected. For example, ΔV of −7 cm3/mol obtained for the reaction of PYP was only ~0.7% of the total protein volume [52]. Using a conventional ΔV measurement, that is, the pressure dependence of Keq, ΔκT should be calculated from the deviation of the linear relation of Keq vs. P. This deviation should be subtle, and it is almost impossible to measure ΔκT for any protein solution. Hence, the experimental measurement of this property of a protein in solution is quite difficult. The reported ΔκT measurements clearly demonstrate the high sensitivities of ΔV and ΔκT. Due to its high sensitivity, a variety of light intensities can be used, and, as a result, the light intensity dependence on the reaction can be studied. Using this merit, some nonlinear light intensity dependence of DSCC has been observed for various photosensor proteins. For example, in addition to BlrP1, shown above, a BLUF protein of decamer PixD (SyPixD) from Synechocystis sp. PCC6803 dissociates into five dimers when two protomers in the decamer are excited but not one [61]. Accompanying this dissociation of SyPixD, a cyanobacterial response regulator PixE was released from the PixD10-PixE5 complex. This suggests that the biological response of PixD is a nonlinear light intensity sensor for light intensity dependent biological functions.
- 3.
- The thermodynamic properties of this method should be exactly along the reaction coordinate. This advantage was demonstrated by the ΔH measurement of the intermediates of octopus rhodopsin, reported above. Furthermore, this advantage becomes apparent for a case in which the photochemical reaction is pressure dependent. In this case, the reaction scheme can be altered by pressure, and “the molecular volume” cannot be determined from the pressure dependence of Keq. However, ΔV from the volume grating intensity should be an intrinsic value for the reaction. Similarly, the temperature dependence measurement Keq for ΔH determination could not be correct because protein conformation and the reactivity are sensitive to the temperature. ΔH, which is measured without changing any external properties, should be intrinsic.
- 4.
- The TG method does not require any modifications or mutations of the target protein. Some spectroscopic methods require a probe molecule to be attached to the protein. Mutation or binding of a probe molecule may alter the reaction. Hence, the reaction monitored for native proteins using the TG method is appropriate.
- 5.
- Water molecules and other nonreactive species in the sample solution do not disturb the measurements. Hence, a variety of sample solutions have been used, such as various pH values, temperatures, solutions containing salts, or crowding macromolecules. The effect of crowding on protein reaction was also examined [62,63]. In some reactions, reactivity was found to be very sensitive to temperature. Based on these findings, it was proposed that this protein may function as a temperature sensor [64].
- 6.
- The TG signal originates only from the reacting species. Hence, the presence of nonreacting species does not disturb or interfere with the measurements. This merit is a big advantage compared with other methods that monitor the ensemble average of the reaction system, such as calorimetry, dynamic light scattering, SAXS, and IR spectroscopy. For example, the IR spectrum in the dark state must be subtracted from the light spectrum to obtain any changes in the reaction. If the reaction yield is small, the analysis of the SAXS signal becomes difficult, because of the superposition of the solvent and nonreactive species. Gel chromatography and other diffusion detection methods monitor the ensemble averages of the species in solution. Consequently, it may be difficult to detect oligomer formation in a sample using gel chromatography, unless the population of the dimer is dominant. Moreover, while covalently linked or stable protein aggregates may be detected by a size exclusion chromatography approach, a noncovalent protein aggregate that is formed by weak hydrophobic or hydrogen bond interactions may not be detected because the aggregate might dissociate during elution through the column. The TG technique can overcome these difficulties, because the measurement is performed in solution, so that even a weak binding complex is maintained in solution.
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Perozzo, R.; Folkers, G.; Scapozza, L. Thermodynamics of protein-ligand interactions: History, presence, and future aspects. J. Recept. Signal Transduct. Res. 2004, 24, 1–52. [Google Scholar] [CrossRef] [PubMed]
- Roman, E.A.; Flecha, F.L.G. Kinetics and Thermodynamics of Membrane Protein Folding. Biomolecules 2014, 4, 354–373. [Google Scholar] [CrossRef] [PubMed]
- Landau, L.D.; Lifshitz, E.M. Statistical Mechanics; Pergamon: London, UK, 1959. [Google Scholar]
- Rock, P.A. Chemical Thermodynamics; University Science Books: Sausalito, CA, USA, 1983. [Google Scholar]
- Cussler, E.L. Diffusion, 2nd ed.; Cambridge University Press: Cambridge, UK, 1997. [Google Scholar]
- Hammes, G.G. Spectroscopy for the Biological Science; John Wiley & Sons: Hoboken, NJ, USA, 2005. [Google Scholar]
- Drlijaca, A.; Hubbard, C.D.; van Eldik, R.; Asano, T.; Basilevsky, M.V.; le Noble, W.J. Activation and Reaction Volumes in Solution. 3. Chem. Rev. 1989, 89, 549–688. [Google Scholar]
- Chalikian, T.V.; Breslauer, K.J. On volume changes accompanying conformational transitions of biopolymers. Biopolymers 1996, 39, 619–626. [Google Scholar] [CrossRef]
- Chalikian, T.V.; Totrov, M.; Abagyan, R.; Breslauer, K.J. The Hydration of Globular Proteins as Derived from Volume and Compressibility Measurements: Cross Correlating Thermodynamic and Structural Data. J. Mol. Biol. 1996, 260, 588–603. [Google Scholar] [CrossRef]
- Eichler, H.J.; Günter, P.; Pohl, D.W. Laser-Induced Dynamic Gratings; Springer: Berlin/Heidelberg, Germany, 1986. [Google Scholar]
- Fayer, M.D. Dynamics of Molecules in Condensed Phases: Picosecond Holographic Grating Experiments. Annu. Rev. Phys. Chem. 1982, 33, 63–87. [Google Scholar] [CrossRef]
- Terazima, M. Energetic and molecular dynamics in solution studied by the third order nonlinear optical spectroscopies. In Transient Lens and Transient Grating. Advances in Multiphoton Processes and Spectroscopy; World Scientific: Singapore, 1996; Volume 10, pp. 1–96. [Google Scholar]
- Terazima, M.; Hirota, N.; Braslavsky, S.E.; Mandelis, A.; Bialkowski, S.E.; Diebold, G.J.; Miller, R.J.D.; Fournier, D.; Palmer, R.A.; Tam, A. Quantities, terminology and symbols in photothermal and related spectroscopies. Pure Appl. Chem 2004, 76, 1083–1118. [Google Scholar] [CrossRef]
- Kogelnik, H. Coupled Wave Theory for Thick Hologram Gratings. Bell. System Tech. J. 1969, 48, 2909–2947. [Google Scholar] [CrossRef]
- Terazima, M.; Hirota, N. Translational diffusion of a transient radical studied by the transient grating method; pyrazinyl radical in 2-propanol. J. Chem. Phys. 1993, 98, 6257–6262. [Google Scholar] [CrossRef]
- Terazima, M. Is the Translational Diffusion of Organic Radicals Different from That of Closed-Shell Molecules? Acc. Chem. Res. 2000, 33, 687–694. [Google Scholar] [CrossRef]
- Nishida, S.; Nada, T.; Terazima, M. Kinetics of intermolecular interaction during protein folding of reduced cytochrome c. Biophys. J. 2004, 87, 2663–2675. [Google Scholar] [CrossRef] [PubMed]
- Terazima, M. Time-Dependent Intermolecular Interaction during Protein Reactions. Phys. Chem. Chem. Phys. 2011, 13, 16928–16940. [Google Scholar] [CrossRef] [PubMed]
- Nakasone, Y.; Ono, T.; Ishii, A.; Masuda, S.; Terazima, M. Transient dimerization and conformational change of a BLUF protein: YcgF. J. Am. Chem. Soc. 2007, 129, 7028–7035. [Google Scholar] [CrossRef] [PubMed]
- Kondoh, M.; Hitomi, K.; Yamamoto, J.; Todo, T.; Iwai, S.; Getzoff, E.D.; Terazima, M. Light-induced conformational change and product release in DNA repair by (6-4) photolyase. J. Am. Chem. Soc. 2011, 133, 2183–2191. [Google Scholar] [CrossRef][Green Version]
- Akiyama, Y.; Nakasone, Y.; Nakatani, Y.; Hisatomi, O.; Terazima, M. Time-resolved detection of light-induced dimerization of monomeric Aureochrome-1 and change in affinity for DNA. J. Phys. Chem. B 2016, 120, 7360–7370. [Google Scholar] [CrossRef]
- Takakado, A.; Nakasone, Y.; Terazima, M. Photoinduced dimerization of a photosensory DNA-binding protein EL222 and its LOV domain. Phys. Chem. Chem. Phys. 2017, 19, 24855–24865. [Google Scholar] [CrossRef]
- Takakado, A.; Nakasone, Y.; Terazima, M. Sequential DNA binding and dimerization processes of the photosensory protein EL222. Biochemistry 2018, 57, 1603–1610. [Google Scholar] [CrossRef]
- Nakasone, Y.; Kikukawa, K.; Masuda, S.; Terazima, M. Time-resolved study of interprotein signaling process of a blue light sensor PapB-PapA complex. J. Phys. Chem. B 2019, 123, 3210–3218. [Google Scholar] [CrossRef]
- Takeda, K.; Terazima, M. Dynamics of conformation changes in full-length Phytochrome from cyanobacterium Synechocystis sp. PCC6803 (Cph1) monitored by time-resolved translational diffusion detection. Biochemistry 2019, 58, 2720–2729. [Google Scholar] [CrossRef]
- Kim, S.; Nakasone, Y.; Takakado, A.; Yamazaki, Y.; Kamikubo, H.; Terazima, M. Wavelength dependent photoreaction of PYP from Rhodobacter capsulatus. Biochemistry 2020, 59, 4810–4821. [Google Scholar] [CrossRef]
- Choi, S.; Nakasone, Y.; Hellingwerf, K.J.; Terazima, M. Photoreaction dynamics of a full-length protein YtvA and intermolecular interaction with RsbRA. Biochemistry 2020, 59, 4703–4710. [Google Scholar] [CrossRef] [PubMed]
- Ikoma, M.; Nakasone, Y.; Terazima, M. Photoreaction of photoactivated adenylate cyclase from cyanobacterium Microcoleus chthonoplastes. J. Photochem. Photobiol. B 2021, 221, 112252. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Nakasone, Y.; Takakado, A.; Yamazaki, Y.; Kamikubo, H.; Terazima, M. A unique photochromic UV-A sensor protein Rc-PYP interacting with PYP-binding protein. Phys. Chem. Chem. Phys. 2021, 21, 17813–17825. [Google Scholar] [CrossRef]
- Huala, E.; Oeller, P.W.; Liscum, E.; Han, I.; Larsen, E.; Briggs, W.R. Arabidopsis NPH1: A Protein Kinase with a Putative Redox-Sensing Domain. Science 1997, 278, 2120–2123. [Google Scholar] [CrossRef]
- Christie, J.M.; Reymond, P.; Powell, G.K.; Bernasconi, P.; Raibekas, A.A.; Liscum, E.; Briggs, W.R. Arabidopsis NPH1: A Flavoprotein with the Properties of a Photoreceptor for Phototropism. Science 1998, 282, 1698–1701. [Google Scholar] [CrossRef] [PubMed]
- Swartz, T.E.; Corchnoy, S.B.; Christie, J.M.; Lewis, J.W.; Szundi, I.; Briggs, W.R.; Bogomolni, R.A. The photocycle of a flavin-binding domain of the blue light photoreceptor phototropin. J. Biol. Chem. 2001, 276, 36493–36500. [Google Scholar] [CrossRef] [PubMed]
- Eitoku, T.; Nakasone, Y.; Matsuoka, D.; Tokutomi, S.; Terazima, M. Conformational dynamics of Phototropin 2 LOV2 domain with the linker upon photoexcitation. J. Am. Chem. Soc. 2005, 127, 13238–13244. [Google Scholar] [CrossRef]
- Nakasone, Y.; Eitoku, T.; Matsuoka, D.; Tokutomi, S.; Terazima, M. Kinetic measurement of transient dimerization and dissociation reactions of Arabidopsis phototropin 1 LOV2 domain. Biophys. J. 2006, 91, 645–653. [Google Scholar] [CrossRef]
- Nakasone, Y.; Eitoku, T.; Matsuoka, D.; Tokutomi, S.; Terazima, M. Dynamics of conformational changes of Arabidopsis phototropin 1 LOV2 with the linker domain. J. Mol. Biol. 2007, 367, 432–442. [Google Scholar] [CrossRef]
- Nakasone, Y.; Eitoku, T.; Zikihara, K.; Matsuoka, D.; Tokutomi, S.; Terazima, M. Photochemical intermediates of Arabidopsis phototropin 2 LOV domains associated with conformational changes. J. Mol. Biol. 2007, 371, 1290–1303. [Google Scholar]
- Nakasone, Y.; Eitoku, T.; Zikihara, K.; Matsuoka, D.; Tokutomi, S.; Terazima, M. Stability of dimer and domain-domain interaction of Arabidopsis phototropin 1 LOV2. J. Mol. Biol. 2008, 383, 904–913. [Google Scholar] [CrossRef] [PubMed]
- Takeda, K.; Nakasone, Y.; Zikihara, K.; Tokutomi, S.; Terazima, M. Dynamics of the amino-terminal and carboxyl-terminal helices of Arabidopsis Phototropin 1 LOV2 studied by the transient grating. J. Phys. Chem. B 2013, 117, 15606–15613. [Google Scholar] [CrossRef] [PubMed]
- Takakado, A.; Nakasone, Y.; Okajima, K.; Tokutomi, S.; Terazima, M. Light-Induced Conformational Changes of the LOV2-Kinase and the Linker Region in Arabidopsis Phototropin2. J. Phys. Chem. B 2017, 121, 4414–4421. [Google Scholar] [CrossRef] [PubMed]
- Shibata, K.; Nakasone, Y.; Terazima, M. Photoreaction of BlrP1: A role of nonlinear photo-intensity sensor. Phys. Chem. Chem. Phys. 2018, 20, 8133–8142. [Google Scholar] [CrossRef] [PubMed]
- Shibata, K.; Nakasone, Y.; Terazima, M. Enzymatic activity of the blue light-regulated phosphodiesterase BlrP1 from Klebsiella pneumoniae shows a nonlinear dependence on light intensity. FEBS Lett. 2021, 595, 1473–1479. [Google Scholar] [CrossRef]
- Terazima, M. Molecular volume and enthalpy changes associated with irreversible photo-reactions. J. Photochem. Photobiol. C 2002, 3, 81–108. [Google Scholar] [CrossRef]
- Terazima, M. Studies of photo-induced protein reactions by spectrally silent reaction dynamics detection methods: Applications to the photoreaction of the LOV2 domain of Phototropin from Arabidopsis thaliana. Biochim. Biophys. Acta 2011, 1814, 1093–1105. [Google Scholar] [CrossRef]
- Nishioku, Y.; Nakagawa, M.; Tsuda, M.; Terazima, M. A spectrally silent transformation in the photolysis of octopus rhodopsin: A protein conformational change without any accompanying change of the chromophore’s absorption. Biophy. J. 2001, 80, 2922–2927. [Google Scholar] [CrossRef]
- Nishioku, Y.; Nakagawa, M.; Tsuda, M.; Terazima, M. Energetics and Volume Changes of the Intermediates in the Photolysis of Octopus Rhodopsin at a physiological temperature. Biophy. J. 2002, 83, 1136–1146. [Google Scholar] [CrossRef]
- Inoue, K.; Tsuda, M.; Terazima, M. Photo-reverse-reaction dynamics of octopus rhodopsin. Biophys. J. 2007, 92, 3643–3651. [Google Scholar] [CrossRef]
- Cooper, A.; Dixon, S.F.; Tsuda, M. Photoenergetics of octopus rhodopsin, Convergent evolution of biological photon counters? Eur. Biophys. J. 1986, 13, 195–201. [Google Scholar] [CrossRef] [PubMed]
- Hirota, S.; Fujimoto, Y.; Choi, J.; Baden, N.; Katagiri, N.; Akiyama, M.; Hulsker, R.; Ubbink, M.; Okajima, T.; Takabe, T.; et al. Conformational changes during Apoplastocyanin folding observed by photocleavable modification and transient grating. J. Am. Chem. Soc. 2006, 128, 7551–7558. [Google Scholar] [CrossRef] [PubMed]
- Yoshidome, T.; Kinoshita, M.; Hirota, S.; Baden, N.; Terazima, M. Thermodynamics of apoplastocyanin folding: Comparison between experimental and theoretical results. J. Chem. Phys. 2008, 128, 225104. [Google Scholar] [CrossRef] [PubMed]
- Prabhu, N.V.; Sharp, K.A. Heat capacity in proteins. Annu. Rev. Phys. Chem. 2005, 56, 521–548. [Google Scholar] [CrossRef]
- Khan, J.S.; Imamoto, Y.; Kataoka, M.; Tokunaga, F.; Terazima, M. Time-resolved thermodynamics: Heat capacity change of transient species during photo-reaction of PYP. J. Am. Chem. Soc. 2006, 128, 1002–1008. [Google Scholar] [CrossRef]
- Takeshita, K.; Hirota, N.; Imamoto, Y.; Kataoka, M.; Tokunaga, F.; Terazima, M. Temperature dependent volume change of initial step of the photoreaction of Photoactive Yellow Protein studied by the transient grating. J. Am. Chem. Soc. 2000, 122, 8524–8528. [Google Scholar] [CrossRef]
- Henry, J.T.; Crosson, S. Ligand-binding PAS domains in a genomic, cellular, and structural context. Annu. Rev. Microbiol. 2011, 65, 261–286. [Google Scholar] [CrossRef]
- Perman, B.; Srajer, V.; Ren, Z.; Teng, T.; Pradervand, C.; Ursby, T.; Bourgeois, D.; Schotte, F.; Wulff, M.; Kort, R.; et al. Energy Transduction on the Nanosecond Time Scale: Early Structural Events in a Xanthopsin Photocycle. Science 1998, 279, 1946–1950. [Google Scholar] [CrossRef]
- Jung, Y.O.; Lee, J.H.; Kim, J.; Schmidt, M.; Moffat, K.; Srajer, V.; Ihee, H. Volume-conserving trans-cis isomerization pathways in photoactive yellow protein visualized by picosecond X-ray crystallography. Nat. Chem. 2013, 5, 212–220. [Google Scholar] [CrossRef]
- Kuroi, K.; Okajima, K.; Ikeuchi, M.; Tokutomi, S.; Terazima, M. Transient conformational fluctuation of TePixD during a reaction. Proc. Natl. Acad. Sci. USA 2014, 111, 14764–14769. [Google Scholar] [CrossRef]
- Kuroi, K.; Sato, F.; Nakasone, Y.; Zikihara, K.; Tokutomi, S.; Terazima, M. Time-resolved fluctuation during the photochemical reaction of a photoreceptor protein: Phototropin1LOV2-Linker. Phys. Chem. Chem. Phys. 2016, 18, 6228–6238. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Hoshihara, Y.; Kimura, Y.; Matsumoto, M.; Nagasawa, M.; Terazima, M. An optical high-pressure cell for transient grating measurements of biological substance with a high reproducibility. Rev. Sci. Instrum. 2008, 79, 034101. [Google Scholar] [CrossRef] [PubMed]
- Terazima, M. Vibrational relaxation from electronically photoexcited states in solution studied by the acoustic peak delay method: Hydrogen bonding effect to Betaine-30. Chem. Phys. Lett. 1999, 305, 189–196. [Google Scholar] [CrossRef]
- Miyata, R.; Terazima, M. Transient thermal expansion of a protein in solution after photo-excitation of the chromophore: Deoxymyoglobin. Bull. Chem. Soc. Jpn. 2003, 76, 1707–1712. [Google Scholar] [CrossRef]
- Tanaka, K.; Nakasone, Y.; Okajima, K.; Ikeuchi, M.; Tokutomi, S.; Terazima, M. A way to sense light intensity: Multiple-excitation of the BLUF photoreceptor TePixD suppresses conformational change. FEBS Lett. 2011, 585, 786–790. [Google Scholar] [CrossRef] [PubMed]
- Toyooka, T.; Tanaka, K.; Okajima, K.; Ikeuchi, M.; Tokutomi, S.; Terazima, M. Macromolecular crowding effects on reactions of TePixD (Tll0078). Photochem. Photobiol. 2011, 87, 584. [Google Scholar] [CrossRef]
- Yoshitake, T.; Toyooka, T.; Nakasone, Y.; Zikihara, K.; Tokutomi, S.; Terazima, M. Macromolecular crowding effect for photoreactions of LOV2 domains of Arabidopsis thaliana phototropin 1. J. Mol. Liq. 2016, 217, 43–50. [Google Scholar] [CrossRef]
- Nakasone, Y.; Ono, T.; Ishii, A.; Masuda, S.; Terazima, M. Temperature sensitive reaction of a photosensor protein YcgF: Possibility of a role of temperature sensor. Biochemistry 2010, 49, 2288–2296. [Google Scholar] [CrossRef]
- Choi, J.; Terazima, M. Denaturation of a protein monitored by diffusion coefficients: Myoglobin. J. Phys. Chem. B 2002, 106, 6587–6593. [Google Scholar] [CrossRef]
- Inoue, K.; Baden, N.; Terazima, M. Diffusion coefficient and the secondary structure of poly-L-glutamic acid in aqueous solution. J. Phys. Chem. B 2005, 109, 22623–22628. [Google Scholar] [CrossRef]
- Choi, J.; Terazima, M. Photoreaction of caged ATP compound studied by the time-resolved transient grating method. Photochem. Photobiol. Sci. 2003, 2, 767–773. [Google Scholar] [CrossRef] [PubMed]
- Kondoh, M.; Baden, N.; Terazima, M. Photochemical reaction and diffusion of caged calcium studied by the transient grating. Chem. Phys. Lett. 2008, 462, 58–63. [Google Scholar] [CrossRef]
- Nakasone, Y.; Takaramoto, S.; Terazima, M. Time-resolved diffusion detection with micro-stopped flow system. Anal. Chem. 2019, 91, 11987–11993. [Google Scholar] [CrossRef] [PubMed]
- Takaramoto, S.; Nakasone, Y.; Sadakane, K.; Maruta, S.; Terazima, M. Time-resolved detection of SDS-induced conformational changes in α-synuclein by a micro-stopped-flow system. RSC Adv. 2021, 11, 1086–1097. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Terazima, M. Applications of Time-Resolved Thermodynamics for Studies on Protein Reactions. J 2022, 5, 186-197. https://doi.org/10.3390/j5010014
Terazima M. Applications of Time-Resolved Thermodynamics for Studies on Protein Reactions. J. 2022; 5(1):186-197. https://doi.org/10.3390/j5010014
Chicago/Turabian StyleTerazima, Masahide. 2022. "Applications of Time-Resolved Thermodynamics for Studies on Protein Reactions" J 5, no. 1: 186-197. https://doi.org/10.3390/j5010014
APA StyleTerazima, M. (2022). Applications of Time-Resolved Thermodynamics for Studies on Protein Reactions. J, 5(1), 186-197. https://doi.org/10.3390/j5010014