
Tryptophan Metabolism Through the Kynurenine Pathway in Glial Cells
 ,
,  ,
, 
 , and
, and
Abstract
1. Introduction
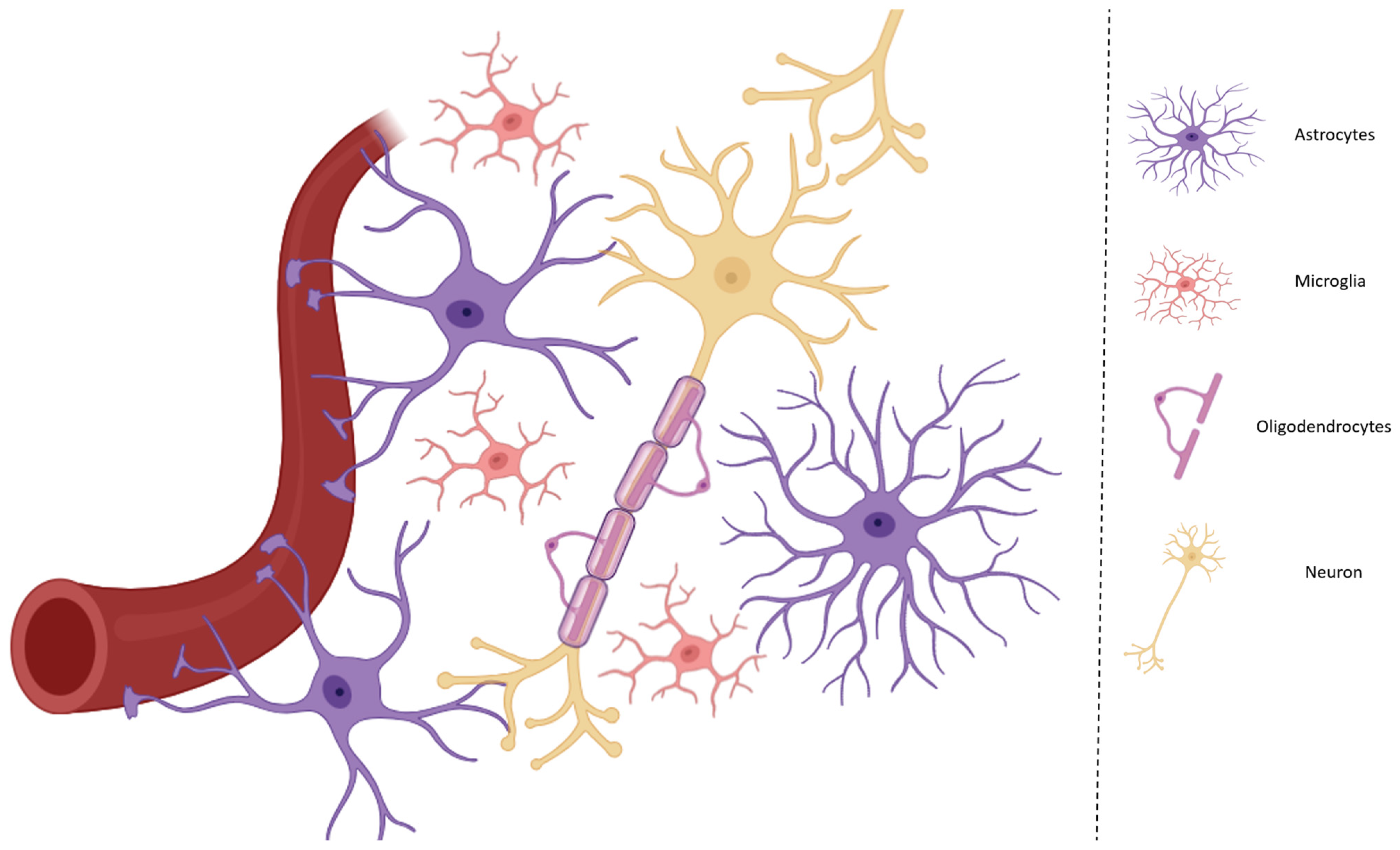
2. Communication Between Microglia and Astrocytes During Neuroinflammation and Activation of the KP
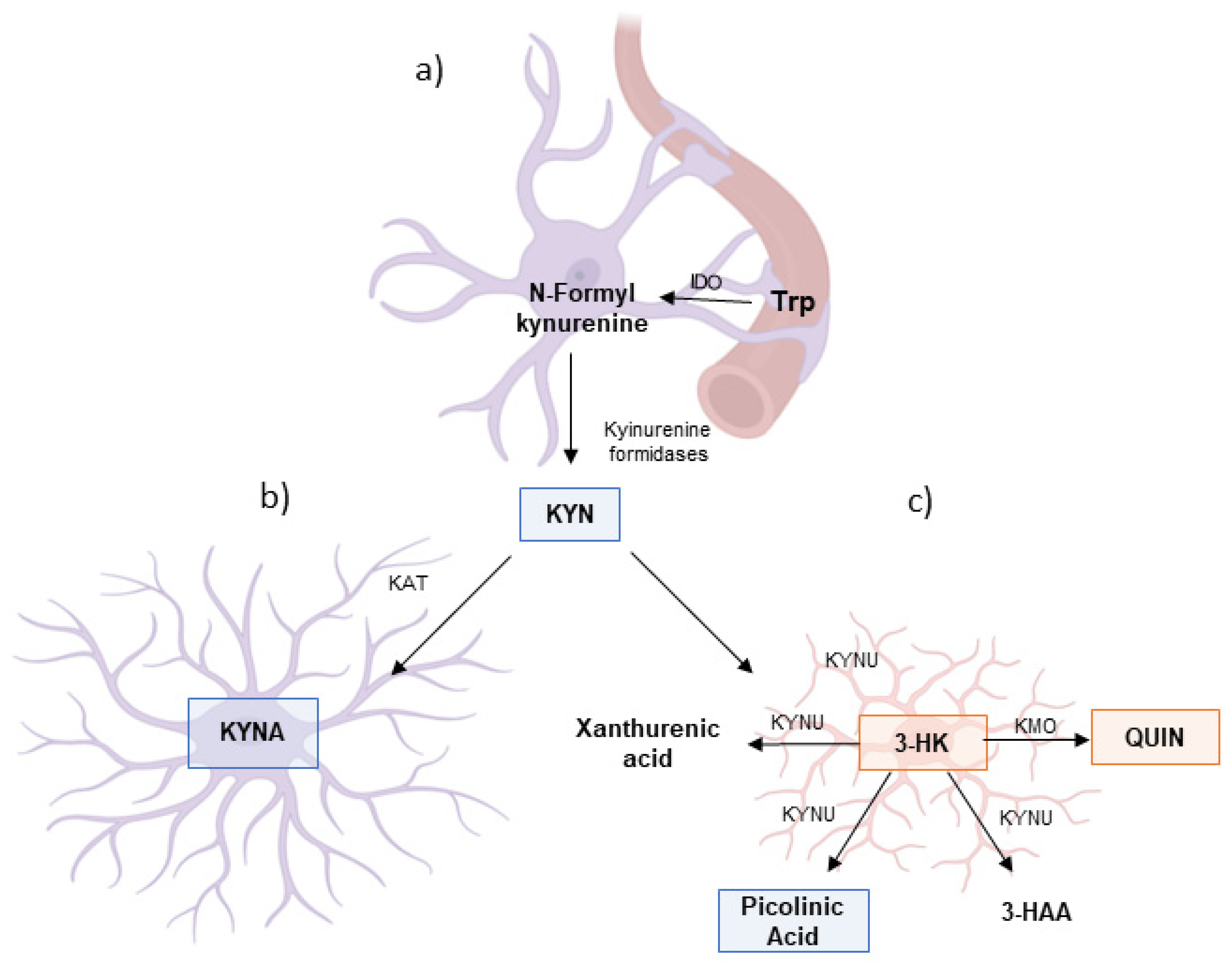
3. Trp Catabolism Through the KP During Neuroinflammatory Conditions
4. KP and Neurodegenerative Diseases
4.1. Trp Catabolism and KP in AD
4.2. Trp Catabolism and KP in PD
4.3. Trp Catabolism in ALS
4.4. Trp Catabolism and KP in MS
5. Biomarkers and Pharmacological Targets in KP for Neurodegenerative Diseases
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Stassart, R.M.; Möbius, W.; Nave, K.A.; Edgar, J.M. The Axon-Myelin Unit in Development and Degenerative Disease. Front. Neurosci. 2018, 12, 467. [Google Scholar] [CrossRef] [PubMed]
- Jäkel, S.; Dimou, L. Glial Cells and Their Function in the Adult Brain: A Journey through the History of Their Ablation. Front. Cell. Neurosci. 2017, 11, 24. [Google Scholar] [CrossRef]
- Harkany, T.; Hökfelt, T. Neuroglia: Function and Pathology by Alexei Verkhratsky and Arthur M. Butt. Acta Physiol. 2023, 239, e14033. [Google Scholar] [CrossRef]
- Herculano-Houzel, S. The glia/neuron ratio: How it varies uniformly across brain structures and species and what that Means for brain physiology and evolution. Glia 2014, 62, 1377–1391. [Google Scholar] [CrossRef]
- Zhang, Y.; Barres, B.A. Astrocyte heterogeneity: An underappreciated topic in neurobiology. Curr. Opin. Neurobiol. 2010, 20, 588–594. [Google Scholar] [CrossRef] [PubMed]
- Escartin, C.; Bonvento, G. Targeted Activation of Astrocytes: A Potential Neuroprotective Strategy. Mol. Neurobiol. 2008, 38, 231–241. [Google Scholar] [CrossRef] [PubMed]
- Verkhratsky, A.; Butt, A.; Li, B.; Illes, P.; Zorec, R.; Semyanov, A.; Tang, Y.; Sofroniew, M.V. Astrocytes in human central nervous system diseases: A frontier for new therapies. Signal Transduct. Target. Ther. 2023, 8, 396. [Google Scholar] [CrossRef]
- Bayraktar, O.A.; Fuentealba, L.C.; Alvarez-Buylla, A.; Rowitch, D.H. Astrocyte Development and Heterogeneity. Cold Spring Harb. Perspect. Biol. 2015, 7, a020362. [Google Scholar] [CrossRef]
- Yoon, H.; Walters, G.; Paulsen, A.R.; Scarisbrick, I.A. Astrocyte Heterogeneity across the brain and spinal cord occurs developmentally, in adulthood and in response to demyelination. PLoS ONE 2017, 12, e0180697. [Google Scholar] [CrossRef]
- Yang, R.; Yang, B.; Liu, W.; Tan, C.; Chen, H.; Wang, X. Emerging role of non-coding RNAs in neuroinflammation mediated by microglia and astrocytes. J. Neuroinflammation 2023, 20, 173. [Google Scholar] [CrossRef]
- Abd-El-Basset, E.M.; Rao, M.S.; Alshawaf, S.M.; Ashkanani, H.K.; Kabli, A.H. Tumor Necrosis Factor (TNF) induces astrogliosis, microgliosis and promotes survival of cortical neurons. AIMS Neurosci. 2021, 8, 558–584. [Google Scholar] [CrossRef]
- Szelényi, J. Cytokines and the central nervous system. Brain Res. Bull. 2001, 54, 329–338. [Google Scholar] [CrossRef]
- Williamson, M.R.; Deneen, B. Astrocytes remember inflammation. Immunity 2024, 57, 938–940. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.H.; Cha, M.; Lee, B.H. Neuroprotective Effect of Antioxidants in the Brain. Int. J. Mol. Sci. 2020, 21, 7152. [Google Scholar] [CrossRef] [PubMed]
- Sharma, M.; Pal, P.; Gupta, S.K.; Potdar, M.B.; Belgamwar, A.V. Microglial-mediated immune mechanisms in autoimmune uveitis: Elucidating pathogenic pathways and targeted therapeutics. J. Neuroimmunol. 2024, 395, 578433. [Google Scholar] [CrossRef] [PubMed]
- Giovannoni, F.; Quintana, F.J. The Role of Astrocytes in CNS Inflammation. Trends Immunol. 2020, 41, 805–819. [Google Scholar] [CrossRef]
- Wang, X.; Hu, J.; Xie, S.; Li, W.; Zhang, H.; Huang, L.; Qian, Z.; Zhao, C.; Zhang, L. Hidden role of microglia during neurodegenerative disorders and neurocritical Care: A mitochondrial perspective. Int. Immunopharmacol. 2024, 142, 113024. [Google Scholar] [CrossRef]
- Mondal, H.; Mondal, S. Microglia and Neuroinflammation. In Physiology and Function of Glial Cells in Health and Disease; IGI Global Scientific Publishing: Hershey, PA, USA, 2024; pp. 100–119. [Google Scholar] [CrossRef]
- Durán Laforet, V.; Schafer, D.P. Microglia: Activity-dependent regulators of neural circuits. Ann. N. Y. Acad. Sci. 2024, 1533, 38–50. [Google Scholar] [CrossRef]
- Kumari, S.; Dhapola, R.; Sharma, P.; Nagar, P.; Medhi, B.; HariKrishnaReddy, D. The impact of cytokines in neuroinflammation-mediated stroke. Cytokine Growth Factor Rev. 2024, 78, 105–119. [Google Scholar] [CrossRef]
- Allaman, I.; Bélanger, M.; Magistretti, P.J. Astrocyte–neuron metabolic relationships: For better and for worse. Trends Neurosci. 2011, 34, 76–87. [Google Scholar] [CrossRef]
- Cervenka, I.; Agudelo, L.Z.; Ruas, J.L. Kynurenines: Tryptophan’s metabolites in exercise, inflammation, and mental health. Science (1979) 2017, 357, eaaf9794. [Google Scholar] [CrossRef]
- Iwaniak, P.; Owe-Larsson, M.; Urbańska, E.M. Microbiota, Tryptophan, and Aryl Hydrocarbon Receptors as the Target Triad in Parkinson’s Disease—A Narrative Review. Int. J. Mol. Sci. 2024, 25, 2915. [Google Scholar] [CrossRef] [PubMed]
- Badawy, A.A.B. Kynurenine Pathway of Tryptophan Metabolism: Regulatory and Functional Aspects. Int. J. Tryptophan Res. 2017, 10, 1178646917691938. [Google Scholar] [CrossRef] [PubMed]
- Badawy, A.A.B.; Guillemin, G. The Plasma [Kynurenine]/[Tryptophan] Ratio and Indoleamine 2,3-Dioxygenase: Time for Appraisal. Int. J. Tryptophan Res. 2019, 12, 1178646919868978. [Google Scholar] [CrossRef] [PubMed]
- Pathak, S.; Nadar, R.; Kim, S.; Liu, K.; Govindarajulu, M.; Cook, P.; Alexander, C.S.W.; Dhanasekaran, M.; Moore, T. The Influence of Kynurenine Metabolites on Neurodegenerative Pathologies. Int. J. Mol. Sci. 2024, 25, 853. [Google Scholar] [CrossRef]
- Fiore, A.; Murray, P.J. Tryptophan and indole metabolism in immune regulation. Curr. Opin. Immunol. 2021, 70, 7–14. [Google Scholar] [CrossRef]
- Lashgari, N.A.; Roudsari, N.M.; Shayan, M.; Niazi Shahraki, F.; Hosseini, Y.; Momtaz, S.; Abdolghaffari, A.H. IDO/Kynurenine: Novel insight for treatment of inflammatory diseases. Cytokine 2023, 166, 156206. [Google Scholar] [CrossRef]
- Guillemin, G.J. Quinolinic acid, the inescapable neurotoxin. FEBS J. 2012, 279, 1356–1365. [Google Scholar] [CrossRef]
- Batabyal, D.; Yeh, S.R. Human Tryptophan Dioxygenase: A Comparison to Indoleamine 2,3-Dioxygenase. J. Am. Chem. Soc. 2007, 129, 15690–15701. [Google Scholar] [CrossRef]
- Heyes, M.P.; Saito, K.; Markey, S.P. Human macrophages convert l-tryptophan into the neurotoxin quinolinic acid. Biochem. J. 1992, 283, 633–635. [Google Scholar] [CrossRef]
- Rossi, F.; Miggiano, R.; Ferraris, D.M.; Rizzi, M. The Synthesis of Kynurenic Acid in Mammals: An Updated Kynurenine Aminotransferase Structural KATalogue. Front. Mol. Biosci. 2019, 6, 7. [Google Scholar] [CrossRef] [PubMed]
- Yu, P.; Li, Z.; Zhang, L.; Tagle, D.A.; Cai, T. Characterization of kynurenine aminotransferase III, a novel member of a phylogenetically conserved KAT family. Gene 2006, 365, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Han, Q.; Robinson, H.; Cai, T.; Tagle, D.A.; Li, J. Biochemical and Structural Properties of Mouse Kynurenine Aminotransferase III. Mol. Cell. Biol. 2009, 29, 784–793. [Google Scholar] [CrossRef]
- Yu, P.; Di Prospero, N.A.; Sapko, M.T.; Cai, T.; Chen, A.; Melendez-Ferro, M.; Du, F.; Whetsell, W.O.; Guidetti, P.; Schwarcz, R.; et al. Biochemical and Phenotypic Abnormalities in Kynurenine Aminotransferase II-Deficient Mice. Mol. Cell. Biol. 2004, 24, 6919–6930. [Google Scholar] [CrossRef]
- Liang, J.; Cheng, Z.Y.; Shan, F.; Cao, Y.; Xia, Q.R. Serum indoleamine 2, 3-dioxygenase and tryptophan-2, 3-dioxygenase: Potential biomarkers for the diagnosis of major depressive disorder. Psychopharmacology 2024, 241, 1093–1099. [Google Scholar] [CrossRef] [PubMed]
- Guillemin, G.J.; Kerr, S.J.; Smythe, G.A.; Smith, D.G.; Kapoor, V.; Armati, P.J.; Croitoru, J.; Brew, B.J. Kynurenine pathway metabolism in human astrocytes: A paradox for neuronal protection. J. Neurochem. 2001, 78, 842–853. [Google Scholar] [CrossRef]
- Sahm, F.; Oezen, I.; Opitz, C.A.; Radlwimmer, B.; von Deimling, A.; Ahrendt, T.; Adams, S.; Bode, H.B.; Guillemin, G.J.; Wick, W.; et al. The Endogenous Tryptophan Metabolite and NAD+ Precursor Quinolinic Acid Confers Resistance of Gliomas to Oxidative Stress. Cancer Res. 2013, 73, 3225–3234. [Google Scholar] [CrossRef]
- Müller, F. Flavin-dependent hydroxylases. Biochem. Soc. Trans. 1985, 13, 443–447. [Google Scholar] [CrossRef]
- Pabarcus, M.K.; Casida, J.E. Cloning, expression, and catalytic triad of recombinant arylformamidase. Protein Expr. Purif. 2005, 44, 39–44. [Google Scholar] [CrossRef]
- Kim, J.; Porciuncula, F.; Yang, H.D.; Wendel, N.; Baker, T.; Chin, A.; Ellis, T.D.; Walsh, C.J. Soft robotic apparel to avert freezing of gait in parkinson’s disease. Nat. Med. 2024, 30, 177–185. [Google Scholar] [CrossRef]
- Guillemin, G.J.; Meininger, V.; Brew, B.J. Implications for the Kynurenine Pathway and Quinolinic Acid in Amyotrophic Lateral Sclerosis. Neurodegener. Dis. 2005, 2, 166–176. [Google Scholar] [CrossRef] [PubMed]
- Du, F.; Okuno, E.; Whetsell, W.O.; Köhler, C.; Schwarcz, R. Immunohistochemical localization of quinolinic acid phosphoribosyltransferase in the human neostriatum. Neuroscience 1991, 42, 397–406. [Google Scholar] [CrossRef]
- Wejksza, K.; Rzeski, W.; Okuno, E.; Kandefer-Szerszen, M.; Albrecht, J.; Turski, W.A. Demonstration of Kynurenine Aminotransferases I and II and Characterization of Kynurenic Acid Synthesis in Oligodendrocyte Cell Line (OLN-93). Neurochem. Res. 2005, 30, 963–968. [Google Scholar] [CrossRef] [PubMed]
- Mor, A.; Tankiewicz-Kwedlo, A.; Krupa, A.; Pawlak, D. Role of Kynurenine Pathway in Oxidative Stress during Neurodegenerative Disorders. Cells 2021, 10, 1603. [Google Scholar] [CrossRef]
- Lovelace, M.D.; Varney, B.; Sundaram, G.; Lennon, M.J.; Lim, C.K.; Jacobs, K.; Guillemin, G.J.; Brew, B.J. Recent evidence for an expanded role of the kynurenine pathway of tryptophan metabolism in neurological diseases. Neuropharmacology 2017, 112, 373–388. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, J.M.; Schardien, K.; Wigdahl, B.; Nonnemacher, M.R. roles of neuropathology-associated reactive astrocytes: A systematic review. Acta Neuropathol. Commun. 2023, 11, 42. [Google Scholar] [CrossRef]
- Okuda, S.; Nishiyama, N.; Saito, H.; Katsuki, H. 3-Hydroxykynurenine, an Endogenous Oxidative Stress Generator, Causes Neuronal Cell Death with Apoptotic Features and Region Selectivity. J. Neurochem. 1998, 70, 299–307. [Google Scholar] [CrossRef]
- Colín-González, A.L.; Maldonado, P.D.; Santamaría, A. 3-Hydroxykynurenine: An intriguing molecule exerting dual actions in the Central Nervous System. Neurotoxicology 2013, 34, 189–204. [Google Scholar] [CrossRef]
- Kadi, L.; Selvaraju, R.; de Lys, P.; Proudfoot, A.E.I.; Wells, T.N.C.; Boschert, U. Differential Effects of chemokines on oligodendrocyte precursor proliferation and myelin formation in vitro. J. Neuroimmunol. 2006, 174, 133–146. [Google Scholar] [CrossRef]
- Jia, L.; Tian, P.; Ding, C. Immunoregulatory effects of indoleamine 2,3-dioxygenase in transplantation. Transpl. Immunol. 2009, 21, 18–22. [Google Scholar] [CrossRef]
- Cheong, J.E.; Sun, L. Targeting the IDO1/TDO2–KYN–AhR Pathway for Cancer Immunotherapy—Challenges and Opportunities. Trends Pharmacol. Sci. 2018, 39, 307–325. [Google Scholar] [CrossRef] [PubMed]
- Anu, R.I.; Shiu, K.K.; Khan, K.H. The immunomodulatory role of IDO1-Kynurenine-NAD+ pathway in switching cold tumor microenvironment in PDAC. Front. Oncol. 2023, 13, 1142838. [Google Scholar] [CrossRef]
- Thomas, S.R.; Stocker, R. Redox reactions related to indoleamine 2,3-dioxygenase and tryptophan metabolism along the kynurenine pathway. Redox Rep. 1999, 4, 199–220. [Google Scholar] [CrossRef]
- Argolo, D.S.; Borges, J.M.P.; Freitas, L.D.S.; Pina, G.A.; Grangeiro, M.S.; da Silva, V.D.A.; Pinheiro, A.M.; Conceição, R.S.; Branco, A.; Guillemin, G.; et al. Activation of the Kynurenine Pathway and Production of Inflammatory Cytokines by Astrocytes and Microglia Infected with Neospora caninum. Int. J. Tryptophan Res. 2022, 15, 117864692110699. [Google Scholar] [CrossRef]
- Del’Arco, A.E.; Argolo, D.S.; Guillemin, G.; Costa, M.D.F.; Costa, S.L.; Pinheiro, A.M. Neurological Infection, Kynurenine Pathway, and Parasitic Infection by Neospora caninum. Front. Immunol. 2022, 12, 714248. [Google Scholar] [CrossRef] [PubMed]
- Munn, D.H.; Zhou, M.; Attwood, J.T.; Bondarev, I.; Conway, S.J.; Marshall, B.; Brown, C.; Mellor, A.L. Prevention of Allogeneic Fetal Rejection by Tryptophan Catabolism. Science (1979) 1998, 281, 1191–1193. [Google Scholar] [CrossRef]
- Sonner, J.K.; Deumelandt, K.; Ott, M.; Thomé, C.M.; Rauschenbach, K.J.; Schulz, S.; Munteanu, B.; Mohapatra, S.; Adam, I.; Hofer, A.-C.; et al. The stress kinase GCN2 does not mediate suppression of antitumor T cell responses by tryptophan catabolism in experimental melanomas. Oncoimmunology 2016, 5, e1240858. [Google Scholar] [CrossRef]
- Dantzer, R.; O’Connor, J.C.; Lawson, M.A.; Kelley, K.W. Inflammation-associated depression: From Serotonin to kynurenine. Psychoneuroendocrinology 2011, 36, 426–436. [Google Scholar] [CrossRef] [PubMed]
- Aaldijk, E.; Vermeiren, Y. The role of serotonin within the microbiota-gut-brain axis in the development of Alzheimer’s disease: A narrative review. Ageing Res. Rev. 2022, 75, 101556. [Google Scholar] [CrossRef]
- Meier, T.B.; Drevets, W.C.; Teague, T.K.; Wurfel, B.E.; Mueller, S.C.; Bodurka, J.; Dantzer, R.; Savitz, J. Kynurenic acid is reduced in females and oral contraceptive users: Implications for depression. Brain Behav. Immun. 2018, 67, 59–64. [Google Scholar] [CrossRef]
- Widner, B.; Werner, E.R.; Schennach, H.; Wachter, H.; Fuchs, D. Simultaneous Measurement of Serum Tryptophan and Kynurenine by HPLC. Clin. Chem. 1997, 43, 2424–2426. [Google Scholar] [CrossRef]
- Reininghaus, E.Z.; Dalkner, N.; Riedrich, K.; Fuchs, D.; Gostner, J.M.; Reininghaus, B. Sex-Specific Changes in Tryptophan Breakdown Over a 6 Week Treatment Period. Front. Psychiatry 2019, 10, 107. [Google Scholar] [CrossRef] [PubMed]
- Seok, S.H.; Ma, Z.X.; Feltenberger, J.B.; Chen, H.; Chen, H.; Scarlett, C.; Lin, Z.; Satyshur, K.A.; Cortopassi, M.; Jefcoate, C.R.; et al. Trace derivatives of kynurenine potently activate the aryl hydrocarbon receptor (AHR). J. Biol. Chem. 2018, 293, 1994–2005. [Google Scholar] [CrossRef]
- Schwarcz, R.; Stone, T.W. The kynurenine pathway and the brain: Challenges, controversies and promises. Neuropharmacology 2017, 112, 237–247. [Google Scholar] [CrossRef] [PubMed]
- Guillemin, G.J.; Smythe, G.; Takikawa, O.; Brew, B.J. Expression of indoleamine 2,3-dioxygenase and production of quinolinic acid by human microglia, astrocytes, and neurons. Glia 2005, 49, 15–23. [Google Scholar] [CrossRef]
- Moroni, F.; Russi, P.; Carlá, V.; Lombardi, G. Kynurenic acid is present in the rat brain and its content increases during development and aging processes. Neurosci. Lett. 1988, 94, 145–150. [Google Scholar] [CrossRef]
- Turski, W.A.; Nakamura, M.; Todd, W.P.; Carpenter, B.K.; Whetsell, W.O.; Schwarcz, R. Identification and quantification of kynurenic acid in human brain tissue. Brain Res. 1988, 454, 164–169. [Google Scholar] [CrossRef] [PubMed]
- Bacciottini, L.; Pellegrini-Giampietro, D.E.; Bongianni, F.; de Luca, G.; Politi, V.; Moroni, F. Biochemical and behavioural studies on indole-pyruvic acid: A keto-analogue of tryptophan. Pharmacol. Res. Commun. 1987, 19, 803–817. [Google Scholar] [CrossRef]
- Zwilling, D.; Huang, S.Y.; Sathyasaikumar, K.V.; Notarangelo, F.M.; Guidetti, P.; Wu, H.Q.; Lee, J.; Truong, J.; Andrews-Zwilling, Y.; Hsieh, E.W.; et al. Kynurenine 3-Monooxygenase Inhibition in Blood Ameliorates Neurodegeneration. Cell 2011, 145, 863–874. [Google Scholar] [CrossRef]
- Lim, C.K.; Bilgin, A.; Lovejoy, D.B.; Tan, V.; Bustamante, S.; Taylor, B.V.; Bessede, A.; Brew, B.J.; Guillemin, G.J. Kynurenine pathway metabolomics predicts and provides mechanistic insight into multiple sclerosis progression. Sci. Rep. 2017, 7, 41473. [Google Scholar] [CrossRef]
- Stone, T.W.; Stoy, N.; Darlington, L.G. An expanding range of targets for kynurenine metabolites of tryptophan. Trends Pharmacol. Sci. 2013, 34, 136–143. [Google Scholar] [CrossRef]
- Bosch, J.; Roca, T.; Armengol, M.; Fernández-Forner, D. Synthesis of 5-(sulfamoylmethyl)indoles. Tetrahedron 2001, 57, 1041–1048. [Google Scholar] [CrossRef]
- Hetherington-Rauth, M.; Johnson, E.; Migliavacca, E.; Parimi, N.; Langsetmo, L.; Hepple, R.T.; Grzywinski, Y.; Corthesy, J.; Ryan, T.E.; Ferrucci, L.; et al. Nutrient Metabolites Associated with Low D3Cr Muscle Mass, Strength, and Physical Performance in Older Men. J. Gerontol. A Biol. Sci. Med. Sci. 2024, 79, glad217. [Google Scholar] [CrossRef] [PubMed]
- Pradhan, R.; Dieterich, W.; Natarajan, A.; Schwappacher, R.; Reljic, D.; Herrmann, H.J.; Neurath, M.F.; Zopf, Y. Influence of Amino Acids and Exercise on Muscle Protein Turnover, Particularly in Cancer Cachexia. Cancers 2024, 16, 1921. [Google Scholar] [CrossRef] [PubMed]
- Grishanova, A.Y.; Perepechaeva, M.L. Kynurenic Acid/AhR Signaling at the Junction of Inflammation and Cardiovascular Diseases. Int. J. Mol. Sci. 2024, 25, 6933. [Google Scholar] [CrossRef] [PubMed]
- Del Tredici, K.; Braak, H. A not entirely benign procedure: Progression of parkinson’s disease. Acta Neuropathol. 2008, 115, 379–384. [Google Scholar] [CrossRef] [PubMed]
- Fujigaki, H.; Yamamoto, Y.; Saito, K. L-Tryptophan-kynurenine pathway enzymes are therapeutic targets for neuropsychiatric diseases: Focus on cell type differences. Neuropharmacology 2017, 112, 264–274. [Google Scholar] [CrossRef]
- Fujigaki, H.; Mouri, A.; Yamamoto, Y.; Nabeshima, T.; Saito, K. Linking phencyclidine intoxication to the tryptophan-kynurenine pathway: Therapeutic implications for schizophrenia. Neurochem. Int. 2019, 125, 94–105. [Google Scholar] [CrossRef]
- De-Paula, V.J.; Radanovic, M.; Diniz, B.S.; Forlenza, O.V. Alzheimer’s disease. Sub-Cell. Biochem. 2012, 65, 329–352. [Google Scholar] [CrossRef]
- Penke, B.; Bogár, F.; Fülöp, L. β-Amyloid and the Pathomechanisms of Alzheimer’s Disease: A Comprehensive View. Molecules 2017, 22, 1692. [Google Scholar] [CrossRef]
- Tomita, S.; Kirino, Y.; Suzuki, T. Cleavage of Alzheimer’s Amyloid Precursor Protein (APP) by Secretases Occurs after O-Glycosylation of APP in the Protein Secretory Pathway. J. Biol. Chem. 1998, 273, 6277–6284. [Google Scholar] [CrossRef]
- Deng, J.; Liu, B.; Tao, Q.; Luo, Y.; Zhu, Y.; Huang, X.; Yue, F. The Co-Oligomers of Aβ42 and Human Islet Amyloid Polypeptide Exacerbate Neurotoxicity and Alzheimer-Like Pathology at Cellular Level. Neuroscience 2024, 547, 37–55. [Google Scholar] [CrossRef]
- Meng, X.; Song, Q.; Liu, Z.; Liu, X.; Wang, Y.; Liu, J. Neurotoxic β-amyloid oligomers cause mitochondrial dysfunction—The trigger for PANoptosis in neurons. Front. Aging Neurosci. 2024, 16, 1400544. [Google Scholar] [CrossRef] [PubMed]
- Sorgdrager, F.J.H.; Vermeiren, Y.; Van Faassen, M.; van der Ley, C.; Nollen, E.A.A.; Kema, I.P.; De Deyn, P.P. Age- and disease-specific changes of the kynurenine pathway in parkinson’s and alzheimer’s disease. J. Neurochem. 2019, 151, 656–668. [Google Scholar] [CrossRef] [PubMed]
- Dong, F.; Perdew, G.H. The aryl hydrocarbon receptor as a mediator of host-microbiota interplay. Gut Microbes 2020, 12, 1859812. [Google Scholar] [CrossRef]
- Verma, A.; Waiker, D.K.; Singh, N.; Roy, A.; Singh, N.; Saraf, P.; Bhardwaj, B.; Krishnamurthy, S.; Trigun, S.K.; Shrivastava, S.K. Design, Synthesis, and Biological Investigation of Quinazoline Derivatives as Multitargeting Therapeutics in Alzheimer’s Disease Therapy. ACS Chem. Neurosci. 2024, 15, 745–771. [Google Scholar] [CrossRef]
- Rascovsky, K.; Grossman, M. Clinical diagnostic criteria and classification controversies in frontotemporal lobar degeneration. Int. Rev. Psychiatry 2013, 25, 145–158. [Google Scholar] [CrossRef] [PubMed]
- Rascovsky, K.; Hodges, J.R.; Knopman, D.; Mendez, M.F.; Kramer, J.H.; Neuhaus, J.; van Swieten, J.C.; Seelaar, H.; Dopper, E.G.; Onyike, C.U.; et al. Sensitivity of revised diagnostic criteria for the behavioural variant of frontotemporal dementia. Brain 2011, 134, 2456–2477. [Google Scholar] [CrossRef]
- The Lancet. What next in parkinson’s disease? Lancet 2024, 403, 219. [Google Scholar] [CrossRef]
- Goldman, J.G.; Postuma, R. Premotor and nonmotor features of Parkinson’s disease. Curr. Opin. Neurol. 2014, 27, 434–441. [Google Scholar] [CrossRef]
- Deng, H.; Wang, P.; Jankovic, J. The genetics of parkinson disease. Ageing Res. Rev. 2018, 42, 72–85. [Google Scholar] [CrossRef]
- Venkatesan, D.; Iyer, M.; Narayanasamy, A.; Siva, K.; Vellingiri, B. Kynurenine pathway in Parkinson’s disease—An update. eNeurologicalSci 2020, 21, 100270. [Google Scholar] [CrossRef]
- Jacobs, K.R.; Lim, C.K.; Blennow, K.; Zetterberg, H.; Chatterjee, P.; Martins, R.N.; Brew, B.J.; Guillemin, G.J.; Lovejoy, D.B. Correlation between plasma and CSF concentrations of kynurenine pathway metabolites in Alzheimer’s disease and relationship to amyloid-β and Tau. Neurobiol. Aging 2019, 80, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Neary, D.; Snowden, J.S.; Gustafson, L.; Passant, U.; Stuss, D.; Black, S.; Freedman, M.; Kertesz, A.; Robert, P.H.; Albert, M.; et al. Frontotemporal lobar degeneration. Neurology 1998, 51, 1546–1554. [Google Scholar] [CrossRef]
- Corcia, P.; Beltran, S.; Bakkouche, S.E.; Couratier, P. Therapeutic news in ALS. Rev. Neurol. 2021, 177, 544–549. [Google Scholar] [CrossRef] [PubMed]
- Ludolph, A.C.; Riepe, M.; Ullrich, K. Excitotoxicity, Energy Metabolism, and Neurodegeneration. Meldrum Garthwaite Excit. Neurodegener. 1993, 16, 123–144. [Google Scholar] [CrossRef] [PubMed]
- Salameh, J.; Brown, R.; Berry, J. Amyotrophic Lateral Sclerosis: Review. Semin. Neurol. 2015, 35, 469–476. [Google Scholar] [CrossRef]
- Ludolph, A.; Drory, V.; Hardiman, O.; Nakano, I.; Ravits, J.; Robberecht, W.; Shefner, J.; The WFN Research Group On ALS/MND. A revision of the El Escorial criteria—2015. Amyotroph. Lateral Scler. Frontotemporal Degener. 2015, 16, 291–292. [Google Scholar] [CrossRef]
- Heylen, A.; Vermeiren, Y.; Kema, I.P.; van Faassen, M.; van der Ley, C.; Van Dam, D.; De Deyn, P.P. Brain Kynurenine Pathway Metabolite Levels May Reflect Extent of Neuroinflammation in ALS, FTD and Early Onset AD. Pharmaceuticals 2023, 16, 615. [Google Scholar] [CrossRef]
- Fifita, J.A.; Chan Moi Fat, S.; McCann, E.P.; Williams, K.L.; Twine, N.A.; Bauer, D.C.; Rowe, D.B.; Pamphlett, R.; Kiernan, M.C.; Tan, V.X.; et al. Genetic Analysis of Tryptophan Metabolism Genes in Sporadic Amyotrophic Lateral Sclerosis. Front. Immunol. 2021, 12, 701550. [Google Scholar] [CrossRef]
- Pollari, E.; Goldsteins, G.; Bart, G.; Koistinaho, J.; Giniatullin, R. The role of oxidative stress in degeneration of the neuromuscular junction in amyotrophic lateral sclerosis. Front. Cell Neurosci. 2014, 8, 131. [Google Scholar] [CrossRef]
- Iłżecka, J.; Kocki, T.; Stelmasiak, Z.; Turski, W.A. Endogenous protectant kynurenic acid in amyotrophic lateral sclerosis. Acta Neurol. Scand. 2003, 107, 412–418. [Google Scholar] [CrossRef]
- Tanaka, M.; Toldi, J.; Vécsei, L. Exploring the Etiological Links Behind Neurodegenerative Diseases: Inflammatory Cytokines and Bioactive Kynurenines. Int. J. Mol. Sci. 2020, 21, 2431. [Google Scholar] [CrossRef] [PubMed]
- Ysrraelit, M.C.; Fiol, M.P.; Gaitán, M.I.; Correale, J. Quality of Life Assessment in Multiple Sclerosis: Different Perception between Patients and Neurologists. Front. Neurol. 2018, 8, 729. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.J.; Chen, W.W.; Zhang, X. Multiple sclerosis: Pathology, diagnosis, and treatments. Exp. Ther. Med. 2017, 13, 3163–3166. [Google Scholar] [CrossRef]
- Lassmann, H. Multiple Sclerosis Pathology. Cold Spring Harb. Perspect. Med. 2018, 8, a028936. [Google Scholar] [CrossRef] [PubMed]
- Gruchot, J.; Weyers, V.; Göttle, P.; Förster, M.; Hartung, H.P.; Küry, P.; Kremer, D. The Molecular Basis for Remyelination Failure in Multiple Sclerosis. Cells 2019, 8, 825. [Google Scholar] [CrossRef]
- Rosenkranz, S.C.; Shaposhnykov, A.A.; Träger, S.; Engler, J.B.; Witte, M.E.; Roth, V.; Vieira, V.; Paauw, N.; Bauer, S.; Schwencke-Westphal, C.; et al. Enhancing mitochondrial activity in neurons protects against neurodegeneration in a mouse model of multiple sclerosis. Elife 2021, 10, e61798. [Google Scholar] [CrossRef]
- Wang, Q.; Lu, M.; Zhu, X.; Gu, X.; Zhang, T.; Xia, C.; Yang, L.; Xu, Y.; Zhou, M. The role of microglia immunometabolism in neurodegeneration: Focus on molecular determinants and metabolic intermediates of metabolic reprogramming. Biomed. Pharmacother. 2022, 153, 113412. [Google Scholar] [CrossRef]
- Castro-Portuguez, R.; Sutphin, G.L. Kynurenine Pathway, NAD+ Synthesis, and mitochondrial function: Targeting tryptophan metabolism to promote longevity and healthspan. Exp. Gerontol. 2020, 132, 110841. [Google Scholar] [CrossRef]
- Ye, J.; Jiang, Z.; Chen, X.; Liu, M.; Li, J.; Liu, N. Electron transport chain inhibitors induce microglia activation through enhancing mitochondrial reactive oxygen species production. Exp. Cell Res. 2016, 340, 315–326. [Google Scholar] [CrossRef]
- Zarzecki, M.S.; Cattelan Souza, L.; Giacomeli, R.; Silva, M.R.P.; Prigol, M.; Boeira, S.P.; Jesse, C.R. Involvement of Indoleamine-2,3-Dioxygenase and Kynurenine Pathway in Experimental Autoimmune Encephalomyelitis in Mice. Neurochem. Res. 2020, 45, 2959–2977. [Google Scholar] [CrossRef]
- Heidker, R.; Emerson, M.; LeVine, S. Metabolic pathways as possible therapeutic targets for progressive multiple sclerosis. Neural Regen. Res. 2017, 12, 1262. [Google Scholar] [PubMed]
- Fathi, M.; Vakili, K.; Yaghoobpoor, S.; Tavasol, A.; Jazi, K.; Mohamadkhani, A.; Klegeris, A.; McElhinney, A.; Mafi, Z.; Hajiesmaeili, M.; et al. Dynamic changes in kynurenine pathway metabolites in multiple sclerosis: A systematic review. Front. Immunol. 2022, 13, 1013784. [Google Scholar] [CrossRef]
- Aeinehband, S.; Brenner, P.; Ståhl, S.; Bhat, M.; Fidock, M.D.; Khademi, M.; Olsson, T.; Engberg, G.; Jokinen, J.; Erhardt, S.; et al. Cerebrospinal fluid kynurenines in multiple sclerosis; relation to disease course and neurocognitive symptoms. Brain Behav. Immun. 2016, 51, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Mancuso, R.; Hernis, A.; Agostini, S.; Rovaris, M.; Caputo, D.; Fuchs, D.; Clerici, M. Indoleamine 2,3-Dioxygenase (IDO) Expression and Activity in Relapsing-Remitting Multiple Sclerosis. PLoS ONE 2015, 10, e0130715. [Google Scholar] [CrossRef] [PubMed]
- Lim, C.K.; Fernández-Gomez, F.J.; Braidy, N.; Estrada, C.; Costa, C.; Costa, S.; Bessede, A.; Fernandez-Villalba, E.; Zinger, A.; Herrero, M.T.; et al. Involvement of the kynurenine pathway in the pathogenesis of Parkinson’s disease. Prog. Neurobiol. 2017, 155, 76–95. [Google Scholar] [CrossRef]
- Isık, S.M.T.; Onmaz, D.E.; Ekmekci, A.H.; Ozturk, S.; Unlu, A.; Abusoglu, S. Relationship of tryptophan metabolites with the type and severity of multiple sclerosis. Mult. Scler. Relat. Disord. 2023, 77, 104898. [Google Scholar] [CrossRef]
- Polyák, H.; Galla, Z.; Nánási, N.; Cseh, E.K.; Rajda, C.; Veres, G.; Spekker, E.; Szabó, Á.; Klivényi, P.; Tanaka, M.; et al. The Tryptophan-Kynurenine Metabolic System Is Suppressed in Cuprizone-Induced Model of Demyelination Simulating Progressive Multiple Sclerosis. Biomedicines 2023, 11, 945. [Google Scholar] [CrossRef]
- Gaetani, L.; Boscaro, F.; Pieraccini, G.; Calabresi, P.; Romani, L.; Di Filippo, M.; Zelante, T. Host and Microbial Tryptophan Metabolic Profiling in Multiple Sclerosis. Front. Immunol. 2020, 11, 157. [Google Scholar] [CrossRef]
- Maddison, D.C.; Giorgini, F. The Kynurenine pathway and neurodegenerative disease. Semin. Cell Dev. Biol. 2015, 40, 134–141. [Google Scholar] [CrossRef]
- Chen, W.W.; Zhang, X.; Huang, W.J. Role of neuroinflammation in neurodegenerative diseases. Mol. Med. Rep. 2016, 13, 3391–3396. [Google Scholar] [CrossRef] [PubMed]
- Skorobogatov, K.; Autier, V.; Foiselle, M.; Richard, J.R.; Boukouaci, W.; Wu, C.L.; Raynal, S.; Carbonne, C.; Laukens, K.; Meysman, P.; et al. kynurenine pathway abnormalities are state-specific but not diagnosis-specific in schizophrenia and bipolar disorder. Brain Behav. Immun. Health 2023, 27, 100584. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.K.; Miller, B.J. Meta-analysis of Cerebrospinal Fluid Cytokine and Tryptophan Catabolite Alterations in Psychiatric Patients: Comparisons Between Schizophrenia, Bipolar Disorder, and Depression. Schizophr. Bull. 2018, 44, 75–83. [Google Scholar] [CrossRef]
- Salinas, J.; Beiser, A.; Himali, J.J.; Satizabal, C.L.; Aparicio, H.J.; Weinstein, G.; Mateen, F.J.; Berkman, L.F.; Rosand, J.; Seshadri, S. Associations between social relationship measures, serum brain-derived neurotrophic factor, and risk of stroke and dementia. Alzheimer’s Dement. Transl. Res. Clin. Interv. 2017, 3, 229–237. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, P.J.; Cryan, J.F.; Dinan, T.G.; Clarke, G. Kynurenine pathway metabolism and the microbiota-gut-brain axis. Neuropharmacology 2017, 112, 399–412. [Google Scholar] [CrossRef]
- Prendergast, G.C.; Malachowski, W.J.; Mondal, A.; Scherle, P.; Muller, A.J. Indoleamine 2,3-Dioxygenase and Its Therapeutic Inhibition in Cancer. In Advances in Cancer Research; Elsevier: Oxford, UK, 2018; pp. 175–203. [Google Scholar]
- Mellor, A.L.; Lemos, H.; Huang, L. Indoleamine 2,3-Dioxygenase and Tolerance: Where Are We Now? Front. Immunol. 2017, 8, 1360. [Google Scholar] [CrossRef]
- Opitz, C.A.; Litzenburger, U.M.; Sahm, F.; Ott, M.; Tritschler, I.; Trump, S.; Schumacher, T.; Jestaedt, L.; Schrenk, D.; Weller, M.; et al. An endogenous tumour-promoting ligand of the human aryl hydrocarbon receptor. Nature 2011, 478, 197–203. [Google Scholar] [CrossRef]
- Badawy, A.A.B.; Namboodiri, A.M.A.; Moffett, J.R. The end of the road for the tryptophan depletion concept in pregnancy and infection. Clin. Sci. 2016, 130, 1327–1333. [Google Scholar] [CrossRef]
- Miller, R.; Mitchell, J.; Lyon, M.; Moore, D. Riluzole for amyotrophic lateral sclerosis (ALS)/motor neuron disease (MND). In Cochrane Database of Systematic Reviews; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2002. [Google Scholar]
- Foster, A.C.; Vezzani, A.; French, E.D.; Schwarcz, R. Kynurenic acid blocks neurotoxicity and seizures induced in rats by the related brain metabolite quinolinic acid. Neurosci. Lett. 1984, 48, 273–278. [Google Scholar] [CrossRef]
- Hilmas, C.; Pereira, E.F.R.; Alkondon, M.; Rassoulpour, A.; Schwarcz, R.; Albuquerque, E.X. The Brain Metabolite Kynurenic Acid Inhibits α7 Nicotinic Receptor Activity and Increases Non-α7 Nicotinic Receptor Expression: Physiopathological Implications. J. Neurosci. 2001, 21, 7463–7473. [Google Scholar] [CrossRef] [PubMed]
- Guillemin, G.J.; Croitoru-Lamoury, J.; Dormont, D.; Armati, P.J.; Brew, B.J. quinolinic acid upregulates chemokine production and chemokine receptor expression in astrocytes. Glia 2003, 41, 371–381. [Google Scholar] [CrossRef]
- Ting, K.K.; Brew, B.J.; Guillemin, G.J. Effect of quinolinic acid on human astrocytes morphology and functions: Implications in Alzheimer’s disease. J. Neuroinflammation 2009, 6, 36. [Google Scholar] [CrossRef]
- Kaur, G.; Han, S.J.; Yang, I.; Crane, C. Microglia and Central Nervous System Immunity. Neurosurg. Clin. N. Am. 2010, 21, 43–51. [Google Scholar] [CrossRef]
- Müller, N.; Myint, A.M.; Schwarz, M.J. The impact of neuroimmune dysregulation on neuroprotection and neurotoxicity in psychiatric disorders—Relation to drug treatment. Dialogues Clin. Neurosci. 2009, 11, 319–332. [Google Scholar] [CrossRef] [PubMed]
- Giorgini, F.; Huang, S.Y.; Sathyasaikumar, K.V.; Notarangelo, F.M.; Thomas, M.A.R.; Tararina, M.; Wu, H.-Q.; Schwarcz, R.; Muchowski, P.J. Targeted Deletion of Kynurenine 3-Monooxygenase in Mice. J. Biol. Chem. 2013, 288, 36554–36566. [Google Scholar] [CrossRef] [PubMed]
- Giil, L.M.; Midttun, Ø.; Refsum, H.; Ulvik, A.; Advani, R.; Smith, A.D.; Ueland, P.M. Kynurenine Pathway Metabolites in Alzheimer’s Disease. J. Alzheimer’s Dis. 2017, 60, 495–504. [Google Scholar] [CrossRef]
- Bratek-Gerej, E.; Ziembowicz, A.; Godlewski, J.; Salinska, E. The Mechanism of the Neuroprotective Effect of Kynurenic Acid in the Experimental Model of Neonatal Hypoxia–Ischemia: The Link to Oxidative Stress. Antioxidants 2021, 10, 1775. [Google Scholar] [CrossRef]
- Whiley, L.; Chappell, K.E.; D’Hondt, E.; Lewis, M.R.; Jiménez, B.; Snowden, S.G.; Soininen, H.; Kłoszewska, I.; Mecocci, P.; AddNeuroMed Consortium; et al. Metabolic phenotyping reveals a reduction in the bioavailability of serotonin and kynurenine pathway metabolites in both the urine and serum of individuals living with Alzheimer’s disease. Alzheimers Res. Ther. 2021, 13, 20. [Google Scholar] [CrossRef]
- Knapskog, A.B.; Edwin, T.H.; Ueland, P.M.; Ulvik, A.; Fang, E.F.; Eldholm, R.S.; Halaas, N.B.; Giil, L.M.; Saltvedt, I.; Watne, L.O.; et al. Sex-specific associations of kynurenic acid with neopterin in Alzheimer’s disease. Alzheimers Res. Ther. 2024, 16, 167. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
| Year | Experimental Conditions | Contributions | Reference |
|---|---|---|---|
| 1984 | Action of KYNA on the metabolism of QUIN in the CNS of rats. | Injection of QUIN (240 nmol/kg) in rats reduced striatal KAT activity. KYNA in the hippocampus antagonized both the neurodegeneration and seizures caused by the local application of QUIN. | Froster et al. [133] |
| 1988 | Role of KYNA during development and aging processes in rats. | KYNA levels vary from 15 pmol/g protein in the first week of life (to 320 pmol/g protein at the third month and to 747 pmol/g protein in the 18th month); QUIN levels do not alter in the liver or kidney with age, but their concentration in blood increases from 28 pmol/mL at 3 months to 65 pmol/mL at 18 months. | Moroni et al. [67] |
| 1988 | Identification and quantification of KYNA in human brain tissue. | The distribution of KYNA in the CNS revealed the highest concentration (1.58 pmol/mg of tissue) in the caudate nucleus and lower levels in the thalamus, globus pallidus, hippocampus, parietal cortex, and frontal cortex, with the lowest concentration of KYNA (0.14 pmol/mg of tissue). | Turski et al. [68] |
| 1992 | Effect of INF-γ on QUIN concentration in human macrophage cultures. | INF-γ increased the QUIN concentration in the macrophage culture medium from 2.8 µM in 24 h to 11.6 µM in 48 h. | Heyes et al. [31] |
| 2001 | KP in culture of human astrocytic cells treated with KYNA and inflammatory agents. | KYNA (85.5 nm), IFN-γ and IFN-γ + TNF-α (100 IU/mL) induced, respectively, the production of 11.7, 10.3 and 0.9 mM of KYN. | Guillemin et al. [37] |
| 2001 | KYNA inhibits α7 nAChR in cultures of neurons from rats. | KYNA (100 nM) inhibits the somatodendritic activation of α7 nAChRs in hippocampal and cortical neurons. | Hilmas et al. [134] |
| 2003 | QUIN induces chemokine Production and receptor expression in human fetal astrocytes. | QUIN (350, 500, or 1200 nM) induces astrocytes to produce large amounts of CCL2, CCL5, IL-8, SDF-1, and CX3CL1, while also increasing the expression of the chemokine receptors CXCR4, CCR5, and CCR3. | Guillemin et al. [135] |
| 2003 | KYNA levels in the serum and CSF from ALS patients. | Serum KYNA was lower (39.9 pmol/mL) in ALS patients with severe clinical status compared with healthy controls (59.6 pmol/mL); KYNA concentration did not vary in CSF in patients and between males and females. | Iłzecka et al. [103] |
| 2005 | KYNA and KAT-1 and -2 in immature rat oligodendrocytes. | KAT isoforms I and II of permanent immature oligodendrocytes (OLN-93) synthesize KYNA from L-KYN (5 µM) added exogenously. | Wejksza et al. [44] |
| 2009 | QUIN and astrogliosis in AD. | Astrocytes isolated and treated with QUIN (50–1200 nM) modify morphology and increase the proliferation of structural proteins. | Ting et al. [136] |
| 2010 | Trp degradation and altered QUIN and KYNA levels in AD patients. | Patients with AD increase Trp degradation, with 22.09 mM elevating QUIN concentration to 334.0 nM and decreasing KYNA to 20.85 nM in plasma. | Kaur et al. [137] |
| 2011 | JM6 elevates brain and serum KYNA by inhibiting blood KMO. | Neuroprotective compound JM6 increases brain and serum KYNA by 180% and 344%, respectively, from basal levels (2.5 nM) by inhibiting blood KMO. | Zwilling et al. [70] |
| 2013 | Altered Trp metabolite levels in patients with psychiatric disorders. | In schizophrenia and depression, opposing patterns of type-7 vs. type-2 immune response seem to be associated with differences in the activation of the enzyme IDO and in the Trp metabolism, resulting in increased production of KYNA in schizophrenia and decreased production of kynurenic acid KYNA in depression. | Schwarz et al. [138] |
| 2013 | KMO gene deletion modulates KP metabolites. | Deletion in the gene for KMO increases KYN and KYNA by about 10%, and decreases QUIN by about 80% in several tissues. | Giorgini et al. [139] |
| 2015 | Serum IDO activity and KYN/Trp in ME. | Serum IDO activity and the KYN/Trp ratio increase in patients with acute disease (64 nM) and decrease in chronic disease (10 nM) relative to neopterin concentration. | Hernis et al. [117] |
| 2017 | KP in AD. | Levels of plasma Trp (57.5 µM), KYN (1.8 µM), HAA (28.7 nM), and QUIN (465 nM) were significantly lower in patients with AD compared to controls. | Giil et al. [140] |
| 2019 | The plasma KYN/Trp ratio and IDO activity. | IDO activity modifies the plasma [KYN]/[Trp] ratio, influencing the concentration of KYN, KMO, KYNU and, to a lesser extent, KAT. | Badawy and Guillemin. [25] |
| 2020 | IDO activity in AIE. | The IDO inhibitor (INCB024360) at 200 mg/kg orally reduced the mRNA expression of IDO, its enzymatic activity and KMO levels and improved clinical signs of AIE in mice. | Zarzecki et al. [113] |
| 2021 | Neuroprotective action against oxidative stress of KYNA | KYNA at concentrations of 300, 150, 50 mg/kg preserves tissue oxidative stress in rat. | Bratek-Gerej et al. [141] |
| 2021 | KP in AD. | In plasma and/or urine of patients with AD, there is a decrease in Trp KYN, XA, KYNA concentration and Kyn/Trp ratio. | Whiley et al. [142] |
| 2024 | Sex differences in CSF Trp and KYN levels in AD | In CSF, levels of Trp, KYN and QUIN are higher in men than in women with AD; KYNA/QUIN ratio is higher in AD women. | Knapskog et al. [143] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Argolo, D.S.; de Oliveira, L.M.G.; Guillemin, G.J.; Barreto, G.E.; Butt, A.M.; Costa, S.L.; Costa, M.d.F.D. Tryptophan Metabolism Through the Kynurenine Pathway in Glial Cells. Neuroglia 2025, 6, 14. https://doi.org/10.3390/neuroglia6010014
Argolo DS, de Oliveira LMG, Guillemin GJ, Barreto GE, Butt AM, Costa SL, Costa MdFD. Tryptophan Metabolism Through the Kynurenine Pathway in Glial Cells. Neuroglia. 2025; 6(1):14. https://doi.org/10.3390/neuroglia6010014
Chicago/Turabian StyleArgolo, Deivison Silva, Lucas Matheus Gonçalves de Oliveira, Gilles J. Guillemin, George E. Barreto, Arthur Morgan Butt, Silvia Lima Costa, and Maria de Fátima Dias Costa. 2025. "Tryptophan Metabolism Through the Kynurenine Pathway in Glial Cells" Neuroglia 6, no. 1: 14. https://doi.org/10.3390/neuroglia6010014
APA StyleArgolo, D. S., de Oliveira, L. M. G., Guillemin, G. J., Barreto, G. E., Butt, A. M., Costa, S. L., & Costa, M. d. F. D. (2025). Tryptophan Metabolism Through the Kynurenine Pathway in Glial Cells. Neuroglia, 6(1), 14. https://doi.org/10.3390/neuroglia6010014