Cooperation between NMDA-Type Glutamate and P2 Receptors for Neuroprotection during Stroke: Combining Astrocyte and Neuronal Protection

 ,
, 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Distinguishing Neurons and Astrocytes in Co-Culture
2.3. Cell Imaging (Viability and [Ca2+]i)
2.4. Biosensors
2.5. Statistics
3. Results
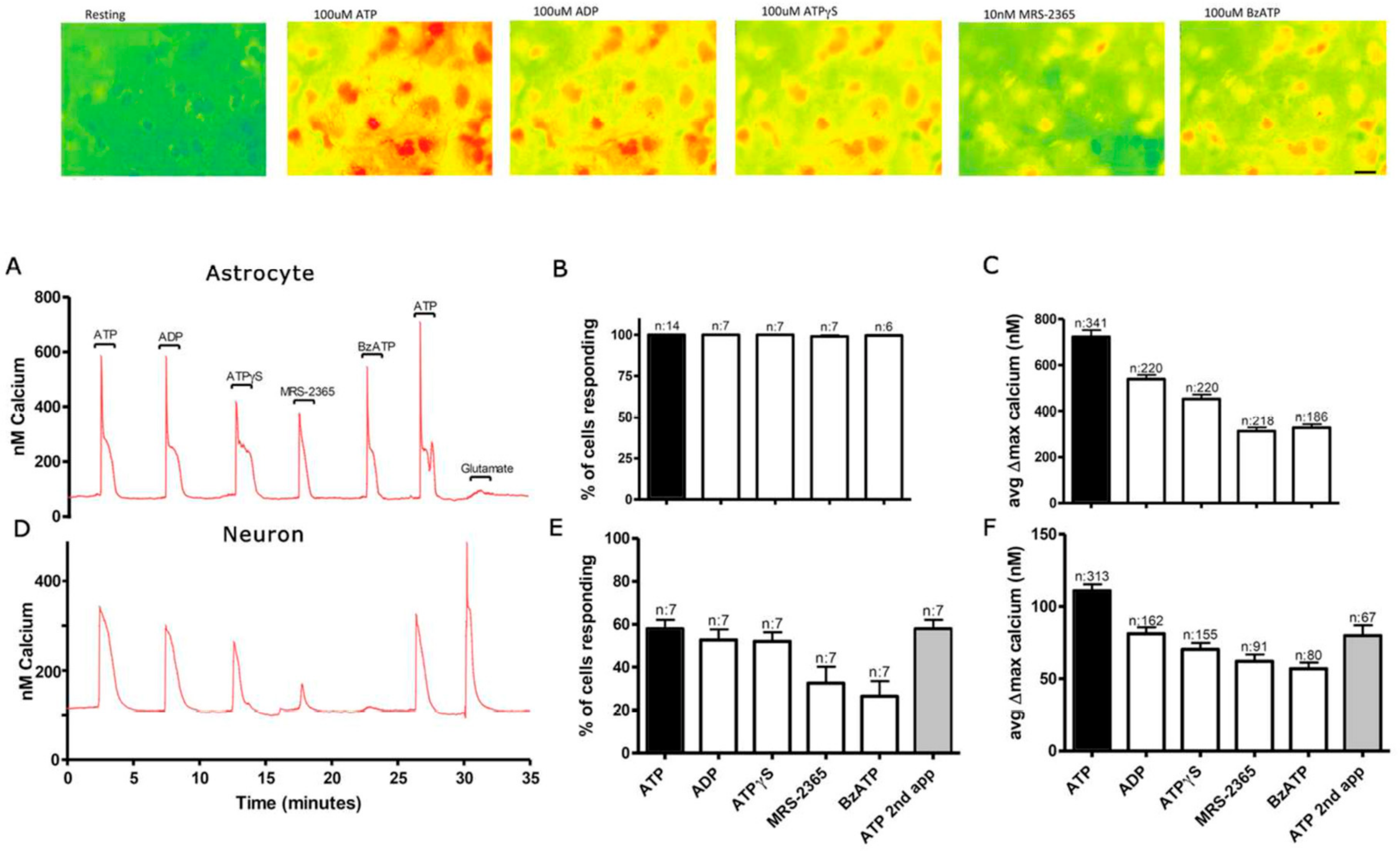
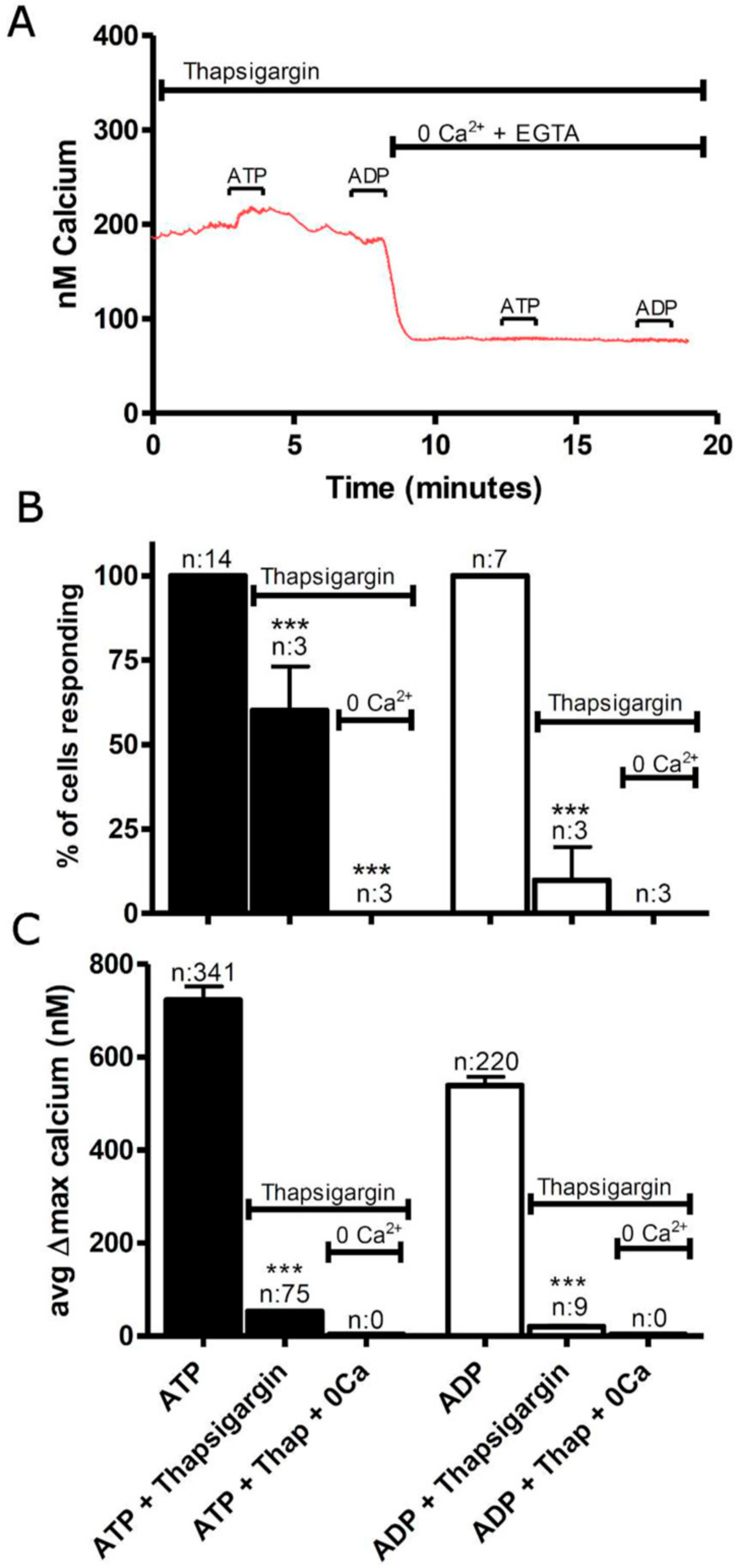
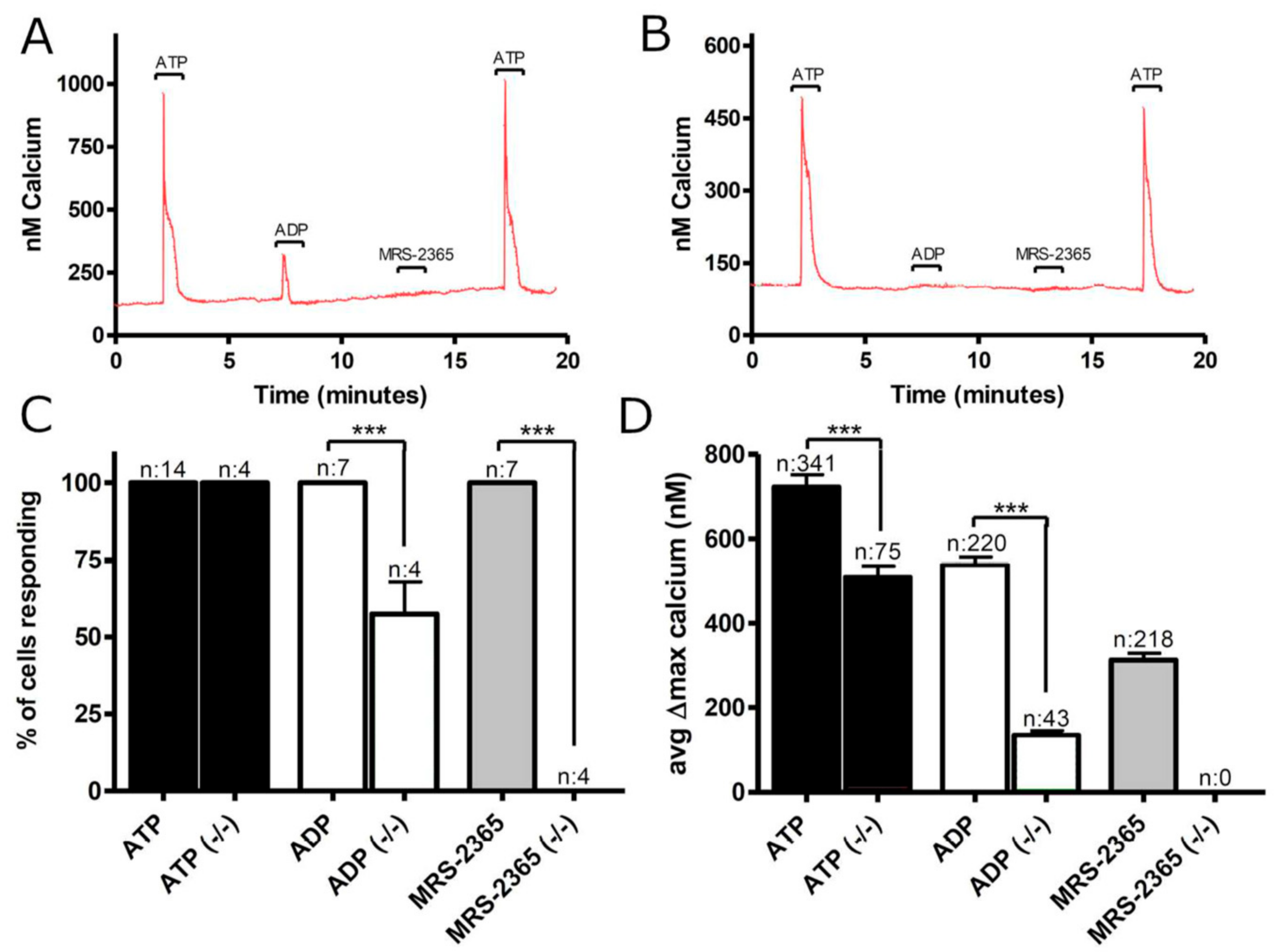
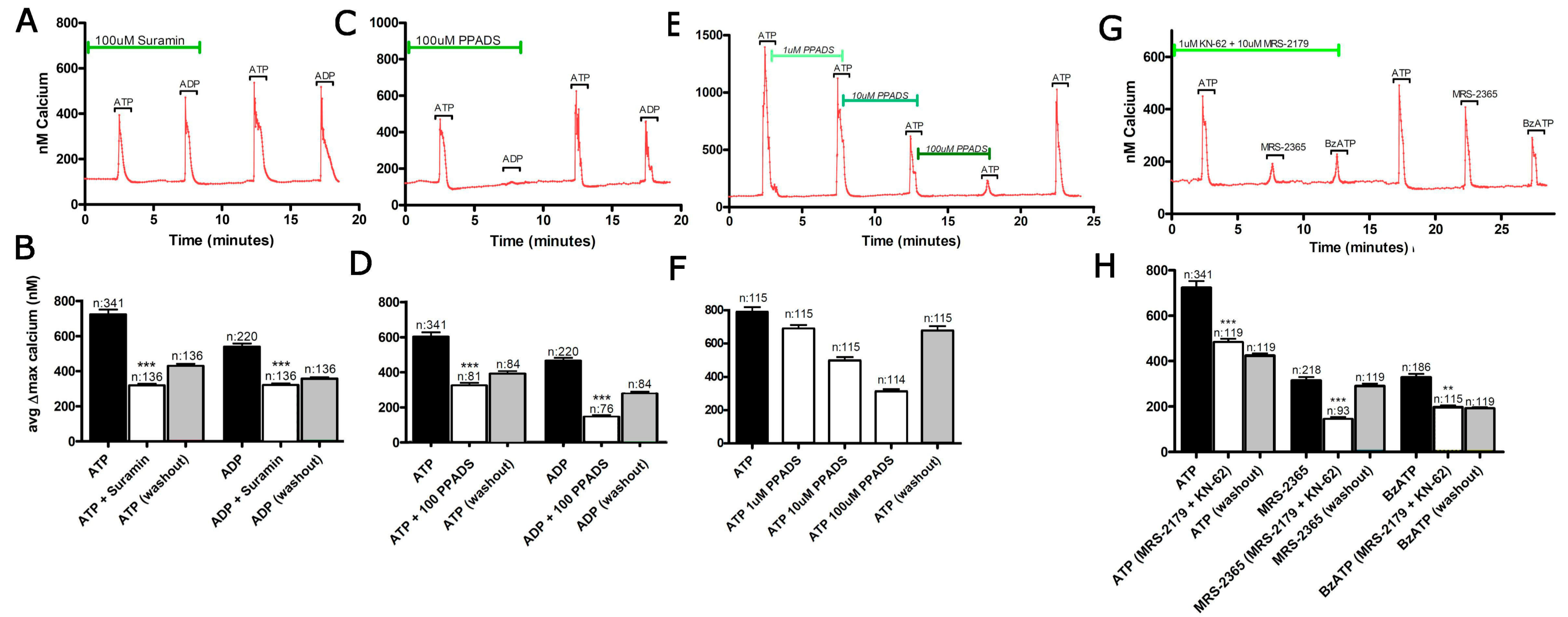
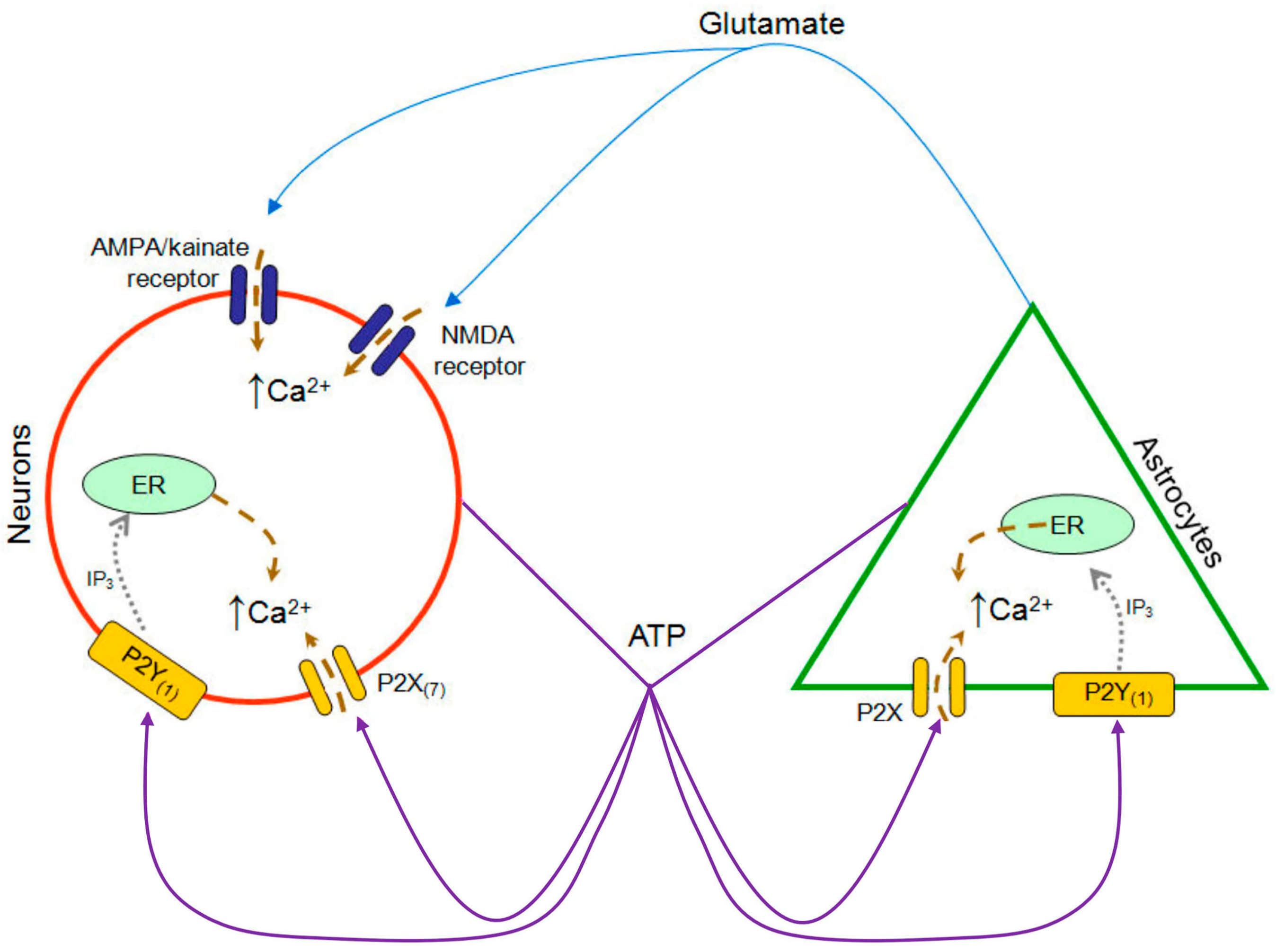
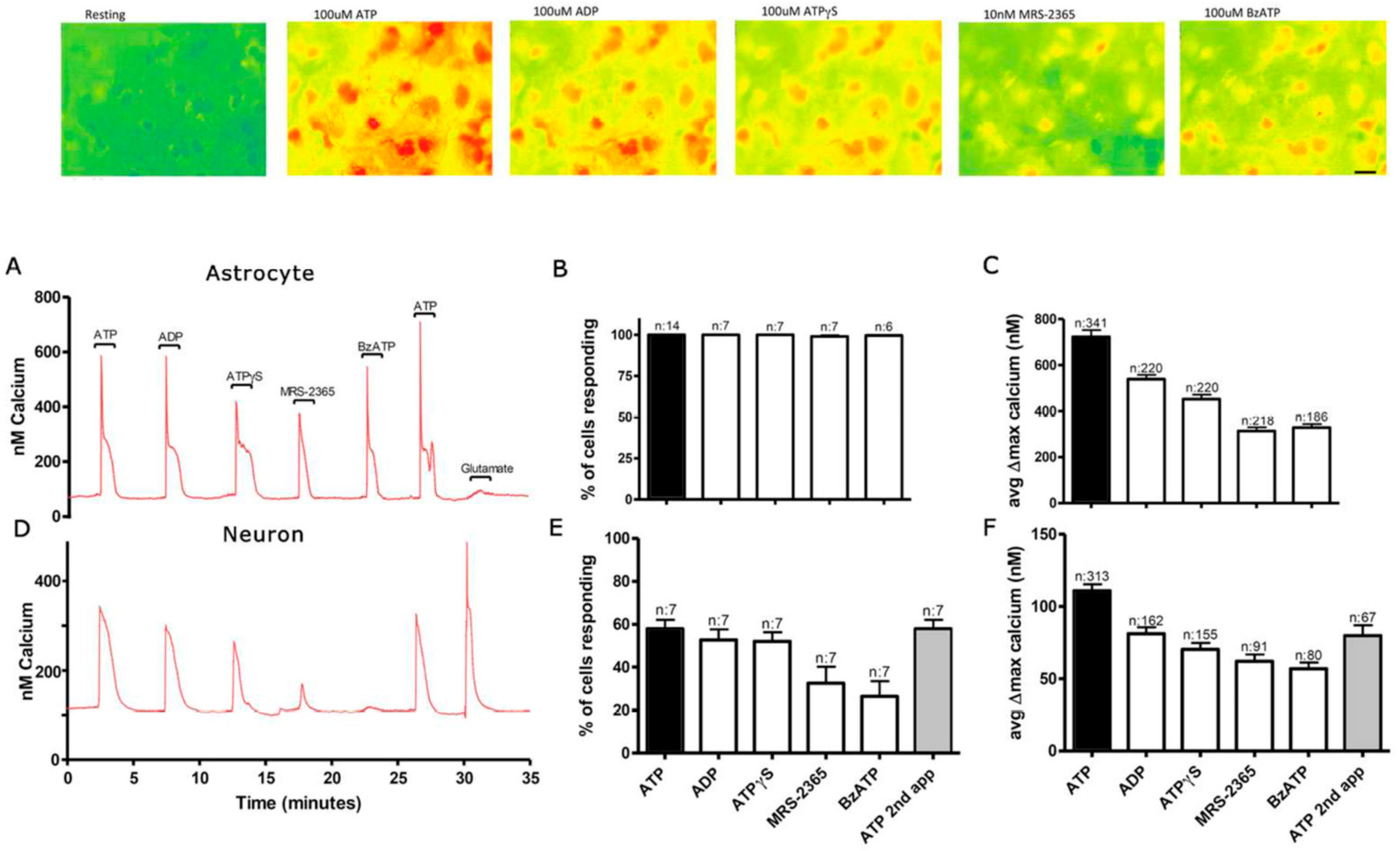
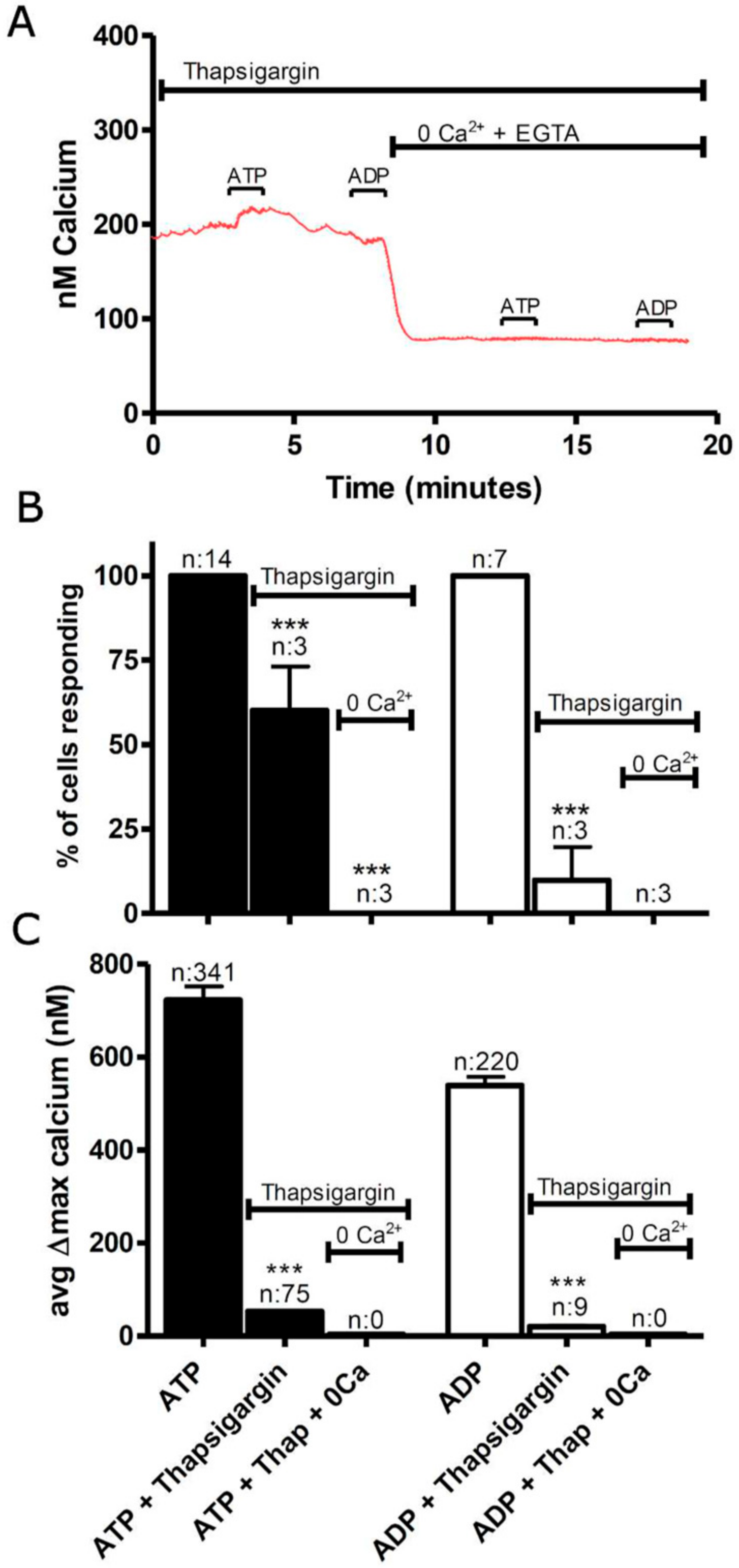
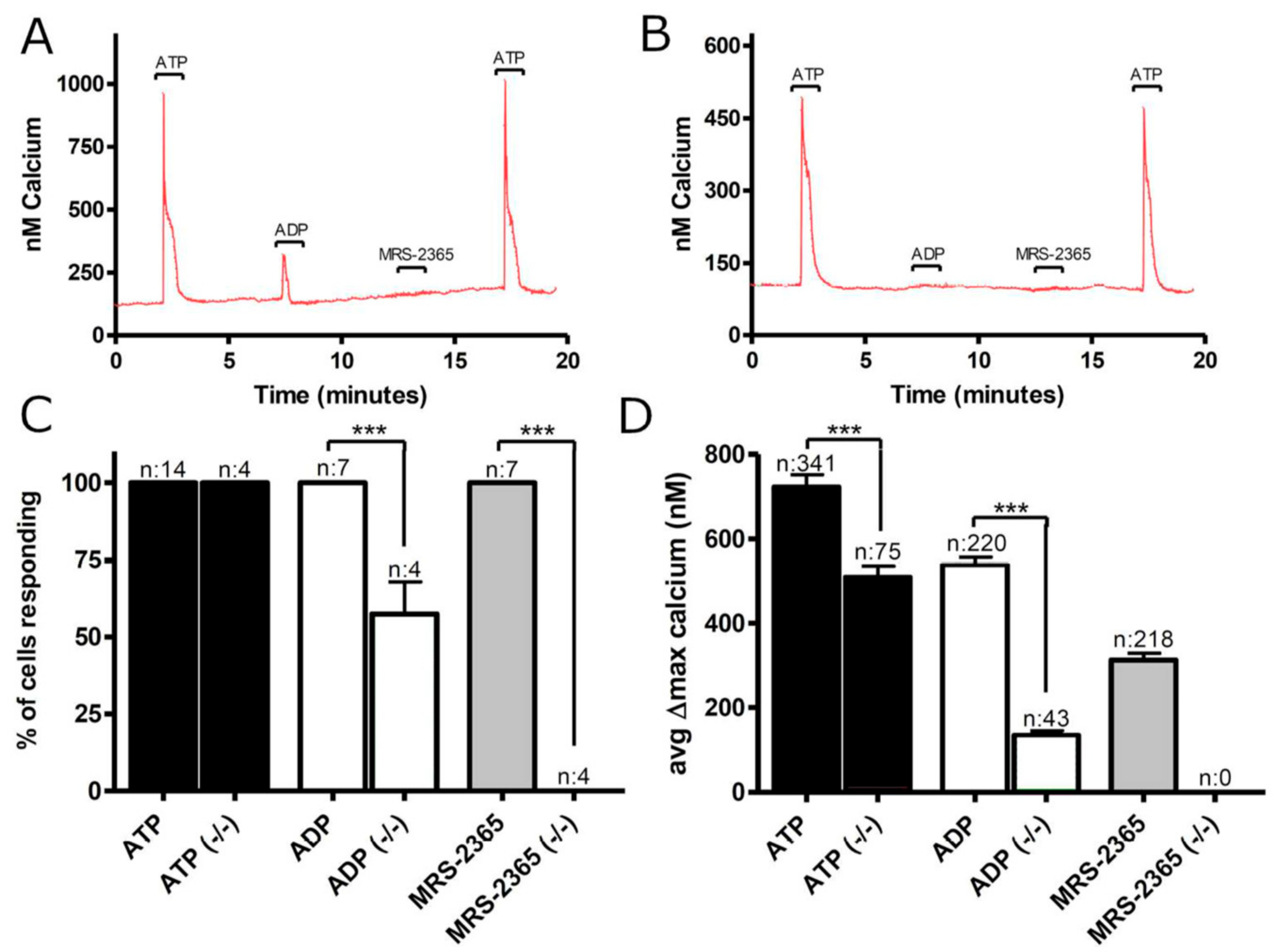
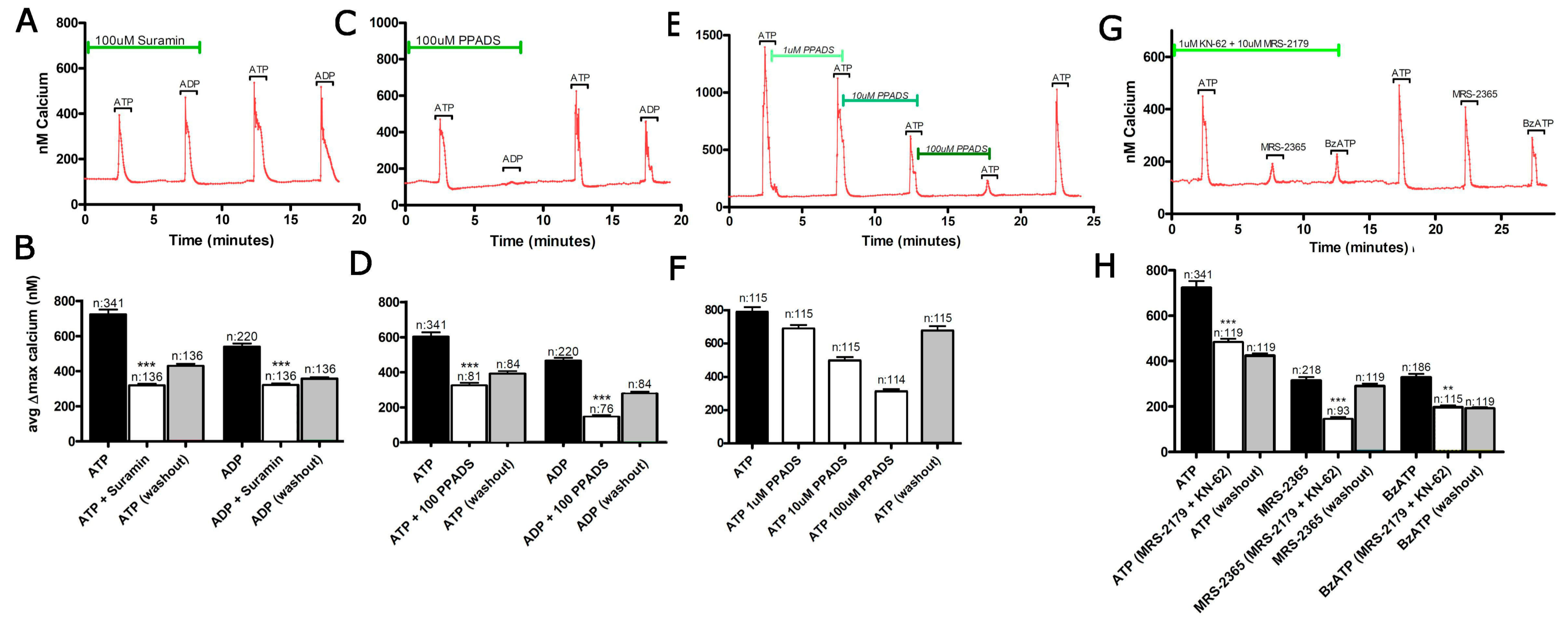
3.1. Functional P2 Receptors and GluR in Astrocyte-Enriched and Neuron-Enriched Cultures
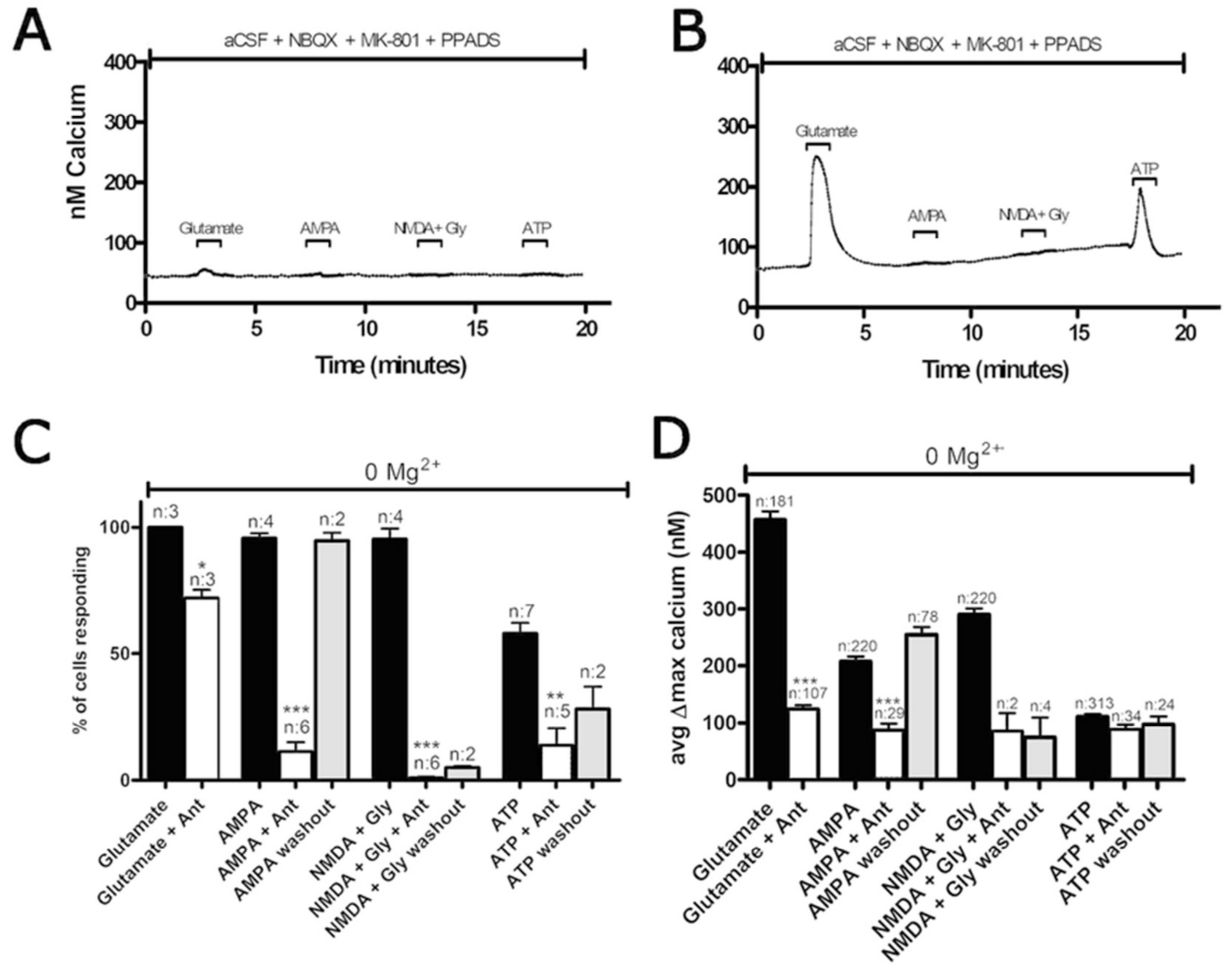
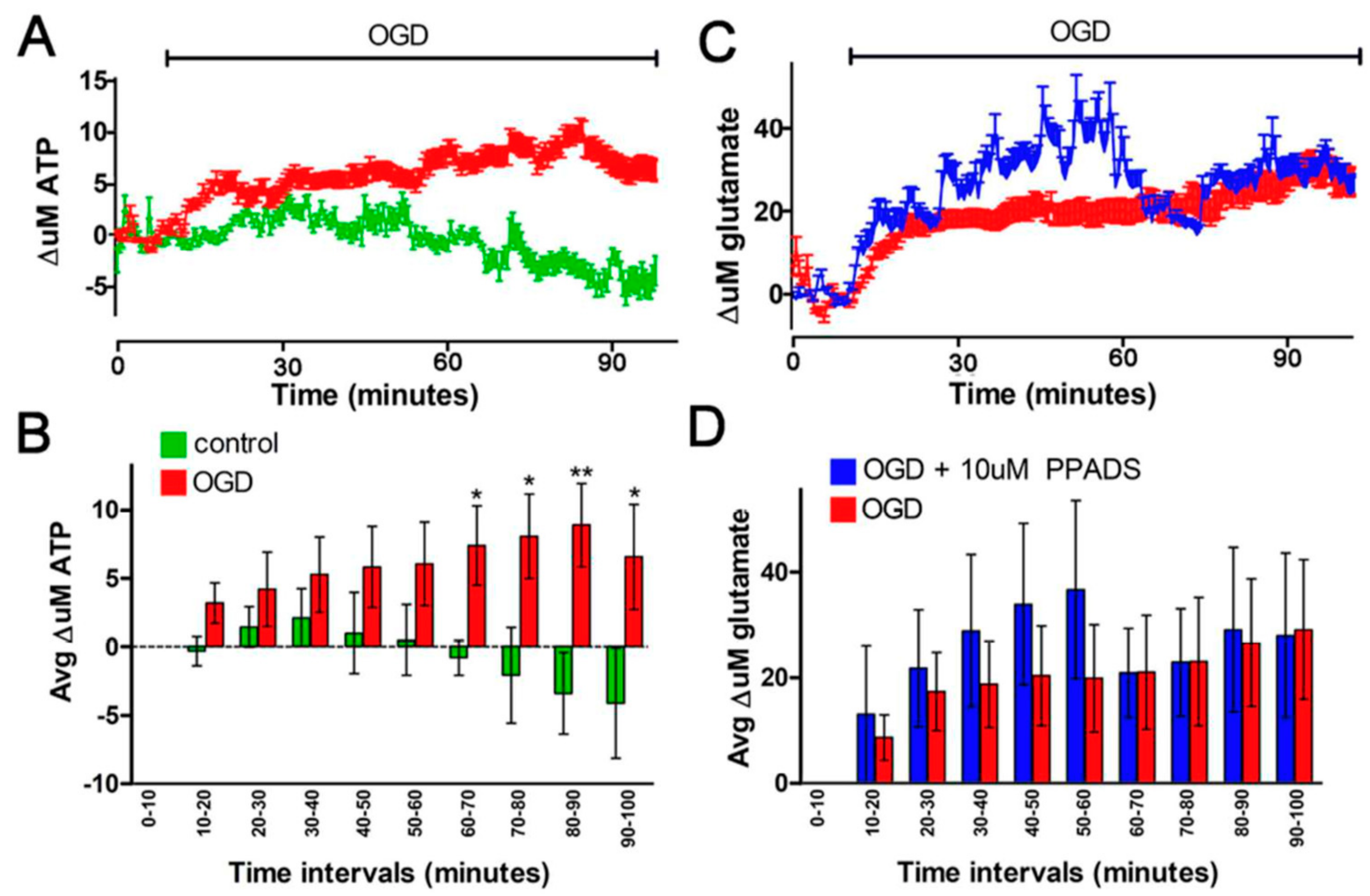
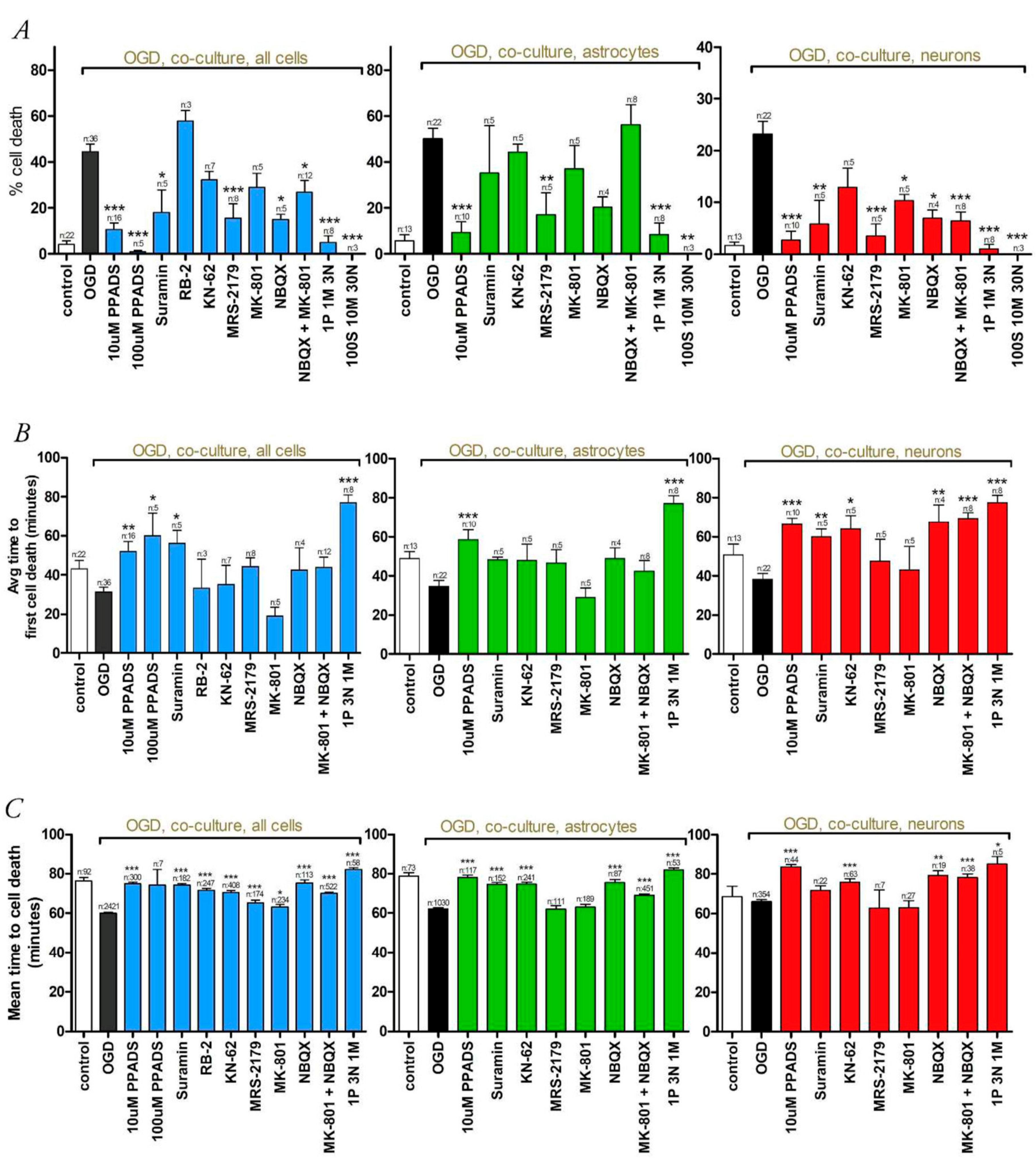
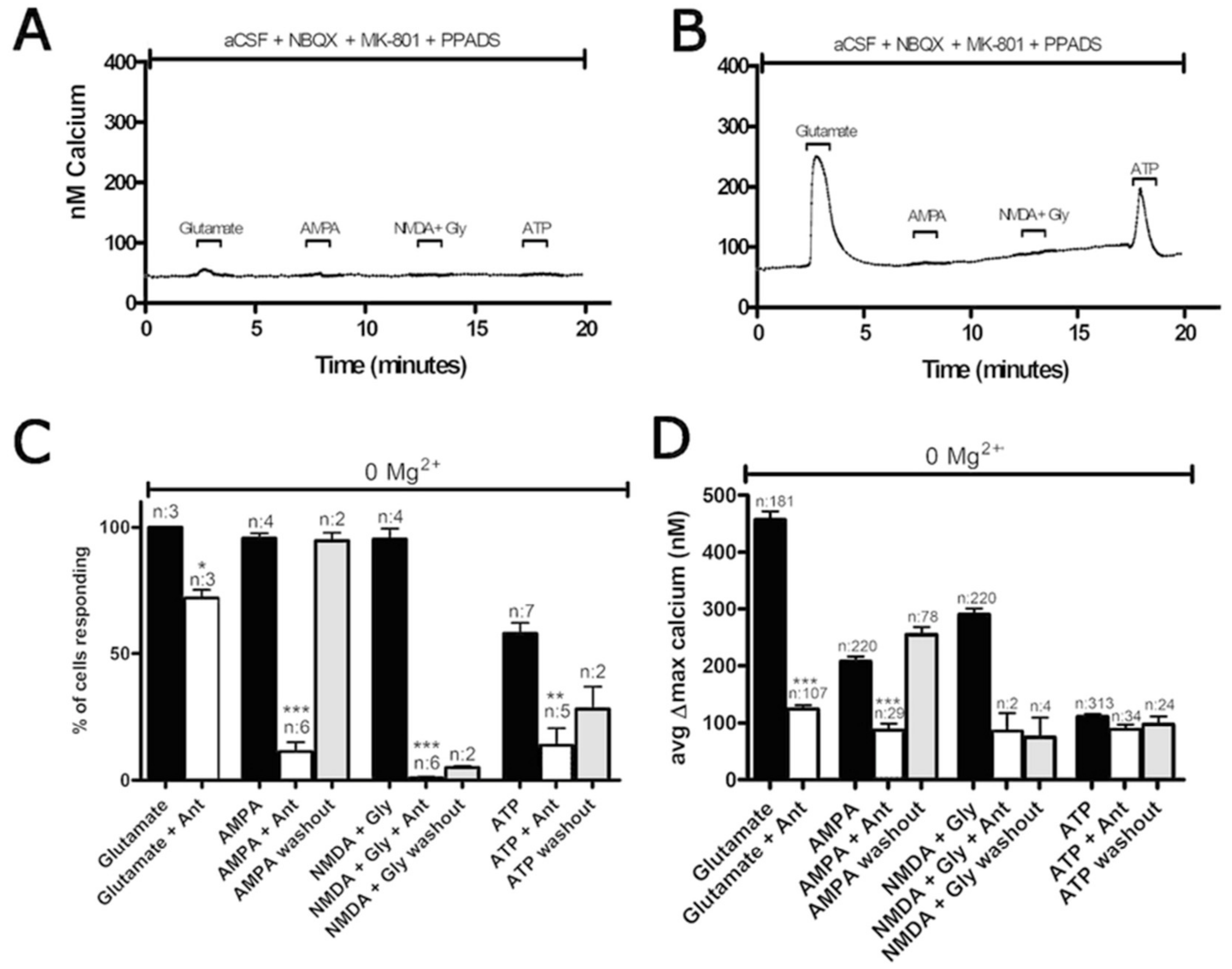
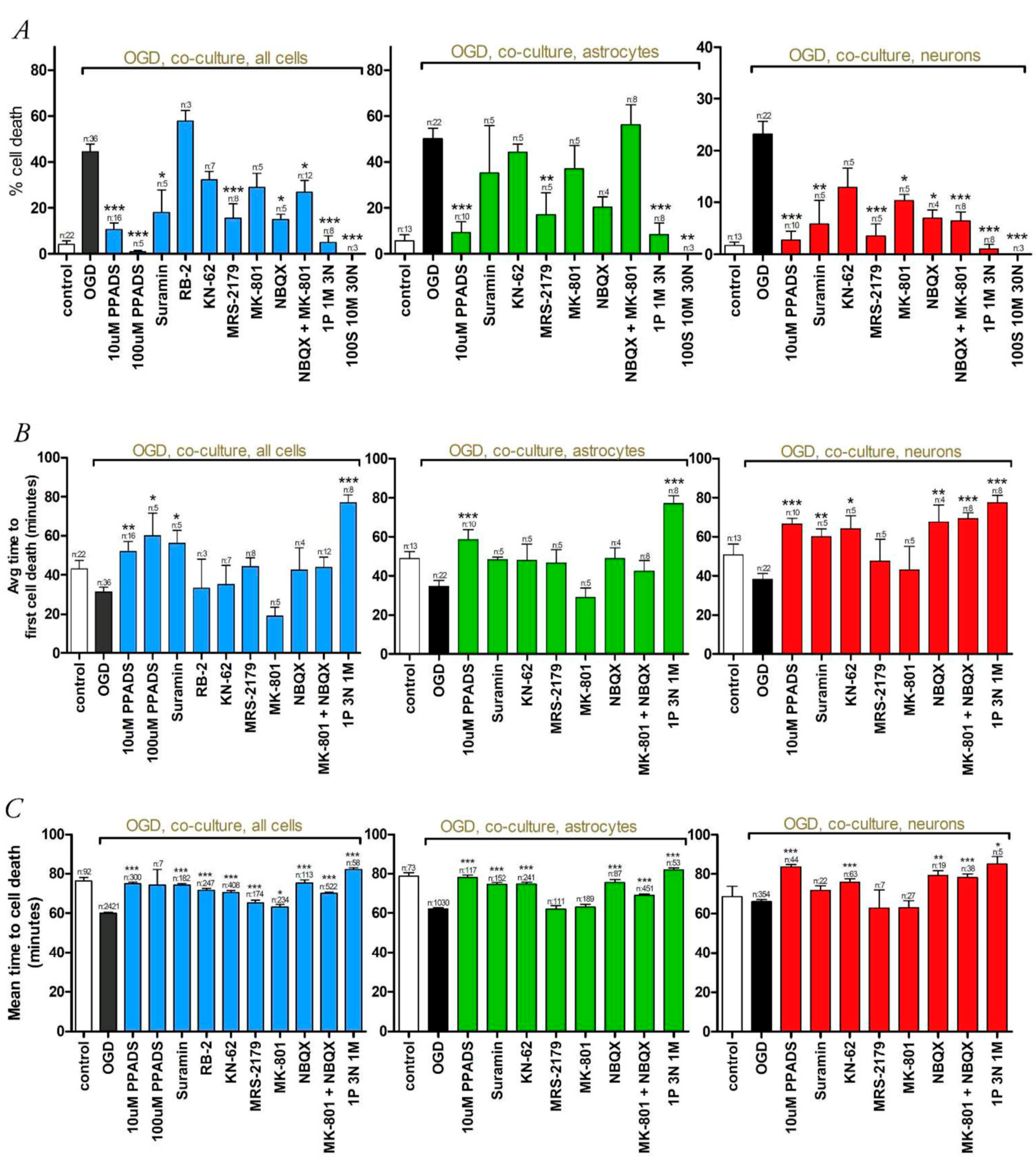
3.2. Oxygen-Glucose Deprivation in High-Density Neuron/Astrocyte Co-Cultures
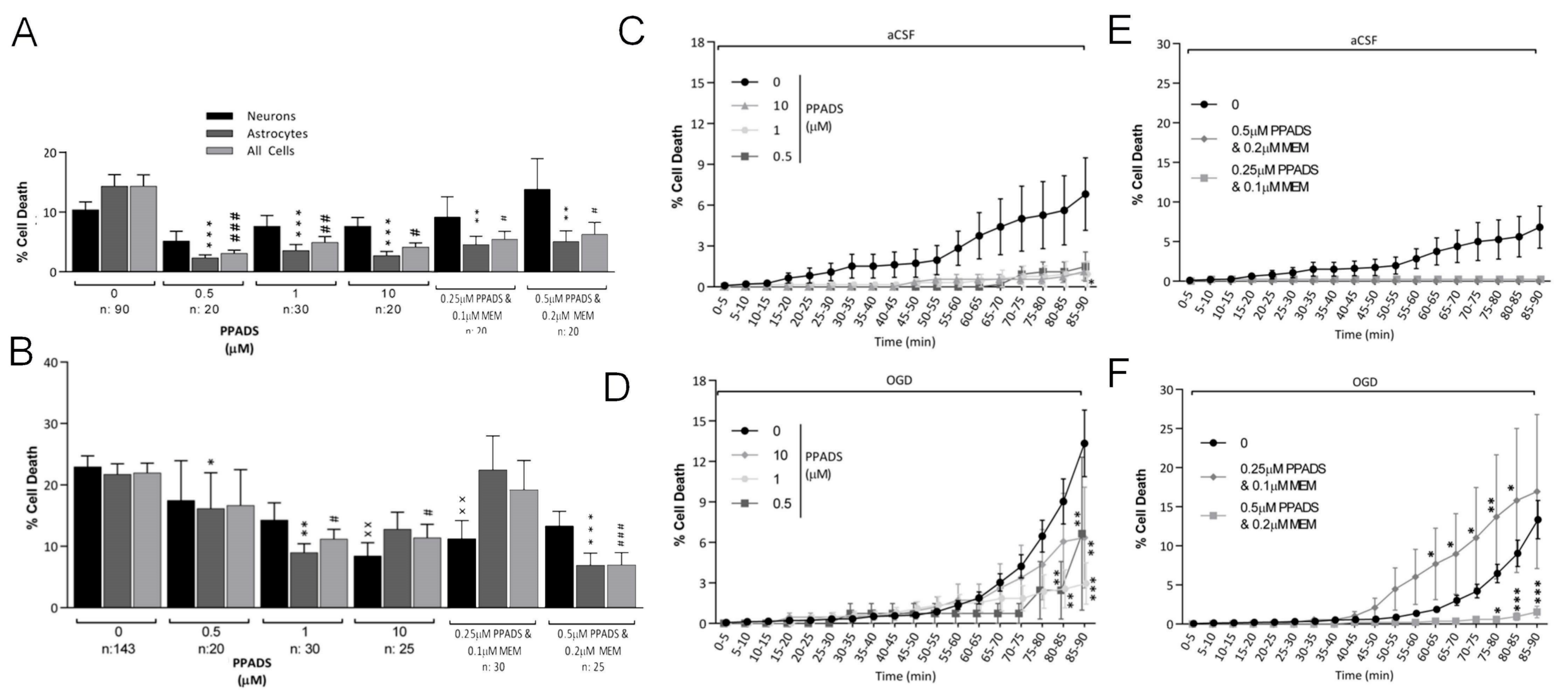
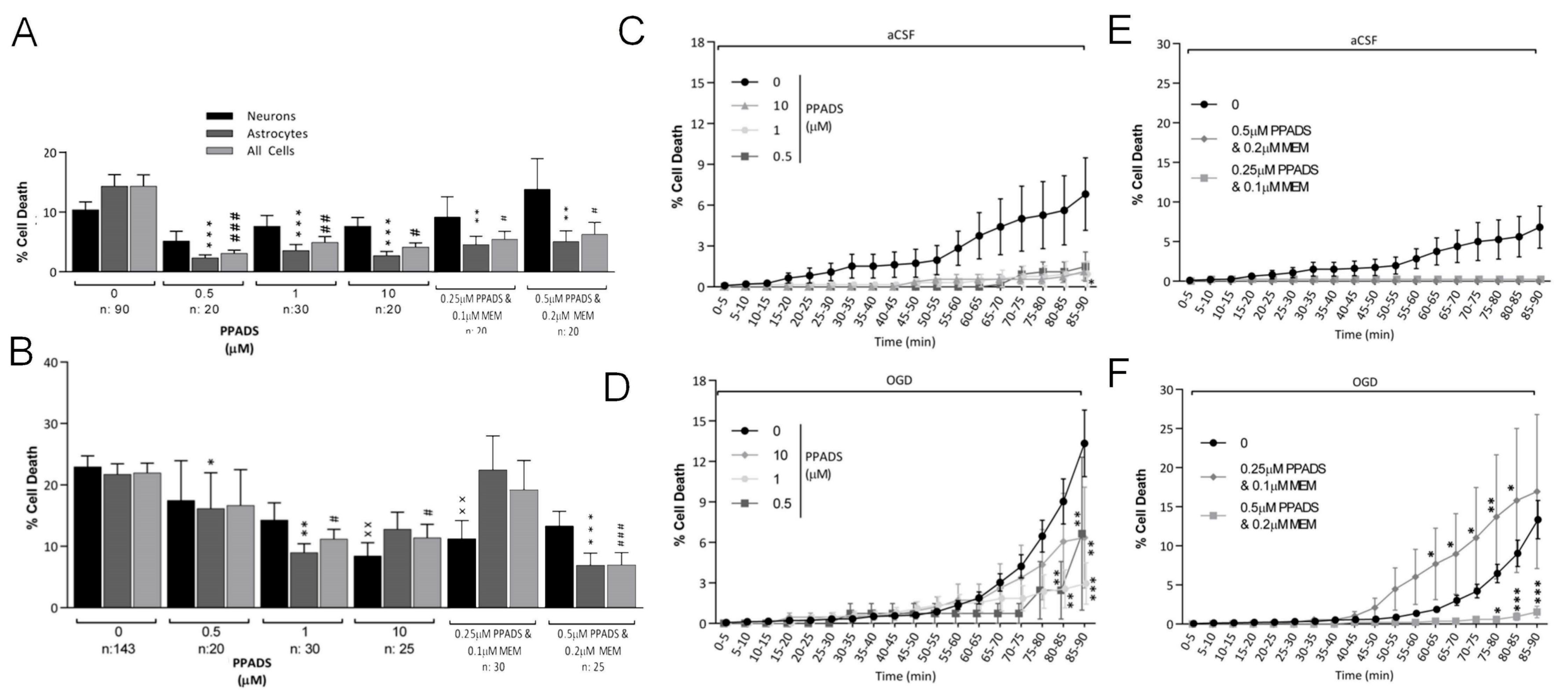
3.3. Oxygen-Glucose Deprivation in Normal-Density Neuron/Astrocyte Co-Cultures
4. Discussion
4.1. GluR Antagonists Protect Neurons but Not Astrocytes
4.2. Broad-Spectrum P2 Antagonists Protect Neurons and Astrocytes
4.3. Synergistic Effect of P2 and Glutamate Receptor Antagonists
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Ginsberg, M.D. Current status of neuroprotection for cerebral ischemia: Synoptic overview. Stroke 2009, 40, S111–S114. [Google Scholar] [CrossRef] [PubMed]
- Dirnagl, U.; Iadecola, C.; Moskowitz, M.A. Pathobiology of ischaemic stroke: An integrated view. Trends Neurosci. 1999, 22, 391–397. [Google Scholar] [CrossRef]
- Pringle, A.K. In, out, shake it all about: Elevation of [Ca2+]i during acute cerebral ischaemia. Cell Calcium 2004, 36, 235–245. [Google Scholar] [CrossRef] [PubMed]
- Trotman, M.; Vermehren, P.; Gibson, C.L.; Fern, R. The dichotomy of memantine treatment for ischemic stroke: Dose-dependent protective and detrimental effects. J. Cereb. Blood Flow Metab. 2015, 35, 230–239. [Google Scholar] [CrossRef] [PubMed]
- Lalo, U.; Pankratov, Y.; Kirchhoff, F.; North, R.A.; Verkhratsky, A. NMDA receptors mediate neuron-to-glia signaling in mouse cortical astrocytes. J. Neurosci. 2006, 26, 2673–2683. [Google Scholar] [CrossRef] [PubMed]
- Butt, A.M. Atp: A ubiquitous gliotransmitter integrating neuron-glial networks. Semin. Cell Dev. Biol. 2011, 22, 205–213. [Google Scholar] [CrossRef] [PubMed]
- Franke, H.; Illes, P. Involvement of P2 receptors in the growth and survival of neurons in the CNS. Pharmacol. Ther. 2006, 109, 297–324. [Google Scholar] [CrossRef] [PubMed]
- Sperlagh, B.; Zsilla, G.; Baranyi, M.; Illes, P.; Vizi, E.S. Purinergic modulation of glutamate release under ischemic-like conditions in the hippocampus. Neuroscience 2007, 149, 99–111. [Google Scholar] [CrossRef] [PubMed]
- Hisanaga, K.; Onodera, H.; Kogure, K. Changes in levels of purine and pyrimidine nucleotides during acute hypoxia and recovery in neonatal rat brain. J. Neurochem. 1986, 47, 1344–1350. [Google Scholar] [CrossRef] [PubMed]
- Phillis, J.W.; O’Regan, M.H.; Perkins, L.M. Adenosine 5′-triphosphate release from the normoxic and hypoxic in vivo rat cerebral cortex. Neurosci. Lett. 1993, 151, 94–96. [Google Scholar] [CrossRef]
- Phillis, J.W.; Smith-Barbour, M.; O’Regan, M.H. Changes in extracellular amino acid neurotransmitters and purines during and following ischemias of different durations in the rat cerebral cortex. Neurochem. Int. 1996, 29, 115–120. [Google Scholar] [CrossRef]
- Lutz, P.L.; Kabler, S. Release of adenosine and ATP in the brain of the freshwater turtle (Trachemys scripta) during long-term anoxia. Brain Res. 1997, 769, 281–286. [Google Scholar] [CrossRef]
- Juranyi, Z.; Sperlagh, B.; Vizi, E.S. Involvement of P2 purinoceptors and the nitric oxide pathway in [3H]purine outflow evoked by short-term hypoxia and hypoglycemia in rat hippocampal slices. Brain Res. 1999, 823, 183–190. [Google Scholar] [CrossRef]
- Parkinson, F.E.; Sinclair, C.J.; Othman, T.; Haughey, N.J.; Geiger, J.D. Differences between rat primary cortical neurons and astrocytes in purine release evoked by ischemic conditions. Neuropharmacology 2002, 43, 836–846. [Google Scholar] [CrossRef]
- Melani, A.; Turchi, D.; Vannucchi, M.G.; Cipriani, S.; Gianfriddo, M.; Pedata, F. ATP extracellular concentrations are increased in the rat striatum during in vivo ischemia. Neurochem. Int. 2005, 47, 442–448. [Google Scholar] [CrossRef] [PubMed]
- Dale, N.; Frenguelli, B.G. Release of adenosine and ATP during ischemia and epilepsy. Curr. Neuropharmacol. 2009, 7, 160–179. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, D.; Chiozzi, P.; Falzoni, S.; Dal Susino, M.; Collo, G.; Buell, G.; Di Virgilio, F. ATP-mediated cytotoxicity in microglial cells. Neuropharmacology 1997, 36, 1295–1301. [Google Scholar] [CrossRef]
- Amadio, S.; D’Ambrosi, N.; Cavaliere, F.; Murra, B.; Sancesario, G.; Bernardi, G.; Burnstock, G.; Volonte, C. P2 receptor modulation and cytotoxic function in cultured CNSs neurons. Neuropharmacology 2002, 42, 489–501. [Google Scholar] [CrossRef]
- Ryu, J.K.; Kim, J.; Choi, S.H.; Oh, Y.J.; Lee, Y.B.; Kim, S.U.; Jin, B.K. ATP-induced in vivo neurotoxicity in the rat striatum via P2 receptors. Neuroreport 2002, 13, 1611–1615. [Google Scholar] [CrossRef] [PubMed]
- Volonte, C.; Amadio, S.; Cavaliere, F.; D’Ambrosi, N.; Vacca, F.; Bernardi, G. Extracellular ATP and neurodegeneration. Curr. Drug Targets CNS Neurol. Disord. 2003, 2, 403–412. [Google Scholar] [CrossRef] [PubMed]
- Matute, C.; Torre, I.; Perez-Cerda, F.; Perez-Samartin, A.; Alberdi, E.; Etxebarria, E.; Arranz, A.M.; Ravid, R.; Rodriguez-Antiguedad, A.; Sanchez-Gomez, M.; et al. P2x7 receptor blockade prevents ATP excitotoxicity in oligodendrocytes and ameliorates experimental autoimmune encephalomyelitis. J. Neurosci. 2007, 27, 9525–9533. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, H. Biochemistry, localization and functional roles of ecto-nucleotidases in the nervous system. Prog. Neurobiol. 1996, 49, 589–618. [Google Scholar] [CrossRef]
- Robson, S.C.; Kaczmarek, E.; Siegel, J.B.; Candinas, D.; Koziak, K.; Millan, M.; Hancock, W.W.; Bach, F.H. Loss of ATP diphosphohydrolase activity with endothelial cell activation. J. Exp. Med. 1997, 185, 153–163. [Google Scholar] [CrossRef] [PubMed]
- Swanson, R.A.; Farrell, K.; Stein, B.A. Astrocyte energetics, function, and death under conditions of incomplete ischemia: A mechanism of glial death in the penumbra. Glia 1997, 21, 142–153. [Google Scholar] [CrossRef]
- Fern, R. Ischemic tolerance in pre-myelinated white matter: The role of astrocyte glycogen in brain pathology. J. Cereb. Blood Flow Metab. 2015, 35, 951–958. [Google Scholar] [CrossRef] [PubMed]
- Fern, R. Intracellular calcium and cell death during ischemia in neonatal rat white matter astrocytes in situ. J. Neurosci. 1998, 18, 7232–7243. [Google Scholar] [PubMed]
- Bondarenko, A.; Chesler, M. Rapid astrocyte death induced by transient hypoxia, acidosis, and extracellular ion shifts. Glia 2001, 34, 134–142. [Google Scholar] [CrossRef] [PubMed]
- Dale, N.; Hatz, S.; Tian, F.; Llaudet, E. Listening to the brain: Microelectrode biosensors for neurochemicals. Trends Biotechnol. 2005, 23, 420–428. [Google Scholar] [CrossRef] [PubMed]
- Centemeri, C.; Bolego, C.; Abbracchio, M.P.; Cattabeni, F.; Puglisi, L.; Burnstock, G.; Nicosia, S. Characterization of the Ca2+ responses evoked by ATP and other nucleotides in mammalian brain astrocytes. Br. J. Pharmacol. 1997, 121, 1700–1706. [Google Scholar] [CrossRef] [PubMed]
- Reifel Saltzberg, J.M.; Garvey, K.A.; Keirstead, S.A. Pharmacological characterization of P2Y receptor subtypes on isolated tiger salamander muller cells. Glia 2003, 42, 149–159. [Google Scholar] [CrossRef] [PubMed]
- Hoyte, L.; Barber, P.A.; Buchan, A.M.; Hill, M.D. The rise and fall of NMDA antagonists for ischemic stroke. Curr. Mol. Med. 2004, 4, 131–136. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, M.P.; Weiss, J.H.; Pham, P.C.; Choi, D.W. N-methyl-d-aspartate receptors mediate hypoxic neuronal injury in cortical culture. J. Pharmacol. Exp. Ther. 1987, 243, 784–791. [Google Scholar] [PubMed]
- Choi, D.W.; Rothman, S.M. The role of glutamate neurotoxicity in hypoxic-ischemic neuronal death. Annu. Rev. Neurosci. 1990, 13, 171–182. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, M.P.; Choi, D.W. Combined oxygen and glucose deprivation in cortical cell culture: Calcium-dependent and calcium-independent mechanisms of neuronal injury. J. Neurosci. 1993, 13, 3510–3524. [Google Scholar] [PubMed]
- Newell, D.W.; Barth, A.; Papermaster, V.; Malouf, A.T. Glutamate and non-glutamate receptor mediated toxicity caused by oxygen and glucose deprivation in organotypic hippocampal cultures. J. Neurosci. 1995, 15, 7702–7711. [Google Scholar] [PubMed]
- Balcar, V.J.; Dias, L.S.; Li, Y.; Bennett, M.R. Inhibition of [3H]CGP 39653 binding to NMDA receptors by a P2 antagonist, suramin. Neuroreport 1995, 7, 69–72. [Google Scholar] [CrossRef] [PubMed]
- Nakazawa, K.; Inoue, K.; Ito, K.; Koizumi, S.; Inoue, K. Inhibition by suramin and reactive blue 2 of GABA and glutamate receptor channels in rat hippocampal neurons. Naunyn-Schmiedeberg's Arch. Pharmacol. 1995, 351, 202–208. [Google Scholar] [CrossRef]
- Price, C.J.; Raymond, L.A. Evans blue antagonizes both alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionate and kainate receptors and modulates receptor desensitization. Mol. Pharmacol. 1996, 50, 1665–1671. [Google Scholar] [PubMed]
- Peoples, R.W.; Li, C. Inhibition of NMDA-gated ion channels by the P2 purinoceptor antagonists suramin and reactive blue 2 in mouse hippocampal neurones. Br. J. Pharmacol. 1998, 124, 400–408. [Google Scholar] [CrossRef] [PubMed]
- Lambrecht, G. Agonists and antagonists acting at P2X receptors: Selectivity profiles and functional implications. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2000, 362, 340–350. [Google Scholar] [CrossRef]
- Zona, C.; Marchetti, C.; Volonte, C.; Mercuri, N.B.; Bernardi, G. Effect of P2 purinoceptor antagonists on kainate-induced currents in rat cultured neurons. Brain Res. 2000, 882, 26–35. [Google Scholar] [CrossRef]
- Suzuki, E.; Kessler, M.; Montgomery, K.; Arai, A.C. Divergent effects of the purinoceptor antagonists suramin and pyridoxal-5′-phosphate-6-(2′-naphthylazo-6′-nitro-4′,8′-disulfonate) (PPNDS) on α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors. Mol. Pharmacol. 2004, 66, 1738–1747. [Google Scholar] [CrossRef] [PubMed]
- Lammer, A.; Gunther, A.; Beck, A.; Krugel, U.; Kittner, H.; Schneider, D.; Illes, P.; Franke, H. Neuroprotective effects of the P2 receptor antagonist PPADS on focal cerebral ischaemia-induced injury in rats. Eur. J. Neurosci. 2006, 23, 2824–2828. [Google Scholar] [CrossRef] [PubMed]
- Cavaliere, F.; D’Ambrosi, N.; Ciotti, M.T.; Mancino, G.; Sancesario, G.; Bernardi, G.; Volonte, C. Glucose deprivation and chemical hypoxia: Neuroprotection by P2 receptor antagonists. Neurochem. Int. 2001, 38, 189–197. [Google Scholar] [CrossRef]
- Cavaliere, F.; Florenzano, F.; Amadio, S.; Fusco, F.R.; Viscomi, M.T.; D’Ambrosi, N.; Vacca, F.; Sancesario, G.; Bernardi, G.; Molinari, M.; et al. Up-regulation of p2x2, p2x4 receptor and ischemic cell death: Prevention by p2 antagonists. Neuroscience 2003, 120, 85–98. [Google Scholar] [CrossRef]
- Cavaliere, F.; Dinkel, K.; Reymann, K. Microglia response and P2 receptor participation in oxygen/glucose deprivation-induced cortical damage. Neuroscience 2005, 136, 615–623. [Google Scholar] [CrossRef] [PubMed]
- Runden-Pran, E.; Tanso, R.; Haug, F.M.; Ottersen, O.P.; Ring, A. Neuroprotective effects of inhibiting N-methyl-d-aspartate receptors, P2X receptors and the mitogen-activated protein kinase cascade: A quantitative analysis in organotypical hippocampal slice cultures subjected to oxygen and glucose deprivation. Neuroscience 2005, 136, 795–810. [Google Scholar] [CrossRef] [PubMed]
- Cavaliere, F.; D’Ambrosi, N.; Sancesario, G.; Bernardi, G.; Volonte, C. Hypoglycaemia-induced cell death: Features of neuroprotection by the P2 receptor antagonist basilen blue. Neurochem. Int. 2001, 38, 199–207. [Google Scholar] [CrossRef]
- Melani, A.; Amadio, S.; Gianfriddo, M.; Vannucchi, M.G.; Volonte, C.; Bernardi, G.; Pedata, F.; Sancesario, G. P2X7 receptor modulation on microglial cells and reduction of brain infarct caused by middle cerebral artery occlusion in rat. J. Cereb. Blood Flow Metab. 2006, 26, 974–982. [Google Scholar] [CrossRef] [PubMed]
- Le Feuvre, R.; Brough, D.; Rothwell, N. Extracellular ATP and P2X7 receptors in neurodegeneration. Eur. J. Pharmacol. 2002, 447, 261–269. [Google Scholar] [CrossRef]
- Le Feuvre, R.A.; Brough, D.; Touzani, O.; Rothwell, N.J. Role of P2X7 receptors in ischemic and excitotoxic brain injury in vivo. J. Cereb. Blood Flow Metab. 2003, 23, 381–384. [Google Scholar] [CrossRef] [PubMed]
- Coppi, E.; Pedata, F.; Gibb, A.J. P2Y1 receptor modulation of Ca2+-activated K+ currents in medium-sized neurons from neonatal rat striatal slices. J. Neurophysiol. 2012, 107, 1009–1021. [Google Scholar] [CrossRef] [PubMed]
- Kuboyama, K.; Harada, H.; Tozaki-Saitoh, H.; Tsuda, M.; Ushijima, K.; Inoue, K. Astrocytic P2Y1 receptor is involved in the regulation of cytokine/chemokine transcription and cerebral damage in a rat model of cerebral ischemia. J. Cereb. Blood Flow Metab. 2011, 31, 1930–1941. [Google Scholar] [CrossRef] [PubMed]
- Chin, Y.; Kishi, M.; Sekino, M.; Nakajo, F.; Abe, Y.; Terazono, Y.; Hiroyuki, O.; Kato, F.; Koizumi, S.; Gachet, C.; et al. Involvement of glial P2Y1 receptors in cognitive deficit after focal cerebral stroke in a rodent model. J. Neuroinflamm. 2013, 10, 95. [Google Scholar] [CrossRef] [PubMed]
- Carmo, M.R.; Simoes, A.P.; Fonteles, A.A.; Souza, C.M.; Cunha, R.A.; Andrade, G.M. ATP P2Y1 receptors control cognitive deficits and neurotoxicity but not glial modifications induced by brain ischemia in mice. Eur. J. Neurosci. 2014, 39, 614–622. [Google Scholar] [CrossRef] [PubMed]
- Domercq, M.; Brambilla, L.; Pilati, E.; Marchaland, J.; Volterra, A.; Bezzi, P. P2Y1 receptor-evoked glutamate exocytosis from astrocytes: Control by tumor necrosis factor-α and prostaglandins. J. Biol. Chem. 2006, 281, 30684–30696. [Google Scholar] [CrossRef] [PubMed]
- Zeng, J.W.; Liu, X.H.; He, W.J.; Du, L.; Zhang, J.H.; Wu, X.G.; Ruan, H.Z. Inhibition of ATP-induced glutamate release by MRS2179 in cultured dorsal spinal cord astrocytes. Pharmacology 2008, 82, 257–263. [Google Scholar] [CrossRef] [PubMed]
- Zeng, J.W.; Liu, X.H.; Zhang, J.H.; Wu, X.G.; Ruan, H.Z. P2Y1 receptor-mediated glutamate release from cultured dorsal spinal cord astrocytes. J. Neurochem. 2008, 106, 2106–2118. [Google Scholar] [CrossRef] [PubMed]
- Tonazzini, I.; Trincavelli, M.L.; Storm-Mathisen, J.; Martini, C.; Bergersen, L.H. Co-localization and functional cross-talk between A1 and P2Y1 purine receptors in rat hippocampus. Eur. J. Neurosci. 2007, 26, 890–902. [Google Scholar] [CrossRef] [PubMed]
- Tonazzini, I.; Trincavelli, M.L.; Montali, M.; Martini, C. Regulation of A1 adenosine receptor functioning induced by P2Y1 purinergic receptor activation in human astroglial cells. J. Neurosci. Res. 2008, 86, 2857–2866. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
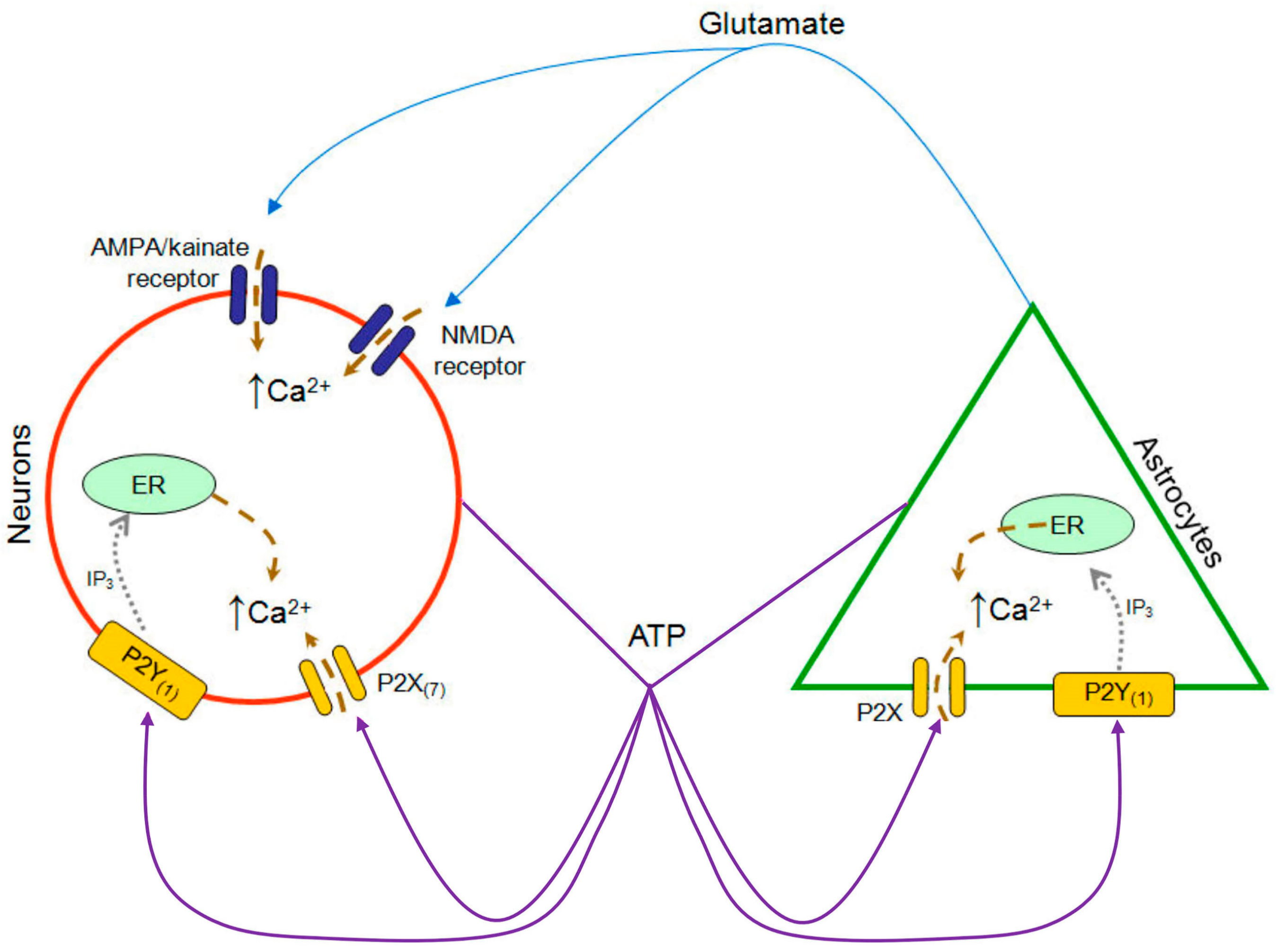{kind=link}
| Suramin | PPADS | KN-62 | Highly Selective Antagonists a,h,p,s | |
|---|---|---|---|---|
| P2X1 | No, >1 mM g (mouse) | 0.4 g (mouse) | No | NF449, NF864, RO-1 |
| 0.01–0.3, 0.851 e,t (human) | 0.01–0.3, 1.29 a,t (human) | |||
| P2X2 | 33.1 e (rat) | 3.8 e (rat) | No | - |
| 1-32 m,p,t (human) | 0.4–20.4 a,m,p,t (human) | |||
| P2X3 | 0.776 e (rat) | 3.63 e (rat) | No | - |
| 14.9, 15.8, >100 e,n,p,t (human) | 1.7–5.13 e,n,t (human) | |||
| P2X4 | >100 q (mouse) | 10.5 q (mouse) | No, >3 q (mouse) | Benzofuro-1,4-diazepin-2-ones |
| 178.1–200 e,o,p (human) | 10–25,27.5, >100 a,o,p (human) | |||
| P2X5 | 4.5 r (rat) | 2 r (rat) | No | - |
| 0.199 p,t (human) | 0.01-0.3, 3.16 p,t (human) | |||
| P2X6 | No, >30 r (rat) | No, >30 r (rat) >0.3 t (human) | No | - |
| P2X7 | 40 f (mouse)No, >100 e,t (human) | Cation pore opening | Cation pore opening | 1A-438079 ** |
| 14.79 a (mouse) | 0.389 a (mouse) | 1A-740003 ** | ||
| 3.24 a (human) | 0.38 a (human) | A-804590 | ||
| Large pore formation | Large pore formation | GSK314181A | ||
| 7, 9, >100 a,b (mouse) | 0.18, 1.17 a,b (mouse) | AZ11645373 | ||
| 0.015 b (human) | 0.011 b (human) | 1AZ10606120 | ||
| P2X2/3 | 33.1 e (rat) | 1.26 e (rat) | No | A-317491 |
| 16.6 a (human) | RO-3 | |||
| P2X1/5 | 1.58 p (rat) | 0.5–8.6 h (mouse) | No | - |
| P2X4/6 | Some activity u (rat) | Some activity u (rat) | No | - |
| Suramin | PPADS | MRS-2179 | Highly Selective Antagonists h | |
|---|---|---|---|---|
| P2Y1 | Effective at 100uM d (mouse) 1.67 t (turkey) 3.16 a (human) | Effective at 30 uM d (mouse) 1.05 t (turkey) 2 b (human) | Effective at 10 uM f,g (mouse) 0.015–0.331 f,h,p,q,u (human) | MRS-2279 MRS-2179 MRS-2500 A3P5P |
| P2Y2 | 47.86 t (human) | No b,t (human) | No a,s,u | AR-C126313 MRS-2576 |
| P2Y4 | No n,o (mouse/human) | 45 n (mouse) 15 o (human) | No a,s,u | MRS-2577 |
| P2Y6 | Effective at 100uM d (mouse) | Effective at 30uM d (mouse) | No a,s,u | MRS-2578 MRS-2575 |
| P2Y12 | 3.6 r (human) | No r (human) | No a,s | MeSAMP AZD6140 INS50589 Clopidogrel |
| P2Y13 | 2.3 e (human) | 11.7 e (human) | >100 e (human) | MRS-2211 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vermehren, P.; Trotman-Lucas, M.; Hechler, B.; Gachet, C.; Evans, R.J.; Gibson, C.L.; Fern, R. Cooperation between NMDA-Type Glutamate and P2 Receptors for Neuroprotection during Stroke: Combining Astrocyte and Neuronal Protection. Neuroglia 2018, 1, 30-47. https://doi.org/10.3390/neuroglia1010005
Vermehren P, Trotman-Lucas M, Hechler B, Gachet C, Evans RJ, Gibson CL, Fern R. Cooperation between NMDA-Type Glutamate and P2 Receptors for Neuroprotection during Stroke: Combining Astrocyte and Neuronal Protection. Neuroglia. 2018; 1(1):30-47. https://doi.org/10.3390/neuroglia1010005
Chicago/Turabian StyleVermehren, Philipp, Melissa Trotman-Lucas, Beatrice Hechler, Christian Gachet, Richard J. Evans, Claire L. Gibson, and Robert Fern. 2018. "Cooperation between NMDA-Type Glutamate and P2 Receptors for Neuroprotection during Stroke: Combining Astrocyte and Neuronal Protection" Neuroglia 1, no. 1: 30-47. https://doi.org/10.3390/neuroglia1010005
APA StyleVermehren, P., Trotman-Lucas, M., Hechler, B., Gachet, C., Evans, R. J., Gibson, C. L., & Fern, R. (2018). Cooperation between NMDA-Type Glutamate and P2 Receptors for Neuroprotection during Stroke: Combining Astrocyte and Neuronal Protection. Neuroglia, 1(1), 30-47. https://doi.org/10.3390/neuroglia1010005