
A Review of Zirconolite Solid Solution Regimes for Plutonium and Candidate Neutron Absorbing Additives


Abstract
1. Status of the United Kingdom’s Plutonium Inventory
2. Criticality Safety and the Role of Neutron Absorbing Additives in Waste Packages
3. Review of Zirconolite Chemistry and Evaluation of Potential Solid Solution Regimes for Pu Immobilisation

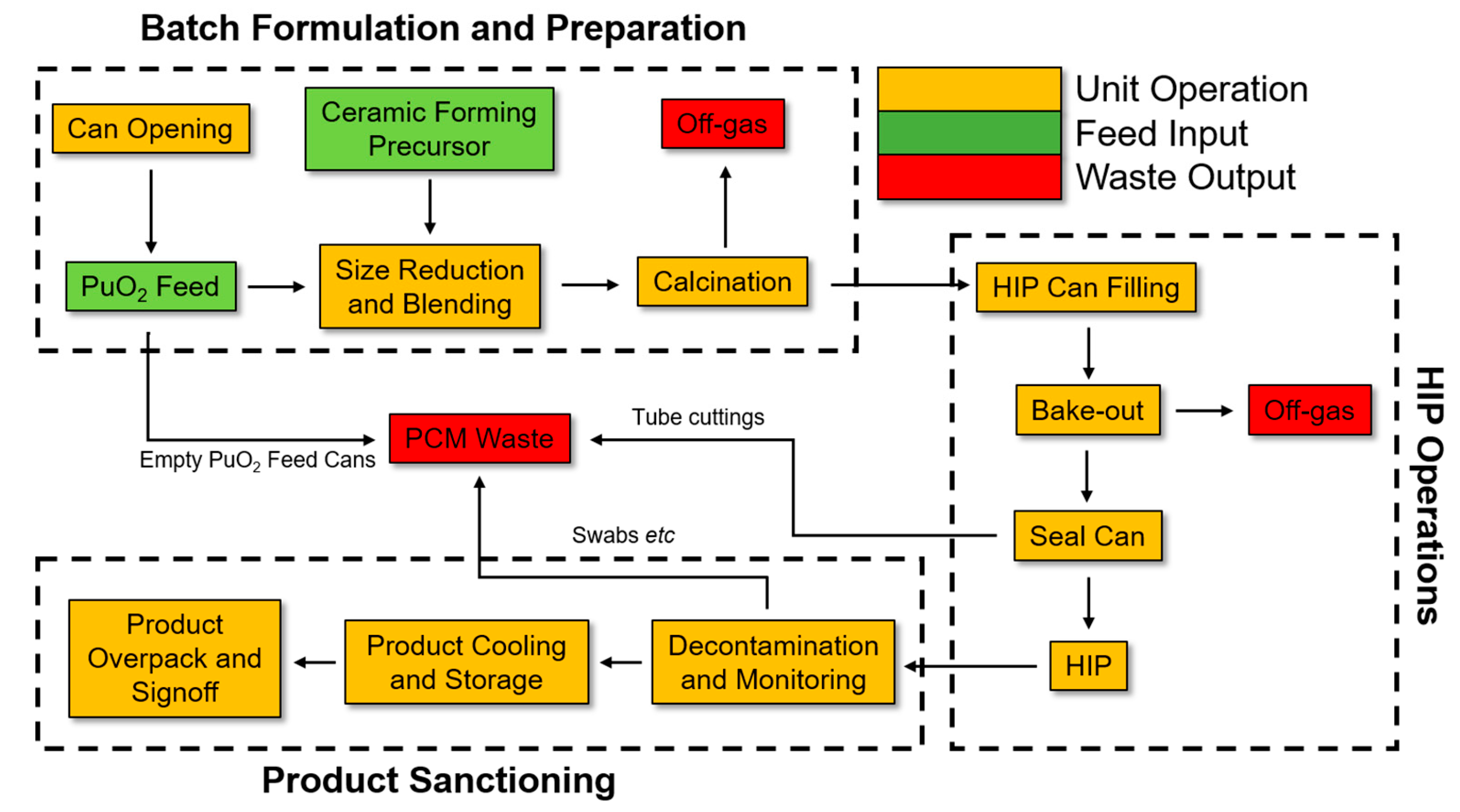{kind=link}
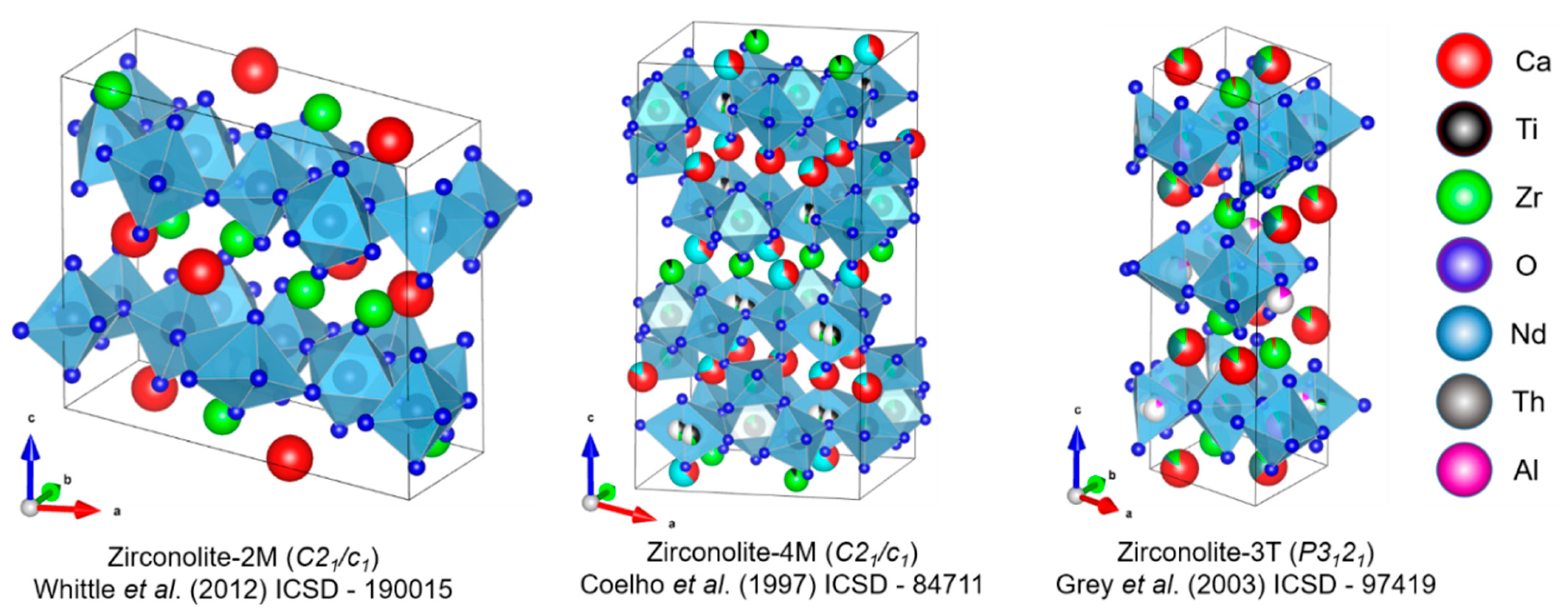{kind=link}
| Structure | Symmetry | a (Å) | b (Å) | c (Å) | β (°) | Space Group | Origin | Ref. |
|---|---|---|---|---|---|---|---|---|
| 2M | Monoclinic | 12.4435 | 7.2735 | 11.3748 | 10.563 | C2/c | Synthetic | [14] |
| 4M | Monoclinic | 12.553 | 7.248 | 23.081 | 84.799 | C2/c | Synthetic | [23] |
| 3T | Trigonal | 7.228 | - | 16.805 | - | P3121 | Synthetic | [27] |
| 3O | Orthorhombic | 10.31 | 14.48 | 7.41 | - | Acam | Natural | [33] |
| Solid Solution Regime | Comments | References |
|---|---|---|
| CaZr1−xPu4+xTi2O7 | This system exhibits a polymorphic transition to 4M in the interval 0.10 ≤ x ≤ 0.20. This has been demonstrated in several key investigations using Ce/U/Pu. A mixture of zirconolite-2M and zirconolite-4M could be expected in the compositional interval 0.20 ≤ x ≤ 0.40. Zirconolite-4M is rarely stabilised as a single phase and exists over a narrow compositional range of 0.40 ≤ x ≤ 0.50, accompanied by pyrochlore. Transformation to the pyrochlore structure is common around x = 0.60, with solid solution limits of CeO2 defined at x ~ 0.80 (this could also be expected for UO2 and PuO2). | [13,16,31,32,37,38] |
| Ca1−xPu4+xZrTi2−2xM3+2xO7 | This solid solution appears to be the most common method of substitution, by which Ce/U/Pu are substituted within the Ca2+ site, and charge balance provided by a small lower valence cation exchanged for Ti4+ (typically Al3+, Fe3+, Cr3+, or Mg2+. Note: in the case of Mg2+, the nominal composition becomes Ca1−xPuxZrTi2−xMgxO7). Zirconolite-2M is typically formed near single phase in the compositional range of 0.00 ≤ x ≤ 0.20, with secondary perovskite commonly observed for Ce surrogate compositions due to partial Ce3+ speciation. At elevated wasteloading (i.e., x = 0.35), free oxides may be observed. Furthermore, the dominant zirconolite polytype at enhanced wasteloading appears to be controlled by choice of M3+ species. For example, Ca0.65Ce0.35ZrTi1.30Cr0.70O7 was reported as zirconolite-2M by Blackburn et al., yet the corresponding Ca0.65Pu0.35ZrTi1.30Fe0.70O7 composition was reported as zirconolite-3T by Gilbert et al. This may be attributed to the different redox and/or site occupancy behaviour between Ce/Pu and/or ionic radii of Cr/Fe. | [14,31,39,40,41,42,43,44] |
| Ca1−xPu3+xZrTi2−xM3+xO7 | Systematic studies in the Ca1−xLnxZrTi2−x(Al,Fe)xO7 system have been reported for Ln = La, Nd, Gd, Ho, and Yb, which may act as Pu3+ surrogates. The phase evolution of these ceramics is seemingly dictated by ionic radii, with increased solubility for smaller cations such as Gd3+. Polytype transformation to zirconolite-3O could be expected at elevated substitution (x = 0.60) for some Ln3+ surrogates (e.g., Nd) but 2M phase stable up to x = 0.80 for others, e.g., Gd3+. | [45,46,47,48] |
| Ca1−xZr1−xPu3+2xTi2O7 | Equimolar substitution of Pu3+ between Ca2+ and Zr4+ could be expected to produce a phase transformation to the pyrochlore structure with elevated Pu content x ≥ 0.60 via formation of intermediate zirconolite-4M phase. This has been demonstrated for the corresponding Nd3+, Y3+, Sm3+, and Gd3+ solid solutions. | [49,50,51,52] |
| Ca1−xPu4+2xZr1−xPu4+xTi2−4xM3+4xO7 | This solid solution mechanism would see Pu4+ cations co-partitioned between Ca2+ and Zr4+ sites. Such targeted solid solutions have been observed to predominantly yield zirconolite-3T. | [53] |
4. Solid Solution Limits of Potential Neutron Absorbing Additives in Zirconolite
4.1. Candidate Neutron Absorbing Additives
4.2. Gadolinium Doped Zirconolite
4.3. Hafnium Doped Zirconolite
4.4. Samarium Doped Zirconolite
4.5. Cadmium Doped Zirconolite
4.6. Indium Doped Zirconolite
4.7. Boron Doped Zirconolite
5. Gap Analysis
6. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Nuclear Decommissioning Authority (NDA). Progress on Plutonium Consolidation, Storage and Disposition; Nuclear Decommissioning Authority (NDA): Moor Row, UK, 2019.
- Hyatt, N.C. Safe management of the UK separated plutonium inventory: A challenge of materials degradation. NPJ Mater. Degrad. 2020, 4, 28. [Google Scholar] [CrossRef]
- Orr, R.; Sims, H.; Taylor, R. A review of plutonium oxalate decomposition reactions and effects of decomposition temperature on the surface area of the plutonium dioxide product. J. Nucl. Mater. 2015, 465, 756–773. [Google Scholar] [CrossRef]
- Nuclear Decommissioning Authority (NDA). NDA Plutonium Options; Nuclear Decommissioning Authority (NDA): Moor Row, UK, 2008.
- ONR—Safeguards—Annual Pu and HEU Holdings in the United Kingdom as of 31 December 2021—Plain Text Version 2022/45638. Available online: https://www.iaea.org/sites/default/files/publications/documents/infcircs/1998/infcirc549a8-25.pdf (accessed on 21 June 2023).
- Nuclear Decommissioning Authority. Geological Disposal: Criticality Safety Status Report; Nuclear Decommissioning Authority: Moor Row, UK, 2016.
- Hicks, T.W. Criticality Safety Assessment for Waste Packages Containing Separated Plutonium; Galson Sciences Limited Report to NDA RWMD, 0560-2; Galson Sciences Limited: Oakham, UK, 2007. [Google Scholar]
- Solano, J.M.; Page, T.; Hicks, T.W.; Thorne, P. The Use of Neutron-Absorbing Materials in ILW and Spent Fuel Packages for Criticality Control; Nuclear Decommissioning Authority: Moor Row, UK, 2012.
- Radioactive Waste Management Ltd. Geological Disposal: Engineered Barrier System Status Report; Radioactive Waste Management Ltd.: Calderbridge, UK, 2016. [Google Scholar]
- Nadykto, B.A.; Timofeeva, L.F. Storage of Plutonium and Nuclear Power Plant Actinide Wastes in the Form of Critical-Mass-Free Ceramics Containing Neutron Poisons. In Proceedings of the Global 2001 International Conference on: Back-End of the Fuel Cycle: From Research to Solutions, Paris, France, 9–13 September 2001; pp. 1–8. [Google Scholar]
- Harrison, M.T.; Scales, C.R.; Maddrell, E.R. Progress in the Assessment of Wasteforms for the Immobilisation of UK Civil Plutonium. In Proceedings of the WM2008 Conference, Phoenix, TX, USA, 24–28 February 2008; Volume 1. [Google Scholar]
- Nuclear Decommissioning Authority (NDA). Plutonium: Credible Options Analysis (Gate A); Nuclear Decommissioning Authority (NDA): Moor Row, UK, 2010.
- Blackburn, L.R.; Gardner, L.J.; Sun, S.K.; Maddrell, E.R.; Stennett, M.C.; Corkhill, C.L.; Hyatt, N.C. Hot Isostatically Pressed Zirconolite Wasteforms for Actinide Immobilisation. IOP Conf. Ser. Mater. Sci. Eng. 2020, 818, 012010. [Google Scholar] [CrossRef]
- Blackburn, L.R.; Sun, S.-K.; Lawson, S.M.; Gardner, L.J.; Ding, H.; Corkhill, C.L.; Maddrell, E.R.; Stennett, M.C.; Hyatt, N.C. Synthesis and characterisation of Ca1-xCexZrTi2-2xCr2xO7: Analogue zirconolite wasteform for the immobilisation of stockpiled UK plutonium. J. Eur. Ceram. Soc. 2020, 40, 5909–5919. [Google Scholar] [CrossRef]
- Blackburn, L.R.; Sun, S.K.; Gardner, L.J.; Maddrell, E.R.; Stennett, M.C.; Hyatt, N.C. Influence of Transition Metal Charge Compensation Species on Phase Assemblage in Zirconolite Ceramics for Pu Immobilisation. MRS Adv. 2020, 5, 93–101. [Google Scholar] [CrossRef]
- Blackburn, L.R.; Sun, S.; Gardner, L.J.; Maddrell, E.R.; Stennett, M.C.; Hyatt, N.C. A systematic investigation of the phase assemblage and microstructure of the zirconolite CaZr1-xCexTi2O7 system. J. Nucl. Mater. 2020, 535, 152137. [Google Scholar] [CrossRef]
- Williams, C.T.; Gieré, R. Zirconolite: A Review of Localities Worldwide, and a Compilation of its Chemical Compositions. Bull. Nat. Hist. Mus. Lond. 1996, 52, 1–24. [Google Scholar]
- Thornber, S.M.; Stennett, M.C.; Vance, E.R.; Chavara, D.T.; Watson, I.; Jovanovic, M.; Davis, J.; Gregg, D.; Hyatt, N.C. A preliminary validation study of PuO2 incorporation into zirconolite glass-ceramics. MRS Adv. 2018, 3, 1065–1071. [Google Scholar] [CrossRef]
- Thornber, S.M.; Stennett, M.C.; Hyatt, N.C. Investigation of Ce incorporation in zirconolite glass-ceramics for UK plutonium disposition. MRS Adv. 2016, 2, 699–704. [Google Scholar] [CrossRef]
- Mason, A.R.; Thornber, S.M.; Stennett, M.C.; Gardner, L.J.; Lützenkirchen-Hecht, D.; Hyatt, N.C. Preliminary investigation of chlorine speciation in zirconolite glass-ceramics for plutonium residues by analysis of Cl K-edge XANES. MRS Adv. 2020, 5, 37–43. [Google Scholar] [CrossRef]
- Thornber, S.M.; Mottram, L.M.; Mason, A.R.; Thompson, P.; Stennett, M.C.; Hyatt, N.C. Solubility, speciation and local environment of chlorine in zirconolite glass–ceramics for the immobilisation of plutonium residues. RSC Adv. 2020, 10, 32497–32510. [Google Scholar] [CrossRef]
- Maddrell, E.; Thornber, S.; Hyatt, N.C. The influence of glass composition on crystalline phase stability in glass-ceramic wasteforms. J. Nucl. Mater. 2015, 456, 461–466. [Google Scholar] [CrossRef]
- Coelho, A.; Cheary, R.; Smith, K. Analysis and Structural Determination of Nd-Substituted Zirconolite-4M. J. Solid State Chem. 1997, 129, 346–359. [Google Scholar] [CrossRef]
- Gatehouse, B.M.; Grey, I.E.; Hill, R.J.; Rossell, H.J. Zirconolite, CaZrxTi3−xO7; Structure Refinements for Near-End-Member Compositions with x = 0.85 and 1.30. Acta Cryst. 1981, B37, 306–312. [Google Scholar] [CrossRef]
- Cheary, R.W.; Coelho, A.A. A site occupancy analysis of zirconolite CaZrxTi3−xO7. Phys. Chem. Miner. 1997, 24, 447–454. [Google Scholar] [CrossRef]
- Whittle, K.R.; Smith, K.L.; Hyatt, N.C.; Lumpkin, G.R. Neutron and Resonant X-ray Diffraction Studies of Zirconolite-2M. Mater. Res. Soc. Symp. Proc. 2008, 1107, 331. [Google Scholar] [CrossRef]
- Grey, I.; Mumme, W.; Ness, T.; Roth, R.; Smith, K. Structural relations between weberite and zirconolite polytypes—Refinements of doped 3T and 4M Ca2Ta2O7 and 3T CaZrTi2O7. J. Solid State Chem. 2003, 174, 285–295. [Google Scholar] [CrossRef]
- Perera, D.S.; Stewart, M.W.A.; Li, H.; Day, R.A.; Vance, E.R. Tentative Phase Relationships in the System CaHfTi2O7-Gd2Ti2O7 with up to 15 mol% Additions of Al2TiO5 and MgTi2O5. J. Am. Ceram. Soc. 2002, 85, 2919–2924. [Google Scholar] [CrossRef]
- White, T.J.; Segall, R.L.; Hutchison, J.L.; Barry, J.C. Polytypic behaviour of zirconolite. Proc. R. Soc. London. Ser. Math. Phys. Sci. 1984, 392, 343–358. [Google Scholar]
- Kessoft, S.; Sinclair, W.; Ringwood, A. Solid solution limits in synroc zirconolite. Nucl. Chem. Waste Manag. 1983, 4, 259–265. [Google Scholar] [CrossRef]
- Vance, E.R.; Lumpkin, G.R.; Carter, M.L.; Cassidy, D.J.; Ball, C.J.; Day, R.A.; Begg, B.D. Incorporation of Uranium in Zirconolite (CaZrTi2O7). J. Am. Ceram. Soc. 2002, 85, 1853–1859. [Google Scholar] [CrossRef]
- Begg, B.D.; Day, R.A.; Brownscombe, A. Structural Effect of Pu Substitutions on the Zr-site in Zirconolite. Mat. Res. Soc. Symp. Proc. 2001, 663, 259. [Google Scholar] [CrossRef]
- Mazzi, F.; Munno, R. Calciobetafite (new mineral of the pyrochlore group) and related minerals from Campi Flegrei, Italy: Crystal structures of polymignyte and zirkelite: Comparison with pyrochlore and zirconolite. Am. Mineral. 1983, 68, 262–276. [Google Scholar]
- Lumpkin, G.R.; Smith, K.L.; Blackford, M.G. Electron microscope study of Synroc before and after exposure to aqueous solutions. J. Mater. Res. 1991, 6, 2218–2233. [Google Scholar] [CrossRef]
- Smith, K.; Lumpkin, G.; Blackford, M.; Day, R.; Hart, K. The durability of synroc. J. Nucl. Mater. 1992, 190, 287–294. [Google Scholar] [CrossRef]
- McGlinn, P.J.; Hart, K.P.; Loi, E.H.; Vance, E.R. pH Dependence of the Aqueous Dissolution Rates of Perovskite and Zirconolite at 90°C. MRS Proc. 1994, 353, 847–854. [Google Scholar] [CrossRef]
- Clark, B.M.; Sundaram, S.K.; Misture, S.T. Polymorphic Transitions in Cerium-Substituted Zirconolite (CaZrTi2O7). Sci. Rep. 2017, 7, 2–10. [Google Scholar] [CrossRef]
- Li, W.; Dong, F.; Bian, L.; Zou, H.; Song, M. Phase relations, microstructure, and valence transition studies on CaZr1−xCexTi2O7 (0.0 ≤ x ≤ 1.0) system. J. Rare Earths 2018, 36, 1184–1189. [Google Scholar] [CrossRef]
- Gilbert, M.R.; Selfslag, C.; Walter, M.; Stennett, M.C.; Somers, J.; Hyatt, N.C.; Livens, F.R. Synthesis and characterisation of Pu-doped zirconolites-(Ca1−xPux)Zr(Ti2−2xFe2x)O7. IOP Conf. Ser. Mater. Sci. Eng. 2010, 9, 012007. [Google Scholar]
- Vance, E.; Ball, C.; Day, R.; Smith, K.; Blackford, M.; Begg, B.; Angel, P. Actinide and rare earth incorporation into zirconolite. J. Alloy. Compd. 1994, 213, 406–409. [Google Scholar] [CrossRef]
- Begg, B.D.; Vance, E.R.; Lumpkin, G.R. Charge Compensation and the Incorporation of Cerium in Zirconolite and Perovskite. Mat. Res. Soc. Symp. Proc. 1998, 506, 79–86. [Google Scholar] [CrossRef]
- Begg, B.D.; Vance, E.R. The Incorporation of Cerium in Zirconolite. Mat. Res. Soc. Symp. Proc. 1997, 465, 333–340. [Google Scholar] [CrossRef]
- Vance, E.R.; Angel, P.J.; Begg, B.D.; Day, R.A. Zirconolite-Rich Titanate Ceramics for High-Level Actinide Wastes. Mat. Res. Soc. Symp. Proc. 1994, 333, 293–298. [Google Scholar] [CrossRef]
- Begg, B.; Vance, E.; Conradson, S. The incorporation of plutonium and neptunium in zirconolite and perovskite. J. Alloys Compd. 1998, 271, 221–226. [Google Scholar] [CrossRef]
- Davoisne, C.; Stennett, M.C.; Hyatt, N.C.; Peng, N.; Jeynes, C.; Lee, W.E. Krypton irradiation damage in Nd-doped zirconolite and perovskite. J. Nucl. Mater. 2011, 415, 67–73. [Google Scholar] [CrossRef]
- Stefanovsky, S.V.; Troole, A.Y.; Lapina, M.I.; Nikonov, B.S.; Sivtsov, A.V.; Yudintsev, S.V. XRD, SEM and TEM study of the Gd-doped zirconolites. Mater. Res. Soc. Symp. Proc. 2002, 713, 345–350. [Google Scholar] [CrossRef]
- Ji, S.; Li, Y.; Ma, S.; Liu, C.; Shih, K.; Liao, C.Z. Synergistic effects of Ln and Fe Co-Doping on phase evolution of Ca1−xLnxZrTi2−xFexO7 (Ln = La, Nd, Gd, Ho, Yb) ceramics. J. Nucl. Mater. 2018, 511, 428–437. [Google Scholar] [CrossRef]
- Ma, S.; Ji, S.; Liao, C.; Liu, C.; Shih, K.; He, W. Effects of ionic radius on phase evolution in Ln-Al co-doped Ca1−xLnxZrTi2−xAlxO7 (Ln = La, Nd, Gd, Ho, Yb) solid solutions. Ceram. Int. 2018, 44, 15124–15132. [Google Scholar] [CrossRef]
- Jafar, M.; Achary, S.N.; Salke, N.P.; Sahu, A.K.; Rao, R.; Tyagi, A.K. X-ray diffraction and Raman spectroscopic investigations on CaZrTi2O7-Y2Ti2O7 system: Delineation of phase fields consisting of potential ceramic host materials. J. Nucl. Mater. 2016, 475, 192–199. [Google Scholar] [CrossRef]
- Jafar, M.; Sengupta, P.; Achary, S.N.; Tyagi, A.K. Phase evolution and microstructural studies in CaZrTi2O7 (zirconolite)-Sm2Ti2O7 (pyrochlore) system. J. Eur. Ceram. Soc. 2014, 34, 4373–4381. [Google Scholar] [CrossRef]
- Jafar, M.; Sengupta, P.; Achary, S.N.; Tyagi, A.K. Phase evolution and microstructural studies in CaZrTi2O7-Nd2Ti2O7 system. J. Am. Ceram. Soc. 2014, 97, 609–616. [Google Scholar] [CrossRef]
- Zhang, Y.B.; Wang, J.; Wang, J.X.; Huang, Y.; Luo, P.; Liang, X.F.; Tan, H.B. Phase evolution, microstructure and chemical stability of Ca1−xZr1−xGd2xTi2O7 (0.0 ≤ x ≤ 1.0) system for immobilizing nuclear waste. Ceram. Int. 2018, 44, 13572–13579. [Google Scholar] [CrossRef]
- Blackburn, L.R.; Crawford, R.; Walling, S.A.; Gardner, L.J.; Cole, M.R.; Sun, S.-K.; Gausse, C.; Mason, A.R.; Stennett, M.C.; Maddrell, E.R.; et al. Influence of accessory phases and surrogate type on accelerated leaching of zirconolite wasteforms. NPJ Mater. Degrad. 2021, 5, 1–11. [Google Scholar] [CrossRef]
- Li, H.; Zhang, Y.; McGlinn, P.J.; Moricca, S.; Begg, B.D.; Vance, E.R. Characterisation of stainless steel-synroc interactions under hot isostatic pressing (HIPing) conditions. J. Nucl. Mater. 2006, 355, 136–141. [Google Scholar] [CrossRef]
- Lumpkin, G.R.; Smith, K.L.; Blackford, M.G. Partitioning of uranium and rare earth elements in synroc: Effect of impurities, metal additive, and waste loading. J. Nucl. Mater. 1995, 224, 31–42. [Google Scholar] [CrossRef]
- Zhang, Y.; Stewart, M.W.A.; Li, H.; Carter, M.L.; Vance, E.R.; Moricca, S. Zirconolite-rich titanate ceramics for immobilisation of actinides—Waste form/HIP can interactions and chemical durability. J. Nucl. Mater. 2009, 395, 69–74. [Google Scholar] [CrossRef]
- ASTM C1285-21; Standard Test Methods for Determining Chemical Durability of Nuclear, Hazardous, and Mixed Waste Glasses and Multiphase Glass Ceramics: The Product Consistency Test (PCT). ASTM International: West Conshohocken, PA, USA, 2014.
- Vance, E.R.; Jostsons, A.; Day, R.A.; Ball, C.J.; Begg, B.D.; Angel, P.J. Excess Pu Disposition in Zirconolite-Rich Synroc. Mat. Res. Soc. Symp. Proc. 1996, 412, 41–47. [Google Scholar] [CrossRef]
- Mikhailenko, N.P.; Ochkin, A.V.; Stefanovsky, S.V.; Kirjanova, O.I. Phase relations and chemical durability of ceramics in the pseudo-binary system: CaZrTi2O7-GdAlO3. Mater. Res. Soc. Symp. Proc. 2004, 807, 327–331. [Google Scholar] [CrossRef]
- Mikhailenko, N.; Stefanovsky, S.; Ochkin, A. Phase relations and elemental distribution among co-existing phases in the ceramics of the pseudobinary system CaZrTi2O7-LnAlO3 (Ln = Nd, Sm). Mat. Res. Soc. Symp. Proc. 2007, 985, 211–216. [Google Scholar]
- Shannon, R.D. Revised Effective Ionic Radii and Systematic Studies of Interatomic Distances in Halides and Chalcogenides. Acta Cryst. 1976, 32, 751–767. [Google Scholar] [CrossRef]
- Putnam, R.L.; Navrotsky, A.; Woodfield, B.F.; Shapiro, J.L.; Stevens, R.; Boerio-Goates, J. Thermochemistry of Hf-Zirconolite, CaHfTi2O7. Mat. Res. Soc. Symp. Proc. 2011, 556, 11. [Google Scholar] [CrossRef]
- Hyatt, N.C.; Stennett, M.C.; Maddrell, E.R.; Lee, W.E. Single Phase Ceramic Wasteforms for Plutonium Disposition. Adv. Sci. Technol. 2006, 45, 2004–2011. [Google Scholar]
- Zhang, K.; Yin, D.; He, Z.; Luo, B.; Zhang, H. Combustion synthesis of Hf-doped zirconolite-rich composite waste forms and the aqueous durability. J. Adv. Ceram. 2019, 8, 448–455. [Google Scholar] [CrossRef]
- Strachan, D.M.; Turcotte, R.P.; Barnes, B.O. MCC-1: A Standard Leach Test for Nuclear Waste Forms. Nucl. Technol. 1982, 56, 306–312. [Google Scholar] [CrossRef]
- Zhang, K.; Yin, D.; Xu, K.; Zhang, H. Self-propagating synthesis and characterization studies of Gd-bearing Hf-zirconolite ceramic waste forms. Materials 2019, 12, 178. [Google Scholar] [CrossRef]
- Caurant, D.; Loiseau, P.; Bardez, I. Structural characterization of Nd-doped Hf-zirconolite Ca1−xNdxHfTi2−xAlxO7 ceramics. J. Nucl. Mater. 2010, 407, 88–99. [Google Scholar] [CrossRef]
- Loiseau, P.; Caurant, D.; Baffier, N.; Fillet, C. Structural characterization of polycrystalline (Nd,Al)-substituted zirconolite. Mat. Res. Soc. Symp. Proc. 2003, 757, 243–250. [Google Scholar] [CrossRef]
- Zhang, K.; Yin, D.; He, Z.; Xue, J.; Zhao, W.; Zhang, H. Combustion synthesis and characterizations of Sm2O3 doped zirconolite-rich waste forms with CuO as oxidant. J. Rare Earths. 2019, 37, 1359–1365. [Google Scholar] [CrossRef]
- Leturcq, G.; Auzemerie, B. Study of the Effects of Potential Impurities from Actinide Flux on the Zirconolite Microstructure. Mat. Res. Soc. Symp. Proc. 2004, 807, 110–115. [Google Scholar] [CrossRef]
- Ali, R.; Yashima, M. Space group and crystal structure of the Perovskite CaTiO3 from 296 to 1720 K. J. Solid State Chem. 2005, 178, 2867–2872. [Google Scholar] [CrossRef]
- Sasaki, S.; Prewitt, C.T.; Bass, J.D. Orthorhombic Perovskite CaTiO3 and CdTiO3: Structure and Space Group. Acta Crystallogr. Sect. Struct. Chem. 1987, 43, 1668–1674. [Google Scholar] [CrossRef]
- Yang, J.; Zhao, M.; Shahid, M.; Feng, J.; Wan, C.; Pan, W. Electronic structure, anisotropic elastic and thermal properties of monoclinic Ca2Nb2O7. Ceram. Int. 2016, 42, 9426–9432. [Google Scholar] [CrossRef]
- Sleight, A.W. High Pressure Substitutions for Cd in Cd2Nb2O7 and Cd2Ta2O7. Mat. Res. Bull. 1974, 9, 1437–1442. [Google Scholar] [CrossRef]
- Botelho, G.; Nogueira, I.C.; Moraes, E.; Longo, E. Study of structural and optical properties of CaMoO4 nanoparticles synthesized by the microwave-assisted solvothermal method. Mater. Chem. Phys. 2016, 183, 110–120. [Google Scholar] [CrossRef]
- Singh, N.P.; Devi, Y.R.; Sh, B.S.; Singh, T.D. Effects of annealing temperature on structural and luminescence properties of CdMoO4:Dy3+ phosphor synthesized at room temperature by co-precipitation method. Solid State Sci. 2020, 102, 106172. [Google Scholar] [CrossRef]
- Begg, B.D.; Vance, E.R.; Hunter, B.A.; Hanna, J.V. Zirconolite transformation under reducing conditions. J. Mater. Res. 1998, 13, 3181–3190. [Google Scholar] [CrossRef]
- Blackburn, L.R.; Townsend, L.T.; Wilkins, M.C.D.; Ina, T.; Kuman, M.; Sun, S.-K.; Mason, A.R.; Gardner, L.J.; Stennett, M.C.; Corkhill, C.L.; et al. Underpinning the use of indium as a neutron absorbing additive in zirconolite by X-ray absorption spectroscopy. Sci. Rep. 2023, 13, 9329. [Google Scholar] [CrossRef]
- Gong, W.; Naz, S.; Lutze, W.; Busch, R.; Prinja, A.; Stoll, W. Safe disposal of surplus plutonium. J. Nucl. Mater. 2001, 295, 295–299. [Google Scholar] [CrossRef]
- Squire, J.; Maddrell, E.R.; Hyatt, N.C.; Stennett, M.C. Influence of lubricants and attrition milling parameters on the quality of zirconolite ceramics, consolidated by hot isostatic pressing, for immobilisation of plutonium. Int. J. Appl. Ceram. Technol. 2015, 12, E92–E104. [Google Scholar] [CrossRef]


| Source | Amount (teHM) |
|---|---|
| Unirradiated separated plutonium in product stores at reprocessing plants | 137.0 |
| Unirradiated separated plutonium in the course of manufacture or fabrication and plutonium contained in unirradiated semi-fabricated or unfinished products at fuel or other fabricating plants elsewhere | 0.4 |
| Plutonium contained in unirradiated MOX fuel or other fabricated products at reactor sites or elsewhere | 2.0 |
| Unirradiated separated plutonium held elsewhere | 1.2 |
| Unirradiated separated plutonium owned by foreign bodies under UK safeguards | 24.1 |
| Total | 164.7 |
| Element | Cost of Component Oxide (£/g) | Merck Product Code | Purity | Thermal Neutron Cross Section (σa/barns) |
|---|---|---|---|---|
| Gd | 2.76 | 278513 | 99.9% trace metals basis | 4.90 × 104 |
| Hf | 151.00 | 203394 | ≥99.95% | 104 |
| Sm | 4.20 | 228672 | 99.9% trace metals basis | 5.92 × 103 |
| Cd | 7.00 | 202894 | ≥99.99% trace metals basis | 2.45 × 104 |
| In | 7.50 | 289418 | 99.99% trace metals basis | 194 |
| B * | 1.76 | 378119 | 98% | 767 |
| Unit Cell Parameters | ||||
|---|---|---|---|---|
| Composition | a (Å) | b (Å) | c (Å) | β (°) |
| CaZrTi2O7 | 12.447 | 7.272 | 11.384 | 100.54 |
| CaHf0.5Zr0.5Ti2O7 | 12.434 | 7.265 | 11.350 | 100.58 |
| CaHfTi2O7 | 12.420 | 7.262 | 11.343 | 100.57 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Blackburn, L.R.; Corkhill, C.L.; Hyatt, N.C. A Review of Zirconolite Solid Solution Regimes for Plutonium and Candidate Neutron Absorbing Additives. Ceramics 2023, 6, 1330-1347. https://doi.org/10.3390/ceramics6030082
Blackburn LR, Corkhill CL, Hyatt NC. A Review of Zirconolite Solid Solution Regimes for Plutonium and Candidate Neutron Absorbing Additives. Ceramics. 2023; 6(3):1330-1347. https://doi.org/10.3390/ceramics6030082
Chicago/Turabian StyleBlackburn, Lewis R., Claire L. Corkhill, and Neil C. Hyatt. 2023. "A Review of Zirconolite Solid Solution Regimes for Plutonium and Candidate Neutron Absorbing Additives" Ceramics 6, no. 3: 1330-1347. https://doi.org/10.3390/ceramics6030082
APA StyleBlackburn, L. R., Corkhill, C. L., & Hyatt, N. C. (2023). A Review of Zirconolite Solid Solution Regimes for Plutonium and Candidate Neutron Absorbing Additives. Ceramics, 6(3), 1330-1347. https://doi.org/10.3390/ceramics6030082