
Zirconolite Matrices for the Immobilization of REE–Actinide Wastes

 ,
,  , and
, and 
Abstract
1. Introduction
2. Zirconolite in Nature
3. Zirconolite in the Family of Polyphase SYNROC Ceramics
4. On the Amounts and Composition of the REE–Actinide Fraction in Waste
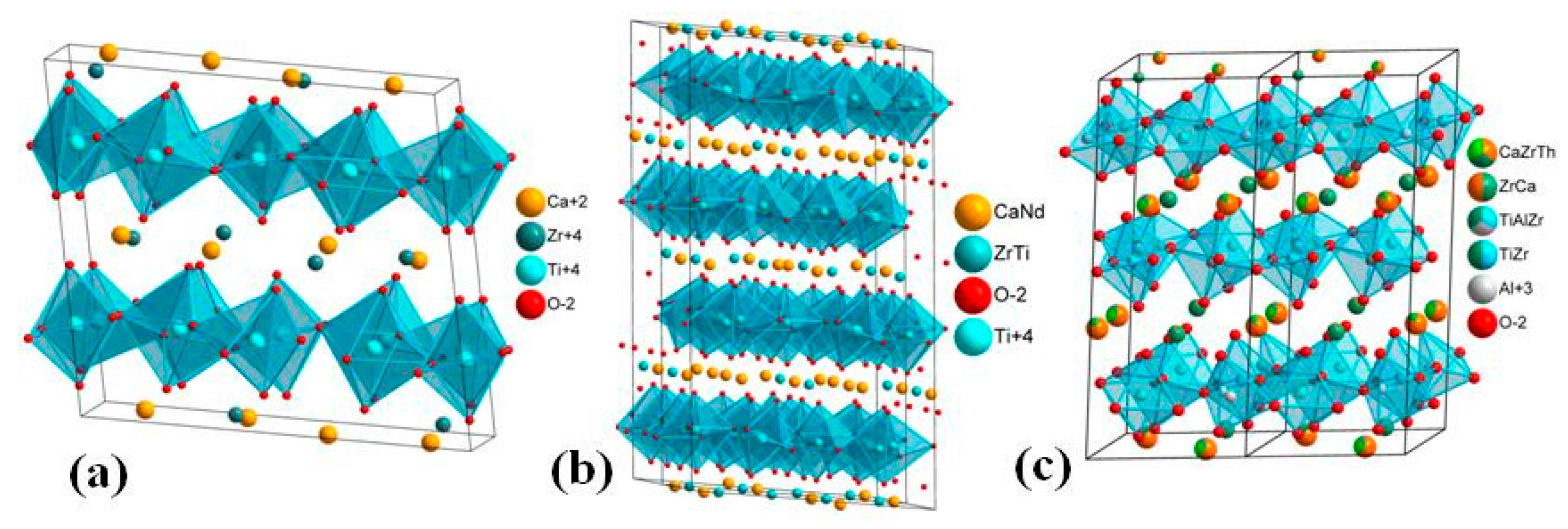
5. Crystal Chemistry of Zirconolite and Its Polytype
6. Influence of Synthesis Conditions on the Structure of Zirconolite-Bearing Matrices
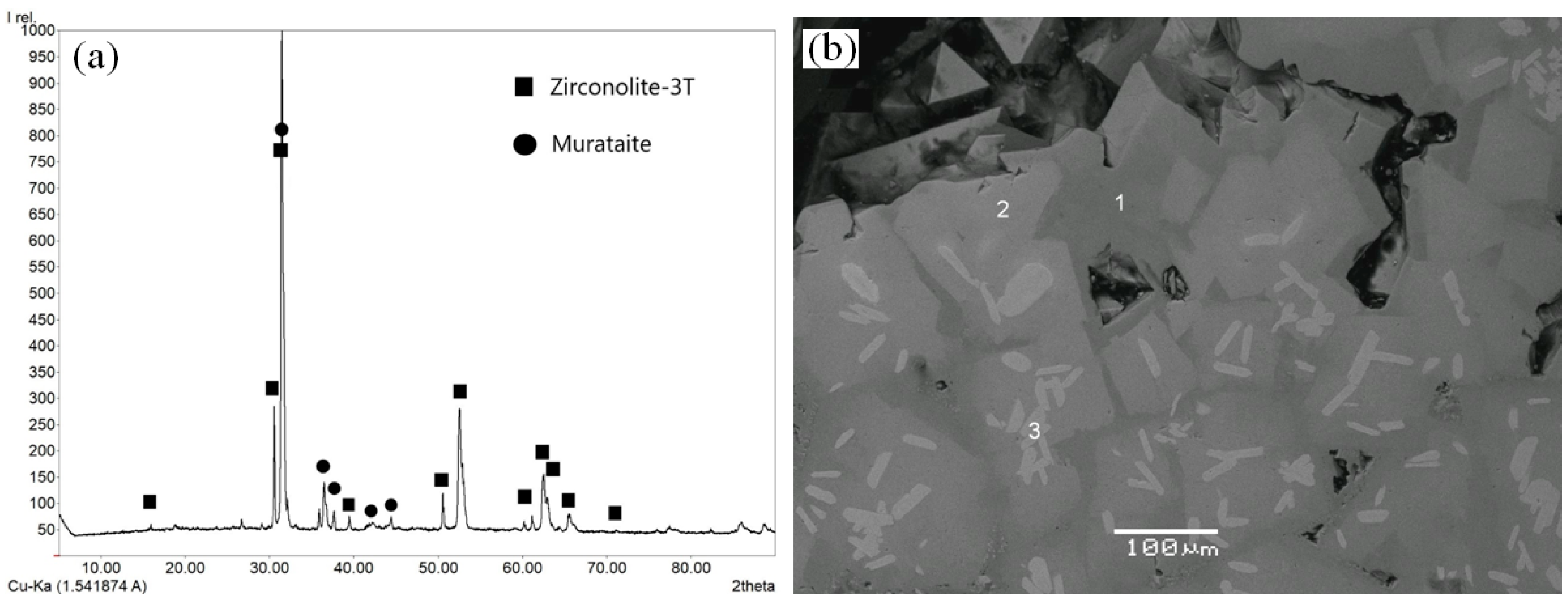
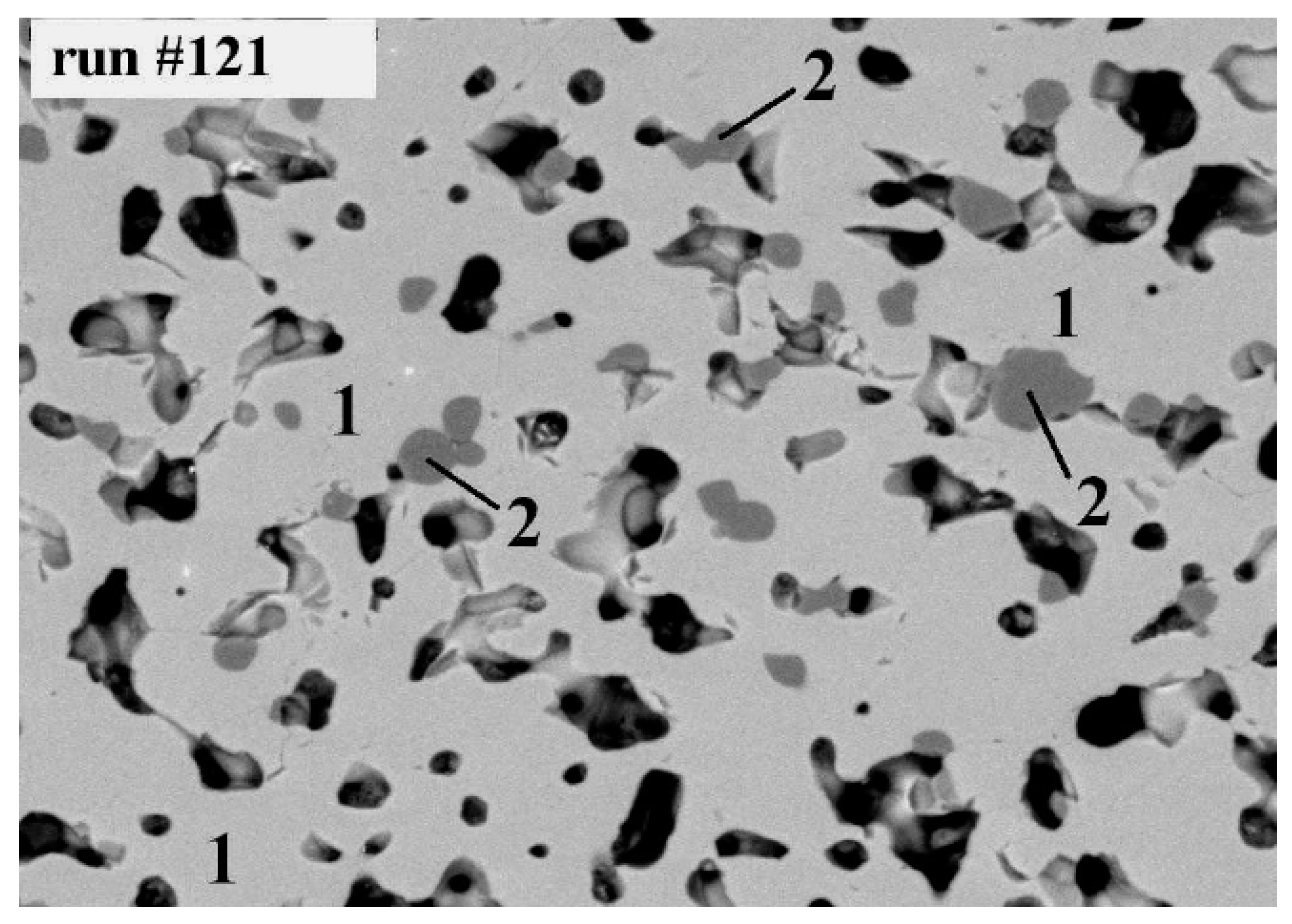
7. Samples Obtained by Melt Crystallization
8. Other Methods for Obtaining Zirconolite-Containing Matrices
9. Influence of Oxidative Conditions of Synthesis on the Structure of Matrices
10. Zirconolite Matrices with Transuranium Actinides
11. Radiation Damage in Zirconolite Matrices
12. Other Applications of Zirconolite Bearing Matrices
13. On the Role of Perovskite in Zirconolite Matrices
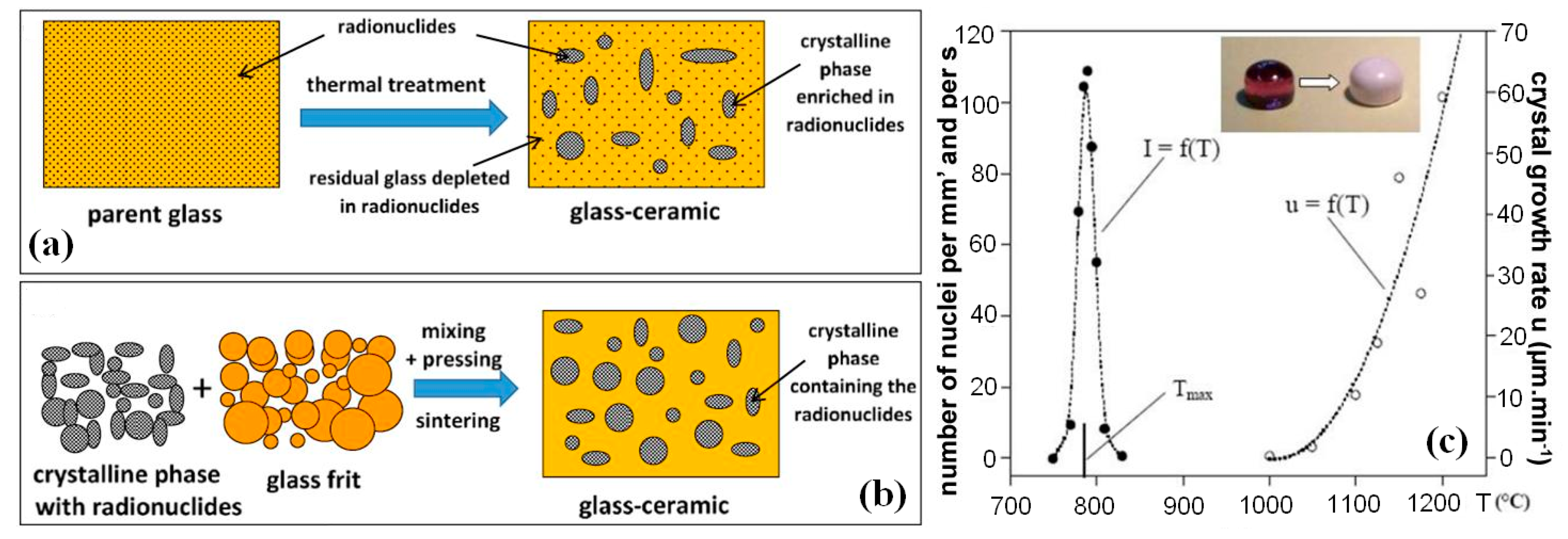
14. Glass Crystalline Materials (GCM) with Zirconolite Crystalline Phase
15. Technologies for Fabrication of Zirconolite Wasteforms
16. Synthesis of Glass Ceramics by Radiation-Induced Heating of REE–MA Glass
17. On Borehole Disposal of Actinide Zirconolite Matrices
18. Discussion and Conclusions
- For trivalent REE, actinides (An = Pu, Am, Cm), and REE–MA fraction:
- For tetravalent REE (Ce) and actinides (An = Th, U, Np, Pu):
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- NEA. Meeting Climate Change Targets. The Role of Nuclear Energy; NEA No. 7628; OECD/NEA Publishing: Paris, France, 2022; p. 49. Available online: https://oecd-nea.org/upload/docs/application/pdf/2022-05/7628_strategic_briefing_climate_change.pdf (accessed on 17 May 2023).
- IAEA. Energy, Electricity and Nuclear Power Estimates for the Period up to 2050; IAEA: Vienna, Austria, 2022; 137p, Available online: https://www.iaea.org/publications/13591/energy-electricity-and-nuclear-power-estimates-for-the-period-up-to-2050 (accessed on 17 May 2023).
- IAEA. Implications of Partitioning and Transmutation in Radioactive Waste Management; Report 435; IAEA: Vienna, Austria, 2004; p. 126. [Google Scholar]
- Zilberman, B.Y.; Puzikov, E.A.; Ryabkov, D.V.; Makarychev-Mikhailov, M.N.; Shadrin, A.Y.; Fedorov, Y.S.; Simonenko, V.A. Development, analysis, and simulation of a technological structure for reprocessing irradiated nuclear fuel from nuclear power plants by water-extraction methods. At. Energy 2009, 107, 333. [Google Scholar]
- Nash, K.L.; Lumetta, G.J. (Eds.) Advanced Separation Techniques for Nuclear Fuel Reprocessing and Radioactive Waste Treatment; Woodhead: Cambridge, UK, 2011. [Google Scholar]
- NEA. Spent Nuclear Fuel Reprocessing Flowsheet; NEA/OECD: Paris, France, 2012; p. 120. [Google Scholar]
- Modolo, G.; Geist, A.; Miguirditchian, M. Minor actinide separations in the reprocessing of spent nuclear fuels: Recent advances in Europe. In Reprocessing and Recycling of Spent Nuclear Fuel; Taylor, R., Ed.; Chapter 10; Woodhead Publishing Series in Energy: Cambridge, UK, 2015; pp. 245–287. [Google Scholar] [CrossRef]
- Veliscek-Carolan, J. Separation of actinides from spent nuclear fuel: A review. J. Hazard. Mater. 2016, 318, 266–281. [Google Scholar] [CrossRef] [PubMed]
- NEA. State-of-the-Art Report on the Progress of Nuclear Fuel Cycle Chemistry; NEA: Paris, France, 2018; p. 299. [Google Scholar]
- Baron, P.; Cornet, S.M.; Collins, E.D.; De Angelis, G.; Del Cul, G.; Fedorov Yu Glatz, J.P.; Ignatiev, V.; Inoue, T.; Khaperskaya, A.; Kim, I.T.; et al. A review of separation processes proposed for advanced fuel cycles based on technology readiness level assessments. Progr. Nucl. Energy 2019, 117, 103091. [Google Scholar] [CrossRef]
- Lutze, W.; Ewing, R.C. (Eds.) Radioactive Waste Forms for the Future; Elsevier: New York, NY, USA, 1988; p. 778. [Google Scholar]
- Donald, I.W.; Metcalfe, B.L.; Taylor, R.N.J. The immobilization of high-level radioactive wastes using ceramics and glasses. J. Mater. Sci. 1997, 32, 5851–5887. [Google Scholar] [CrossRef]
- Caurant, D.; Loiseau, P.; Majérus, O.; Aubin-Chevaldonnet, V.; Bardez, I.; Quintas, A. Glasses, Glass-Ceramics and Ceramics for Immobilization of Highly Radioactive Nuclear Wastes; Nova Science Publishers: New York, NY, USA, 2009; p. 445. [Google Scholar]
- Donald, I.W. Waste Immobilization in Glass and Ceramic Based hosts: Radioactive, Toxic, and Hazardous Wastes; John Wiley & Sons Ltd.: Chichester, West Sussex, UK, 2010. [Google Scholar]
- Yudintsev, S.V. Isolation of separated waste of nuclear industry. Radiochemistry 2021, 63, 527–555. [Google Scholar] [CrossRef]
- Orlova, A.I.; Ojovan, M.I. Ceramic Mineral Waste-Forms for Nuclear Waste Immobilization. Materials 2019, 12, 2638. [Google Scholar] [CrossRef]
- Caurant, D.; Majérus, O. Glasses and Glass-Ceramics for Nuclear Waste Immobilization. In Encyclopedia of Materials: Technical Ceramics and Glasses; Pomeroy, M., Ed.; Elsevier: Kidlington, Oxford, 2021; Volume 2, pp. 762–790. Available online: https://hal.science/hal-03375652/document (accessed on 15 May 2023). [CrossRef]
- Ojovan, M.I.; Petrov, V.A.; Yudintsev, S.V. Glass Crystalline Materials as Advanced Nuclear Wasteforms. Sustainability 2021, 13, 4117. [Google Scholar] [CrossRef]
- McCloy, J.S.; Schuller, S. Vitrification of wastes: From unwanted to controlled crystallization, a review. Comptes Rendus. Géoscience Sci. Planète 2022, 354, 121–160. [Google Scholar] [CrossRef]
- Ojovan, M.I.; Steinmetz, H.J. Approaches to Disposal of Nuclear Waste. Energies 2022, 15, 7804. [Google Scholar] [CrossRef]
- Ewing, R.C.; Weber, W.J. Actinide waste forms and radiation effects. In The Chemistry of the Actinide and Transactinide Elements; Morss, L.R., Edelstein, N.M., Fuger, J., Eds.; Springer: Dordrecht, The Netherlands, 2011; Volume 6, Chapter 35; pp. 3813–3889. [Google Scholar] [CrossRef]
- Vance, E.R.; Zhang, Y.; Gregg, D.J. Ceramic Waste Forms. In Comprehensive Nuclear Materials, 2nd ed.; Konings, R., Stoller, R., Eds.; Elsevier: Amsterdam, The Netherlands, 2020. [Google Scholar]
- Omel’yanenko, B.I.; Livshits, T.S.; Yudintsev, S.V.; Nikonov, B.S. Natural and artificial minerals as matrices for immobilization of actinides. Geol. Ore Depos. 2007, 49, 173–193. [Google Scholar] [CrossRef]
- Lumpkin, G.R. Ceramic waste forms for actinides. Elements 2006, 2, 365–372. [Google Scholar] [CrossRef]
- Lumpkin, G.R.; Geisler-Wierwille, T. Minerals and Natural Analogues. In Comprehensive Nuclear Materials; Elsevier: Amsterdam, The Netherlands, 2012; Volume 5, Chapter 5.22; pp. 563–600. [Google Scholar] [CrossRef]
- Lumpkin, G.R.; Gao YGiere, R. The role of Th-U minerals in assessing the performance of nuclear waste forms. Mineral. Mag. 2014, 78, 1071–1095. [Google Scholar] [CrossRef]
- Lumpkin, G.R. Ceramic Host Phases for Nuclear Waste Remediation. In Experimental and Theoretical Approaches to Actinide Chemistry, 1st ed.; Gibson, J.K., de Jong, W.A., Eds.; John Wiley & Sons Ltd.: Hoboken, NJ, USA, 2018; pp. 333–377. [Google Scholar] [CrossRef]
- Ringwood, A.E. Safe Disposal of High-Level Nuclear Reactor Wastes: A New Strategy; ANU Press: Canberra, Australia, 1978; p. 64. [Google Scholar]
- Ringwood, A.E.; Kesson, S.E.; Ware, N.G.; Hibberson, W.O.; Major, A. The SYNROC process: A geochemical approach to nuclear waste immobilization. Geochem. J. 1979, 13, 141–169. [Google Scholar] [CrossRef]
- Ringwood, A.E.; Kesson, S.E.; Ware, N.G.; Hibberson, W.O.; Major, A. Immobilisation of high-level nuclear reactor wastes in SYNROC. Nature 1979, 278, 219–223. [Google Scholar] [CrossRef]
- Fielding, P.E.; White, T.J. Crystal chemical incorporation of high-level waste species in alumino-titanate-based ceramics: Valence, location, radiation damage, and hydrothermal durability. J. Mater. Res. 1987, 2, 388–414. [Google Scholar] [CrossRef]
- Laverov, N.P.; Omel’yanenko, B.I.; Yudintsev, S.V.; Nikonov, B.S. Zirconolite as a matrix for immobilization of high-level radioactive wastes (HLW). Geol. Ore Depos. 1996, 38, 345–352. [Google Scholar]
- Mazzi, F.; Munno, R. Calciobetafite (new mineral of the pyrochlore group) and related minerals from Campi Flegrei, Italy; crystal structures of polymignyte and zirkelite: Comparison with pyrochlore and zirconolite. Am. Mineral. 1983, 68, 262–276. [Google Scholar]
- Williams, C.T.; GiereR. Zirconolite: A review of localities worldwide, and a compilation of its chemical compositions. Bull. Nat. Hist. Mus. Lond. 1996, 52, 1–24. [Google Scholar]
- Gieré, R.; Williams, C.T.; Lumpkin, G.R. Chemical characteristics of natural zirconolite. Schweiz. Mineral. Und Petrogr. Mitteilungun 1998, 78, 433–459. [Google Scholar]
- Della Ventura, G.; Bellatreccia, F.; Williams, C.T. Zirconolite with significant REEZrNb(Mn,Fe)O7 from a xenolith of the Laacher See eruptive center, Eifel volcanic region, Germany. Can. Mineral. 2000, 38, 57–65. [Google Scholar] [CrossRef]
- Zubkova, N.V.; Chukanov, N.V.; Pekov, I.V.; Ternes, B.; Schüller, W.; Ksenofontov, D.A.; Pushcharov- sky, D.Y. The crystal structure of nonmetamict Nb-rich zirconolite-3T from the Eifel paleovolcanic region, Germany. Z. Für Krist. -Cryst. Mater. 2018, 233, 463–468. [Google Scholar] [CrossRef]
- Haifler, J.; Škoda, R.; Filip, J.; Larsen, A.O.; Rohlíček, J. Zirconolite from Larvik Plutonic Complex, Norway, its relationship to stefanweissite and nöggerathite, and contribution to the improvement of zirconolite end-member systematics. Am. Mineral. 2021, 106, 1255–1272. [Google Scholar] [CrossRef]
- Smith, J.V. Lunar mineralogy: A heavenly detective story. Am. Mineral. 1974, 59, 231–243. [Google Scholar]
- Frondel, J.W. Lunar Mineralogy; Wiley: New York, NY, USA, 1975; p. 323. [Google Scholar]
- Wang, N.; Mao, Q.; Zhang, T.; Hao, J.; Lin, Y. NanoSIMS and EPMA dating of lunar zirconolite. Prog. Earth Planet. Sci. 2021, 8, 51. [Google Scholar] [CrossRef]
- Gieré, R. Zirconolite, allanite and hoegbomite in a marble skarn from the Bergell contact aureole: Implications for mobility of Ti, Zr and REE. Contrib. Mineral. Petrol. 1986, 93, 459–470. [Google Scholar] [CrossRef]
- Borodin, L.S.; Nazarenko, I.I.; Richter, T.L. The new mineral zirconolite—A complex oxide of AB3O7 type. Dokl. Akad. Nauk. SSSR 1956, 110, 845–848. (In Russian) [Google Scholar]
- Borodin, L.S.; Bykova, A.V.; Kapitonova, T.A.; Pyatenko, Y.A. New data on zirconolite and its niobium variety. Dokl. Akad. Nauk. SSSR 1961, 134, 1022–1024. (In Russian) [Google Scholar]
- Pyatenko, Y.A.; Pudovkina, Z.V. The lattice metric of CaZrTi2O7 crystals. Kristallografiya 1964, 9, 98–100. (In Russian) [Google Scholar]
- Pudovkina, Z.V.; Pyatenko, Y.A. On Zirconolite and Its Crystallographic Characteristics; Proceedings of the Fersman Mineralogical Museum: Moscow, Russia, 1966; pp. 124–133. (In Russian) [Google Scholar]
- Campbell, J.; Hoenig, C.; Bazan, F.; Ryerson, F.; Guinan, M.; Van Konynenburg, R.; Rozsa, R. Properties of SYNROC-D Nuclear Waste Form: A State-of-the-Art Review; LLNL: Livermore, CA, USA, 1982. [Google Scholar]
- Wu, F.-Y.; Yang, Y.-H.; Mitchell, R.H.; Bellatreccia, F.; Li, Q.-L.; Zhao, Z.-F. In situ U–Pb and Nd–Hf– (Sr) isotopic investigations of zirconolite and calzirtite. Chem. Geol. 2010, 277, 178–195. [Google Scholar] [CrossRef]
- Farges, F.; Ewing, R.C.; Brown, G.E. The structure of aperiodic, metamict (Ca,Th)ZrTi2O7 (zirconolite): An EXAFS study of the Zr, Th, and U sites. J. Mater. Res. 1993, 8, 1983–1995. [Google Scholar] [CrossRef]
- Lumpkin, G.R.; Hart, K.P.; McGlinn, P.J.; Payne, T.E. Retention of actinides in natural pyrochlores and zirconolites. Radiochim. Acta 1994, 66/67, 469–474. [Google Scholar] [CrossRef]
- Gieré, R. Nuclear Wast Forms. In Proceedings of the International School Earth and Planetary Science, Siena, Italy, 24 September–3 October 1999; pp. 67–78. [Google Scholar]
- Giere, R. Minerals as natural analogue for crystalline nuclear waste forms. A Geochemical and Mineralogical Approach to Environmental Protection. In Proceedings of the Intern, School Earth and Planetary Science, Siena, Italy, 25 September–2 October 2000; pp. 83–101. [Google Scholar]
- Lumpkin, G.R. Alpha-decay damage and aqueous durability of actinide host phases in natural systems. J. Nucl. Mater. 2001, 289, 136–166. [Google Scholar] [CrossRef]
- Kesson, S.E.; Sinclair, W.J.; Ringwood, A.E. Solid solution limits in Synroc zirconolite. Nucl. Chem. Waste Manag. 1983, 4, 259–265. [Google Scholar] [CrossRef]
- Lumpkin, G.R.; Smith, K.L.; Blackford, M.G.; Giere, R.; Williams, C.T. The crystalline-amorphous transformation in natural zirconolite: Evidence for long-term annealing. Mater. Res. Soc. Symp. Proc. 1998, 556, 215–222. [Google Scholar] [CrossRef]
- Lumpkin, G.R.; Smith, K.L.; Giere, R.; Williams, C.T. Geochemical behavior of host phases for actinides and fission products in crystalline ceramic nuclear waste forms. In Energy, Waste, and the Environment: A Geochemical Perspective; Special Publication 236; Geological Society: London, UK, 2004; pp. 89–111. [Google Scholar] [CrossRef]
- Ewing, R.C.; Weber, W.J.; Clinard, F.W., Jr. Radiation effects in nuclear waste forms for high-level radioactive waste. Prog. Nucl. Energy 1995, 29, 63–127. [Google Scholar] [CrossRef]
- Ewing, R.C.; Weber, W.J.; Lutze, W. Waste form for the disposal of weapons plutonium. In Disposal of Weapon Plutonium; Merz, E.R., Walter, C.E., Eds.; Kluwer Academic Publishers: Dordrecht, The Netherlands, 1996; pp. 65–83. [Google Scholar]
- Weber, W.J.; Ewing, R.C.; Catlow, C.R.A.; Diaz de la Rubia, T.; Hobbs, L.W.; Kinoshita, C.; Matzke Hj Motta, A.T.; Nastasi, M.; Salje, E.H.K.; Vance, E.R.; et al. Radiation effects in crystalline ceramics for the immobilization of high-level nuclear waste and plutonium. J. Mater. Res. 1998, 13, 1434–1479. [Google Scholar] [CrossRef]
- Weber, W.J.; Ewing, R.C. Radiation effects in crystalline oxide host phases for the immobilization of actinides. Mater. Res. Soc. Symp. Proc. 2002, 713, 443. [Google Scholar] [CrossRef]
- Weber, W.J.; Ewing, R.C. Ceramic waste forms for uranium and transuranium elements. In Uranium: Cradle to Grave; Burns, P.C., Sigmon, G.E., Eds.; Short Course Series; Mineralogical Association of Canada: Quebec, QC, Canada, 2013; Volume 43, pp. 317–336. [Google Scholar]
- Laverov, N.P.; Yudintsev, S.V.; Yudintseva, T.S.; Stefanovsky, S.V.; Ewing, R.C.; Lian, J.; Utsunomiya, S.; Wang, L.M. Effect of radiation on properties of confinement matrices for immobilization of actinide-bearing wastes. Geol. Ore Depos. 2003, 45, 423–451. [Google Scholar]
- Yudintsev, S.V.; Stefanovsky, S.V.; Kalenova MYu Nikonov, B.S.; Nikol’skii, M.S.; Koshcheev, A.M.; Shchepin, A.S. Matrices for immobilization of the rare earth–actinide waste fraction, synthesized by cold crucible induction melting. Radiochemistry 2015, 57, 321–333. [Google Scholar] [CrossRef]
- Ringwood, A.E. Disposal of high-level nuclear wastes: A geological perspective. Mineral. Mag. 1985, 49, 159–176. [Google Scholar] [CrossRef]
- Gregg, D.J.; Farzana, R.; Dayal, P.; Holmes, R.; Gerry, T. Synroc technology: Perspectives and current status (Review). J. Am. Ceram. Soc. 2020, 103, 5424–5441. [Google Scholar] [CrossRef]
- Ebbinghaus, B.B.; Cicero-Herman, C.; Gray, L.; Shaw, H.F. Plutonium Immobilization Project Baseline Formulation; UCRL-ID-133089; LLNL: Livermore, CA, USA, 1999; p. 61. [Google Scholar]
- Ewing, R.C.; Weber, W.J.; Lian, J. Nuclear waste disposal—pyrochlore (A2B2O7): Nuclear waste form for the immobilization of plutonium and “minor” actinides. J. Appl. Phys. 2004, 95, 5949–5971. [Google Scholar] [CrossRef]
- Malmström, J.; Reusser, E.; Giere, R.; Lumpkin, G.R.; Düggelin, M.; Mathys, D.; Guggenheim, R. Zirconolite corrosion in dilute acidic and basic fluids at 180-700°C and 50 MPa. Mater. Res. Soc. Symp. Proc. 1999, 556, 165–172. [Google Scholar] [CrossRef]
- Vance, E.R.; Dytlewski, N.; Prince, K.E.; Hart, K.P.; Loi, E. Surface alteration of titanate ceramics in aqueous media. Mater. Res. Soc. Symp. Proc. 2000, 608, 379–385. [Google Scholar] [CrossRef]
- Giere, R.; Malmström, J.; Reusser, E.; Lumpkin, G.R.; Düggelin, M.; Mathys, D.; Guggenheim, R.; Günther, D. Durability of zirconolite in hydrothermal fluids: Implications for nuclear waste disposal. Mater. Res. Soc. Symp. Proc. 2001, 663, 267–276. [Google Scholar] [CrossRef]
- Nikoloski, A.N.; Gilligan, R.; Squire, J.; Maddrell, E.R. Chemical stability of zirconolite for proliferation resistance under conditions typically required for the leaching of highly refractory uranium minerals. Metals 2019, 9, 1070. [Google Scholar] [CrossRef]
- Zhang, Z.; Vance, E.R.; Blackford, M.G. Surface alteration of Nd-bearing zirconolite following hydrothermal treatment. Mater. Res. Soc. Symp. Proc. 2001, 663, 285–292. [Google Scholar] [CrossRef]
- Smith, K.L.; Zhang, Z.; McGlinn, P.; Attard, D.; Li, H.; Lumpkin, G.R.; Colella, M.; McLeod, T.; Aly, Z.; Loi, E.; et al. The effect of radiation damage on zirconolite dissolution. Mater. Res. Soc. Symp. Proc. 2003, 757, 289–296. [Google Scholar] [CrossRef]
- Weber, W.J.; Wald, J.W.; Matzke, H. Effects of self-irradiation damage in Cm-doped Gd2Ti2O7 and CaZrTi2O7. J. Nucl. Mater. 1986, 138, 196–209. [Google Scholar] [CrossRef]
- Carter, J.T.; Luptak, A.J.; Gastelum, J.; Stockman, C.; Miller, A. Fuel Cycle Potential Waste Inventory for Disposition. FCR&D-USED-2010-000031. Rev 5. SRNL-INL-PNNL-SNL. 2012. 328p. Available online: https://www.energy.gov/ne/articles/fuel-cycle-potential-waste-inventory-disposition-rev-5 (accessed on 17 January 2013).
- Stewart, M.W.A.; Vance, E.R.; Moricca, S.A.; Brew, D.R.; Cheung, C.; Eddowes, T.; Bermudez, W. Immobilization of higher activity wastes from nuclear reactor production of 99Mo. Sci. Technol. Nucl. Install. 2013, 2013, 926026. [Google Scholar] [CrossRef]
- NEA. Minor Actinide Burning in Thermal Reactors; Rep. 6997; OECD NEA: Paris, France, 2013; p. 78. [Google Scholar]
- Collins, E.D.; Jubin, R.T.; DelCul, G.D. Advanced fuel cycle treatment, recycling, and disposal of nuclear waste. In Proceedings of the Conference “Global 2009”, Paris, France, 6–11 September 2009; pp. 2595–2602. [Google Scholar]
- Pudovkina, Z.V.; Pyatenko, Y.A. X-ray study of zirconolite and its synthetic analogue. In X-ray of Mineral Raw Materials; Nedra: Moscow, Russia, 1964. (In Russian) [Google Scholar]
- Rossell, H.J. Zirconolite—A fluorite-related superstructure. Nature 1980, 283, 282–283. [Google Scholar] [CrossRef]
- Gatehouse, B.M.; Grey, I.E.; Hill RJRossell, H.J. Zirconolite, CaZrxTi3−xO7; structure refinements for near-end-member compositions with x = 0.85 and 1.30. Acta Cryst. 1981, 3, 306–312. [Google Scholar] [CrossRef]
- White, T.J. The microstructure and microchemistry of synthetic zirconolite, zirkelite and related phases. Am. Mineral. 1984, 69, 1156–1172. [Google Scholar]
- Smith, K.L.; Lumpkin, G.R. Structural features of zirconolite, hollandite and perovskite, the major waste-bearing phases in Synroc. In Defects and Processes in the Solid State: Geoscience Applications; Boland, J.N., Fitzgerald, J.D., Eds.; Elsevier: Amsterdam, The Netherlands, 1993; pp. 401–422. [Google Scholar]
- Yudintsev, S.V.; Nickolsky, M.S.; Ojovan, M.I.; Stefanovsky, O.I.; Nikonov, B.S.; Ulanova, A.S. Zirconolite polytypes and murataite polysomes in matrices for the REE—Actinide fraction of HLW. Materials 2022, 15, 6091. [Google Scholar] [CrossRef]
- Coelho, A.A.; Cheary, R.W.; Smith, K.L. Analysis and structural determination of Nd-substituted zirconolite-4M. J. Solid State Chem. 1997, 129, 346–359. [Google Scholar] [CrossRef]
- Stefanovsky, S.V.; Chizhevskaya, S.V.; Mironov, A.S.; Kiryanova, O.I.; Yudintsev, S.V. Synthetic calcium-free REE-substituted zirconolites. Perspekt. Mater. 2003, 6, 61–68. (In Russian) [Google Scholar]
- Ma, S.; Ji, S.; Liao, C.; Liu, C.; Shih, K. Effects of ionic radius on phase evolution in Ln-Al co-doped Ca1−xLnxZrTi2-xAlxO7 (Ln = La, Nd, Gd, Ho, Yb) solid solutions. Ceram. Int. 2018, 44, 15124–15132. [Google Scholar] [CrossRef]
- Ji, S.; Li, Y.; Ma, S.; Liu, C.; Shih, K.; Liao, C.-Z. Synergistic effects of Ln and Fe co-doping on phase evolution of Ca1−xLnxZrTi2−xFexO7 (Ln = La, Nd, Gd, Ho, Yb) ceramics. J. Nucl. Mater. 2018, 511, 428–437. [Google Scholar] [CrossRef]
- Wang, S.X.; Lumpkin, G.R.; Wang, L.M.; Ewing, R.C. Ion-irradiation-induced amorphization of six zirconolite compositions. Nucl. Instrum. Methods Phys. Res. 2000, 166–167, 293–298. [Google Scholar] [CrossRef]
- Caurant, D.; Loiseau, P.; Bardez, I. Structural characterization of Nd-doped Hf-zirconolite Ca1−xNdxHfTi2−xAlxO7 ceramics. J. Nucl. Mater. 2010, 407, 88–99. [Google Scholar] [CrossRef]
- Chukanov, N.V.; Zubkova, N.V.; Britvin, S.N.; Pekov, I.V.; Vigasina, M.F.; Schäfer, C.; Ternes, B.; Schüller, W.; Polekhovsky, Y.S.; Ermolaeva, V.N.; et al. Nöggerathite-(Ce), (Ce,Ca)2Zr2(Nb,Ti)(Ti,Nb)2Fe2+O14, a New Zirconolite-Related Mineral from the Eifel Volcanic Region, Germany. Minerals 2018, 8, 449. [Google Scholar] [CrossRef]
- Giere, R. Natural analogues for crystalline nuclear waste forms. A Geochemical and Mineralogical Approach to Environmental Protection. In Proceedings of the International School Earth and Planetary Science, Siena, Italy, 24 September–3 October 1999; pp. 175–192. [Google Scholar]
- Caurant, D.; Bardez, I.; Loiseau, P. Crystallization of CaHf1−xZrxTi2O7 (0 < x < 1) zirconolite in SiO2–Al2O3–CaO–Na2O–TiO2– HfO2–ZrO2–Nd2O3 glasses. J. Mater. Sci. 2007, 42, 10203–10218. [Google Scholar] [CrossRef]
- Caurant, D.; Loiseau, P.; Bardez, I.; Gervais, C. Effect of Al2O3 concentration on zirconolite Ca(Zr,Hf)Ti2O7 crystallization in (TiO2,ZrO2,HfO2)-rich SiO2–Al2O3–CaO–Na2O glasses. J. Mater. Sci. 2007, 42, 8558–8570. [Google Scholar] [CrossRef]
- Vance, E.R.; Lumpkin, G.R.; Carter, M.L.; Cassidy, D.J.; Ball, C.J.; Day, R.A.; Begg, B.D. Incorporation of uranium in zirconolite (CaZrTi2O7). J. Am. Ceram. Soc. 2002, 85, 1853–1859. [Google Scholar] [CrossRef]
- Begg, B.D.; Day, R.A.; Brownscombe, A. Structural effect of Pu substitutions on the Zr-site in zirconolite. Mater. Res. Soc. Symp. Proc. 2001, 663, 259–269. [Google Scholar] [CrossRef]
- Zhong, M.X.; Walkley, B.; Bailey, D.J.; Blackburn, L.R.; Ding, H.; Wang, S.Q.; Bao, W.C.; Gardner, L.J.; Sun, S.K.; Stennett, M.C. Synthesis of Ca1−xCexZrTi2-2xAl2xO7 zirconolite ceramics for plutonium disposition. J. Nucl. Mater. 2021, 556, 153198. [Google Scholar] [CrossRef]
- Shannon, R.D. Revised effective ionic radii and systematic studies of interatomic distances in halides and chalcogenides. Acta Crystallogr. Sect. A 1976, 32, 751–767. [Google Scholar] [CrossRef]
- Blackburn, L.R.; Sun, S.-K.; Gardner, L.J.; Maddrell, E.R.; Stennett, M.C.; Corkhill, C.L.; Hyatt, N.C. Synthesis, structure, and characterization of the thorium zirconolite CaZr1−xThxTi2O7 system. J. Am. Ceram. Soc. 2021, 104, 2937–2951. [Google Scholar] [CrossRef]
- Begg, B.D.; Vance, E.R.; Day, R.A.; Hambley, M.; Conradson, S.D. Plutonium and neptunium incorporation in zirconolite. MRS Online Proc. Libr. 1997, 465, 325–332. [Google Scholar] [CrossRef]
- Gilbert, M.R.; Selfslag, C.; Walter, M.; Stennett, M.C.; Somers, J.; Hyatt, N.C. Synthesis and characterization of Pu-doped zirconolites—(Ca1−xPux)Zr(Ti2−2xFe2x)O7. IOP Conf. Series: Mater. Sci. Eng. 2010, 9, 012007. [Google Scholar] [CrossRef]
- Thornber, S.M.; Stennett, M.C.; Vance, E.R.; Chavara, D.T.; Watson, I.; Jovanovic, M.; Davis, J.; Gregg, D.; Hyatt, N.C. A preliminary validation study of PuO2 incorporation into zirconolite glass-ceramics. MRS Adv. 2018, 3, 1065–1071. [Google Scholar] [CrossRef]
- Ebbinghaus, B.B.; Van Konynenburg, R.A.; Ryerson, F.J.; Vance, E.R.; Stewart, M.W.A.; Jostsons, A.; Allender, J.S.; Rankin, T.; Congdon, J. Ceramic formulation for the immobilization of plutonium. In Proceedings of the WM’98 Conference, Tucson, AZ, USA, 1–5 March 1998. Paper 65-04. [Google Scholar]
- Ebbinghaus, B.B.; Van Konynenburg, R.A.; Vance, E.R.; Stewart, M.W.; Jostsons, A.; Allender, J.S.; Rankin, D.T. Ceramic Composition for Immobilization of Actinides. U.S. Patent 6,137,025, 24 October 2000. [Google Scholar]
- Ebbinghaus, B.B.; Van Konynenburg, R.A.; Vance, E.R.; Stewart, M.W.; Walls, P.A.; Brummond, W.A.; Armantrout, G.A.; Herman, C.C.; Hobson, B.F.; Herman, D.T.; et al. Process for Making a Ceramic Composition for Immobilization of Actinides. U.S. Patent 6,320,091, 21 November 2001. [Google Scholar]
- Ewing, R.C.; Meldrum, A.; Wang, L.-M.; Wang, S.-X. Radiation induced amorphization. In Transformation Processes in Minerals; Chapter 12; Reviews in Mineralogy & Geochemistry; Redfern, S.A., Carpenter, M.A., Eds.; DeGruyter: Berlin, Germany, 2000; Volume 39, pp. 319–361. [Google Scholar]
- National Academies of Sciences, Engineering, and Medicine. Review of the Department of Energy's Plans for Disposal of Surplus Plutonium in the Waste Isolation Pilot Plant; The National Academies Press: Washington, DC, USA, 2020. [Google Scholar] [CrossRef]
- Zhang, Y.; Stewart, M.W.A.; Li, H.; Carter, M.L.; Vance, E.R.; Moricca, S. Zirconolite-rich titanate ceramics for immobilisation of actinides—waste form / HIP can interactions and chemical durability. J. Nucl. Mater. 2009, 395, 69–74. [Google Scholar] [CrossRef]
- Zhang, Y.; Gregg, D.J.; Kong, L.; Jovanovich, M.; Triani, G. Zirconolite glass-ceramics for plutonium immobilization: The effects of processing redox conditions on charge compensation and durability. J. Nucl. Mater. 2017, 490, 238–241. [Google Scholar] [CrossRef]
- Blackburn, L.R.; Sun, S.; Gardner, L.J.; Maddrell, E.R.; Stennett, M.C.; Hyatt, N.C. A systematic investigation of the phase assemblage and microstructure of the zirconolite CaZr1−xCexTi2O7 system. J. Nucl. Mater. 2020, 535, 152137. [Google Scholar] [CrossRef]
- Blackburn, L.R.; Bailey, D.J.; Sun, S.-K.; Gardner, L.J.; Stennett, M.C.; Corkhill, C.L.; Hyatt, N.C. Review of zirconolite crystal chemistry and aqueous durability. Adv. Appl. Ceram. 2021, 120, 69–83. [Google Scholar] [CrossRef]
- Blackburn, L.R.; Crawford, R.; Walling, S.A.; Gardner, L.J.; Cole, M.R.; Shi, S.-K.; Gausse, C.; Mason, A.R.; Stennett, M.C.; Maddrell, E.R.; et al. Influence of accessory phases and surrogate type on accelerated leaching of zirconolite wasteforms. NPJ Mater. Degrad. 2021, 5, 24. [Google Scholar] [CrossRef]
- Ji, S.; Su, M.; Liao, C.; Ma, S.; Wang, Z.; Shih, K.; Chang, C.K.; Lee, J.F.; Chan, T.S.; Li, Y. Synchrotron X-ray spectroscopy investigation of the Ca1−xLnxZrTi2−x(Al,Fe)xO7 zirconolite ceramics (Ln = La, Nd, Gd, Ho, Yb). J. Am. Ceram. Soc. 2020, 103, 1463–1475. [Google Scholar] [CrossRef]
- Loiseau, P.; Caurant, D.; Baffier, N.; Fillet, C. Neodymium incorporation in zirconolite-based glass-ceramics. Mater. Res. Soc. Symp. Proc. 2001, 663, 169–178. [Google Scholar] [CrossRef]
- Loiseau, P.; Caurant, D.; Baffier, N.; Mazerolles, L.; Fillet, C. Development of zirconolite-based glass-ceramics for the conditioning of actinides. Mater. Res. Soc. Symp. Proc. 2001, 663, 179–188. [Google Scholar] [CrossRef]
- Kong, L.; Karatchevtseva, I.; Zhang, Y.; Wei, T. The incorporation of Nd or Ce in CaZrTi2O7 zirconolite: Ceramic versus glass-ceramic. J. Nucl. Mater. 2021, 543, 152583. [Google Scholar] [CrossRef]
- Bardez-Giboire, I.; Kidari, A.; Magnin, M.; Dussossoy, J.-L.; Peuget, S.; Caraballo, R.; Tribet, M.; Doreau, F.; Jégou, C. Americium and trivalent lanthanides incorporation in high-level waste glass-ceramics. J. Nucl. Mater. 2017, 492, 231–238. [Google Scholar] [CrossRef]
- Yin, D.; Zhang, K.; Peng, L.; He, Z.; Liu, Y.; Zhang, H.; Lu, X. Solid-state reaction synthesis and chemical stability studies in Nd-doped zirconolite-rich ceramics. J. Rare Earths 2018, 36, 492–498. [Google Scholar] [CrossRef]
- Loiseau, P.; Caurant, D.; Baffier, N.; Fillet, C. Structural characterization of polycrystalline (Nd,Al)-substituted zirconolite. MRS Symp. Proc. 2003, 757, 243–250. [Google Scholar] [CrossRef]
- Jafar, M.; Sengupta, P.; Achary, S.N.; Tyagi, A.K. Phase evolution and microstructural studies in CaZrTi2O7 (zirconolite)—Sm2Ti2O7 (pyrochlore) system. J. Eur. Ceram. Soc. 2014, 34, 4373–4381. [Google Scholar] [CrossRef]
- Blackburn, L.R.; Townsend, L.T.; Lawson, S.M.; Mason, A.R.; Stennett, M.C.; Sun, S.-K.; Gardner, L.J.; Maddrell, E.R.; Corkhill, C.L.; Hyatt, N.C. Phase evolution in the CaZrTi2O7 − Dy2Ti2O7 system: A potential host phase for minor actinide immobilization. Inorg. Chem. 2022, 61, 5744–5756. [Google Scholar] [CrossRef]
- Ji, S.; Liao, C.Z.; Chen, S.; Zhang, K.; Shih, K.; Chang, C.K.; Sheu, H.; Yan, S.; Li, Y.; Wang, Z. Higher valency ion substitution causing different fluorite-derived structures in CaZr1−xNdxTi2-xNbxO7 (0.05 ≤ x ≤ 1) solid solution. Ceram. Int. 2021, 47, 2694–2704. [Google Scholar] [CrossRef]
- Meng, C.; Ding, X.; Li, W.; Zhao, J.; Yang, H. Phase structure evolution and chemical durability studies of Ce-doped zirconolite–pyrochlore Synroc for radioactive waste storage. J. Mater. Sci. 2016, 51, 5207–5215. [Google Scholar] [CrossRef]
- Loiseau, P.; Caurant, D.; Baffier, N.; Mazerolles, L.; Fillet, C. Glass-ceramic nuclear waste forms obtained from SiO2–Al2O3–CaO–ZrO2–TiO2 glasses comtaining lanthanides (Ce, Nd, Eu, Gd, Yb) and actinides (Th): Study of internal crystallization. J. Nucl. Mater. 2004, 335, 14–32. [Google Scholar] [CrossRef]
- Zhang, K.; Luo, B.; Zhang, H. Immobilization of CeO2 using single-phase zirconolite and the chemical stability analysis. Mater. Res. Express 2019, 6, 115526. [Google Scholar] [CrossRef]
- Yudintsev, S.V.; Nickolsky, M.S.; Stefanovsky, O.I.; Nikonov, B.S. Crystal chemistry of titanates and zirconates of rare earths–possible matrices for actinide isolation. Radiochemistry 2022, 64, 667–679. [Google Scholar] [CrossRef]
- Zirconolite. mindat.org. Hudson Institute of Mineralogy. Available online: https://www.mindat.org/min-6657.html (accessed on 9 May 2023).
- Grey, I.E.; Mumme, W.G.; Ness, T.J.; Roth, R.S.; Smith, K.L. Structural relations bet ween weberite and zi rconolite polytypes - refinements of dop ed 3T and 4M Ca2Ta2O7 and CaZrTi2O7. J. Solid State Chem. 2003, 174, 285–295. [Google Scholar] [CrossRef]
- Maddrell, E.R.; Paterson, H.C.; May, S.E.; Burns, K.M. Phase evolution in zirconolite glass-ceramic wasteforms. J. Nucl. Mater. 2017, 493, 380–387. [Google Scholar] [CrossRef]
- Zhu, H.; Wang, F.; Liao, Q.; Wang YZhu, Y. Effect of CeO2 and Nd2O3 on phases, microstructure and aqueous chemical durability of borosilicate glass-ceramics for nuclear waste immobilization. Mater. Chem. Phys. 2020, 249, 122936. [Google Scholar] [CrossRef]
- Cachia, J.-N.; Deschanels, X.; Den Auwer, C.; Pinet, O.; Phalippou, J.; Henning, C.; Scheinost, A. Enhancing cerium and plutonium solubility by reduction in borosilicate glass. J. Nucl. Mater. 2006, 352, 182–189. [Google Scholar] [CrossRef]
- Gin, S.; Jollivet, P.; Tribet, M.; Peuget, S.; Schuller, S. Radionuclides containment in nuclear glasses: An overview. Radiochim. Acta 2017, 105, 927–959. [Google Scholar] [CrossRef]
- Loiseau, P.; Caurant, D.; Baffier, N.; Fillet, C. Neodymium partitioning in zirconolite-based glass-ceramics designed for minor actinides immobilization. In Proceedings of the International Conference Atalante-2000, Avignon, France, 24–26 October 2000. [Google Scholar]
- Advocat, T.; McGlinn, P.J.; Fillet, C.; Leturcq, G.; Schuller, S.; Bonnetier, A.; Hart, K. Melted synthetic zirconolite-based matrices: Effect of cooling rate and heat treatment on ceramic microstructure and chemical durability. Mater. Res. Soc. Symp. Proc. 2001, 663, 277–284. [Google Scholar] [CrossRef]
- Caurant, D.; Majerus, O.; Loiseau, P.; Bardez, I.; Baffier, N.; Dussossoy, J.L. Crystallization of neodymium-rich phases in silicate glasses developed for nuclear waste immobilization. J. Nucl. Mater. 2006, 354, 143–162. [Google Scholar] [CrossRef]
- Liao, C.-Z.; Liu, C.; Su, M.; Shih, K. Quantification of the partitioning ratio of minor actinide surrogates between zirconolite and glass in glass-ceramic for nuclear waste disposal. Inorg. Chem. 2017, 56, 9913–9921. [Google Scholar] [CrossRef]
- Advocat, T.; Fillet, C.; Marillet, J.; Leturcq, G.; Boubals, J.M.; Bonnetier, A. Nd-doped zirconolite ceramic and glass ceramic synthesized by melting and controlled cooling. Mater. Res. Soc. Symp. Proc. 1998, 506, 55–62. [Google Scholar] [CrossRef]
- Stewart, M.W.A.; Begg, B.D.; Day, R.A. Low-risk alternative waste forms for actinide immobilization. In Proceedings of the WM Conference, Tucson, AZ, USA, 27 February–3 March 2004. [Google Scholar]
- Poluektov, P.P.; Sukhanov, L.P.; Matyunin, Y.I. Scientific approaches and technical solutions in the field of liquid high-level waste management. Russ. Chem. J. 2005, XLIX, 29–41. (In Russian) [Google Scholar]
- Zhang, Y.; Kong, L.; Ionescu, M.; Gregg, D.J. Current advances on titanate glass-ceramic composite materials as waste forms for actinide immobilization: A technical review. J. Eur. Ceram. Soc. 2022, 42, 1852–1876. [Google Scholar] [CrossRef]
- Hatch, L.P. Ultimate disposal of radioactive waste. Am. Sci. 1953, 41, 410–421. [Google Scholar]
- Yudintsev, S.V.; Stefanovskii, S.V.; Jang, Y.N.; Che, S. X-ray diffraction analysis of phase formation in synthesis of actinide matrices. Glass Ceram. 2002, 59, 237–241. [Google Scholar] [CrossRef]
- Laverov, N.P.; Yudintsev, S.V.; Stefanovskii, S.V.; Jang, Y.N.; Bae, I.K.; Chae, S. Phase formation during the synthesis of actinide matrices. Dokl. Earth Sci. 2002, 383, 190–193. [Google Scholar]
- Laverov, N.P.; Yudintsev, S.V.; Stefanovsky, S.V.; Jang, Y.; Lapina, M.I.; Sivtsov, A.V.; Ewing, R. Phase transformations during synthesis of actinide matrices. Dokl. Earth Sci. 2002, 385A, 671–675. [Google Scholar]
- Laverov, N.P.; Yudintsev, S.V.; Lapina, M.I.; Stefanovsky, S.V.; Chae, S.C.; Ewing, R.C. Phases Formation rate at synthesis of actinide waste forms. Mater. Res. Soc. Symp. Proc. 2003, 757, 321–328. [Google Scholar] [CrossRef]
- Chen, Y.B.; Bao, W.C.; Sun, S.K.; Blackburn, L.R.; Wei, Z.J.; Guo, W.M.; Lin, H.T. Phase and structural evolution of zirconolite ceramics prepared by solid-state reaction sintering. Ceram. Int. 2023, 49, 419–424. [Google Scholar] [CrossRef]
- Brummond, W.; Armantrout, G.; Maddux, P. Ceramic process equipment for the immobilization of plutonium. In Proceedings of the 3rd Topical Meeting of DOE Spent Nuclear Fuel and Fissile Material Management, Charleston, SC, USA, 8–11 September 1998; American Nuclear Society: La Grange Park, IL, USA, 1998; pp. 380–384. [Google Scholar]
- Stewart, M.W.A.; Vance, E.R.; Jostsons, A.; Finnie, K.; Day, R.A.; Ebbinghause, B.B. Atmosphere processing effect on titanate ceramics designed for plutonium disposition. Mater. Res. Soc. Symp. Proc. 2002, 713, 381–388. [Google Scholar] [CrossRef]
- Greenquist, I.; Tonks, M.R.; Zhang, Y. Review of sintering and densification in nuclear fuels: Physical mechanisms, experimental results, and computational models. J. Nucl. Mater. 2018, 507, 381–395. [Google Scholar] [CrossRef]
- Lebreton, F.; Prieur, D.; Jankowiak, A.; Tribet, M.; Leorier, C.; Delahaye, T.; Donnet, L.; Dehaudt, P. Fabrication and characterization of americium, neptunium and curium bearing MOX fuels obtained by powder metallurgy process. J. Nucl. Mater. 2012, 420, 213–217. [Google Scholar] [CrossRef]
- Lebreton, F.; Prieur, D.; Horlait, D.; Delahaye, T.; Jankowiak, A.; Léorier, C.; Jorion, F.; Gavilan, E.; Desmoulière, F. Recent progress on minor-actinide-bearing oxide fuel fabrication at CEA Marcoule. J. Nucl. Mater. 2013, 438, 99–107. [Google Scholar] [CrossRef]
- Strachan, D.M.; Scheele, R.D.; Buchmiller, W.C.; Vienna, J.D.; Sell, R.L.; Elovich, R.J. Preparation and Characterization of 238Pu-Ceramics for Radiation Damage Experiments; PNNL-13251; Pacific Northwest National Laboratory: Richland, WD, USA, 2000. [Google Scholar]
- Strachan, D.M.; Kozelisky, A.E.; Scheele, R.D.; Sell, R.L.; Icenhower, J.P.; Elovich, R.J.; Buck, E.C.; Buckmiller, W.C. Radiation Damage Effects in Candidate Ceramics for Plutonium Immobilization: Final Report; PNNL-14588; Pacific Northwest National Laboratory: Richland, WD, USA, 2004. [Google Scholar]
- Strachan, D.M.; Scheele, R.D.; Buck, E.C.; Kozelisky, A.E.; Sell, R.L.; Elovich, R.J.; Buchmiller, W.C. Radiation damage effects in candidate titanates for Pu disposition: Zirconolite. J. Nucl. Mater. 2008, 372, 16–31. [Google Scholar] [CrossRef]
- Chizhevskaya, S.V.; Chekmarev, A.M.; Stefanovsky, S.V.; Medvedev, D.G. Synthesis of zirconolite by cold pressing and sintering. Issues Radiat. Saf. 1999, 4, 34–41. (In Russian) [Google Scholar]
- Wen, G.; Zhang, K.; Yin, D.; Zhang, H. Solid-state reaction synthesis and aqueous durability of Ce-doped zirconolite-rich ceramics. J. Nucl. Mater. 2015, 466, 113–119. [Google Scholar] [CrossRef]
- Squire, J.; Maddrell, E.R.; Hyatt, N.C.; Stennett, M.C. Influence of lubricants and attrition milling parameters on the quality of zirconolite ceramics, consolidated by hot isostatic pressing, for immobilization of plutonium. Int. J. Appl. Ceram. Technol. 2014, 12, E92–E104. [Google Scholar] [CrossRef]
- Dayal, P.; Farzana, R.; Zhang, Y.; Lumpkin, G.R.; Holmes, R.; Triani, G.; Gregg, D.J. Profiling hot isostatically pressed canister–wasteform interaction for Pu-bearing zirconolite-rich wasteforms. J. Am. Ceram. Soc. 2022, 105, 5359–5372. [Google Scholar] [CrossRef]
- Hsieh, Y.H.; Humphry-Baker, S.A.; Horlait, D.; Gregg, D.J.; Vance, E.R.; Lee, W.E. Durability of hot uniaxially pressed Synroc derivative wasteform for EURO-GANEX wastes. J. Nucl. Mater. 2018, 509, 43–53. [Google Scholar] [CrossRef]
- Knyazev, O.A.; Stefanovsky, S.V.; Ioudintsev, S.V.; Nikonov, B.S.; Omelianenko, B.I.; Mokhov, A.V.; Yakushev, A.I. Phase equilibria and elements partitioning in zirconolite-rich region of Ca-Zr-Ti-Al-Gd-Si-O system. Mater. Res. Soc. Symp. Proc. 1997, 465, 401–408. [Google Scholar] [CrossRef]
- Yudintsev, S.V.; Lizin, A.A.; Livshits, T.S.; Stefanovsky, S.V.; Tomilin, S.V.; Ewing, R.C. Ion-beam irradiation and 244Cm-doping investigations of the radiation response of actinide-bearing crystalline waste forms. J. Mater. Res. 2015, 30, 1516–1528. [Google Scholar] [CrossRef]
- Yudintsev, S.V. Lanthanide titanates as promising matrices for immobilization of actinide wastes. Dokl. Earth Sci. 2015, 460, 130–136. [Google Scholar] [CrossRef]
- Xu, H.; Wang, Y. Microstructure evolution and weathering reactions of Synroc samples crystallized from CaZrTi2O7 melts: Tem/AEM investigation and geochemical modeling. Mater. Res. Soc. Symp. Proc. 1997, 556, 47–54. [Google Scholar] [CrossRef]
- Sun, S.-K.; Stennett, M.C.; Corkhill, C.L.; Hyatt, N.C. Reactive spark plasma synthesis of CaZrTi2O7 zirconolite ceramics for plutonium disposition. J. Nucl. Mater. 2018, 500, 11–14. [Google Scholar] [CrossRef]
- Aldean, I.; Sun, S.-K.; Wilkins, M.C.D.; Gardner, L.J.; Mason, A.R.; Stennett, M.C.; Corkhill, C.L.; Hyatt, N.C.; Blackburn, L.R. Synthesis and characterisation of Ce-doped zirconolite Ca0.80Ce0.20ZrTi1.60 M0.40O7 (M = Fe, Al) formed by reactive spark plasma sintering (RSPS). MRS Adv. 2022, 7, 75–80. [Google Scholar] [CrossRef]
- Barinova, T.V.; Borovinskaya, I.P.; Ratnikov, V.I.; Ignat’eva, T.I. Self-propagating high-temperature synthesis for immobilization of high-level waste in mineral-like ceramics: 1. Synthesis and study of titanate ceramics based on perovskite and zirconolite. Radiochemistry 2008, 50, 316–320. [Google Scholar] [CrossRef]
- Barinova, T.V.; Alymov, M.I. Self-propagating high-temperature synthesis for disposal of radioactive waste. Int. J. Self-Propagating High-Temp. Synth. 2022, 31, 179–187. [Google Scholar] [CrossRef]
- Zhang, K.; He, S.; Yin, D.; Peng, L.; Wu, J. Self-propagating synthesis and aqueous durability of Nd-bearing zirconolite-rich composites using Ca(NO3)2 as the oxidant. J. Nucl. Mater. 2016, 478, 315–321. [Google Scholar] [CrossRef]
- Zhang, K.; Yin, D.; Peng, L.; Wu, J. Self-propagating synthesis and CeO2 immobilization of zirconolite-rich composites using CuO as the oxidant. Ceram. Int. 2017, 43, 1415–1423. [Google Scholar] [CrossRef]
- Zhang, K.; Yin, D.; Lu, X.; Zhang, H. Self-propagating high-temperature synthesis, phase composition and aqueous durability of Nd–Al bearing zirconolite-rich composites as nuclear waste form. Adv. Appl. Ceram. 2018, 117, 78–84. [Google Scholar] [CrossRef]
- Vance, E.R.; Begg, B.D.; Day, R.A.; Ball, C.J. Zirconolite-rich ceramics for actinide wastes. Mater. Res. Soc. Symp. Proc. 1995, 353, 767–774. [Google Scholar] [CrossRef]
- Begg, B.D.; Vance, E.R. The incorporation of cerium in zirconolite. Mater. Res. Soc. Symp. Proc. 1997, 465, 333–340. [Google Scholar] [CrossRef]
- Begg, B.D.; Vance, E.R.; Lumpkin, G.R. Charge compensation and the incorporation of cerium in zirconolite and perovskite. Mater. Res. Soc. Symp. Proc. 1998, 506, 79–86. [Google Scholar] [CrossRef]
- Begg, B.D.; Vance, E.R.; Hunter, B.A.; Hanna, J.V. Zirconolite transformation under reducing conditions. J. Mater. Res. 1998, 13, 3181–3190. [Google Scholar] [CrossRef]
- Li, H.; Zhang, Y.; McGlinn, P.J.; Moricca, S.; Begg, B.D.; Vance, E.R. Characterisation of stainless steel–Synroc interactions under hot isostatic pressing (HIPing) conditions. J. Nucl. Mater. 2006, 355, 136–141. [Google Scholar] [CrossRef]
- Thornber, S.M.; Mottram, L.M.; Mason, A.R.; Thompson, P.; Stennett, M.C.; Neil, C.H. Solubility, speciation and local environment of chlorine in zirconolite glass–ceramics for the immobilisation of plutonium residues. R. Soc. Chem. Adv. 2020, 10, 32497–32510. [Google Scholar] [CrossRef]
- Macfarlane, A. Immobilization of excess weapon plutonium: A better alternative to glass. Sci. Glob. Secur. 1998, 7, 271–309. [Google Scholar] [CrossRef]
- Clinard, F.; Hobbs, L.W.; Land, C.C.; Peterson, D.E.; Rohr, D.L.; Roof, R.B. Alpha decay self-irradiation damage in 238Pu- substituted zirconolite. J. Nucl. Mater. 1982, 105, 248–256. [Google Scholar] [CrossRef]
- Deschanels, X.; Picot VGlorieux, B. Plutonium incorporation in phosphate and titanate ceramics for minor actinide containment. J. Nucl. Mater. 2006, 352, 233–240. [Google Scholar] [CrossRef]
- Sobolev, I.A.; Myasoedov, B.F.; Stefanovskii, S.V.; Yudintsev, S.V.; Kulyako, Y.M. Phase composition and structure of uranium and plutonium-containing ceramics based on zirconolite and pyrochlore as influenced by preparation conditions. Radiochemistry 2001, 43, 124–130. [Google Scholar] [CrossRef]
- Pöml, P.; Geisler, T.; Cobos-Sabaté, J.; Wiss, T.; Raison, P.E.; Schmid-Beurmann, P.; Deschanels, X.; Jégou, C.; Heimink, J.; Putnis, A. The mechanism of the hydrothermal alteration of cerium- and plutonium-doped zirconolite. J. Nucl. Mater. 2011, 410, 10–23. [Google Scholar] [CrossRef]
- Clinard, F.W.; Hobbs, L.W.; Land, C.C.; Peterson, D.E.; Rohr, D.L.; Roof, R.B. Self-irradiation effects in 238Pu-substituted zirconolite: I. Temperature dependence of damage. J. Nucl. Mater. 1984, 126, 245–254. [Google Scholar] [CrossRef]
- Clinard, F.W.; Rohr, D.L.; Roof, R.B. Structural damage in a self-irradiated zirconolite-based ceramic. Nucl. Instrum. Methods Phys. Res. 1984, 1, 581–586. [Google Scholar] [CrossRef]
- Clinard, F.W. Review of self-irradiation effects in Pu-substituted zirconolite. Am. Ceram. Soc. Bull. 1985, 65, 1181–1187. [Google Scholar]
- Matzke, H.-J.; Ray, I.L.F.; Seatonberry, B.W.; Thiele, H.; Trisoglio, C.; Walker, C.T.; White, T.J. Incorporation of transuranic elements in titanate nuclear waste ceramics. J. Am. Ceram. Soc. 1990, 73, 370–378. [Google Scholar] [CrossRef]
- Matzke, H.; van Geel, J. Incorporation of Pu and other actinides in borosilicate glass and in waste ceramics. In Disposal of Weapons Plutonium; Mertz, E.R., Walter, C.E., Eds.; Kluwer Academic: Amsterdam, The Netherlands, 1996; pp. 93–105. [Google Scholar]
- Jorion, F.; Deschanels, X.; Advocat, T.; Desmouliere, F.; Cachia, J.N.; Peuget, S.; Roudil, D.; Leturcq, G. Zirconolite for minor actinide containment and alpha irradiation resistance. Nucl. Sci. Eng. 2006, 153, 262–271. [Google Scholar] [CrossRef]
- Davoisne, C.; Stennett, M.C.; Hyatt, N.C.; Peng, N.; Jeynes, C.; Lee, W.E. Krypton irradiation damage in Nd-doped zirconolite and perovskite. J. Nucl. Mater. 2011, 415, 67–73. [Google Scholar] [CrossRef]
- Sun, Z.; Lv, P.; Zhang, J.; Zhao, J.; Deng, W.; Zhao, Y.; Cao, Y.; Jia, Y.; Si, S.; Zhang, L.; et al. Morphology and chemical composition of Si-ion-irradiated zirconolite glass-ceramic. J. Eur. Ceram. Soc. 2023, 43, 3610–3620. [Google Scholar] [CrossRef]
- Wei, T.; Zhang, Y.; Xu, A.; Gregg, D.J.; Karatchevtseva, I.; Kong, L.; Ionescu, M.; Vance, E.R. Surface evolution and radiation damage of a zirconolite glass-ceramic by Au ion implantation. Appl. Surf. Sci. 2019, 478, 373–382. [Google Scholar] [CrossRef]
- Gupta, M.; Kulriya, P.K.; Meena, R.C.; Neumeier, S.; Ghumman, S.S. Probing swift heavy ion irradiation damage in Nd-doped zirconolite. Nucl. Instrum. Methods Phys. Res. Sect. B Beam Interact. Mater. At. 2019, 453, 22–27. [Google Scholar] [CrossRef]
- Gupta, M.; Kulriya, P.K.; Kumar, R.; Ghumman, S.S. Structural investigation of Nd-zirconolite irradiated with He+ions. J. Radioanal. Nucl. Chem. 2020, 323, 1413–1418. [Google Scholar] [CrossRef]
- Xu, A.; Wei, T.; Gregg, D.J.; Vance, E.R.; Zhang, Y.; Lumpkin, G.R. Micro-compression testing of gold ion irradiated zirconolite glass-ceramics as nuclear waste forms. J. Nucl. Mater. 2019, 527, 151813. [Google Scholar] [CrossRef]
- Smith, K.L.; Zaluzec, N.J.; Lumpkin, G.R. In situ studies of ion irradiated zirconolite, pyrochlore and perovskite. J. Nucl. Mater. 1997, 250, 36–52. [Google Scholar] [CrossRef]
- O’Holleran, T.P.; Johnson, S.G.; Frank, S.M.; Meyer, M.K.; Noy, M.; Wood, E.L.; Knecht, D.A.; Vinjamuri, K.; Staples, B.A. Glass-ceramic waste forms for immobilizing plutonium. Mater. Res. Soc. Symp. Proc. 1997, 463, 1251–1258. [Google Scholar] [CrossRef]
- Knecht, D.A.; Vinjamuri, K.; Raman, S.V.; Staples, B.A.; Grandy, J.D.; Johnson, S.; O'Holleran, T.P.; Frank, S. Summary of HLW Glass-Ceramic Waste Forms Developed in IDAHO for Immobilizing Plutonium. Proc. Conf., Waste Management–1998, AZ, USA, Paper 34-04. Available online: https://archivedproceedings.econference.io/wmsym/1998/html/sess34/34-04/34-04.htm (accessed on 17 January 2023).
- Bailey, D.J.; Lawson, S.M.; Sun, S.K.; Stennett, M.C.; Lee, T.-H.; Ravel, B.; Corkhill, C.L.; Heo, J.; Hyatt, N.C. A new approach to the immobilisation of technetium and transuranics: Co-disposal in a zirconolite ceramic matrix. J. Nucl. Mater. 2020, 528, 151885. [Google Scholar] [CrossRef]
- Wan, W.; Zhu, Y.; Zhang, X.; Yang, D.; Huo, Y.; Xu, C.; Yu, H.; Zhao, J.; Huo, J.; Meng, B. Borosilicate glass-ceramics containing zirconolite and powellite for RE- and Mo-rich nuclear waste immobilization. Materials 2021, 14, 5747. [Google Scholar] [CrossRef] [PubMed]
- Cai, X.; Teng, Y.; Wu, L.; Zhang, K.; Huang, Y. The synthesis and chemical durability of Nd-doped single-phase zirconolite solid solutions. J. Nucl. Mater. 2016, 479, 455–460. [Google Scholar] [CrossRef]
- Yudintsev, S.V.; Danilov, S.S.; Vinokurov, S.E.; Stefanovskaya, O.I.; Nikonov, B.S.; Nikol’sky, M.S.; Skvortsov, M.V.; Myasoedov, B.F. Phase composition and hydrothermal stability of ceramics based on murataite. Radiochemistry 2020, 62, 744–751. [Google Scholar] [CrossRef]
- White, T.J.; Mitamura, H.; Tsuboi, T. Rietveld analysis of phase separation in annealed and leach tested Cm-doped perovskite. Mater. Res. Soc. Symp. Proc. 1995, 353, 871–878. [Google Scholar] [CrossRef]
- Mitamura, H.; Matsumoto, S.; Tsuboi, T.; Vance, E.R.; Begg, B.D.; Hart, K.P. Alpha-decay damage of Cm-doped perovskite. Mater. Res. Soc. Symp. Proc. 1995, 353, 1405–1412. [Google Scholar] [CrossRef]
- Ringwood, A.E.; Kesson, S.E.; Reeve, K.D.; Levins, D.M.; Ramm, E.J. Synroc. In Radioactive Waste Forms for the Future; Lutze, W., Ewing, R.C., Eds.; Elsevier: New York, NY, USA, 1988; pp. 233–334. [Google Scholar]
- Lumpkin, G.R.; Smith, K.L.; Blackford, M.G. Development of secondary phases on Synroc leached et 150 °C. Mater. Res. Soc. Symp. Proc. 1995, 353, 855–862. [Google Scholar] [CrossRef]
- Kastrissios, T.; Stephenson, M.; Turner, P.S.; White, T.J. Hydrothermal dissolution of perovskite: Implications for Synroc formulation. J. Am. Ceram. Soc. 1987, 70, 144–146. [Google Scholar] [CrossRef]
- Ojovan, M.I.; Yudintsev, S.V. Glass, ceramic, and glass-crystalline matrices for HLW immobilisation. Open Ceram. 2023, 14, 100355. [Google Scholar] [CrossRef]
- McGlinn, P.J.; Advocat, T.; Loi, E.; Leturcq, G.; Mestre, J.P. Nd- and Ce-doped ceramic-glass composites: Chemical durability under aqueous conditions and surface alteration in a moist clay medium at 90 °C. Mater. Res. Soc. Symp. Proc. 2001, 663, 249–258. [Google Scholar] [CrossRef]
- Sun, Z.; Lv, P.; Wang, Z.; Jiang, Y.; Zhao, Y.; Deng, W.; Zhang, S.; Zhang, L.; Wang, T.; Chen, L. Phase evolution and valence state change in Gd-Ce co-doped zirconolite glass-ceramics. J. Nucl. Mater. 2022, 568, 153879. [Google Scholar] [CrossRef]
- Day, R.A.; Ferenczy, J.; Drabarek, E.; Advocat, T.; Fillet, C.; Lacombe, J.; Ladirat, C. Glass-ceramics in a cold-crucible melter: The optimum combination for greater waste processing efficiency. In Proceedings of the WM’03 Conference, Tucson, AZ, USA, 23–27 February 2003. [Google Scholar]
- Lv, P.; Chen, L.; Zhang, B.; Zhang, D.; Yuan, W.; Duan, B.; Guan, Y.; Pan, C.; Chen, Z.; Zhang, L.; et al. The effects of temperature and Ce-dopant concentration on the synthesis of zirconolite glass-ceramic. Ceram. Int. 2019, 45, 11819–11825. [Google Scholar] [CrossRef]
- Paknahad, E.; Grosvenor, A.P. Investigation of CeTi2O6- and CaZrTi2O7-containing glass-ceramic composite materials. Can. J. Chem. 2017, 95, 109–117. [Google Scholar] [CrossRef]
- Wilkins, M.C.J.D.; Gausse, C.; Townsend, L.T.; Gardner, L.J.; Corkhill, C.L. Characterisation of a complex CaZr0.9Ce0.1Ti2O7 glass-ceramic produced by Hot Isostatic Pressing. Ceramics 2022, 5, 1035–1050. [Google Scholar] [CrossRef]
- Kim, M.; Kang, J.; Yoon, J.-H.; Lee, S.-G.; Um, W.; Kim, H.G. Partitioning effects and corrosion characteristics of oxyapatite glass-ceramic wasteforms sequestering rare-earth elements. Nucl. Eng. Technol. 2022, 54, 997–1002. [Google Scholar] [CrossRef]
- Kong, L.; Wei, T.; Zhang, Y.; Karatchevtseva, I. Phase evolution and microstructure analysis of CaZrTi2O7 zirconolite in Glass. Ceram. Int. 2018, 44, 6285–6292. [Google Scholar] [CrossRef]
- Crum, J.; Maio, V.; McCloy, J.; Scott, C.; Riley, B.; Benefiel, B.; Vienna, J.; Archibald, K.; Rodriguez, C.; Rutledge, V.; et al. Cold crucible induction melter studies for making glass ceramic waste forms: A feasibility assessment. J. Nucl. Mater. 2014, 444, 481–492. [Google Scholar] [CrossRef]
- Wei, Z.-J.; Blackburn, L.R.; Gardner, L.J.; Tan, S.-H.; Sun, S.-K.; Guo, W.-M.; Hyatt, N.C.; Lin, H.-T. Rapid synthesis of zirconolite ceramic wasteform by microwave sintering for disposition of plutonium. J. Nucl. Mater. 2020, 539, 152332. [Google Scholar] [CrossRef]
- Clark, B.M.; Sundaram, S.K.; Misture, S.T. Polymorphic transitions in cerium-substituted zirconolite (CaZrTi2O7). Sci. Rep. 2017, 7, 5920. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.-H.; Eun, H.-C.; Lee, T.-K.; Lee, K.-R.; Han, S.-Y.; Jeon, M.-K.; Hwan, P.-S.; Ahn, D.-H. Estimation of centerline temperature of the waste form for the rare earth waste generated from pyrochemical process. J. Nucl. Mater. 2017, 483, 82–89. [Google Scholar] [CrossRef]
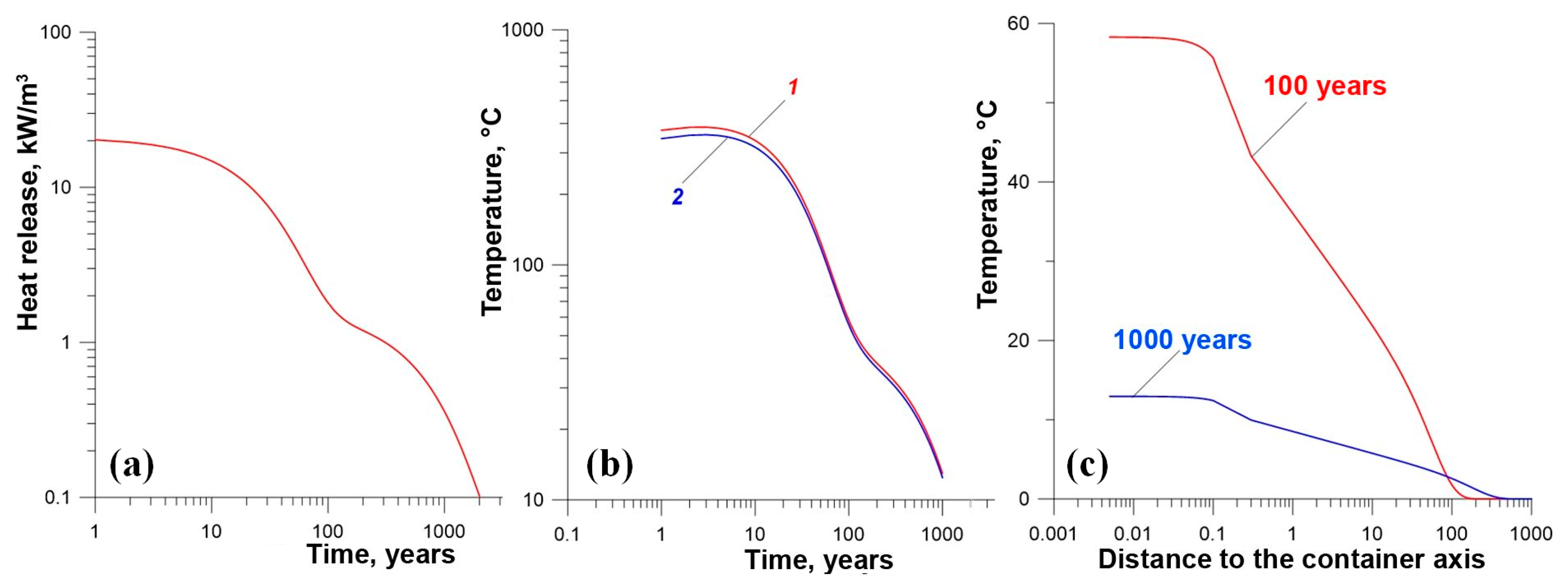
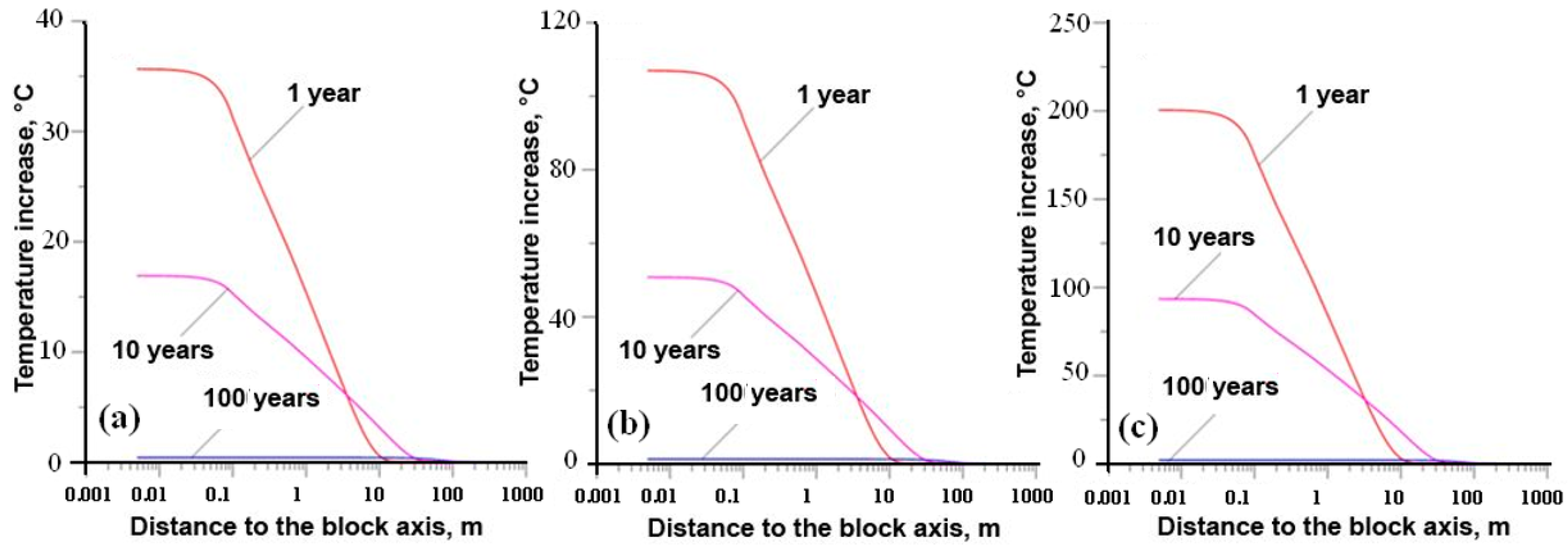
- Yudintsev, S.V.; Malkovsky, V.I.; Kalenova, M.Y. The thermal field around a borehole repository of radioactive waste. Dokl. Earth Sci. 2021, 498, 525–532. [Google Scholar] [CrossRef]
- Kim, M.; Hong, K.-S.; Lee, J.; Byeon, M.; Jeong, Y.; Kim, J.H.; Um, W.; Kim, H.G. Evaluating thermal stability of rare-earth containing wasteforms at extraordinary nuclear disposal conditions. Nucl. Eng. Technol. 2021, 53, 2576–2581. [Google Scholar] [CrossRef]
- Loiseau, P.; Caurant, D.; Majerus, O.; Baffier, N.; Mazerolles, L.; Fillet, C. Crystallisation of zirconolite (CaZrTi2O7) in SiO2–Al2O3–CaO glasses containing simulated actinides nuclear wastes. Phys. Chem. Glasses. 2002, 43C, 195–200. [Google Scholar]
- Caurant, D. Des Matrices de Confinement Pour Déchets Nucléaires Aux Verres du Patrimoine. Chimie; Sorbonne Université: Paris, France, 2018; Available online: https://hal.science/tel-02374975/document (accessed on 17 January 2023).
- Lopez, C.; Deschanels, X.; Bart, J.M.; Boubals, J.M.; Den Auwer, C.; Simoni, E. Solubility of actinide surrogates in nuclear glasses. J. Nucl. Mater. 2003, 312, 76–80. [Google Scholar] [CrossRef]
- Fabian, M.; Gergely, F.; Osan, J.; Cendak, T.; Kesari, S.; Rao, R. Structural investigation of borosilicate glasses containing lanthanide ions. Sci. Rep. 2020, 10, 7835. [Google Scholar] [CrossRef]
- Fabian, M.; Pinakidou, F.; Tolnai, I.; Czompoly, O.; Osan, J. Lanthanide (Ce, Nd, Eu) environments and leaching behavior in borosilicate glasses. Sci. Rep. 2021, 11, 13272. [Google Scholar] [CrossRef]
- Jantzen, C.M. Development of glass matrices for high level radioactive wastes. In Handbook of Advanced Radioactive Waste Conditioning Technologies; Ojovan, M.I., Ed.; Woodhead Publishing Limited: Derbyshire, UK, 2011; Volume 488, pp. 230–292. [Google Scholar] [CrossRef]
- Schwartz, F.W.; Kim, Y.; Chae, B.-G. Deep borehole disposal of nuclear wastes: Opportunities and challenges. J. Nucl. Fuel Cycle Waste Technol. 2017, 15, 15–301. [Google Scholar] [CrossRef]
- Kochkin, B.; Malkovsky, V.; Yudintsev, S.; Petrov, V.; Ojovan, M. Problems and perspectives of borehole disposal of radioactive waste. Prog. Nucl. Energy 2021, 139, 103867. [Google Scholar] [CrossRef]
- Buser, M. Nuclear waste disposal: An exploratory historical overview. Atw -Int. J. Nucl. Power 2021, 66, 9–14. [Google Scholar]
- Kochkin, B.T.; Bogatov, S.A. Prospects for using the well concept for RW removal in Russia. Radioact. Waste 2022, 2, 85–99. (In Russian) [Google Scholar] [CrossRef]
- Swift, P.; Newman, A. Deep Borehole Disposal of Radioactive Waste: Next Steps and Applicability to National Programs. Center on Global Energy Policy; Columbia SIPA: New York, NY, USA, 2022; p. 26. [Google Scholar]
- Mallants, D.; Griffiths, H.; Bonano, E.J. Does deep borehole disposal of nuclear waste allow for termination from safeguards. In Symposium on International Safeguards; IAEA: Vienna, Austria, 2022. [Google Scholar]
- Fernández, A.M.; Marco, J.F.; Nieto, P.; León, F.J.; Robredo, L.M.; Clavero, M.Á.; Cardona, A.I.; Fernández, S.; Svensson, D.; Sellin, P. Characterization of bentonites from the in situ ABM5 heater experiment at Äspö hard rock laboratory, Sweden. Minerals 2022, 12, 471. [Google Scholar] [CrossRef]
- Zandanel, A.; Sauer, K.B.; Rock, M.; Caporuscio, F.A.; Telfeyan, K.; Matteo, E.N. Impacts of crystalline host rock on repository barrier materials at 250 °C: Hydrothermal co-alteration of Wyoming bentonite and steel in the presence of Grimsel granodiorite. Minerals 2022, 12, 1556. [Google Scholar] [CrossRef]
- Laverov, N.P.; Omel’yanenko, B.I.; Yudintsev, S.V. Isolating properties of a bentonite buffer in conditions of an underground repository for high-level radioactive wastes. Geol. Ore Depos. 2004, 46, 22–35. [Google Scholar]
- Mori, A.; Alexander, W.R.; Geckeis, H.; Hauser, W.; Schafer, T.; Eikenberg, J.; Fierz Th Degueldre, C.; Missana, T. The colloid and radionuclide retardation experiment at the Grimsel Test Site: Influence of bentonite colloids on radionuclide migration in a fractured rock. Colloids Surf. A Physicochem. Eng. Asp. 2003, 217, 33–47. [Google Scholar] [CrossRef]
- Zänker, H.; Hennig, C. Colloid-borne forms of tetravalent actinides: A brief review. J. Contam. Hydrol. 2014, 157, 87–105. [Google Scholar] [CrossRef] [PubMed]
- Vance, E.R.; Agraval, D.K. Incorporation of radionuclides in crystalline titanates. Nucl. Chem. Waste Manag. 1982, 3, 229–234. [Google Scholar] [CrossRef]
- Cheary, R.W.; Coelho, A.A. A site occupancy analysis of zirconolite CaZrxTi3-xO7. Phys. Chem. Miner. 1997, 24, 447–454. [Google Scholar] [CrossRef]
- Blackburn, L.R.; Gardner, L.J.; Sun, S.K.; Maddrell, E.R.; Stnnett, M.C.; Corkhill, C.L.; Hyatt, N.C. Hot isostatically pressed zirconolite wasteforms for actinide immobilisation. IOP Conf. Ser. Mater. Sci. Eng. 2020, 818, 012010. [Google Scholar] [CrossRef]
- Vance, E.R.; Ball, C.J.; Day, R.A.; Smith, K.L.; Blackford, M.G.; Begg, B.D.; Angel, P.J. Actinide and rare earth incorporation into zirconolite. J. Alloys Compd. 1994, 213–214, 406–409. [Google Scholar] [CrossRef]
- Bellatreccia, F.; Ventura, G.; Williams, C.T.; Lumpkin, G.; Smith, K.; Colella, M. Non-metamict zirconolite polytypes from the feldspathoid-bearing alkalisyenitic ejecta of the Vico volcanic complex (Latium, Italy). Eur. J. Mineral 2002, 14, 809–820. [Google Scholar] [CrossRef]
- Stefanovsky, S.V.; Yudintsev, S.V. Titanates, zirconates, aluminates and ferrites as waste forms for actinide immobilization. Russ. Chem. Rev. 2016, 85, 962–994. [Google Scholar] [CrossRef]
- Equipment Being Installed in ANSTO’s Synroc Radioactive Waste Processing Facility. WNN. 2022. Available online: https://www.world-nuclear-news.org/Articles/Equipment-being-installed-in-ANSTO-s-Synroc (accessed on 17 January 2023).
- Ojovan, M.I.; Gibb, F.G.F.; Lee, W.E. In situ sintering of waste forms in an underground disposal environment. Mat. Res. Soc. Symp. Proc. 2004, 807, 949–954. [Google Scholar] [CrossRef]
- National Academies of Sciences, Engineering, and Medicine. Merits and Viability of Different Nuclear Fuel Cycles and Technology Options and the Waste Aspects of Advanced Nuclear Reactors; The National Academies Press: Washington, DC, USA, 2023; p. 314. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Element | After 5 Years of SNF Storage | After 30 Years of SNF Storage | ||||||
|---|---|---|---|---|---|---|---|---|
| 45 GW × d/t | 60 GW × d/t | 45 GW × d/t | 60 GW × d/t | |||||
| a | b | a | b | a | b | a | b | |
| Gd | 150 | Stable 1 | 310 | Stable | 180 | Stable | 346 | Stable |
| Eu | 190 | 60 | 260 | 90 | 170 | 8 | 230 | 12 |
| Sm 1 | 1060 | Stable | 1370 | Stable | 1120 | Stable | 1430 | Stable |
| Pm | 63 | 21 | 62 | 21 | 0 | 0 | 0 | 0 |
| Ce | 3210 | 10 | 4230 | 10 | 3210 | Stable | 4220 | Stable |
| Pr | 1540 | 114 | 2010 | 113 | 1540 | Stable | 2010 | Stable |
| Nd 2 | 5570 | Stable | 7310 | Stable | 5570 | Stable | 7310 | Stable |
| La | 1670 | Stable | 2190 | Stable | 1670 | Stable | 2190 | Stable |
| Σ REE | 13,453 | 205 | 17,742 | 234 | 13,460 | 8 | 17,736 | 12 |
| U | 941,000 | 0.06 | 923,000 | 0.06 | 941,000 | 0.06 | 923,000 | 0.06 |
| Pu | 11,200 | 164 | 12,600 | 283 | 10,200 | 138 | 11,500 | 236 |
| Np | 570 | 0.01 | 780 | 0.02 | 570 | 0.01 | 780 | 0.02 |
| Am | 510 | 47 | 740 | 58 | 1380 | 146 | 1780 | 178 |
| Cm | 33 | 88 | 113 | 292 | 14 | 34 | 50 | 112 |
| Am + Cm (MA) | 543 | 135 | 853 | 350 | 1394 | 180 | 1830 | 290 |
| MA share 3 | 4% | 40% | 5% | 60% | 9% | 96% | 9% | 96% |
| Radionuclide (T1/2, Years) | Content, wt% | Daughter Nuclide, T1/2, Years | Type and Probability of Decay | Heat Release, W/kg |
|---|---|---|---|---|
| 241Am (433) | 63.85 | 237Np (2.14 × 106) | α (≈1.0), SF (3.77 × 10−12) | 114.7 |
| 243Am (7.3 × 103) | 25.35 | 239Pu (2.41 × 104) | α (≈1.0), SF (3.7 × 10−11) | 6.4 |
| 243Cm (29.1) | 0.09 | 239Pu (2.41 × 104) | α (0.9976), β + (0.0024) | 1860.7 |
| 244Cm (18.1) | 9.78 | 240Pu (6537) | α (≈1.0), SF (1.35 × 10−6) | 2841.8 |
| 245Cm (8.5 × 103) | 0.82 | 241Pu (14.4) | α (1.0) | 5.8 |
| 246Cm (4.76 × 103) | 0.11 | 242Pu (3.76 × 105) | α (≈1.0), SF (2.61 × 10−4) | 10.2 |
| Nuclide, g/t SNF | After 1 Year | After 5 Years | After 30 Years |
|---|---|---|---|
| 241Am | 135 | 407 | 1272 |
| 243Am | 105 | 105 | 105 |
| Total, Am | 240 | 512 | 1377 |
| 242Cm | 3.8 | 0.1 | <0.01 |
| 244Cm | 35.3 | 30.3 | 11.6 |
| 245Cm | 2.2 | 2.2 | 2.2 |
| Total, Cm 1 | 41.9 | 33.0 | 14.4 |
| Cm/(Am + Cm), % | 14.9 | 6.1 | 1.0 |
| (Am + Cm)/REE 2, % | 1.8 | 3.5 | 8.5 |
| Cation | x = 0.10 | x = 0.20 | x = 0.30 | x = 0.40 | x = 0.50 | x = 0.60 |
|---|---|---|---|---|---|---|
| Ce4+ | 2M | 2M + 4M | 2M + 4M | 2M + 4M | 2M + 4M + Pyr | 4M + Pyr |
| U4+ | 2M | 2M + 4M | 2M + 4M | 4M + Pyr | 4M + Pyr | 4M + Pyr |
| Th4+ | 2M + Pyr | 2M + Pyr | 2M + Pyr | 2M + Pyr | 2M + Pyr | Pyr |
| Pu4+ | 2M | 2M + 4M | 2M + 4M | 4M + Pyr | 4M + Pyr | Pyr |
| Element | Al | Ga | Cr | Fe | Mg | Ni | Mo | W | Ta |
|---|---|---|---|---|---|---|---|---|---|
| Kd | 16 | >30 | 15 | 3 | 3 | 1.5 | 0.1 | 0.02 | <0.1 |
| Phase | Basic Composition | Possible Variation | Acceptable Range |
|---|---|---|---|
| Pyrochlore | 80 | 62–90 | >50 |
| Brannerite | 12 | 0–22 | 0–50 |
| Zirconolite | no | 0–25 | 0–50 |
| Rutile | 8 | 0–16 | 0–20 |
| (Pu,U)O2 | 0.5 | 0.04–0.6 | 0–1 |
| Other phases | no | 0–6 | 0–10 |
| A.m. | 89 | 139 | 140 | 141 | 142 | 143 | 144 | 145 | 146 | 147 | 148 | 149 | 150 | 151 | 152 | 153 | 154 | 155 | 156 | 158 | 159 | 160 | 241 | 241 | 243 | 244 | 245 | Σ (Share) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Y | 546 100% | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | 546 (4.2%) |
| La | - | 1470 100% | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | 1470 (11.3%) |
| Ce | - | - | 1490 52% | - | 1360 48% | - | 4 <0.1% | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | 2854 (21.9%) |
| Pr | - | - | - | 1340 100% | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | 1340 (10.3%) |
| Nd | - | - | - | - | 35 1% | 940 19% | 1590 33% | 793 16% | 845 18% | - | 445 9% | - | 214 4% | - | - | - | - | - | - | - | - | - | - | - | - | - | - | 4862 (37.4%) |
| Pm | - | - | - | - | - | - | - | - | - | 37 100% | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | 37 (0.3%) |
| Sm | - | - | - | - | - | - | - | - | - | 186 19% | 232 23% | 4 <1% | 369 37% | 18 2% | 145 14% | - | 46 5% | - | - | - | - | - | - | - | - | - | - | 1000 (7.7%) |
| Eu | - | - | - | - | - | - | - | - | - | - | - | - | - | 1 <1% | - | 150 78% | 32 17% | 9 5% | - | - | - | - | - | - | - | - | - | 192 (1.5%) |
| Gd | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | 20 14% | 9 6% | 87 61% | 25 18% | - | 2 1% | - | - | - | - | - | 143 (1.1%) |
| Tb | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | 3 100% | - | - | - | - | - | - | 3 (<0.1%) |
| Am | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | 378 73% | 3 <1% | 139 23% | - | - | 520 (4.0%) |
| Cm | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | 43 96% | 2 4% | 45 (0.3%) |
| Type of Exchange | 0.1 | 0.2 | 0.3 | 0.4 | 0.5 | 0.6 | 0.7 | 0.8 | 0.9 | 1.0 | Reference | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Ca2+ + Ti4+ = Nd3+ + Al3+ | 2M, Per (traces) | 3O, 2M, Per | 3O, Per | [87,119] | ||||||||
| Nd2O3, wt% | 4.8 | 9.5 | 13.9 | 18.1 | 22.1 | 26.0 | 29.6 | 33.2 | 36.6 | 39.8 | NdZrTiAlO7 | |
| Ca2+ + Ti4+ = Nd3+ + Al3+ | 2M | No data | [116] | |||||||||
| Ca2+ + Ti4+ = Nd3+ + Fe3+ | 2M, Per (traces) | 2M, 3O | 3O | [88] | ||||||||
| Ca2+ + Ti4+ = Nd3+ + 0.5Fe3+ +0.5Al3+ | 2M, Per (traces) | 2M, 3O, Per | 3O, Per (traces) | [113] | ||||||||
| Ca2+ + Zr4+ = 2Nd3+ | 2M | 2M, 4M | 4M | No data | [85,116] | |||||||
| ` Ca2+ + Zr4+ = 2Nd3+ | 2M | 2M, 4M | 4M | 4M, Pyr | Pyr | Pyr, NT | NT | [120,121] | ||||
| Ca2+ + Zr4+ + Ti4+ = Nd3+ + Hf4+ + Al3+ | 2M | No data | [90] | |||||||||
| Zr4+ + Ti4+ = Nd3+ + Nb5+ | 2M Per | 2M, 3T | 2M, 4M, Pyr, Per | Pyr, Per (traces) | [122] | |||||||
| Oxide, wt% | Nd | Sm | Gd | ||||||
|---|---|---|---|---|---|---|---|---|---|
| 1450 °C | 1550 °C | 1450 °C | 1450 °C | ||||||
| Z | Py | C | Z | P | C + P | Z | F | Z | |
| Al2O3 | 11.3 | 0.5 | 99.0 | 7.1 | 8.5 | 71.0 | 9.6 | 2.5 | 6.7 |
| TiO2 | 18.7 | 11.3 | 0.4 | 21.0 | 21.6 | 2.1 | 18.0 | 8.8 | 17.2 |
| Fe2O3 2 | 0.8 | No | No | 0.2 | No | 1.6 | No | No | 5.0 |
| ZrO2 | 27.0 | 34.2 | No | 29.5 | 4.6 | 3.5 | 30.6 | 55.1 | 30.4 |
| REE2O3 | 42.2 | 54.0 | 0.6 | 42.2 | 65.3 | 21.8 | 41.8 | 33.6 | 40.7 |
| Phase Formula According to SEM Data | T, °C | a, Å | b, Å | c, Å | β, deg. |
|---|---|---|---|---|---|
| Nd1.07Zr0.95Ti0.99Al0.95Fe0.04O7 | 1450 | 10.060 | 13.957 | 7.299 | 90 |
| Nd1.08Zr1.04Ti1.14Al0.61Fe0.02O7 | 1550 | 10.116 | 13.985 | 7.316 | 90 |
| Sm1.06Zr1.07Ti1.00Al0.86Fe0.04O7 | 1450 | 10.020 | 13.854 | 7.245 | 90 |
| Gd1.01Zr1.05Ti0.98Al0.60Fe0.32O7 | 1450 | 10.020 | 13.854 | 7.245 | 90 |
| Zirconolite-3O JCPDS 85-0928 | Reference | 10.14 | 14.14 | 7.278 | 90 |
| No | Parameters | Intensities of the Strongest Reflection of Phase at XRD Pattern, rel.% | ||||
|---|---|---|---|---|---|---|
| T °C | t, h | TiO2 | ZrO2, Baddeleyite | CaTiO3, Perovskite | CaZrTi2O7, Zirconolite | |
| 51 | 1200 | 10 | 88 | 100 | 13 | 90 |
| 45 | 1200 | 15 | 58 | 84 | 92 | 100 |
| 75 | 1200 | 20 | 57 | 62 | 70 | 100 |
| 53 | 1300 | 10 | 29 | 30 | 38 | 100 |
| 67 | 1300 | 20 | - | <5 rel.% | <5 rel.% | 100 |
| 168 | 1400 | 1 | 14 | 24 | 69 | 100 |
| 166 | 1400 | 3 | <5% | 11 | 43 | 100 |
| 164 | 1400 | 5 | <5% | 11 | 16 | 100 |
| 94 | 1400 | 20 | - | - | <5 rel.% | 100 |
| 20 | 1450 | 5 | - | - | <5 rel.% | 100 |
| 172 | 1500 | 1 | - | <5 rel.% | <5 rel.% | 100 |
| 121 | 1500 | 20 | - | - | <5 rel.% | 100 |
| 18 | 1550 | 5 | Melting | |||
| T °C | Zirconolite | Perovskite, CaTiO3 | Baddeleyite, ZrO2 | Rutile, TiO2 | Density, rel.% |
|---|---|---|---|---|---|
| 900 | 4.9 | 38.0 | 35.3 | 21.8 | 55.0 |
| 1000 | 36.9 | 27.7 | 22.9 | 12.5 | 55.9 |
| 1100 | 78.7 | 11.2 | 10.1 | - | 56.8 |
| 1200 | 95.1 | 2.6 | 2.3 | - | 59.58 |
| 1300 | 99.0 | 1.0 | - | - | 72.7 |
| 1400 | 99.0 | 1.0 | - | - | 97.0 |
| 1450 | 99.0 | 1.0 | - | - | 98.3 |
| Oxide, wt% | Sample–I | Sample–II | Sample–III | Sample–IV |
|---|---|---|---|---|
| Al2O3 | 8.70/6.3 | 8.8/6.5 | 11.3/9.4 | 2.9/1.3 |
| SiO2 | 6.1/5.6 | 5.4/7.9 | 5.6/4.9 | 1.0/2.6 |
| CaO | 5.3/4.9 | 5.4/5.1 | 6.2/5.7 | 13.2/14.3 |
| TiO2 | 26.7/24.1 | 32.0/27.3 | 38.1/38.5 | 33.6/38.3 |
| ZrO2 | 23.5/25.4 | 19.0/19.5 | 31.0/25.5 | 41.3/32.5 |
| Gd2O3 | 30.0/n.a. | 29.2/24.9 | 7.6/6.1 | 7.7/n.a. |
| Phase | Sample–I | Sample–II | Sample–III | Sample–IV |
|---|---|---|---|---|
| Zirconolite (Zir) | 80 1 | 75 1 | 65 | 50 |
| Gd-Zr oxide (GZO) | 10 | 10 | 10 | 25 |
| Ti oxide (TO) | <1 | <5 | 15 | 5 |
| Perovskite (Per) | n.o. | n.o. | n.o. | 20 |
| Al2Ti2O5 (AT) | n.o. | n.o. | 5 | n.o. |
| Glass (Gl) | 10 | 10 | 5 | <5 |
| Oxide | S-I | Zir-1 | Zir-2 | GZO | TO | Gl | S-IV | Zir | GZO | TO | Per | Gl |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Gd2O3 | 29.2 | 31.6 | 33.5 | 23.2 | 1.2 | 12.6 | 7.0 | 6.8 | 9.8 | Bdl | 7.7 | 2.4 |
| ZrO2 | 19.0 | 30.6 | 10.0 | 69.0 | 3.4 | Bdl | 40.4 | 44.9 | 81.5 | 3.6 | 1.2 | 0.7 |
| TiO2 | 32.0 | 26.3 | 42.9 | 6.0 | 91.8 | 8.3 | 34.5 | 33.8 | 5.4 | 95.5 | 54.9 | 12.2 |
| CaO | 5.4 | 5.0 | 4.8 | 1.1 | 0.9 | 14.0 | 14.3 | 12.9 | 2.9 | 0.5 | 35.5 | 22.4 |
| Al2O3 | 8.8 | 6.4 | 8.0 | Bdl | 1.3 | 24.0 | 2.8 | 1.7 | 0.3 | 0.3 | 0.7 | 26.6 |
| SiO2 | 5.4 | Bdl | Bdl | Bdl | Bdl | 41.0 | 3.0 | Bdl | Bdl | Bdl | Bdl | 35.0 |
| Sample Number | Generation of Zirconolite | Formulae Normalized to 7 O2− |
|---|---|---|
| Sample I | Zirconolite-1 (early) | Ca0.34Gd0.69Zr1.06Ti1.37Al0.49O7.0 |
| Zirconolite-2 (late) | Ca0.30Gd0.74Zr0.33Ti2.01Al0.60O7.0 | |
| Sample II | Zirconolite-1 (early) | Ca0.35Gd0.72Zr1.03Ti1.42Al0.45O7.0 |
| Zirconolite-2 (late) | Ca0.31Gd0.71Zr0.32Ti2.02Al0.59O7.0 | |
| Sample III | Zirconolite | Ca0.52Gd0.39Zr0.91Ti1.69Al0.44O7.0 |
| Sample IV | Zirconolite | Ca0.84Gd0.14Zr1.26Ti1.60Al0.14O7.0 |
| N | Phase | Ideal Formula | Initial Sample | After Heat Treatment |
|---|---|---|---|---|
| 1 | Zirconolite-1 | CaZrTi2O7 | (Ca0.40La0.03Ce0.10Nd0.30Eu0.14) Zr1.03Ti1.84O7 | (Ca0.36La0.03Ce0.10Nd0.29Eu0.12) Zr1.01Ti1.90O7 |
| 2 | Zirconolite-2 | (Ca0.35La0.10Ce0.45Nd0.53Eu0.11) Zr0.29Ti2.14O7 | (Ca0.38La0.11Ce0.44Nd0.56Eu0.13) Zr0.28Ti2.09O7 | |
| 3 | Zr titanate | ZrTiO4 | Zr0.92Ti1.08O4 | (Ca0.02Zr0.95)Ti1.04O4 |
| 4 | Rutile | TiO2 | Not analyzed | Ti0.91Zr0.09O2 |
| Oxide | NZO 1 | Zir-1 | Zir-2 | Per | TO | Hib | 5/1 | 5/2 | mt | 3O |
|---|---|---|---|---|---|---|---|---|---|---|
| Al2O3 | 2.1 | 5.8 | 5.5 | 2.4 | - | 55.2 | - | 0.8 | - | 3.1 |
| CaO | 6.8 | 6.9 | 5.9 | 8.8 | - | 2.3 | - | - | - | - |
| TiO2 | 21.3 | 30.3 | 46.0 | 41.9 | 95.6 | 25.1 | 32.9 | 28.5 | 31.4 | 33.1 |
| FeO | - | 1.1 | 3.0 | - | 1.1 | 0.9 | - | - | - | - |
| ZrO2 | 47.9 | 33.4 | 11.6 | - | 3.3 | - | - | 6.2 | 0.8 | 25.2 |
| Nd2O3 | 21.9 | 22.5 | 28.0 | 46.9 | - | 16.5 | 67.1 | 64.5 | 67.8 | 38.5 |
| Al3+ | 0.04 | 0.44 | 0.40 | 0.08 | - | 8.37 | - | - | 0.27 | |
| Ca2+ | 0.13 | 0.48 | 0.38 | 0.28 | - | 0.32 | - | - | - | |
| Ti4+ | 0.28 | 1.46 | 2.11 | 0.93 | 0.97 | 2.43 | 2.0 | 1.96 | 1.82 2 | |
| Fe2+ | - | 0.05 | 0.14 | - | 0.01 | 0.09 | - | - | - | |
| Zr4+ | 0.41 | 1.05 | 0.34 | - | 0.02 | - | - | 0.03 | 0.90 | |
| Nd3+ | 0.14 | 0.52 | 0.61 | 0.49 | 0.76 | 2.0 | 2.01 | 1.01 | ||
| Σ cat. | 1.0 | 3.99 | 3.98 | 1.78 | 1.0 | 11.96 | 4.0 | 4.0 | 4.0 | |
| Σ O2− | 1.79 | 7.0 | 7.0 | 3.0 | 2.0 | 19.0 | 7.0 | 6.99 | 7.37 |
| Oxide | CGd (in) | CGd (1200 °C) | CGd (1050 °C) | CTh (in) | CTh (1200 °C) | CTh (1050 °C) |
|---|---|---|---|---|---|---|
| SiO2 | 40.6 | 43.8 | 47.9 | 40.6 | 44.3 | 46.3 |
| Al2O3 | 12.0 | 15.9 | 17.4 | 12.0 | 14.6 | 15.1 |
| CaO | 19.6 | 22.2 | 22.9 | 19.6 | 21.3 | 21.7 |
| TiO2 | 12.4 | 8.1 | 4.8 | 12.4 | 8.4 | 6.7 |
| ZrO2 | 8.5 | 3.8 | 1.9 | 8.5 | 4.1 | 3.6 |
| Na2O | 0.9 | 1.0 | 1.0 | 0.9 | 1.2 | 1.2 |
| Gd2O3 | 6.0 | 5.2 | 4.1 | - | - | - |
| ThO2 | - | - | - | 6.0 | 6.1 | 5.4 |
| Method (and Synthesis Parameters) | Matrix, Content of REE–MA Fraction, wt% | Prototypes and Application Experience | Peculiarities |
|---|---|---|---|
| Hot pressing (T = 1200–1300 °C, P = 30–50 MPa) | Glass ceramics with zirconolite, britholite, pyrochlore (20–40%). | Pyrochlore ceramics with Pu. Nuclear fuel with Pu and minor actinides (Np, Am). | No experience in synthesizing large radioactive samples with real minor actinides. |
| Sintering (1 atm, 1300–1500 °C) | Ceramics with zirconolite, pyrochlore, brannerite (30–50%). | ||
| Electric furnace (up to 1200 °C) | Low-melting B–Si and Al–B–P glasses, glass ceramics with zirconolite (20–40%). | Industrial vitrification of fractionated waste (HLW, ILW). Laboratory tests with MA simulators. | Highly corrosive melts reduce lifetime of the ceramic melter. |
| CCIM (1200–1600 °C) | Refractory Al–B–Si glasses or low-melting glasses, glass ceramics with zirconolite (15–25%). | No experience in synthesizing matrices with actinides. | |
| CCIM (1400–1800 °C) | Ceramics with zirconolite, pyrochlore, murataite and perovskite (10–50%). | Laboratory tests with simulators (REE, U, Th) of actinides. | No experience in synthesizing matrices with real minor actinides. |
| Element | Content, g/t | Radionuclide | Content, g/t | Half-Life |
|---|---|---|---|---|
| La | 1205 | No | No | Stable |
| Ce | 2352 | 144Ce | 23 | 284 days |
| Pr | 1109 | No | No | Stable |
| Nd 1 | 4000 | No | No | Stable |
| Pm | 86 | 147Pm | 86 | 2.6 years |
| Sm 1 | 777 | 151Sm | 16 | 93 years |
| Eu | 133 | 154Eu | 20 | 8.6 years |
| 155Eu | 12 | 4.8 years | ||
| Gd | 76 | No | No | Stable |
| Am | 369, including: | 241Am | 290 | 433 years |
| 243Am | 79 | 7370 years | ||
| Cm | 20, including: | 243Cm | 0.2 | 29 years |
| 244Cm | 18.3 | 18 years | ||
| 245Cm | 1.0 | 8500 years | ||
| 246Cm | 0.1 | 4760 years |
| Heat Release, W/t SNF | 1 | 10 | 30 | 50 | 70 | 100 | 300 | 500 |
|---|---|---|---|---|---|---|---|---|
| Fraction Cs/Sr/Ba/Rb | 2765 | 1054 | 566 | 354 | 222 | 110 | 1 | 0 |
| Ag/Pd/Ru/Rh | 2752 | 11 | 0 | 0 | 0 | 0 | 0 | 0 |
| La/Ce/Pr/Nd/Pm/Sm/Eu | 3593 | 64 | 10 | 2 | 0 | 0 | 0 | 0 |
| Ac/Th/Pa/U | 0.1 | 0.1 | 0.1 | 0.1 | 0.1 | 0.1 | 0.1 | 0.1 |
| Np/Pu/Am/Cm/Bk | 819 | 348 | 332 | 309 | 287 | 258 | 159 | 116 |
| Other radioisotopes | 515 | 15 | 2 | 1 | <0.1 | <0.1 | <0.1 | <0.1 |
| TOTAL | 10,444 | 1492 | 910 | 666 | 509 | 368 | 160 | 116 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yudintsev, S.V.; Nickolsky, M.S.; Ojovan, M.I.; Stefanovsky, O.I.; Malkovsky, V.I.; Ulanova, A.S.; Blackburn, L.R. Zirconolite Matrices for the Immobilization of REE–Actinide Wastes. Ceramics 2023, 6, 1573-1622. https://doi.org/10.3390/ceramics6030098
Yudintsev SV, Nickolsky MS, Ojovan MI, Stefanovsky OI, Malkovsky VI, Ulanova AS, Blackburn LR. Zirconolite Matrices for the Immobilization of REE–Actinide Wastes. Ceramics. 2023; 6(3):1573-1622. https://doi.org/10.3390/ceramics6030098
Chicago/Turabian StyleYudintsev, Sergey V., Maximilian S. Nickolsky, Michael I. Ojovan, Olga I. Stefanovsky, Victor I. Malkovsky, Amina S. Ulanova, and Lewis R. Blackburn. 2023. "Zirconolite Matrices for the Immobilization of REE–Actinide Wastes" Ceramics 6, no. 3: 1573-1622. https://doi.org/10.3390/ceramics6030098
APA StyleYudintsev, S. V., Nickolsky, M. S., Ojovan, M. I., Stefanovsky, O. I., Malkovsky, V. I., Ulanova, A. S., & Blackburn, L. R. (2023). Zirconolite Matrices for the Immobilization of REE–Actinide Wastes. Ceramics, 6(3), 1573-1622. https://doi.org/10.3390/ceramics6030098