Hydrothermal Synthesis, Characterization, and Sintering Behavior of Core-Shell Particles: A Principle Study on Lanthanum Strontium Cobaltite Coated with Nanosized Gadolinium Doped Ceria

 , ,
, ,
Abstract
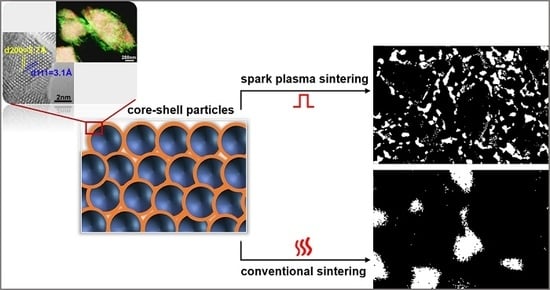
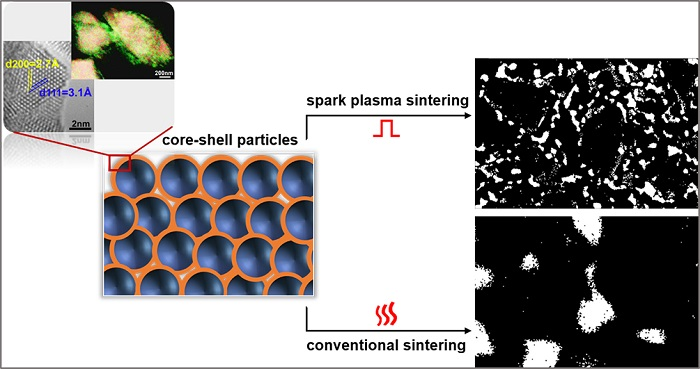{kind=link}
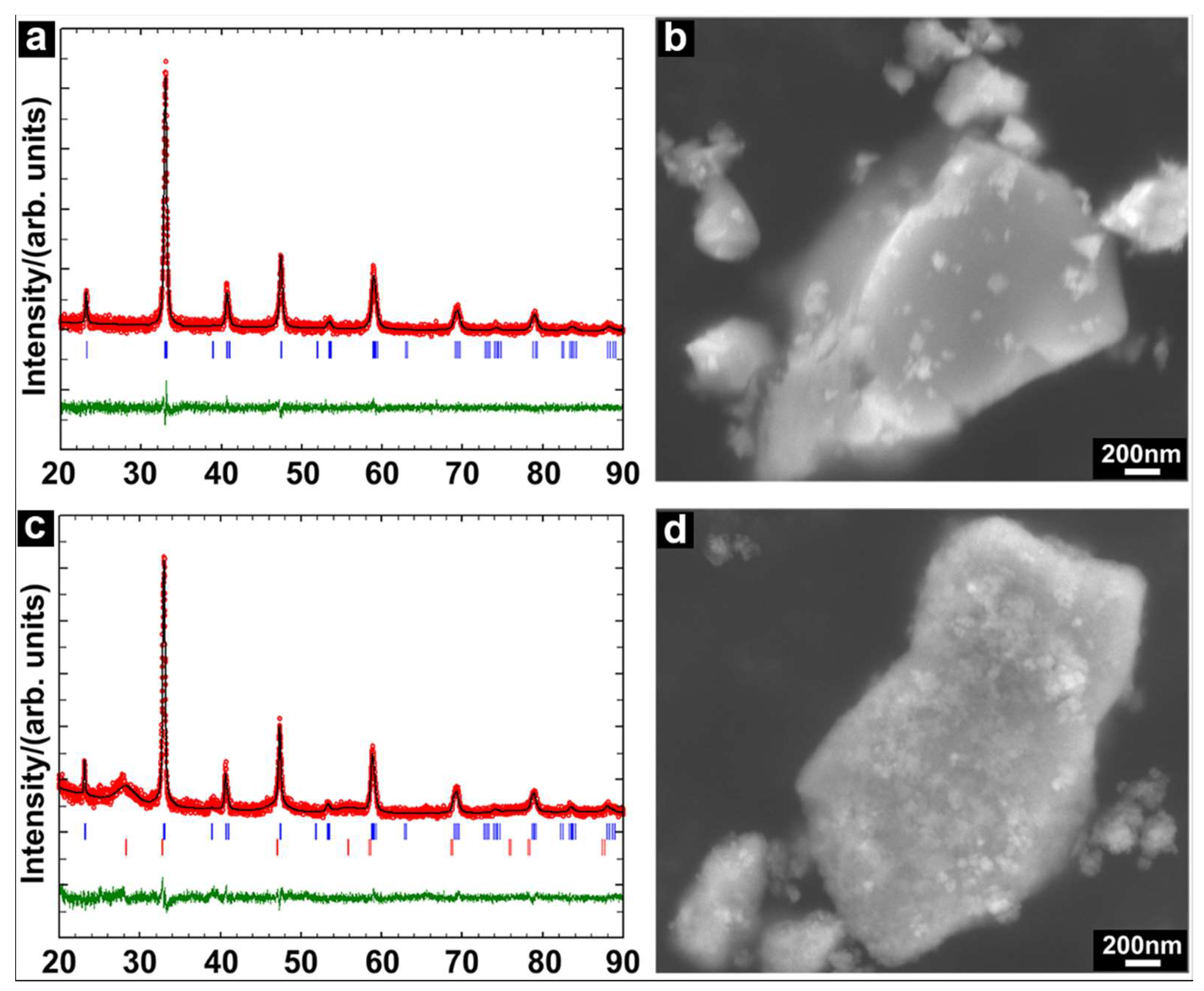{kind=link}
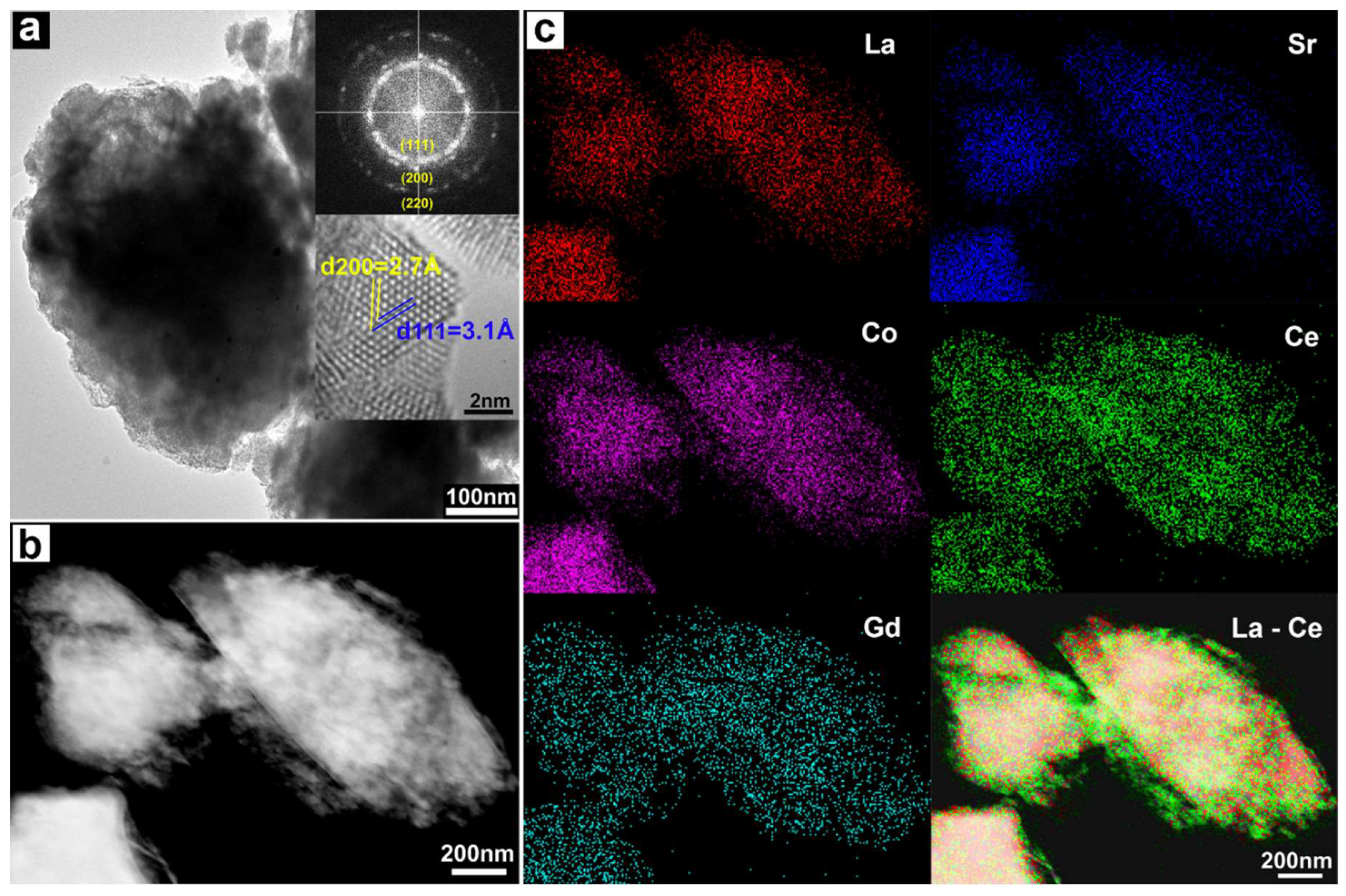{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Experimental
2.1. Preparation of LSC-CGO Core-Shell Particles in a Hydrothermal Autoclave
2.2. Densification of LSC-CGO Particles
2.2.1. Spark Plasma Sintering (SPS)
2.2.2. Conventional Sintering in a Muffle Furnace in Air
2.3. Investigation of the SPS-Densified LSC-CGO Composite in a Thermal Cycling Process in Air
2.4. Characterization
3. Results and Discussion
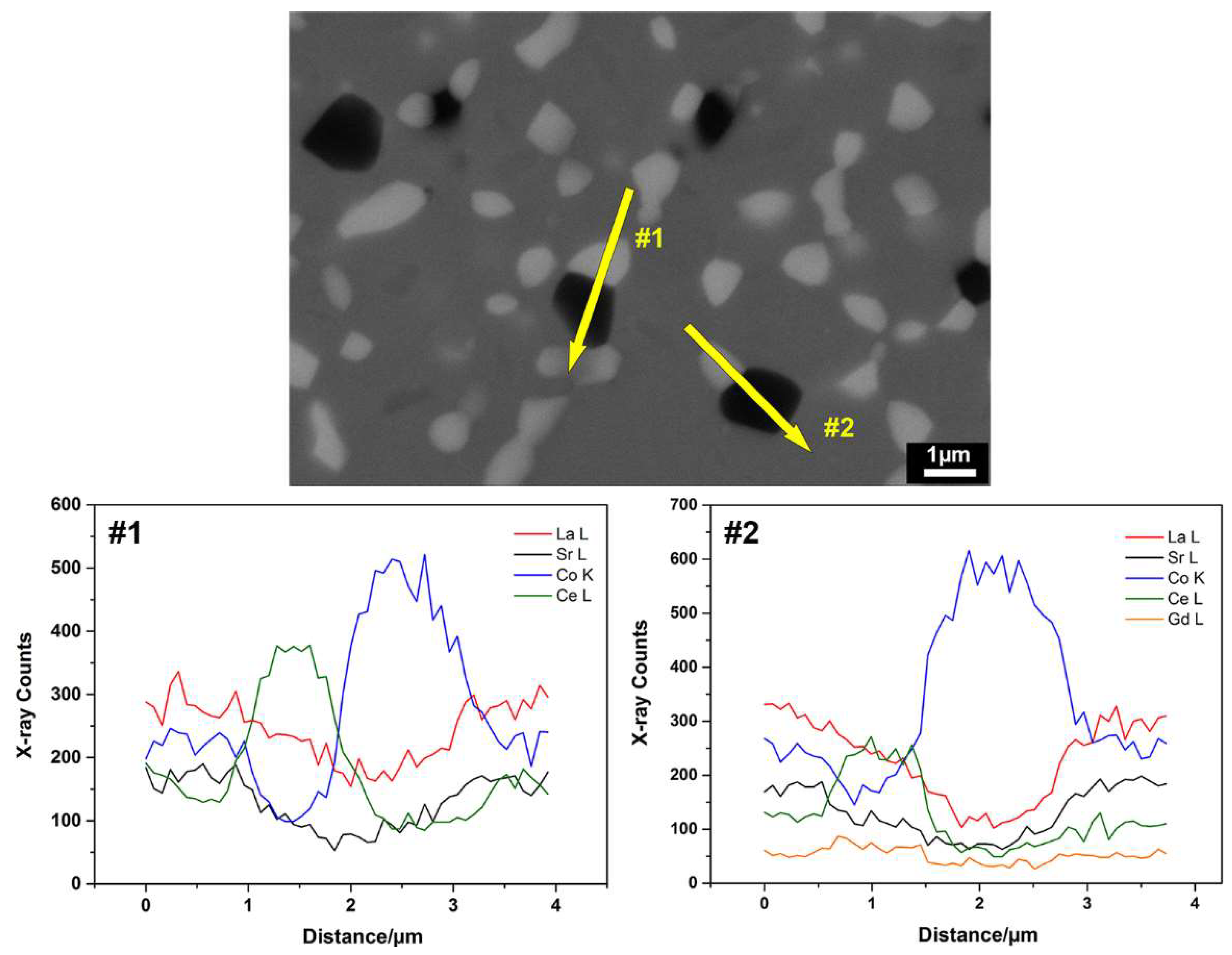
3.1. Structure and Morphology of Nano-Structured LSC-CGO Core-Shell Particles
3.2. Behavior of LSC-CGO Core-Shell Particles during SPS and Conventional Sintering
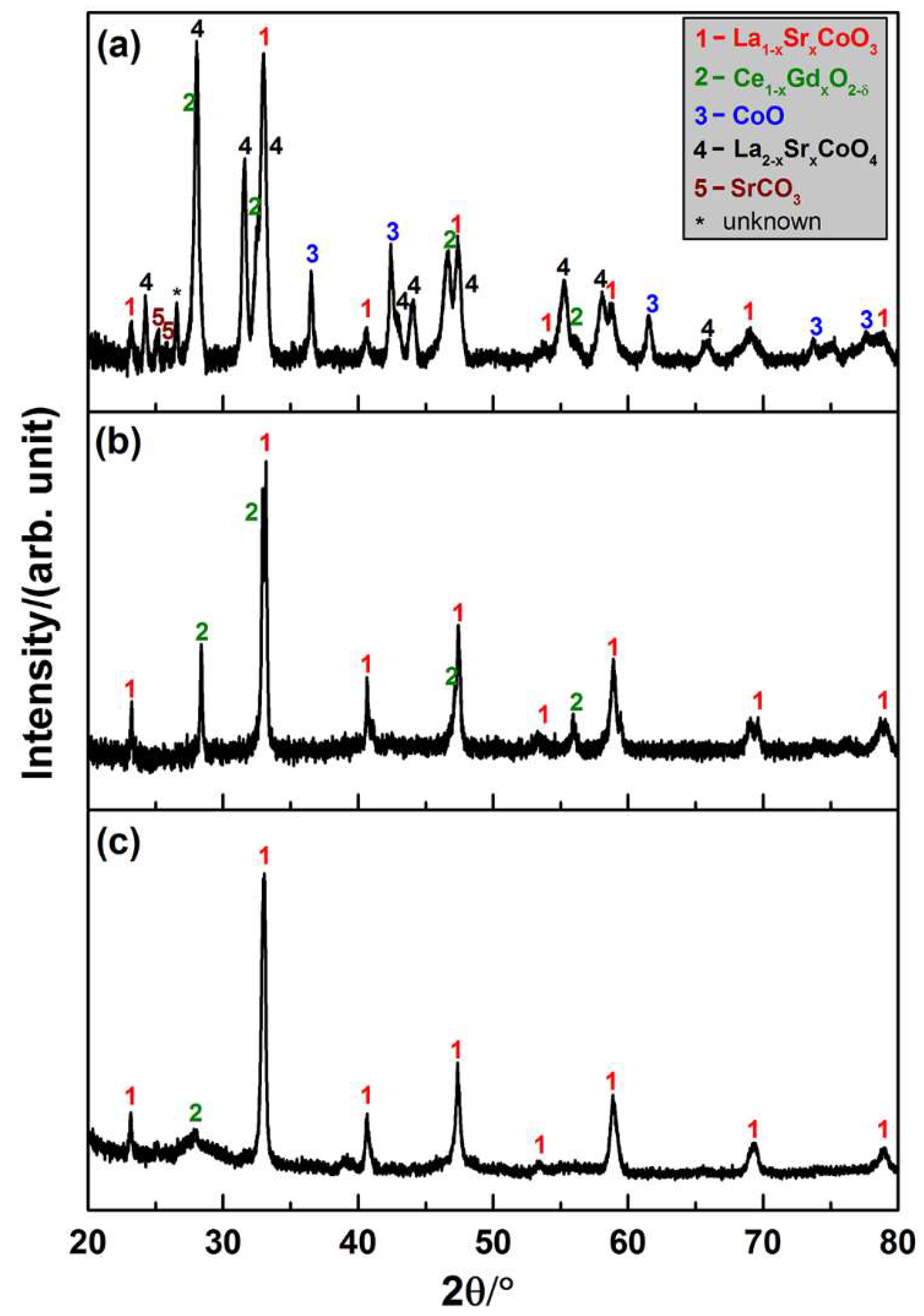
3.2.1. Phase Stability in the Sintering Processes
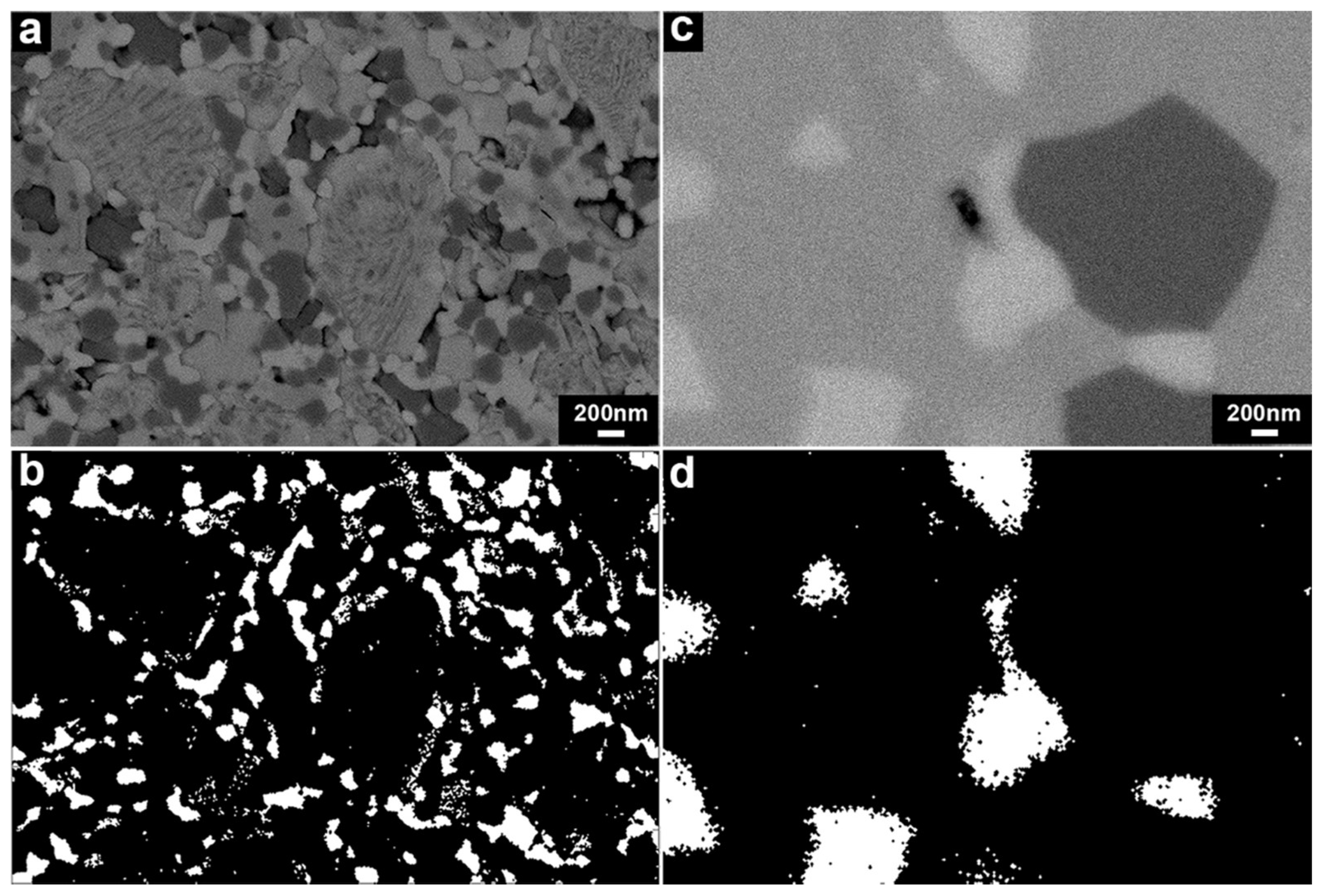
3.2.2. Microstructure
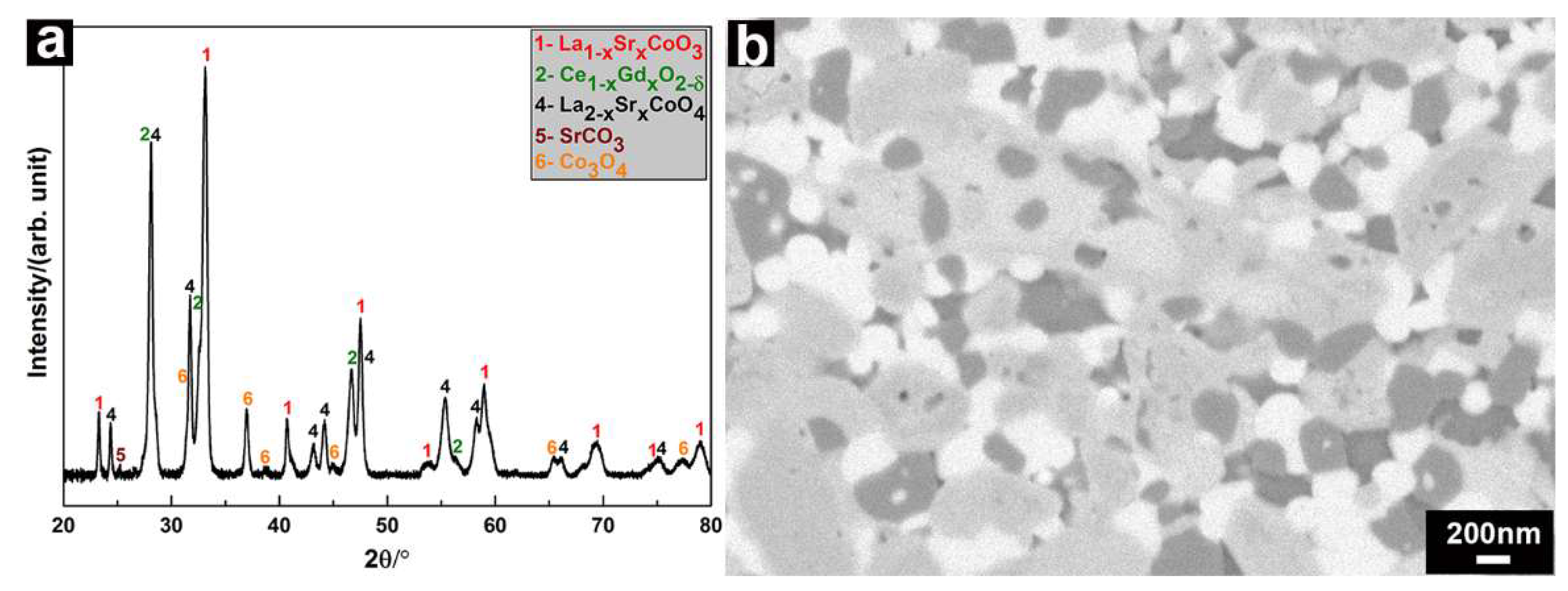
3.3. Analysis of the SPS-densified LSC-CGO Composite after Thermal Cycling
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Schärtl, W. Current directions in core–shell nanoparticle design. Nanoscale 2010, 2, 829–843. [Google Scholar] [CrossRef] [PubMed]
- Wei, S.; Wang, Q.; Zhu, J.; Sun, L.; Lin, H.; Guo, Z. Multifunctional composite core–shell nanoparticles. Nanoscale 2011, 3, 4474–4502. [Google Scholar] [CrossRef] [PubMed]
- Ghosh Chaudhuri, R.; Paria, S. Core/shell nanoparticles: Classes, properties, synthesis mechanisms, characterization, and applications. Chem. Rev. 2012, 112, 2373–2433. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.; Xia, C.; Chen, F. Nano-structured composite cathodes for intermediate-temperature solid oxide fuel cells via an infiltration/impregnation technique. Electrochim. Acta 2010, 55, 3595–3605. [Google Scholar] [CrossRef]
- Manthiram, A.; Kim, J.-H.; Kim, Y.N.; Lee, K.-T. Crystal chemistry and properties of mixed ionic-electronic conductors. J. Electroceram. 2011, 27, 93–107. [Google Scholar] [CrossRef]
- Ebbesen, S.D.; Jensen, S.H.; Hauch, A.; Mogensen, M.B. High temperature electrolysis in alkaline cells, solid proton conducting cells, and solid oxide cells. Chem. Rev. 2014, 114, 10697–10734. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.-L.; Chen, I.-W. Grain growth in CeO2: Dopant effects, defect mechanism, and solute drag. J. Am. Ceram. Soc. 1996, 79, 1793–1800. [Google Scholar] [CrossRef]
- Datta, P. Doped ceria based solid oxide fuel cell electrolytes and their sintering aspects: An overview. Mater. Sci. Forum 2016, 835, 199–236. [Google Scholar] [CrossRef]
- Sun, C.; Hui, R.; Roller, J. Cathode materials for solid oxide fuel cells: A review. J. Solid State Electrochem. 2010, 14, 1125–1144. [Google Scholar] [CrossRef]
- Fang, S.; Chen, C.; Winnubst, L. Effect of microstructure and catalyst coating on the oxygen permeability of a novel CO2-resistant composite membrane. Solid State Ion. 2011, 190, 46–52. [Google Scholar] [CrossRef]
- Joo, J.H.; Yun, K.S.; Kim, J.-H.; Lee, Y.; Yoo, C.-Y.; Yu, J.H. Substantial oxygen flux in dual-phase membrane of ceria and pure electronic conductor by tailoring the surface. ACS Appl. Mater. Interfaces 2015, 7, 14699–14707. [Google Scholar] [CrossRef] [PubMed]
- Ibáñez, M.; Zamani, R.; Gorsse, S.; Fan, J.; Ortega, S.; Cadavid, D.; Morante, J.R.; Arbiol, J.; Cabot, A. Core–shell nanoparticles as building blocks for the bottom-up production of functional nanocomposites: PbTe–PbS thermoelectric properties. ACS Nano 2013, 7, 2573–2586. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.; Liu, Y.; Liu, P.; Zhang, W.; Zhu, J. Microwave dielectric properties of (1 − x)SiO2-xTiO2 composite ceramics derived from core-shell structured microspheres. Mater. Res. Bull. 2014, 53, 54–57. [Google Scholar] [CrossRef]
- Mojić-Lanté, B.; Vukmirović, J.; Giannakopoulos, K.P.; Gautam, D.; Kukovecz, A.; Srdić, V.V. Influence of synthesis conditions on formation of core–shell titanate–ferrite particles and processing of composite ceramics. Ceram. Int. 2015, 41, 1437–1445. [Google Scholar] [CrossRef]
- Suarez, M.; Fernandez, A.; Menendez, J.L.; Torrecillas, R.; Kessel, H.; Hennicke, J.; Kirchner, R.; van Kessel, H. Challenges and Opportunities for Spark Plasma Sintering: A Key Technology for a New Generation of Materials. In Sintering Applications; Ertug, B., Ed.; InTech: Munich, Germany, 2013; ISBN 978-953-51-0974-7. [Google Scholar]
- Munir, Z.A.; Anselmi-Tamburini, U.; Ohyanagi, M. The effect of electric field and pressure on the synthesis and consolidation of materials: A review of the spark plasma sintering method. J. Mater. Sci. 2006, 41, 763–777. [Google Scholar] [CrossRef]
- Buscaglia, M.T.; Viviani, M.; Zhao, Z.; Buscaglia, V.; Nanni, P. Synthesis of BaTiO3 core-shell particles and fabrication of dielectric ceramics with local graded structure. Chem. Mater. 2006, 18, 4002–4010. [Google Scholar] [CrossRef]
- Basu, B.; Venkateswaran, T.; Kim, D.-Y. Microstructure and properties of spark plasma-sintered ZrO2–ZrB2 nanoceramic composites. J. Am. Ceram. Soc. 2006, 89, 2405–2412. [Google Scholar] [CrossRef]
- Rodríguez-Carvajal, J. Recent advances in magnetic structure determination by neutron powder diffraction. Phys. B Condens. Matter 1993, 192, 55–69. [Google Scholar] [CrossRef]
- Cote, L.J.; Teja, A.S.; Wilkinson, A.P.; Zhang, Z.J. Continuous hydrothermal synthesis of CoFe2O4 nanoparticles. Fluid Phase Equilibria 2003, 210, 307–317. [Google Scholar] [CrossRef]
- Sear, R.P. The non-classical nucleation of crystals: Microscopic mechanisms and applications to molecular crystals, ice and calcium carbonate. Int. Mater. Rev. 2012, 57, 328–356. [Google Scholar] [CrossRef]
- Sear, R.P. Quantitative studies of crystal nucleation at constant supersaturation: Experimental data and models. CrystEngComm 2014, 16, 6506–6522. [Google Scholar] [CrossRef]
- Tealdi, C.; Ferrara, C.; Mustarelli, P.; Saiful Islam, M. Vacancy and interstitial oxide ion migration in heavily doped La2−xSrxCoO4±δ. J. Mater. Chem. 2012, 22, 8969–8975. [Google Scholar] [CrossRef]
- Sase, M.; Hermes, F.; Yashiro, K.; Sato, K.; Mizusaki, J.; Kawada, T.; Sakai, N.; Yokokawa, H. Enhancement of oxygen surface exchange at the hetero-interface of (La,Sr)CoO3/(La,Sr)2CoO4 with PLD-layered films. J. Electrochem. Soc. 2008, 155, B793–B797. [Google Scholar] [CrossRef]
- Petrov, A.N.; Cherepanov, V.A.; Zuev, A.Y. Thermodynamics, defect structure, and charge transfer in doped lanthanum cobaltites: An overview. J. Solid State Electrochem. 2006, 10, 517–537. [Google Scholar] [CrossRef]
- Ovenstone, J.; White, J.S.; Misture, S.T. Phase transitions and phase decomposition of La1−xSrxCoO3−δ in low oxygen partial pressures. J. Power Sources 2008, 181, 56–61. [Google Scholar] [CrossRef]
- Samson, A.J.; Søgaard, M.; Vang Hendriksen, P. (Ce,Gd)O2−δ-based dual phase membranes for oxygen separation. J. Membr. Sci. 2014, 470, 178–188. [Google Scholar] [CrossRef]
- Chen, I.-W.; Wang, X.-H. Sintering dense nanocrystalline ceramics without final-stage grain growth. Nature 2000, 404, 168–171. [Google Scholar] [CrossRef] [PubMed]
- Rufner, J.; Anderson, D.; van Benthem, K.; Castro, R.H.R. Synthesis and sintering behavior of ultrafine (<10 nm) magnesium aluminate spinel nanoparticles. J. Am. Ceram. Soc. 2013, 96, 2077–2085. [Google Scholar] [CrossRef]
- Anselmi-Tamburini, U.; Garay, J.E.; Munir, Z.A. Fast low-temperature consolidation of bulk nanometric ceramic materials. Scr. Mater. 2006, 54, 823–828. [Google Scholar] [CrossRef]







© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, Y.; Zielke, P.; Van Nong, N.; Pirou, S.; Reolon, R.; Si, X.; Simonsen, S.B.; Norby, P.; Lühmann, H.; Bensch, W.; et al. Hydrothermal Synthesis, Characterization, and Sintering Behavior of Core-Shell Particles: A Principle Study on Lanthanum Strontium Cobaltite Coated with Nanosized Gadolinium Doped Ceria. Ceramics 2018, 1, 246-260. https://doi.org/10.3390/ceramics1020020
Xu Y, Zielke P, Van Nong N, Pirou S, Reolon R, Si X, Simonsen SB, Norby P, Lühmann H, Bensch W, et al. Hydrothermal Synthesis, Characterization, and Sintering Behavior of Core-Shell Particles: A Principle Study on Lanthanum Strontium Cobaltite Coated with Nanosized Gadolinium Doped Ceria. Ceramics. 2018; 1(2):246-260. https://doi.org/10.3390/ceramics1020020
Chicago/Turabian StyleXu, Yu, Philipp Zielke, Ngo Van Nong, Stéven Pirou, Raquel Reolon, Xiaoqing Si, Søren Bredmose Simonsen, Poul Norby, Henning Lühmann, Wolfgang Bensch, and et al. 2018. "Hydrothermal Synthesis, Characterization, and Sintering Behavior of Core-Shell Particles: A Principle Study on Lanthanum Strontium Cobaltite Coated with Nanosized Gadolinium Doped Ceria" Ceramics 1, no. 2: 246-260. https://doi.org/10.3390/ceramics1020020
APA StyleXu, Y., Zielke, P., Van Nong, N., Pirou, S., Reolon, R., Si, X., Simonsen, S. B., Norby, P., Lühmann, H., Bensch, W., & Kiebach, R. (2018). Hydrothermal Synthesis, Characterization, and Sintering Behavior of Core-Shell Particles: A Principle Study on Lanthanum Strontium Cobaltite Coated with Nanosized Gadolinium Doped Ceria. Ceramics, 1(2), 246-260. https://doi.org/10.3390/ceramics1020020