Role of the Counterions in the Surface Tension of Aqueous Surfactant Solutions. A Computer Simulation Study of Alkali Dodecyl Sulfate Systems




Abstract
1. Introduction
2. Calculation of the Surface Tension Contribution of the Molecules through the Lateral Pressure
3. Computational Details
4. Results and Discussion
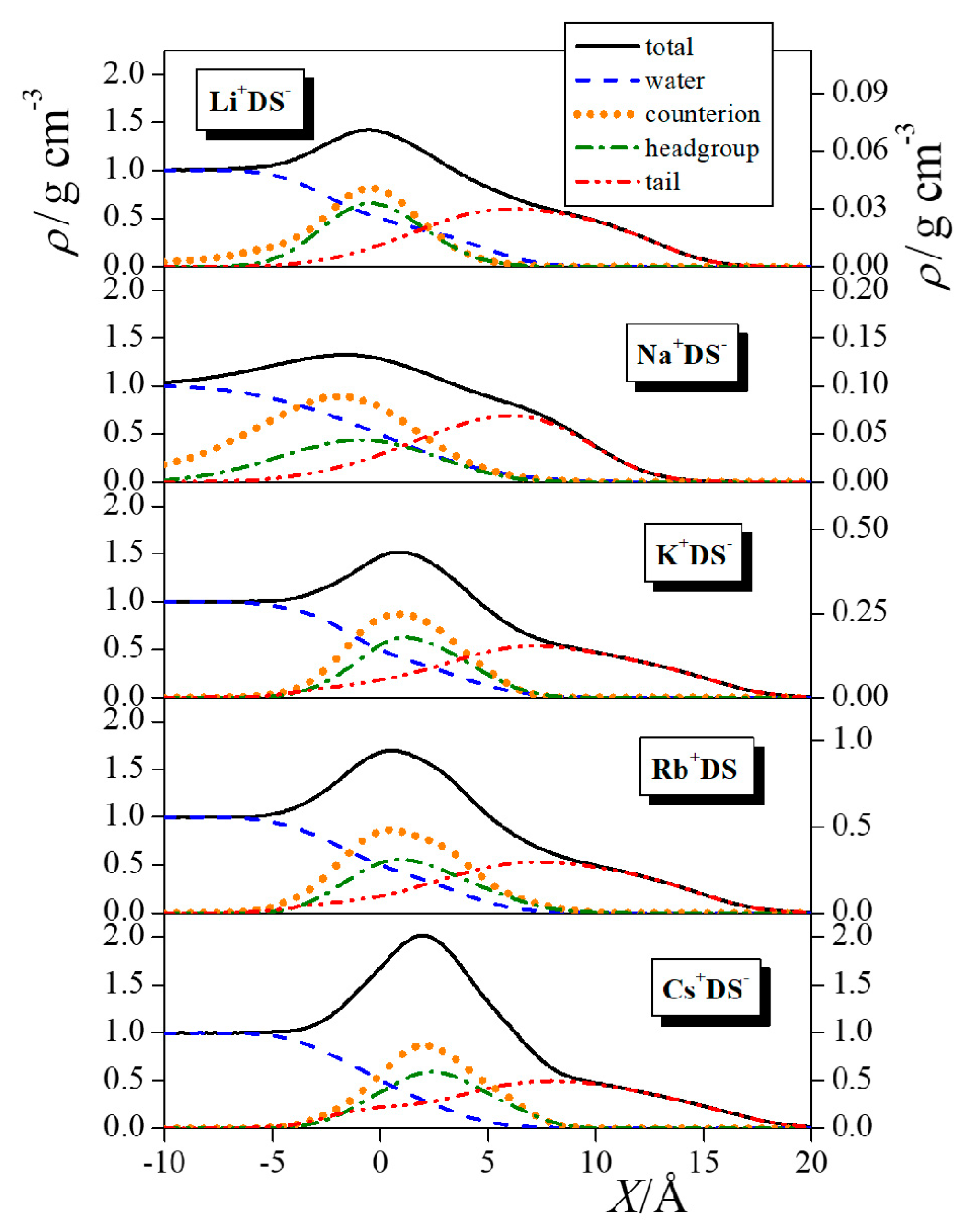
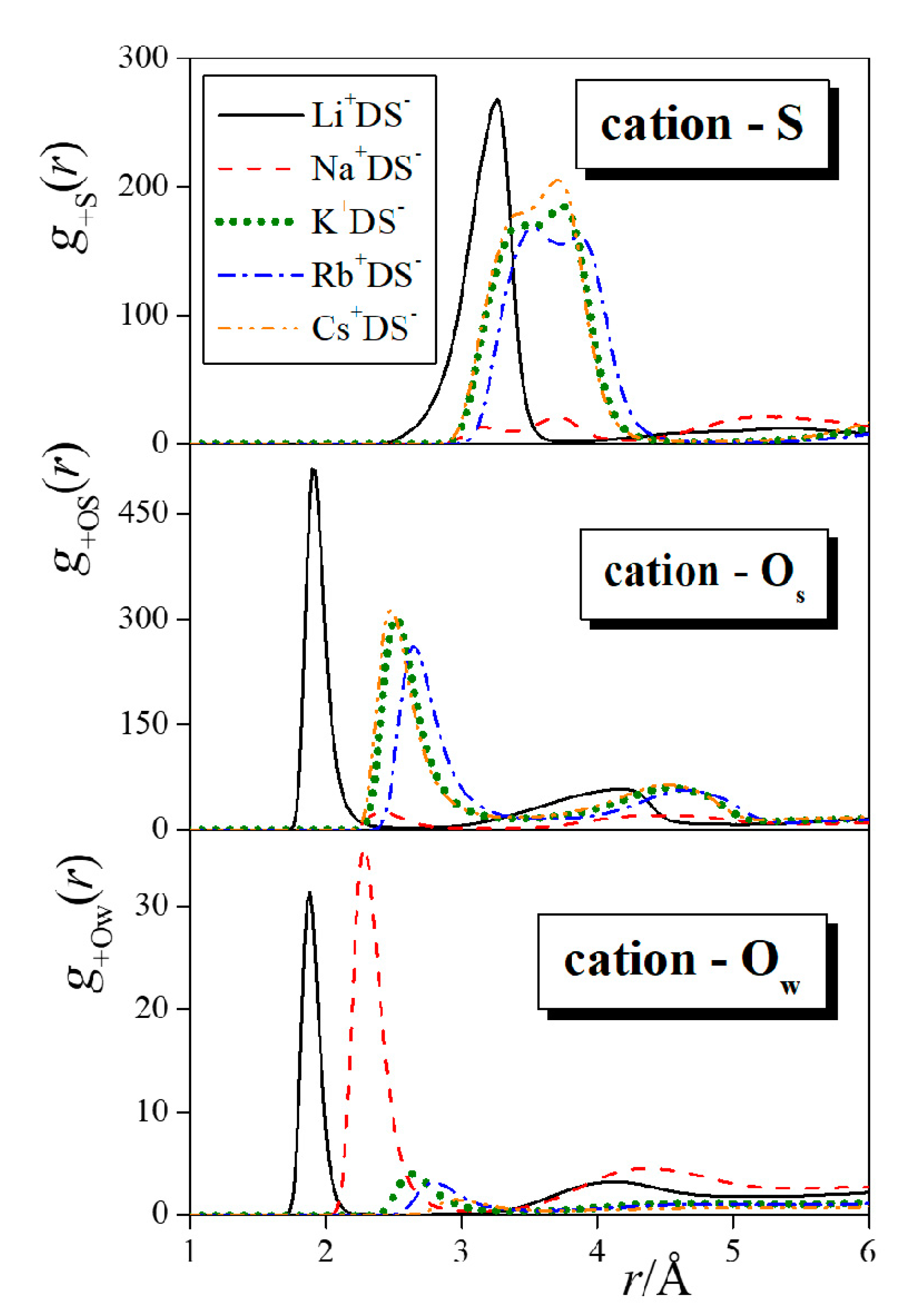
4.1. Surface Tension and Radial Distribution Functions
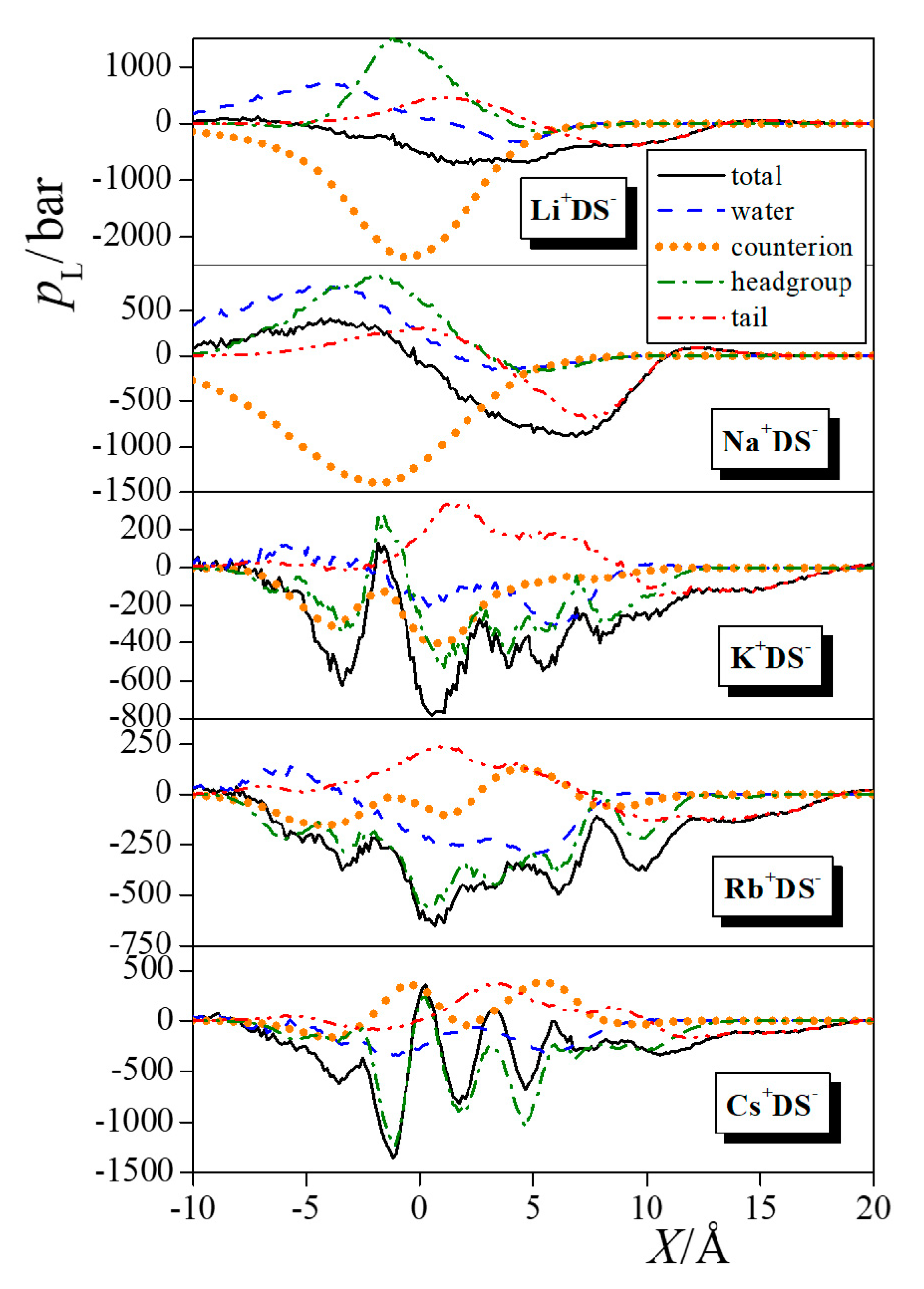
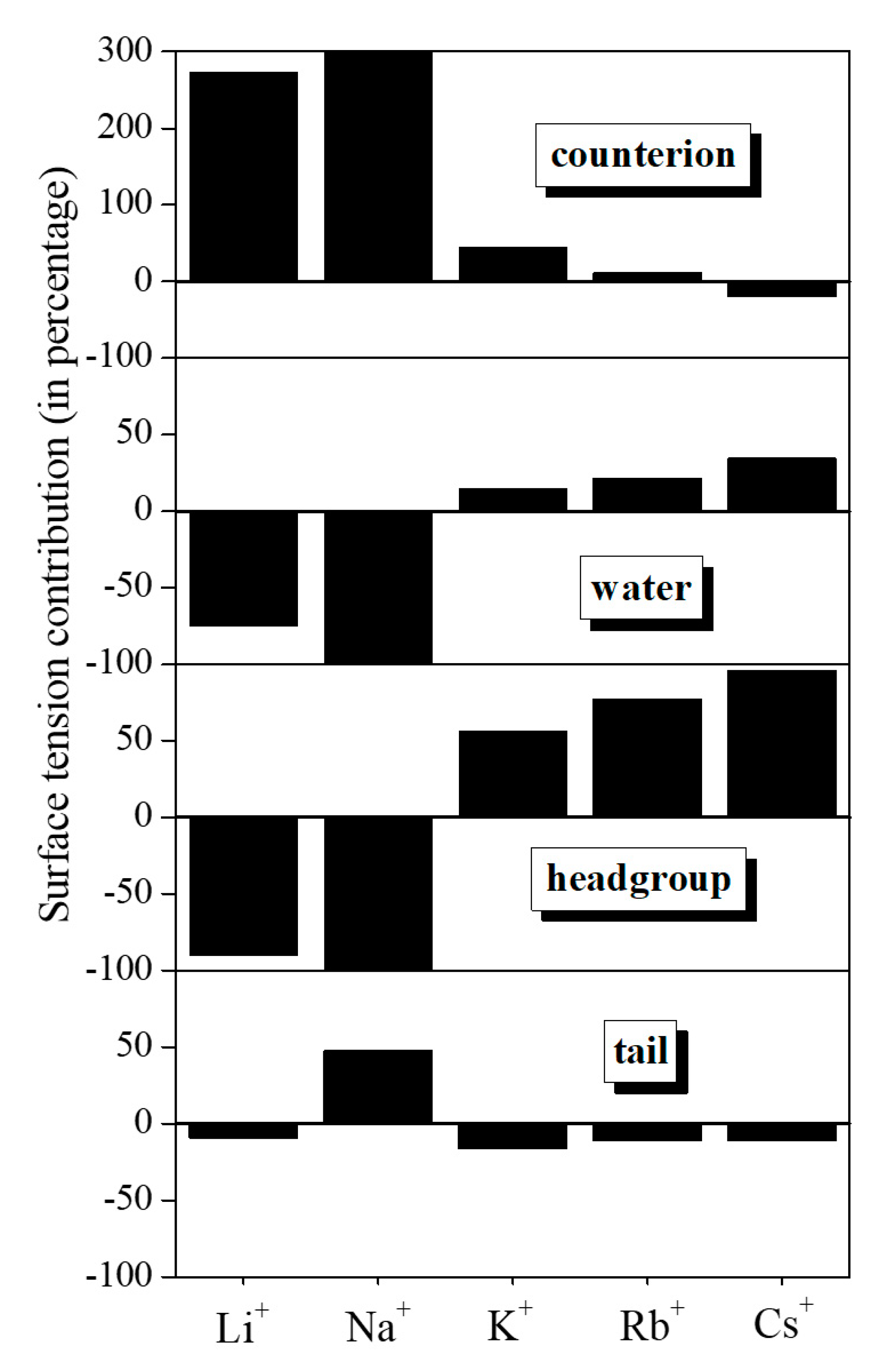
4.2. Lateral Pressure Profiles and Surface Tension Contributions
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Rowlinson, J.S.; Widom, B. Molecular Theory of Capillarity, 1st ed.; Clarendon Press: Oxford, UK, 1982. [Google Scholar]
- Sega, M.; Fábián, B.; Horvai, G.; Jedlovszky, P. How Is the Surface Tension of Various Liquids Distributed along the Interface Normal? J. Phys. Chem. C 2016, 120, 27468–27477. [Google Scholar] [CrossRef]
- Allen, M.P.; Tildesley, D.J. Computer Simulation of Liquids, 1st ed.; Clarendon Press: Oxford, UK, 1987. [Google Scholar]
- Hua, X.Y.; Rosen, M.J. Dynamic Surface Tension of Aqueous Surfactant Solutions. I. Basic Parameters. J. Coll. Interface Sci. 1988, 124, 652–659. [Google Scholar]
- Lu, J.R.; Simister, E.A.; Lee, E.M.; Thomas, R.K.; Rennie, A.R.; Penfold, J. Direct Determination by Neutron Reflection of the Penetration of Water into Surfactant Layers at the Air/Water Interface. Langmuir 1992, 8, 1837–1844. [Google Scholar] [CrossRef]
- Lu, J.R.; Simister, E.A.; Thomas, R.K.; Penfold, J. Structure of an Octadecyltrimethylammonium Bromide Layer at the Air/Water Interface Determined by Neutron Reflection: Systematic Errors in Reflectivity Measurements. J. Phys. Chem. 1993, 97, 6024–6602. [Google Scholar] [CrossRef]
- Messmer, M.C.; Conboy, J.C.; Richmond, G.L. Observation of Molecular Ordering at the Liquid-Liquid Interface by Resonant Sum Frequency Generation. J. Am. Chem. Soc. 1995, 117, 8039–8040. [Google Scholar] [CrossRef]
- Conboy, J.C.; Messmer, M.C.; Richmond, G.L. Investigation of Surfactant Conformation and Order at the Liquid–Liquid Interface by Total Internal Reflection Sum-Frequency Vibrational Spectroscopy. J. Phys. Chem. 1996, 100, 7617–7622. [Google Scholar] [CrossRef]
- Gragson, D.E.; McCarty, B.M.; Richmond, G.L. Surfactant/Water Interactions at the Air/Water Interface Probed by Vibrational Sum Frequency Generation. J. Phys. Chem. 1996, 100, 14272–14275. [Google Scholar] [CrossRef]
- Bain, C.D. Studies of Adsorption at Interfaces by Optical Techniques: Ellipsometry, Second Harmonic Generation and Sum-Frequency Generation. Curr. Opin. Colloid Interface Sci. 1998, 3, 287–292. [Google Scholar] [CrossRef]
- Li, Z.X.; Dong, C.C.; Thomas, R.K. Neutron Reflectivity Studies of the Surface Excess of Gemini Surfactants at the Air–Water Interface. Langmuir 1999, 15, 4392–4396. [Google Scholar] [CrossRef]
- Stubenrauch, C.; Albouy, P.A.; von Klitzing, R.; Langevin, D. Polymer/Surfactant Complexes at the Water/Air Interface: A Surface Tension and X-ray Reflectivity Study. Langmuir 2000, 16, 3206–3213. [Google Scholar] [CrossRef]
- Lu, J.R.; Thomas, R.K.; Penfold, J. Surfactant Layers at the Air/Water Interface: Structure and Composition. Adv. Coll. Interface Sci. 2000, 84, 143–304. [Google Scholar] [CrossRef]
- Benderskii, A.V.; Eisenthal, K.B. Aqueous Solvation Dynamics at the Anionic urfactant Air/Water Interface. J. Phys. Chem. B 2001, 105, 6698–6703. [Google Scholar] [CrossRef][Green Version]
- Gilányi, T.; Varga, I.; Mészáros, R. Specific Counterion Effect on the Adsorption of Alkali Decyl Sulfate Surfactants at Air/Solution Interface. Phys. Chem. Chem. Phys. 2004, 6, 4348–4346. [Google Scholar] [CrossRef]
- Ma, G.; Allen, H.C. Real-Time Investigation of Lung Surfactant Respreading with Surface Vibrational Spectroscopy. Langmuir 2006, 22, 11267–11274. [Google Scholar] [CrossRef]
- Kirsch, M.J.; D’Auria, R.; Brown, M.A.; Tobias, D.J.; Hemminger, J.C.; Ammann, M.; Starr, D.E.; Bluhm, H. The Effect of an Organic Surfactant on the Liquid-Vapor Interface of an Electrolyte Solution. J. Phys. Chem. C 2007, 111, 13497–13509. [Google Scholar] [CrossRef]
- Sloutskin, E.; Sapir, Z.; Bain, C.D.; Lei, Q.; Wilkinson, K.M.; Tamam, L.; Deutsch, M.; Ocko, B.M. Wetting, Mixing, and Phase Transitions in Langmuir-Gibbs Films. Phys. Rev. Lett. 2007, 99, 136102. [Google Scholar] [CrossRef]
- Zhang, Z.; Zheng, D.; Guo, Y.; Wang, H. Water Penetration/Accommodation and Phase Behaviour of the Neutral Langmuir Monolayer at the Air/Water Interface Probed with Sum Frequency Generation Vibrational Spectroscopy (SFG-VS). Phys. Chem. Chem. Phys. 2009, 11, 991–1002. [Google Scholar] [CrossRef]
- de Aguiar, H.B.; Scheu, R.; Jena, K.C.; de Beer, A.G.F.; Roke, S. Comparison of Scattering and Reflection SFG: A Question of Phase-Matching. Phys. Chem. Chem. Phys. 2012, 14, 6826–6832. [Google Scholar] [CrossRef]
- Angus-Smyth, A.; Campbell, R.A.; Bain, C.D. Dynamic Adsorption of Weakly Interacting Polymer/Surfactant Mixtures at the Air/Water Interface. Langmuir 2012, 28, 12479–12492. [Google Scholar] [CrossRef]
- Gong, H.; Xu, G.; Liu, T.; Xu, L.; Zhai, X.; Zhang, J.; Lv, X. Aggregation Behaviors of PEO-PPO-ph-PPO-PEO and PPO-PEO-ph-PEO-PPO at an Air/Water Interface: Experimental Study and Molecular Dynamics Simulation. Langmuir 2012, 28, 13590–13600. [Google Scholar] [CrossRef]
- Scheu, R.; Chen, Y.; de Aguiar, H.B.; Rankin, B.M.; Ben-Amotz, D.; Roke, S. Specific Ion Effects in Amphiphile Hydration and Interface Stabilization. J. Am. Chem. Soc. 2014, 136, 2040–2047. [Google Scholar] [CrossRef] [PubMed]
- Phan, C.M.; Nguyen, C.V.; Yusa, S.; Yamada, N.L. Synergistic Adsorption of MIBC/CTAB Mixture at the Air/Water Interface and Applicability of Gibbs Adsorption Equation. Langmuir 2014, 30, 5790–5796. [Google Scholar] [CrossRef] [PubMed]
- Shahir, A.A.; Arabadzhieva, D.; Petkova, H.; Karakashev, S.I.; Nguyen, A.V.; Mileva, E. Effect of Under-Monolayer Adsorption on Foamability, Rheological Characteristics, and Dynamic Behavior of Fluid Interfaces: Experimental Evidence for the Guggenheim Extended Interface Model. J. Phys. Chem. C 2017, 121, 11472–11487. [Google Scholar] [CrossRef]
- Shahir, A.A.; Nguyen, K.T.; Nguyen, A.V. A Sum-Frequency Generation Spectroscopic Study of the Gibbs Analysis Paradox: Monolayer or Sub-Monolayer Adsorption? Phys. Chem. Chem. Phys. 2016, 18, 8794–8805. [Google Scholar] [CrossRef] [PubMed]
- Böcker, J.; Schlenkirch, M.; Bopp, P.; Brickmann, J. Molecular Dynamics Simulation of a n-Hexadecyltrimethylammonium Chloride Monolayer. J. Phys. Chem. 1992, 96, 9915–9922. [Google Scholar] [CrossRef]
- Tarek, M.; Tobias, D.J.; Klein, M.L. Molecular Dynamics Simulation of Tetradecyltrimethylammonium Bromide Monolayers at the Air/Water Interface. J. Phys. Chem. 1995, 99, 1393–1402. [Google Scholar] [CrossRef]
- Schweighofer, K.; Essman, U.; Berkowitz, M. Simulation of Sodium Dodecyl Sulfate at the Water-Vapor and Water-Carbon Tetrachloride Interfaces at Low Surface Coverage. J. Phys. Chem. B 1997, 101, 3793–3799. [Google Scholar] [CrossRef]
- Schweighofer, K.; Essman, U.; Berkowitz, M. Structure and Dynamics of Water in the Presence of Charged Surfactant Monolayers at the Water-CCl4 Interface. A Molecular Dynamics Study. J. Phys. Chem. B 1997, 101, 10775–10780. [Google Scholar] [CrossRef]
- Schweighofer, K.; Benjamin, I. Transfer of a Tetramethylammonium Ion across the Water–Nitrobenzene Interface: Potential of Mean Force and Nonequilibrium Dynamics. J. Phys. Chem. A 1999, 103, 10274–10279. [Google Scholar] [CrossRef]
- Domínguez, H.; Berkowitz, M. Computer Simulations of Sodium Dodecyl Sulfate at Liquid/Liquid and Liquid/Vapor Interfaces. J. Phys. Chem. B 2000, 104, 5302–5308. [Google Scholar] [CrossRef]
- Domínguez, H. Computer Simulations of Surfactant Mixtures at the Liquid/Liquid Interface. J. Phys. Chem. B 2002, 106, 5915–5924. [Google Scholar] [CrossRef]
- Jedlovszky, P.; Varga, I.; Gilányi, T. Adsorption of 1-Octanol at the Free Water Surface as Studied By Monte Carlo Simulation. J. Chem. Phys. 2004, 120, 11839–11851. [Google Scholar] [CrossRef] [PubMed]
- Domínguez, H. Computer Simulation Studies of Surfactant Monolayer Mixtures at the Water/Oil Interface: Charge Distribution Effects. J. Colloid Interface Sci. 2004, 274, 665–672. [Google Scholar] [CrossRef] [PubMed]
- Paszternák, A.; Kiss, É.; Jedlovszky, P. Structure of the nonionic Surfactant Triethoxy Monooctylether C8E3 Adsorbed at the Free Water Surface, As Seen from Surface Tension Measurements and Monte Carlo Simulations. J. Chem. Phys. 2005, 122, 124704. [Google Scholar] [CrossRef] [PubMed]
- Domínguez, H.; Rivera, M. Mixtures of Sodium Dodecyl Sulfate/Dodecanol at the Air/Water Interface by Computer Simulations. Langmuir 2005, 21, 7257–7262. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, J.; Clavero, E.; Laria, D. Computer Simulations of Catanionic Surfactants Adsorbed at Air/Water Interfaces. J. Phys. Chem. B 2005, 109, 24427–24433. [Google Scholar] [CrossRef] [PubMed]
- Chanda, J.; Bandyopadhyay, S. Molecular Dynamics Study of Surfactant Monolayers Adsorbed at the Oil/Water and Air/Water Interfaces. J. Phys. Chem. B 2006, 110, 23482–23488. [Google Scholar] [CrossRef]
- Hantal, G.; Pártay, L.B.; Varga, I.; Jedlovszky, P.; Gilányi, T. Counterion and Surface Density Dependence of the Adsorption Layer of Ionic Surfactants at the Vapor-Aqueous Solution Interface: A Computer Simulation Study. J. Phys. Chem. B 2007, 111, 1769–1774. [Google Scholar] [CrossRef]
- Darvas, M.; Gilányi, T.; Jedlovszky, P. Adsorption of Poly(ethylene oxide) at the Free Water Surface. A Computer Simulation Study. J. Phys. Chem. B 2010, 114, 10995–11001. [Google Scholar] [CrossRef]
- Martínez, H.; Chacón, E.; Tarazona, P.; Bresme, F. The Intrinsic Interfacial Structure of Ionic Surfactant Monolayers at Water-Oil and Water-Vapour Interfaces. Proc. R. Soc. A 2011, 467, 1939–1958. [Google Scholar] [CrossRef]
- Bresme, F.; Chacón, E.; Martínez, H.; Tarazona, P. Adhesive Transitions in Newton Black Films: A Computer Simulation Study. J. Chem. Phys. 2011, 134, 214701. [Google Scholar] [CrossRef]
- Darvas, M.; Gilányi, T.; Jedlovszky, P. Competitive Adsorption of Surfactants and Polymers at the Free Water Surface. A Computer Simulation Study of the Sodium Dodecyl Sulfate-Poly(ethylene oxide) System. J. Phys. Chem. B 2011, 115, 933–944. [Google Scholar] [CrossRef] [PubMed]
- Pang, J.; Wang, Y.; Xu, G.; Han, T. Molecular Dynamics Simulation of SDS, DTAB, and C12E8 Monolayers Adsorbed at the Air/Water Surface in the Presence of DSEP. J. Phys. Chem. B 2011, 115, 2518–2526. [Google Scholar] [CrossRef] [PubMed]
- Rideg, N.A.; Darvas, M.; Varga, I.; Jedlovszky, P. Lateral Dynamics of Surfactants at the Free Water Surface. A Computer Simulation Study. Langmuir 2012, 28, 14944–14953. [Google Scholar] [CrossRef] [PubMed]
- Vacha, R.; Roke, S. Sodium Dodecyl Sulfate at Water–Hydrophobic Interfaces: A Simulation Study. J. Phys. Chem. B 2012, 116, 11936–11942. [Google Scholar] [CrossRef] [PubMed]
- Abrankó-Rideg, N.; Darvas, M.; Horvai, G.; Jedlovszky, P. Immersion Depth of Surfactants at the Free Water Surface: A Computer Simulation and ITIM Analysis Study. J. Phys. Chem. B 2013, 117, 8733–8746. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Lu, X.; Liu, X.; Hou, Q.; Zhu, Y.; Zhou, H. Specific Counterion Effects on the Atomistic Structure and Capillary-Waves Fluctuation of the Water/Vapor Interface Covered by Sodium Dodecyl Sulfate. J. Phys. Chem. C 2014, 118, 19205–19213. [Google Scholar] [CrossRef]
- Chen, M.; Lu, X.; Liu, X.; Hou, Q.; Zhu, Y.; Zhou, H. Temperature-Dependent Phase Transition and Desorption Free Energy of Sodium Dodecyl Sulfate at the Water/Vapor Interface: Approaches from Molecular Dynamics Simulations. Langmuir 2014, 30, 10600–10607. [Google Scholar] [CrossRef]
- Tesei, G.; Paradossi, G.; Chiessi, E. Influence of Surface Concentration on Poly(vinyl alcohol) Behavior at the Water–Vacuum Interface: A Molecular Dynamics Simulation Study. J. Phys. Chem. B 2014, 118, 6946–6955. [Google Scholar] [CrossRef]
- Bernardino, K.; de Moura, A.F. Surface Electrostatic Potential and Water Orientation in the presence of Sodium Octanoate Dilute Monolayers Studied by Means of Molecular Dynamics Simulations. Langmuir 2015, 31, 10995–11004. [Google Scholar] [CrossRef]
- Abrankó-Rideg, N.; Horvai, G.; Jedlovszky, P. Structure of the Adsorption Layer of Various Ionic and Non-Ionic Surfactants at the Free Water Surface, As Seen from Computer Simulation and ITIM Analysis. J. Mol. Liq. 2015, 205, 9–15. [Google Scholar] [CrossRef]
- Chong, L.; Lai, Y.; Gray, M.; Soong, Y.; Shi, F.; Duan, Y. Molecular Dynamics Study of the Bulk and Interface Properties of Frother and Oil with Saltwater and Air. J. Phys. Chem. B 2017, 121, 2788–2796. [Google Scholar] [CrossRef]
- Sresht, V.; Lewandowski, E.P.; Blankschtein, D.; Jusufi, A. Combined Molecular Dynamics Simulation–Molecular-Thermodynamic Theory Framework for Predicting Surface Tensions. Langmuir 2017, 33, 8319–8329. [Google Scholar] [CrossRef]
- Hantal, G.; Sega, M.; Horvai, G.; Jedlovszky, P. Contribution of Different Molecules and Moieties to the Surface Tension in Aqueous Surfactant Solutions. J. Phys. Chem. C 2019, 123, 16660–16670. [Google Scholar] [CrossRef]
- Sega, M.; Fábián, B.; Jedlovszky, P. Layer-by-Layer and Intrinsic Analysis of Molecular and Thermodynamic Properties across Soft Interfaces. J. Chem. Phys. 2015, 143, 114709. [Google Scholar] [CrossRef] [PubMed]
- Hofmeister, F. Zur Lehre von der Wirkung der Salze. Archiv Experiment. Pathol. Pharmakol. 1888, 24, 247–260. [Google Scholar] [CrossRef]
- Jungwirth, P.; Tobias, D.J. Molecular Structure of Salt Solutions: A New View of the Interface with Implications for Heterogeneous Atmospheric Chemistry. J. Phys. Chem. B 2001, 105, 10468–10472. [Google Scholar] [CrossRef]
- Vrbka, L.; Mucha, M.; Minofar, B.; Jungwirth, P.; Brown, E.C.; Tobias, D.J. Propensity of Soft Ions for the Air/Water Interface. Curr. Opin. Coll. Interface Sci. 2004, 9, 67–73. [Google Scholar] [CrossRef]
- Boström, M.; Kunz, W.; Ninham, B.W. Hofmeister Effects in Surface Tension of Aqueous Electrolyte Solution. Langmuir 2005, 21, 2619–2623. [Google Scholar] [CrossRef] [PubMed]
- Jungwirth, P.; Tobias, D.J. Specific Ion Effects at the Air/Water Interface. Chem. Rev. 2006, 106, 1259–1281. [Google Scholar] [CrossRef]
- Pegram, L.M.; Record, M.T., Jr. Hofmeister Salt Effects on Surface Tension Arise from Partitioning of Anions and Cations between Bulk Water and the Air-Water Interface. J. Phys. Chem. B 2007, 111, 5411–5417. [Google Scholar] [CrossRef]
- dos Santos, A.P.; Diehl, A.; Levin, Y. Surface Tensions, Surface Potentials, and the Hofmeister Series of Electrolyte Solutions. Langmuir 2010, 26, 10778–10783. [Google Scholar] [CrossRef]
- dos Santos, A.P.; Levin, Y. Surface and Interfacial Tensions of Hofmeister Electrolytes. Faraday Discuss. 2013, 160, 75–87. [Google Scholar] [CrossRef]
- Schofield, P.; Henderson, J.R. Statistical Mechanics of Inhomogeneous Fluids. Proc. R. Soc. Lond. A 1982, 379, 231–246. [Google Scholar]
- Irving, J.H.; Kirkwood, J.G. The Statistical Mechanical Theory of Transport Processes. IV. The Equations of Hydrodynamics. J. Chem. Phys. 1950, 18, 817–829. [Google Scholar] [CrossRef]
- Harasima, A. Molecular Theory of Surface Tension. Adv. Chem. Phys. 1958, 1, 203–237. [Google Scholar]
- Sonne, J.; Hansen, F.Y.; Peters, G.H. Methodological Problems in Pressure Profile Calculations for Lipid Bilayers. J. Chem. Phys. 2005, 122, 124903. [Google Scholar] [CrossRef] [PubMed]
- Schuler, L.D.; Daura, X.; van Gunsteren, W.F. An Improved GROMOS96 Force Field for Aliphatic Hydrocarbons in the Condensed Phase. J. Comp. Chem. 2001, 22, 1205–1218. [Google Scholar] [CrossRef]
- van Gunsteren, W.F.; Billeter, S.R.; Eising, A.A.; Hünenberger, P.H.; Krüger, P.; Mark, A.E.; Scott, W.R.P.; Tironi, I.G. Biomolecular Simulation: The GROMOS96 Manual and User Guide; Vdf Hochschulverlag AG an der ETH Zürich: Zürich, Switzerland, 1996; pp. 1–1042. [Google Scholar]
- Gee, M.B.; Cox, N.R.; Jiao, Y.; Bentenitis, N.; Weerasinghe, S.; Smith, P.E. A Kirkwood-Buff Derived Force Field for Aqueous Alkali Halides. J. Chem. Theory Comput. 2011, 7, 1369–1380. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; Grigera, J.R.; Straatsma, T. The Missing Term in Effective pair Potentials. J. Phys. Chem. 1987, 91, 6269–6271. [Google Scholar] [CrossRef]
- Essman, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A Smooth Particle Mesh Ewald Method. J. Chem. Phys. 1995, 103, 8577–8594. [Google Scholar] [CrossRef]
- Ryckaert, J.P.; Ciccotti, G.; Berendsen, H.J.C. Numerical Integration of the Cartesian Equations of Motion of a System with Constraints; Molecular Dynamics of n-Alkanes. J. Comp. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef]
- GitHub–Marcello Sega. Available online: https://github.com/Marcello-Sega/gromacs/tree/virial/ (accessed on 25 March 2019). The code is freely available.
- Pronk, S.; Páll, S.; Schulz, R.; Larsson, P.; Bjelkmar, P.; Apostolov, R.; Shirts, M.R.; Smith, J.C.; Kasson, P.M.; van der Spoel, D.; et al. GROMACS 4.5: A High-Throughput and Highly Parallel Open Source Molecular Simulation Toolkit. Bioinformatics 2013, 29, 845–854. [Google Scholar] [CrossRef] [PubMed]
- Sega, M.; Fábián, B.; Jedlovszky, P. Pressure Profile Calculation with Particle Mesh Ewald Methods. J. Chem. Theory Comput. 2016, 12, 4509–4515. [Google Scholar] [CrossRef] [PubMed]
- Nosé, S. A Molecular Dynamics Method for Simulations in the Canonical Ensemble. Mol. Phys. 1984, 52, 255–268. [Google Scholar] [CrossRef]
- Hoover, W.G. Canonical Dynamics: Equilibrium Phase-Space Distributions. Phys. Rev. A 1985, 31, 1695–1697. [Google Scholar] [CrossRef]
- Sega, M.; Dellago, C. Long-Range Dispersion Effects on the Water/Vapor Interface Simulated Using the Most Common Models. J. Phys. Chem. B 2017, 121, 3798–3803. [Google Scholar] [CrossRef]
- Weerasinghe, S.; Smith, P.E. A Kirkwood–Buff Derived Force Field for Sodium Chloride in Water. J. Chem. Phys. 2003, 119, 11342–11349. [Google Scholar] [CrossRef]
- Klauda, J.B.; Venable, R.M.; Freites, J.A.; O’Connor, J.W.; Tobias, D.J.; Mondragon-Ramirez, C.; Vorobyov, I.; MacKerrell, A.D., Jr.; Pastor, R.W. Update of the CHARMM All-Atom Additive Force Field for Lipids: Validation on Six Lipid Types. J. Phys. Chem. B 2010, 114, 7830–7843. [Google Scholar] [CrossRef]
- Ríos-López, M.; Mendez-Bermúdez, J.G.; Domínguez, H. New Force Field Parameters for the Sodium Dodecyl Sulfate and Alpha Olefin Sulfonate Anionic Surfactants. J. Phys. Chem. B 2018, 122, 4558–4565. [Google Scholar]
- Joung, I.S.; Cheatham, T.E., III. Determination of Alkali and Halide Monovalent Ion Parameters for Use in Explicitly Solvated Biomolecular Simulations. J. Phys. Chem. B 2008, 112, 9020–9041. [Google Scholar] [CrossRef] [PubMed]
- Deublein, S.; Vrabec, J.; Hasse, H. A Set of Molecular Models for Alkali and Halide Ions in Aqueous Solution. J. Chem. Phys. 2012, 136, 084501. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Neat Water | Li+DS− | Na+DS− | K+DS− | Rb+DS− | Cs+DS− |
|---|---|---|---|---|---|
| 59.0 | 68.3 ± 0.6 | 36.7 ± 0.2 | 71.1 ± 1.0 | 68.3 ± 1.0 | 76.4 ± 1.5 |
| Counterion | Water | Headgroup | Tail | |
|---|---|---|---|---|
| Li+DS− | 187.0 | −51.3 | −61.4 | −6 |
| Na+DS− | 233.6 | −129.4 | −84.9 | 17.4 |
| K+DS− | 31.8 | 10.6 | 39.9 | −11.2 |
| Rb+DS− | 7.9 | 14.8 | 52.7 | −7.1 |
| Cs+DS− | −15.4 | 26.5 | 73.5 | −8.2 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hantal, G.; Sega, M.; Horvai, G.; Jedlovszky, P. Role of the Counterions in the Surface Tension of Aqueous Surfactant Solutions. A Computer Simulation Study of Alkali Dodecyl Sulfate Systems. Colloids Interfaces 2020, 4, 15. https://doi.org/10.3390/colloids4020015
Hantal G, Sega M, Horvai G, Jedlovszky P. Role of the Counterions in the Surface Tension of Aqueous Surfactant Solutions. A Computer Simulation Study of Alkali Dodecyl Sulfate Systems. Colloids and Interfaces. 2020; 4(2):15. https://doi.org/10.3390/colloids4020015
Chicago/Turabian StyleHantal, György, Marcello Sega, George Horvai, and Pál Jedlovszky. 2020. "Role of the Counterions in the Surface Tension of Aqueous Surfactant Solutions. A Computer Simulation Study of Alkali Dodecyl Sulfate Systems" Colloids and Interfaces 4, no. 2: 15. https://doi.org/10.3390/colloids4020015
APA StyleHantal, G., Sega, M., Horvai, G., & Jedlovszky, P. (2020). Role of the Counterions in the Surface Tension of Aqueous Surfactant Solutions. A Computer Simulation Study of Alkali Dodecyl Sulfate Systems. Colloids and Interfaces, 4(2), 15. https://doi.org/10.3390/colloids4020015