Surface Complexation Modeling of Biomolecule Adsorptions onto Titania

Abstract
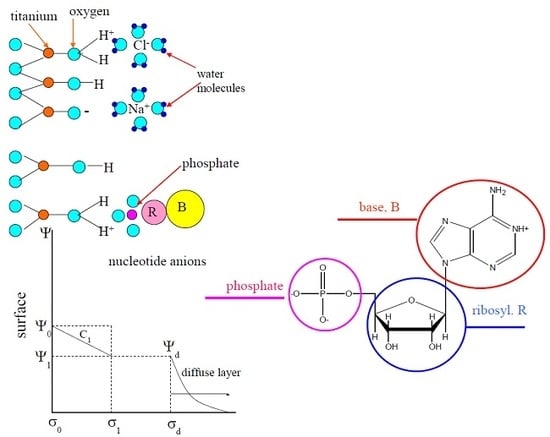
:
1. Introduction
2. Materials and Methods
2.1. Potentiometric Titration
2.2. Sorption Experiments
2.3. Model Calculations
3. Results and Discussion
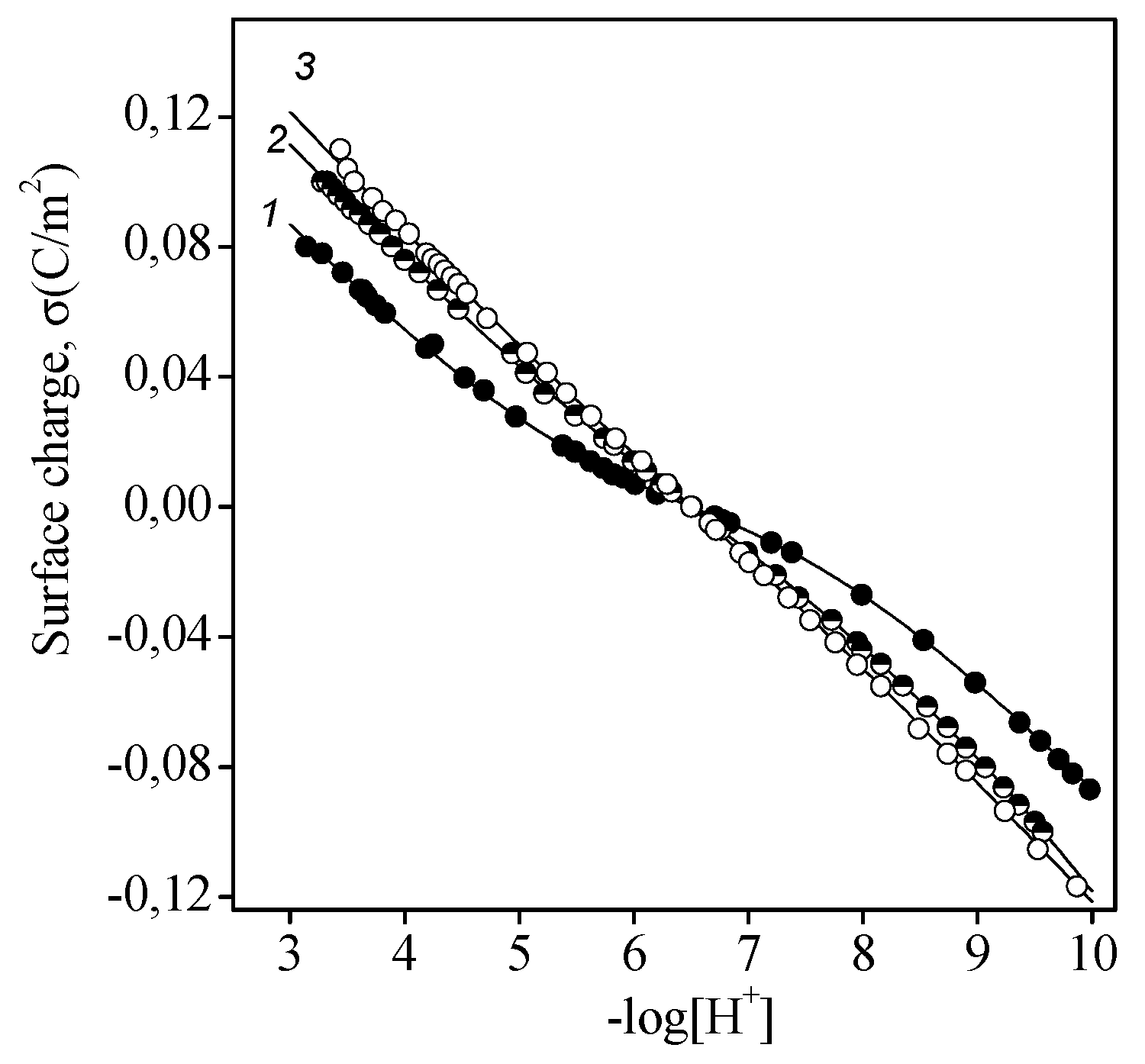
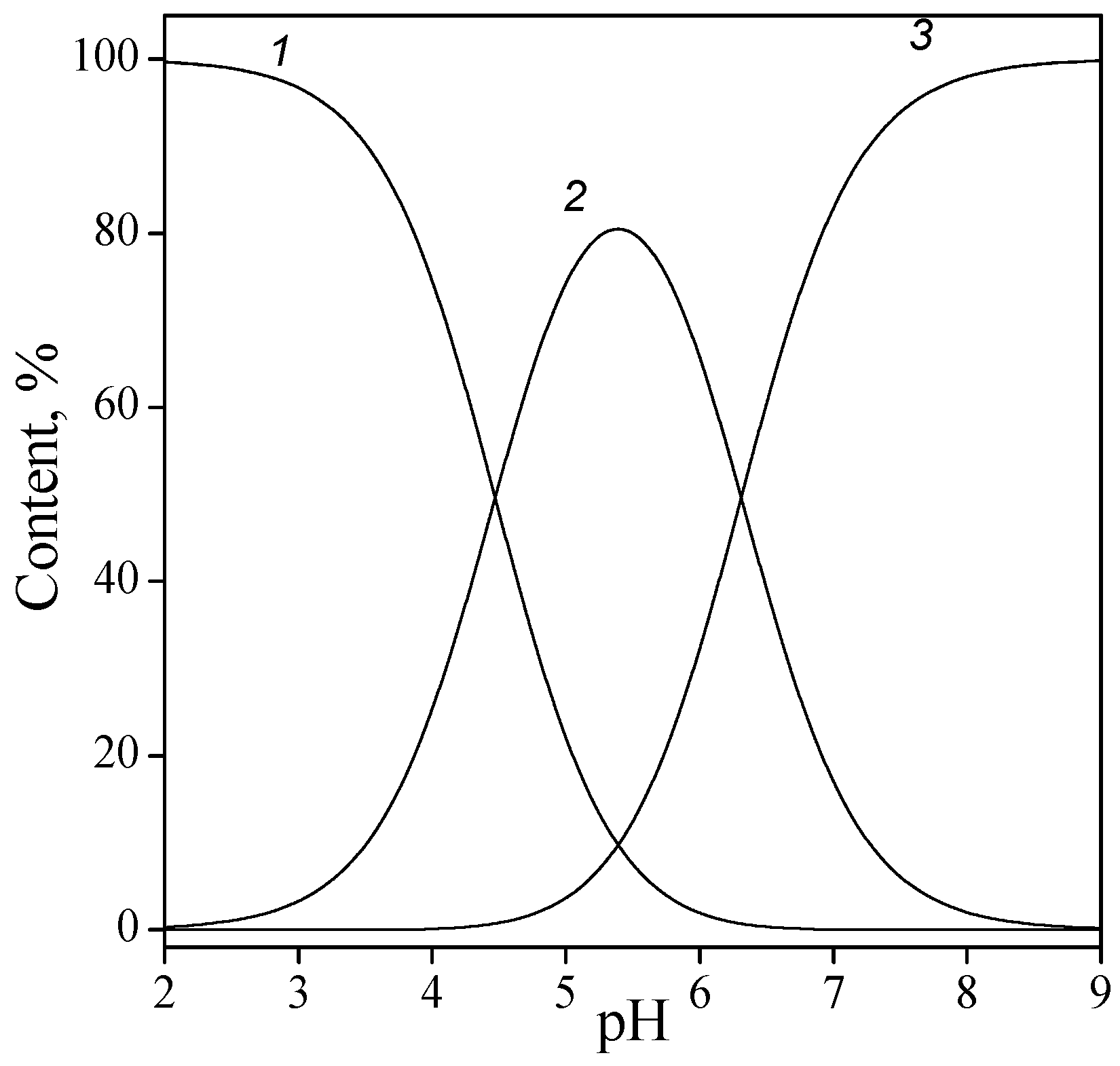
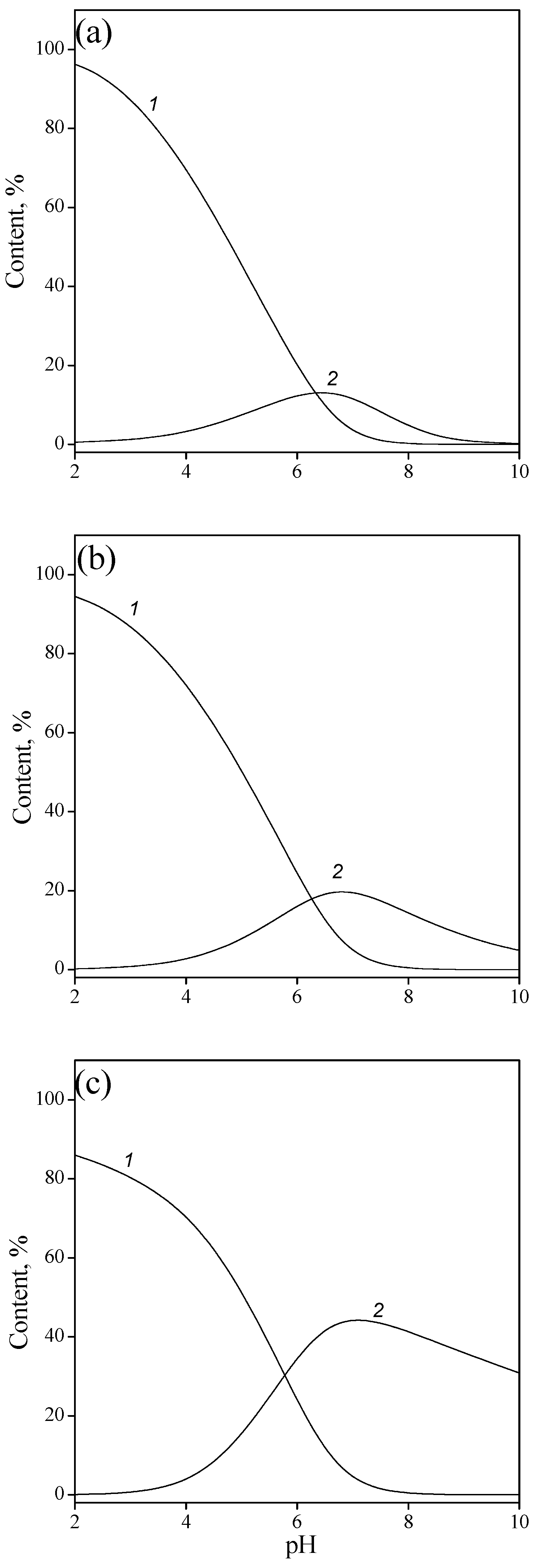
3.1. Titania Surface Acidity
- The concentration of hydroxyl groups on the surface was 0.5 mmol/g, which corresponded to the density of active site per surface unit of 5 groups/nm2. The value for different crystallographic faces of titanium dioxide was found to be 5.2–7.0 groups/nm2 [29], although somewhat smaller values are usually determined experimentally [30,31,32,33].
- The specific capacitance of EDL was estimated to be 0.76 F/m2. As has been shown in [34], such a low capacitance is quite feasible and has a physical sense. This fact indicates that the first layer of physically adsorbed water has a relatively low dielectric permittivity, which is virtually independent of the dielectric properties of the solid [35].
- The equilibrium reaction constants (with an accuracy ±0.05) for the aforementioned reactions were as follows: protonation (2), logK = 5.2; ionization (3), logK = −7.8; anion binding (4), logK = 6.2; and cation binding (5), logK = −6.8.
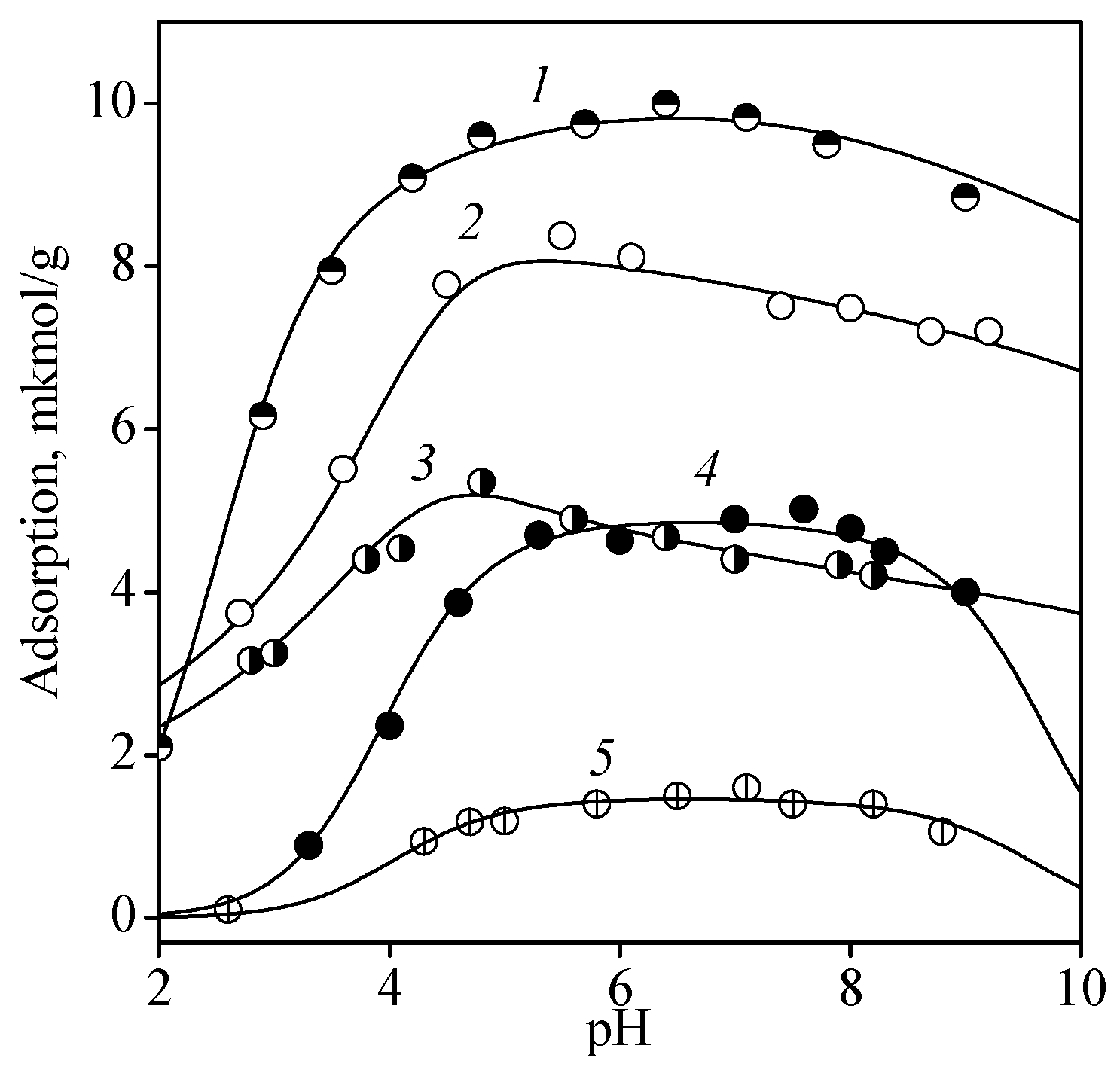
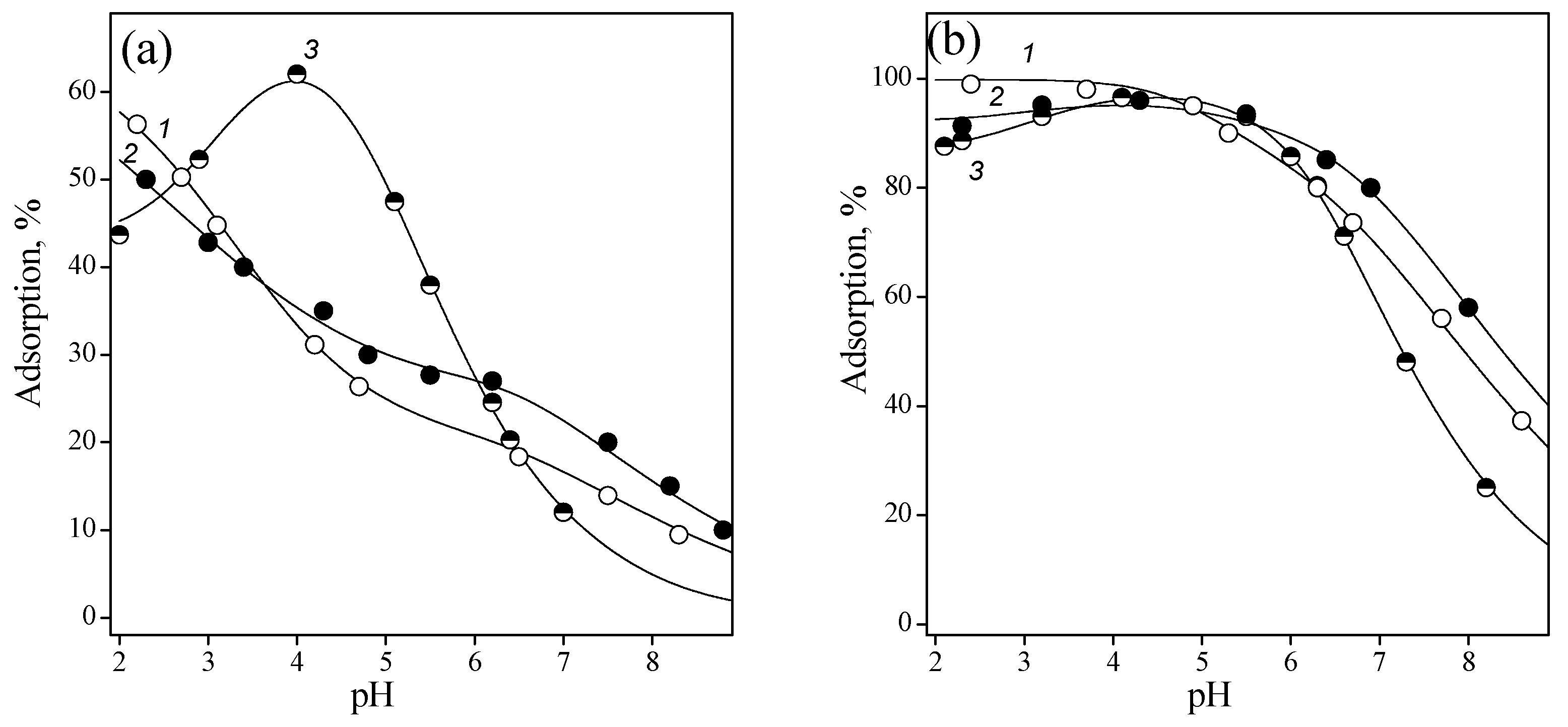
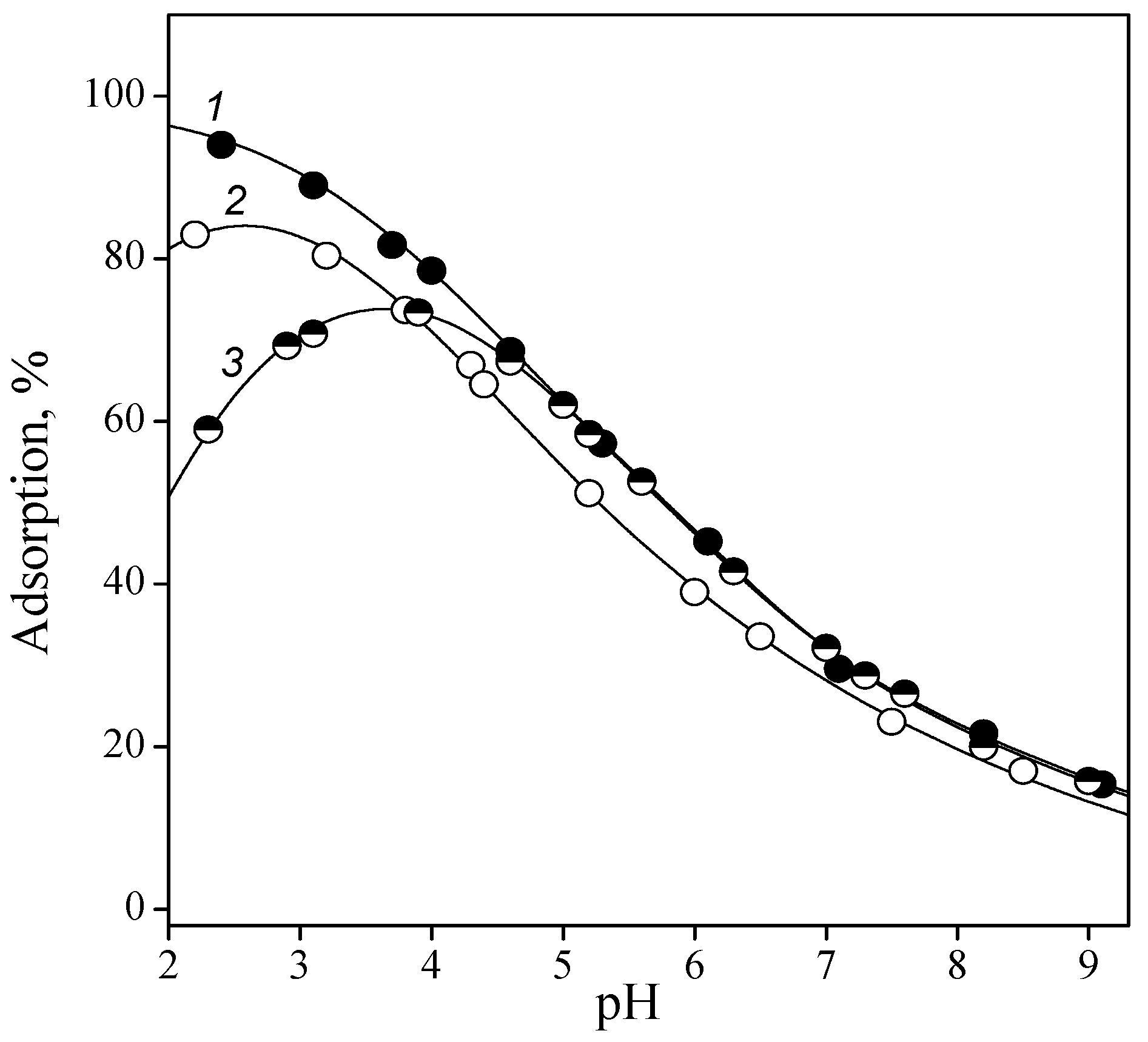
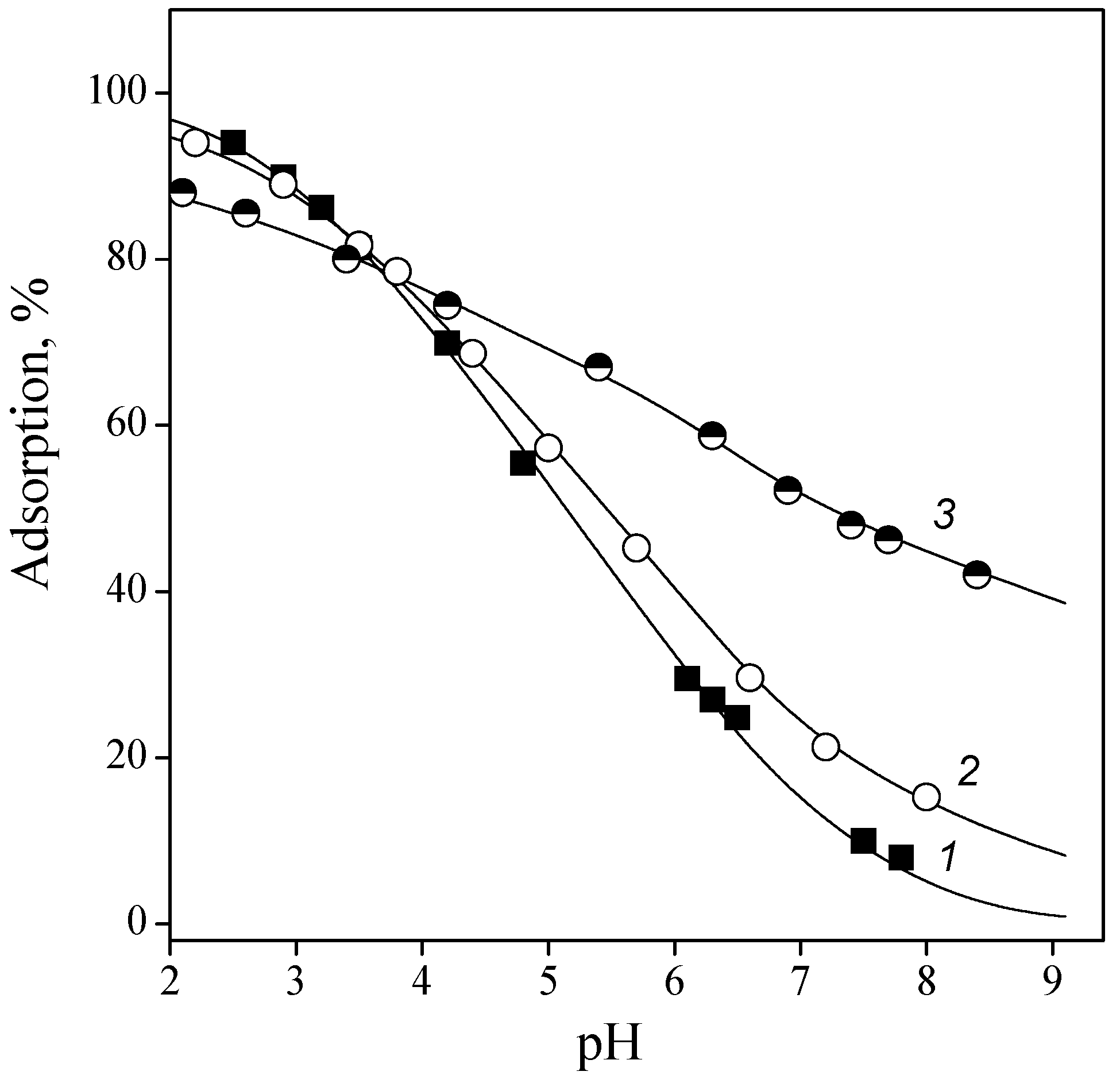
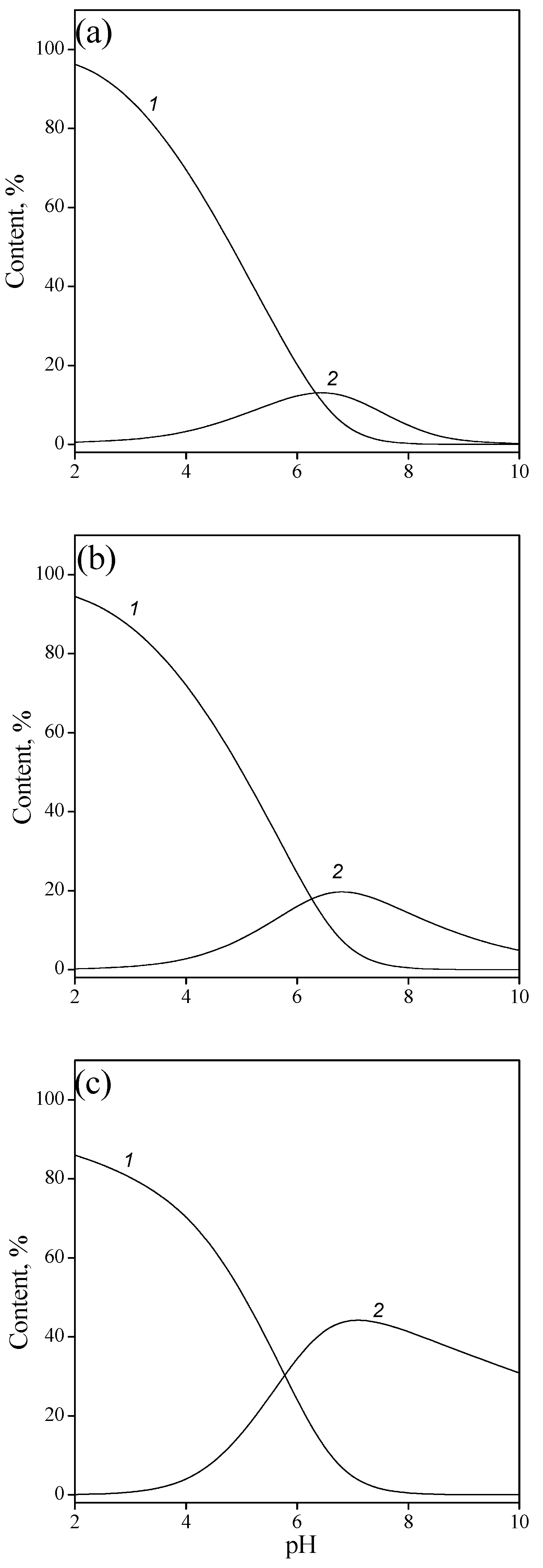
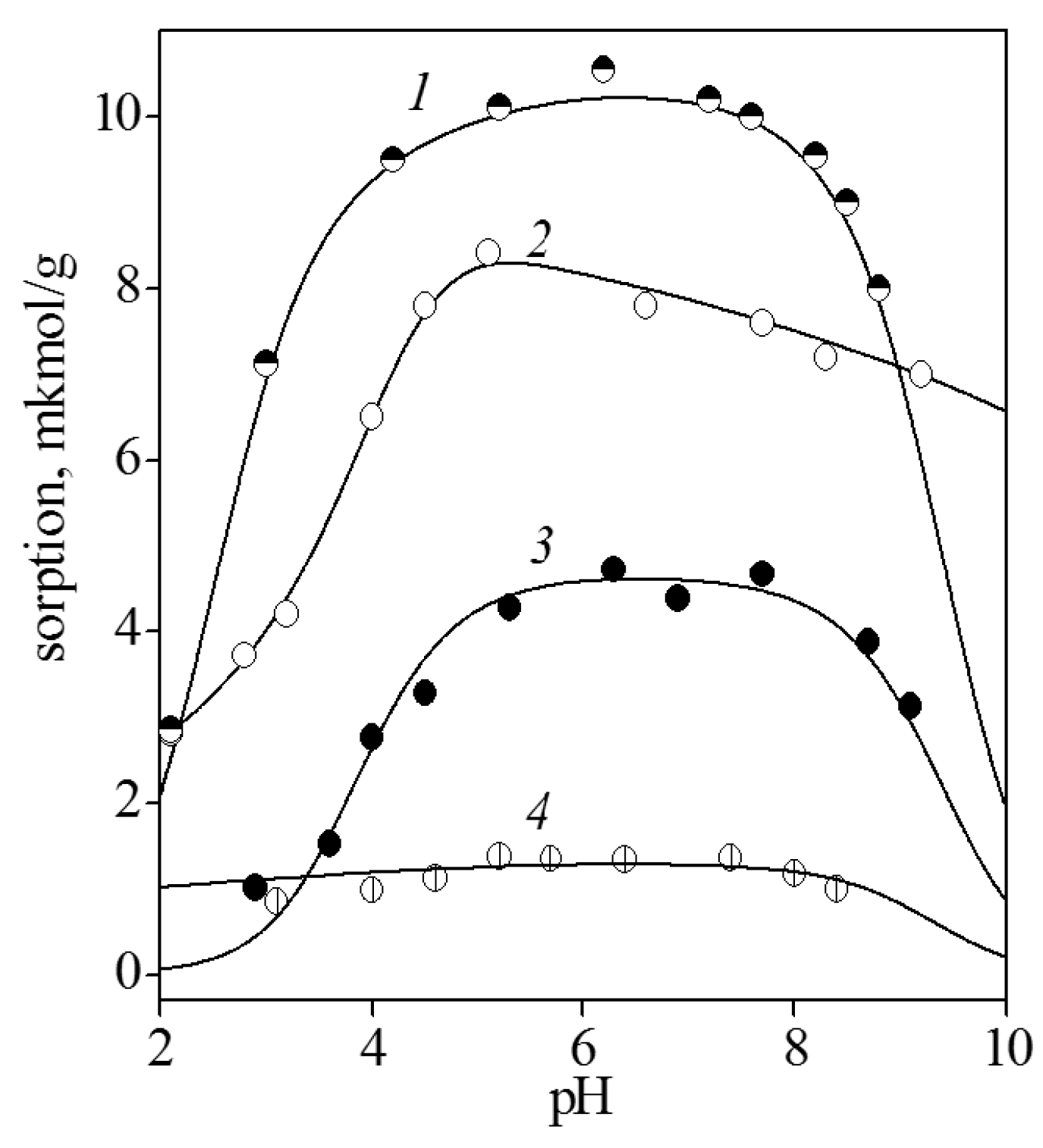
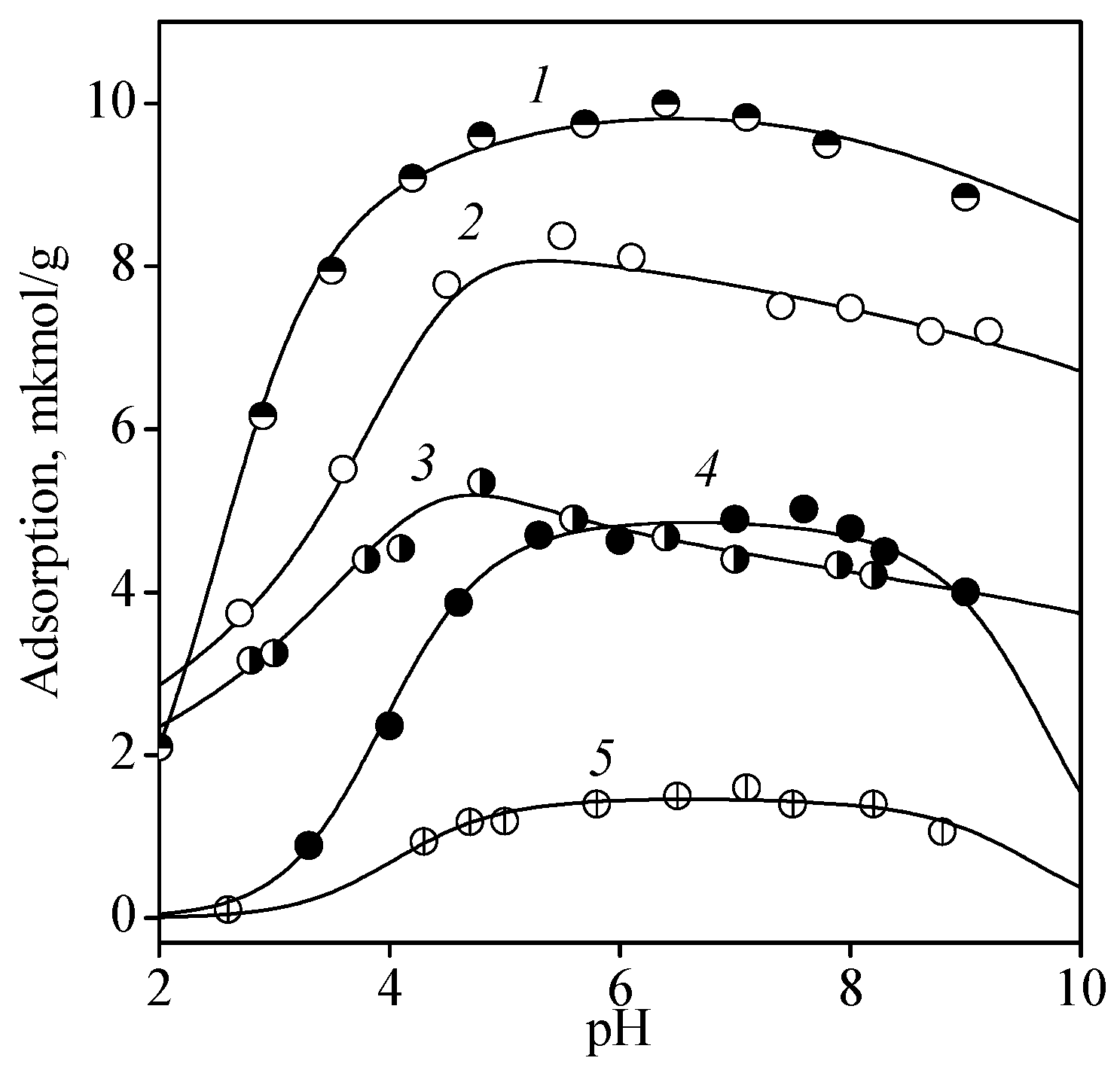
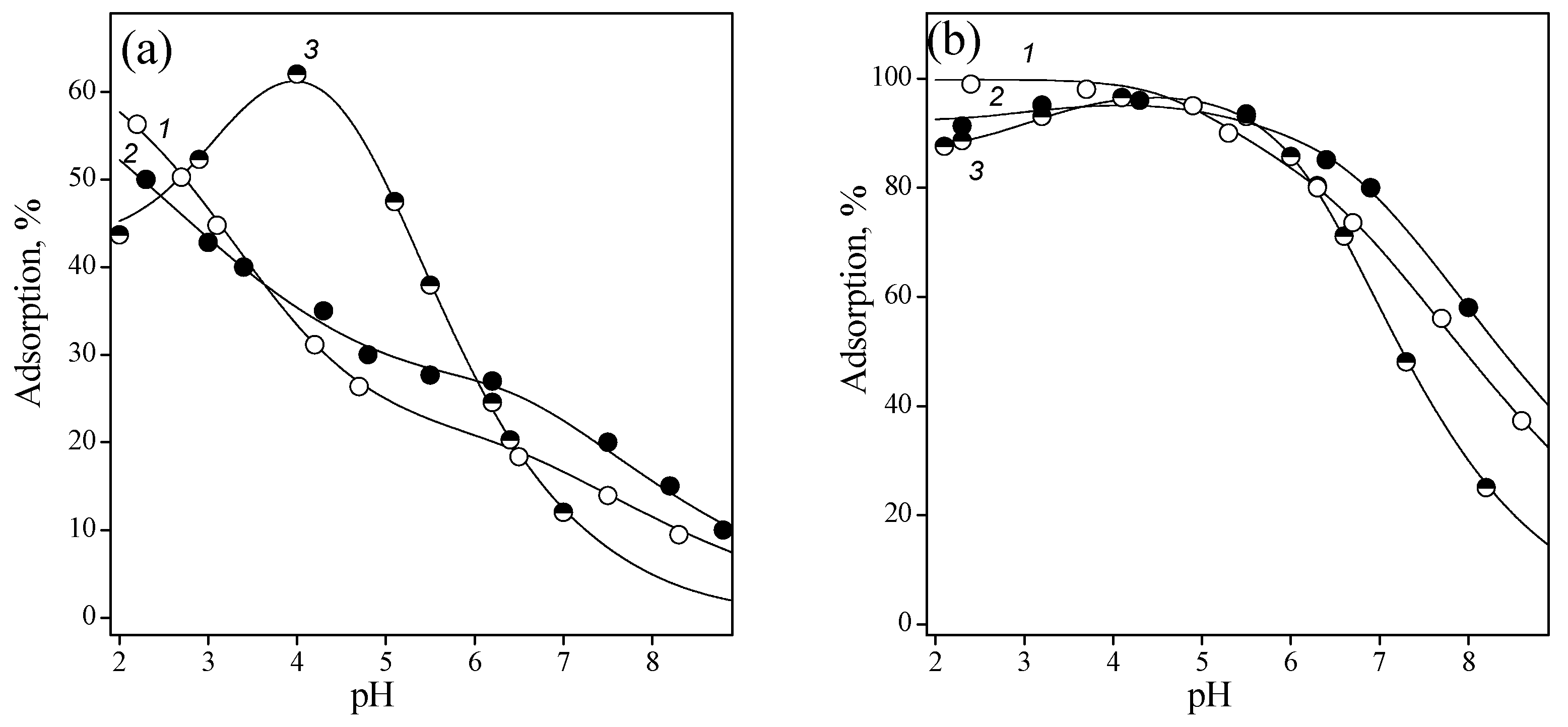
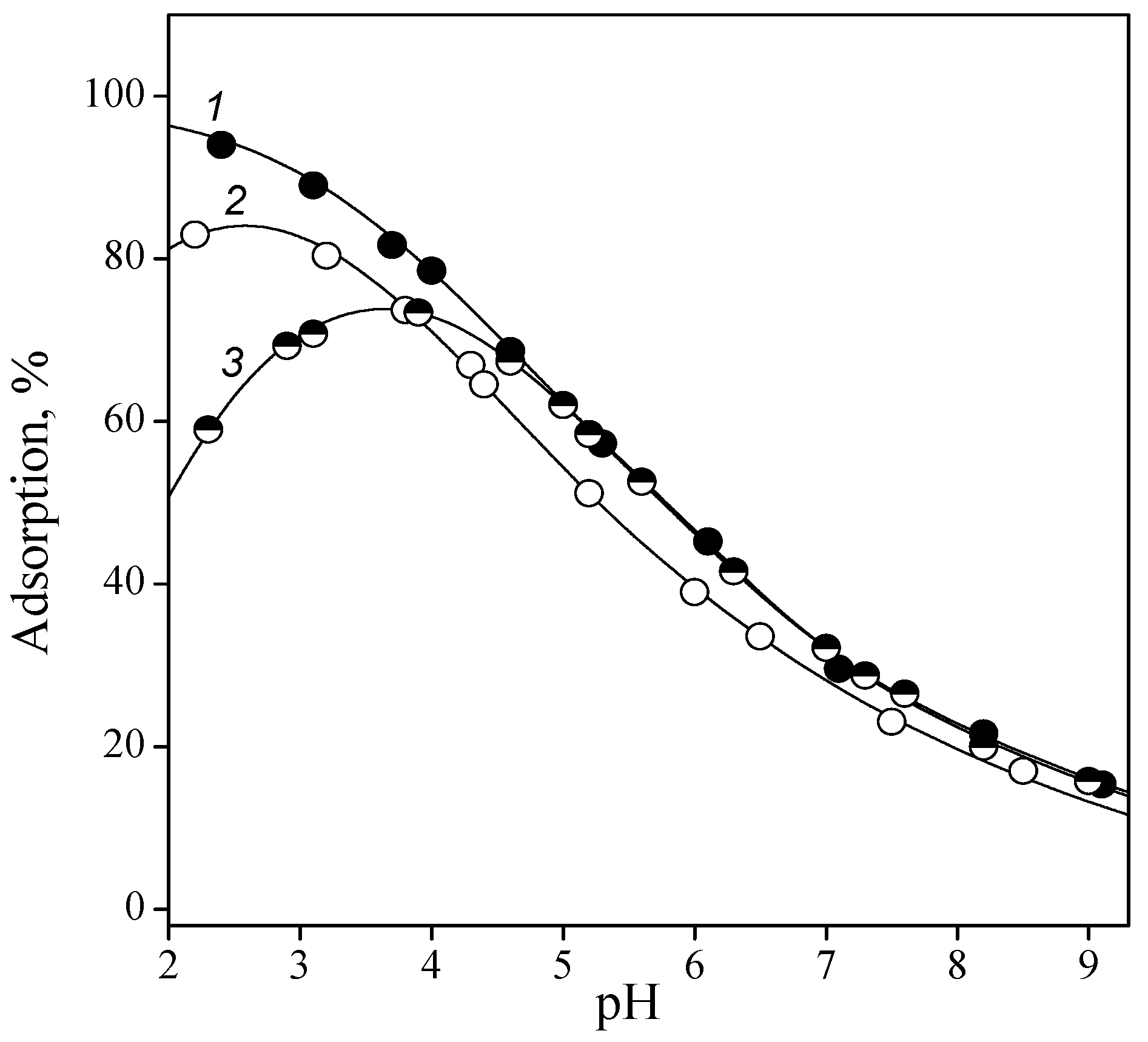
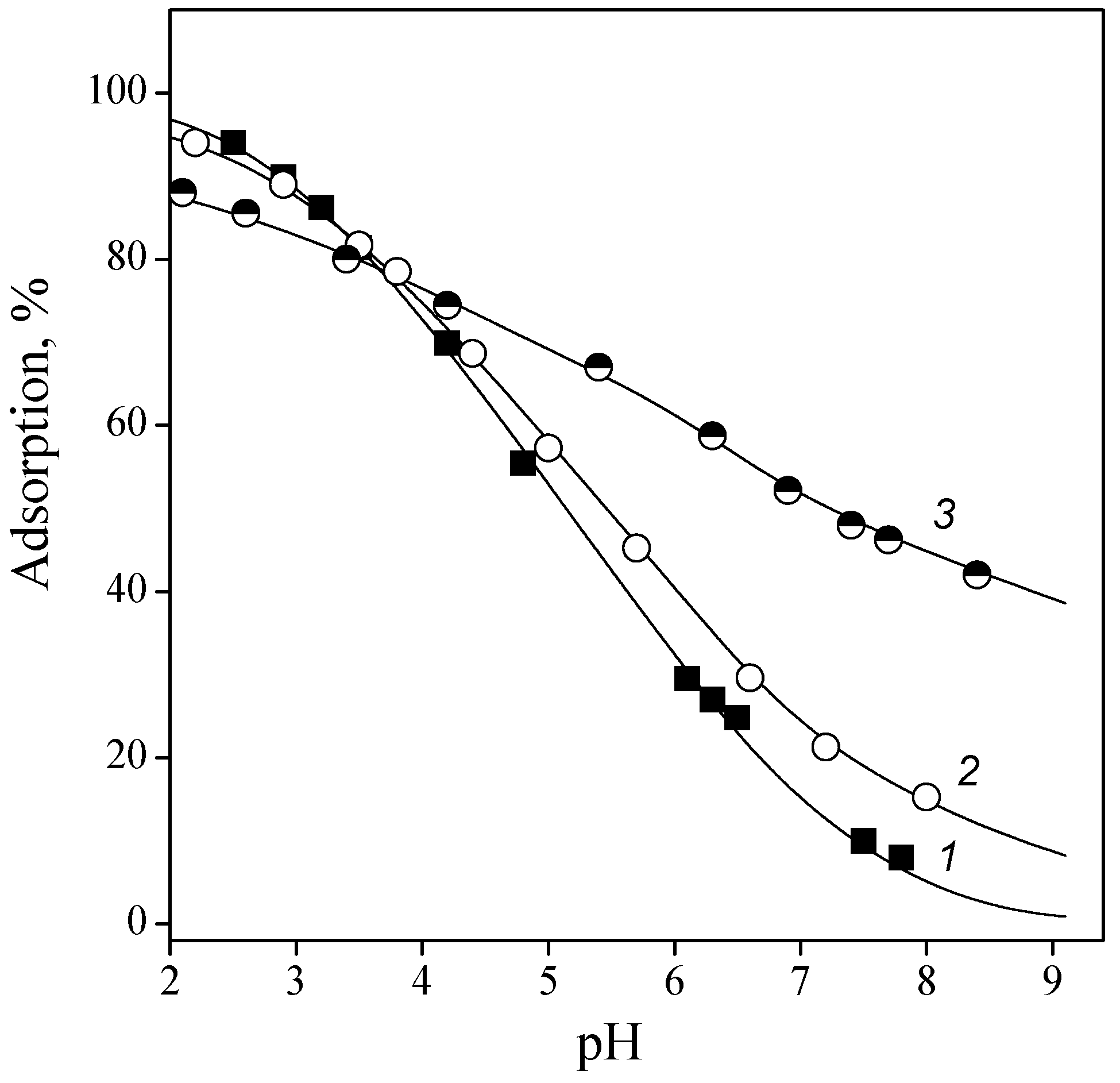
3.2. Adsorption of Nucleic Acid Components
3.2.1. Nucleobases
3.2.2. Nucleosides

3.2.3. Nucleotides
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Nel, A.E.; Madler, L.; Velegol, D.; Xia, T.; Hoek, E.M.V.; Somasundaran, P.; Klaessig, F.; Castronova, V.; Thompson, M. Understanding biochemical interactions at the nano-bio interface. Nat. Mater. 2009, 8, 543–557. [Google Scholar] [CrossRef] [PubMed]
- Vallee, A.; Humblot, V.; Pradier, C.-M. Peptide interactions with metal and oxide surfaces. Acc. Chem. Res. 2010, 43, 1297–1306. [Google Scholar] [CrossRef] [PubMed]
- Stark, W.J. Nanoparticles in biological systems. Angew. Chem. Int. Ed. 2011, 50, 1242–1258. [Google Scholar] [CrossRef] [PubMed]
- Shemetov, A.A.; Nabiev, I.; Sukhanova, A. Molecular interaction of proteins and peptides with nanoparticles. ACS Nano 2012, 6, 4585–4602. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.; Lau, B.L.T. Biomolecule-nanoparticel interactions: Eludicidation of the thermodymanics by isothermal titration calorimetry. Biochim. Biophys. Acta 2016, 1860, 945–956. [Google Scholar] [CrossRef] [PubMed]
- Gunnarsson, S.B.; Bernfur, K.; Mikkelsen, A.; Cedervall, T. Analysis of nanoparticle biomolecule complexes. Nanoscale 2018, 10, 4246–4257. [Google Scholar] [CrossRef]
- Fisher, J.; Egerton, T.A. Titanitum Compounds. In Inorganic Kirk-Othmer Encyclopedia of Chemical Technology; Wiley: New York, NY, USA, 2001. [Google Scholar]
- Bourikas, K.; Kordulis, C.; Lycourghiotis, A. Titanium dioxide (anatase and rutile): Surface chemistry, liquid-solid interface chemistry, and scientific synthesis of supported catalysts. Chem. Rev. 2014, 114, 9754–9823. [Google Scholar] [CrossRef] [PubMed]
- Ai, J.; Bizzar, E.; Montazeri, M.; Majdi, A.; Aminifard, S.; Zafari, M.; Akbari, H.R.; Rad, H.G. Nanotoxicology and nanoparticle safety in biomedical design. Int. J. Nanomed. 2011, 6, 1117–1127. [Google Scholar]
- Roddick-Lanzilotta, A.D.; Connor, P.A.; McQillan, A.J. An situ infrared spectroscopic study of the adsorption of lysine to TiO2 from an aqueous solution. Langmuir 1998, 14, 6479–6484. [Google Scholar] [CrossRef]
- Tran, T.H.; Nosaka, A.Y.; Nosaka, Y. Adsorption and photocatalytic decomposition of amino acids in TiO2 photocatalytic systems. J. Phys. Chem. B 2006, 110, 25525–25531. [Google Scholar] [CrossRef] [PubMed]
- Sverjensky, D.A.; Jonsson, C.M.; Jonsson, C.L.; Cleaves, H.J.; Hazen, R.M. Glutamate surface speciation on amorphous titanium dioxide and hydrous ferric oxide. Environ. Sci. Technol. 2008, 42, 6034–6039. [Google Scholar] [CrossRef] [PubMed]
- Jonsson, C.M.; Jonsson, C.L.; Sverjensky, D.A.; Cleaves, H.J.; Hazen, R.M. Attachment of L-glutamate to rutile (α-TiO2): A potemtiometric, adsorption, and surface complexation study. Langmuir 2009, 25, 12127–12135. [Google Scholar] [CrossRef] [PubMed]
- Thomas, A.G.; Syres, K.L. Adsorption of organic molecules on rutile and anatase single crystal surfaces. Chem. Soc. Rev. 2012, 41, 4207–4217. [Google Scholar] [CrossRef] [PubMed]
- Sultan, A.M.; Hughes, Z.E.; Walsh, T.R. Binding affinities of amino acids analogues at the charged aqueous titania interface. Langmuir 2014, 30, 13321–13329. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Li, K.; Liang, X.; Zheng., Y.; Li, L. Amino acid adsorption on anatase (101) surface at vacuum and aqueous solution: A density functional study. J. Mol. Model. 2018, 24, 107. [Google Scholar] [CrossRef] [PubMed]
- Burton, F.G.; Neuman, M.W.; Neuman, W.F. On the possible role of crystals in the origin of life. I. The adsorption of nucleosides, nucleotides, and pyrophosphate by apatite crystals. Curr. Mol. Biol. 1969, 3, 20–26. [Google Scholar] [CrossRef]
- Cortez, J.; Schnitzer, M. Reactions of nucleic acid bases with inorganic soil constituents. Soil Biol. Biochem. 1981, 13, 173–178. [Google Scholar] [CrossRef]
- Banin, A.; Lawless, J.G.; Mazzurco, J.; Church, F.M.; Margulies, L.; Orenberg, J.B. pH profile of the adsorption of nucleotides onto montmorillonite. Orig. Life Evol. Biosph. 1985, 15, 89–101. [Google Scholar] [CrossRef]
- Holm, N.G.; Ertem, G.; Ferris, J.P. The binding and reactions of nucleotides and polynucleotides on iron oxide hydroxide polymorphs. Orig. Life Evol. Biosph. 1993, 23, 195–215. [Google Scholar] [CrossRef] [PubMed]
- Arrora, A.K. Interaction of ribose nucleotides with zinc oxide and relevance in chemical evolution. Colloids Surf. A Physicochem. Eng. Asp. 2007, 298, 186–191. [Google Scholar] [CrossRef]
- Arrora, A.K. Role of metal oxide in chemical evolution: Interaction of ribose nucleotides with alumina. Astrobiology 2009, 9, 165–171. [Google Scholar] [CrossRef] [PubMed]
- Cleaves, H.J.; Jonsson, C.M.; Jonsson, C.L.; Sverjensky, D.A.; Hazen, R.M. Adsorption of nucleic acid components on rutile (TiO2) surfaces. Astrobiology 2010, 10, 311–323. [Google Scholar] [CrossRef] [PubMed]
- Westall, J.C.; Hohl, H. A comparison of electrostatic models for the oxide/solution interface. Adv. Colloid Interface Sci. 1980, 12, 265–294. [Google Scholar] [CrossRef]
- Ludwig, C. GRFIT, a Program for Solving Speciation Problems, Evaluation of Equilibrium Constants, Concentrations, and Other Physical Parameters; Internal Report; University of Bern: Bern, Switzerland, 1992. [Google Scholar]
- Stumm, W. Aquatic Surface Chemistry; Wiley-Interscience: New York, NY, USA, 1987. [Google Scholar]
- Dzombak, D.A.; Morel, F.M.M. Surface Complexation Modeling: Hydrous Ferric Oxides; Wiley-Interscience: New York, NY, USA, 1990. [Google Scholar]
- Davis, J.A.; Kent, D.B. Surface complexation modeling in aqueous geochemistry. Rev. Mineral. 1990, 23, 177–260. [Google Scholar]
- Kosmulski, M. Chemical Properties of Material Surfaces; Marcel Dekker: New-York, NY, USA, 2001. [Google Scholar]
- Hiemstra, T.; Venema, P.; Van Riemsdijk, W.H. Intrinsic proton affinity of reactive surface groups of metal (hydr)oxides: The bond valence principle. J. Colloid Interface Sci. 1996, 184, 680–692. [Google Scholar] [CrossRef] [PubMed]
- Van Veen, R.J.A.; Veltmaat, F.T.G.; Jonkers, G. A method for quantitative determination of the basic, acidic, and total surface hydroxyl content of TiO2. J. Chem Soc. Chem. Commun. 1985, 1656–1958. [Google Scholar] [CrossRef]
- Ludwig, C.; Schindler, P.W. Surface complexation on TiO2: Adsorption of H+ and Cu2+ ions onto TiO2 (anatase). J. Colloid Interface Sci. 1995, 169, 284–290. [Google Scholar] [CrossRef]
- Rodriguez, R.; Blesa, M.A.; Regazzoni, A.E. Surface complexation at the TiO2 (anatase)/aqueous solution interface: Chemisorption of catechol. J. Colloid Interface Sci. 1996, 177, 122–131. [Google Scholar] [CrossRef] [PubMed]
- Bourikas, K.; Hiemstra, T.; Van Riemsdijk, W.H. Ion pair formation and primary charging behavior of titanium oxide (anatase and rutile). Langmuir 2001, 17, 749–756. [Google Scholar] [CrossRef]
- Brown, G.E.; Henrich, V.E.; Casey, W.H.; Clark, D.L.; Eggleston, C.; Felmy, A.; Goodman, D.V.; Grätzel, M.; Maciel, G.; McCarthy, M.I.; et al. Metal oxide surfaces and their interactions with aqueous solutions and microbial organisms. Chem. Rev. 1999, 99, 77–174. [Google Scholar] [CrossRef] [PubMed]
- Saenger, W. Principles of Nucleic Acid Structure; Springer: Berlin, Germany, 1984; Chapters 2,4,5. [Google Scholar]
- Morozov, Y.V.; Bazhulina, N.P. Electron Structure, Spectroscopy, and Reactivity of Molecules; Nauka: Moscow, Russia, 1989. Chapter 3. (In Russian) [Google Scholar]
- Yatsimirskii, K.B.; Kriss, E.E.; Gvyazdovskaya, V.L. Stability Constants of Complexes of Biometals with Ligands: A Handbook; Naukova Dumka: Kiev, Russia, 1979. (In Russian) [Google Scholar]
- Smith, R.M.; Martell, A.E.; Chen, Y. Critical evaluation of stability constants for nucleotide complexes with protons and metal ions and the accompanying enthalpy changes. Pure Appl. Chem. 1991, 63, 1015–1080. [Google Scholar] [CrossRef]
- Hingston, F.J.; Atkinson, R.J.; Posner, A.M.; Quirk, J.P. Specific adsorption of anions. Nature 1967, 215, 1459–1461. [Google Scholar] [CrossRef]
- Childs, C.W. Potentiometric study of equilibriums in aqueous divalent metal orthophosphate solutions. Inorg. Chem. 1970, 9, 2465–2469. [Google Scholar] [CrossRef]
- Galal-Gorchev, H.; Stumm, W. The reaction of ferric ions with orthophosphate. J. Inorg. Nucl. Chem. 1963, 65, 567–574. [Google Scholar] [CrossRef]
- Lahav, N.; Chang, S. The possible role of solid surface area in condensation reactions during chemical evolution: Reevaluation. J. Mol. Evol. 1976, 8, 357–380. [Google Scholar] [CrossRef] [PubMed]
- Lambert, J.-F. Adsorption and polymerization of amino acids on mineral surfaces: A review. Orig. Life Evol. Biosph. 2008, 38, 211–242. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
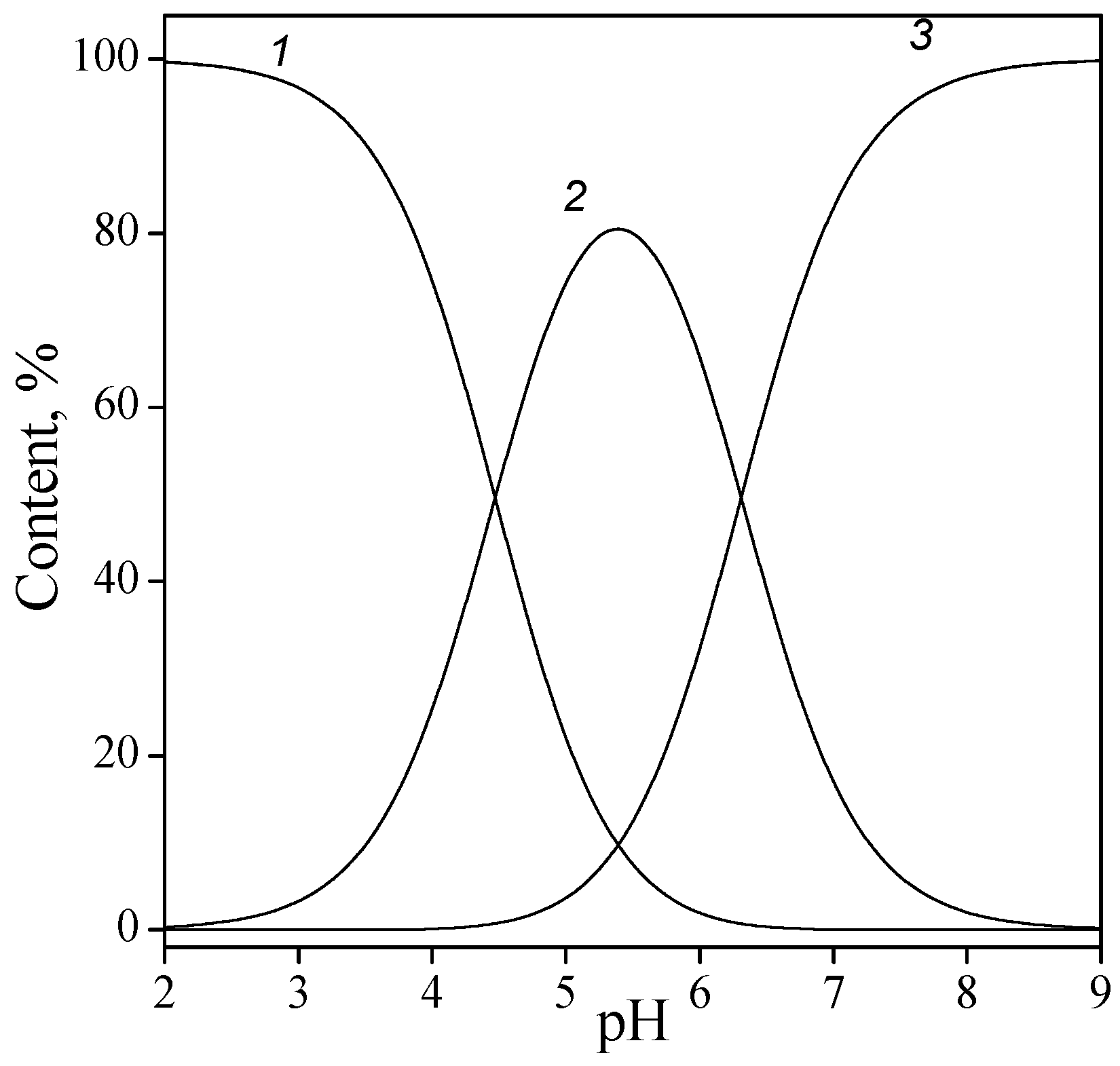
| Base | Ionization Reaction (pK) [38] (0.01 M) | |
|---|---|---|
| BH+ ↔ B + H+ | B ↔ B− + H+ | |
 adenine adenine | 3.92 (N1H+) | 9.63 (N9H) |
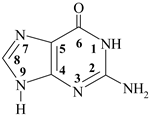 guanine guanine | 2. 20 (N1H+) | 9.38 (N9H) |
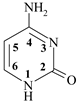 cytosine cytosine | 4.23 (N3H+) | 12.20 (N1H) |
 uracil uracil | 10.13 (N1H) | |
| Biomolecule | Surface Complex | |
|---|---|---|
| ≡TiOH⋯BH+ | ≡TiOH⋯B | |
| adenine | 2.38 | |
| adenosine | 2.35 | |
| 2′-deoxyadenosine | 1.75 | |
| guanine | 2.76 | |
| guanosine | 2.71 | |
| cytosine | 3.60 | 2.50 |
| cytidine | 3.50 | 2.43 |
| 2′-deoxycytidine | 3.38 | 2.13 |
| uracil | 1.71 | |
| Nucleotide | Ionization Constant pKn (0.01 M) |
|---|---|
 Cytidine-5′-monophosphate (H2L±), CMP Cytidine-5′-monophosphate (H2L±), CMP | 4.31 (N3–H+) 6.15 (–PO3H−) |
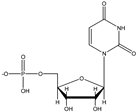 Uridine-5′-monophosphate (HL−), UMP Uridine-5′-monophosphate (HL−), UMP | 6.04 (–PO3H−) 9.39 (N3–H) |
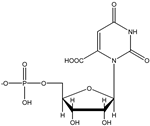 Orotidine-5′-monophosphate (H2L−), OMP Orotidine-5′-monophosphate (H2L−), OMP | 2.40 (–COOH) 6.10 (–PO3H−) |
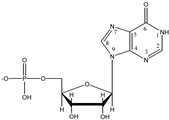 Inosine-5′-monophosphate (HL−), IMP Inosine-5′-monophosphate (HL−), IMP | 6.47 (–PO3H−) |
 Guanosine-5′-monophosphate (H2L±), GMP Guanosine-5′-monophosphate (H2L±), GMP | 2.48 (–N7H+) 6.48 (–PO3H−) |
 Adenosine-5′-monophosphate (H2L±), AMP Adenosine-5′-monophosphate (H2L±), AMP | 3.96 (–N1H+) 6.46(–PO3H−) |
| Surface Reaction | Nucleotide | |||||
|---|---|---|---|---|---|---|
| CMP | UMP | OMP | AMP | GMP | IMP | |
| ≡TiOH + HL− + H+ ↔ ≡TiOHHL− | 10.14 | 9.59 | 10.60 | 10.31 | 10.11 | |
| ≡TiOH + HL− ↔ ≡TiOHHL− | 4.94 | 4.39 | 5.40 | 5.11 | 4.91 | |
| ≡TiOH + HL− ↔ ≡TiOHL2− | 4.29 | 4.06 | 5.36 | 5.19 | 4.90 | |
| ≡TiOH + L2− ↔ ≡TiOHL2− | 5.24 | 4.90 | 6.62 | 6.46 | 6.18 | |
| ≡TiOH + HL2− + H+ ↔ ≡TiOHHL2− | 9.75 | |||||
| ≡TiOH + HL2− ↔ ≡TiOHHL2− | 4.55 | |||||
| ≡TiOH + HL2− ↔ ≡TiOHL3− | 6.32 | |||||
| ≡TiOH + L3− ↔ ≡TiOHL3− | 6.22 | |||||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vlasova, N.N.; Markitan, O.V. Surface Complexation Modeling of Biomolecule Adsorptions onto Titania. Colloids Interfaces 2019, 3, 28. https://doi.org/10.3390/colloids3010028
Vlasova NN, Markitan OV. Surface Complexation Modeling of Biomolecule Adsorptions onto Titania. Colloids and Interfaces. 2019; 3(1):28. https://doi.org/10.3390/colloids3010028
Chicago/Turabian StyleVlasova, Nataliya N., and Olga V. Markitan. 2019. "Surface Complexation Modeling of Biomolecule Adsorptions onto Titania" Colloids and Interfaces 3, no. 1: 28. https://doi.org/10.3390/colloids3010028
APA StyleVlasova, N. N., & Markitan, O. V. (2019). Surface Complexation Modeling of Biomolecule Adsorptions onto Titania. Colloids and Interfaces, 3(1), 28. https://doi.org/10.3390/colloids3010028