Abstract
Surfaces and interfaces are ubiquitous in nature and are involved in many biological processes. Due to this, natural organisms have evolved a number of methods to control interfacial and surface properties. Many of these methods involve the use of specialised protein biosurfactants, which due to the competing demands of high surface activity, biocompatibility, and low solution aggregation may take structures that differ from the traditional head–tail structure of small molecule surfactants. As well as their biological functions, these proteins have also attracted interest for industrial applications, in areas including food technology, surface modification, and drug delivery. To understand the biological functions and technological applications of protein biosurfactants, it is necessary to have a molecular level description of their behaviour, in particular at surfaces and interfaces, for which molecular simulation is well suited to investigate. In this review, we will give an overview of simulation studies of a number of examples of protein biosurfactants (hydrophobins, surfactin, and ranaspumin). We will also outline some of the key challenges and future directions for molecular simulation in the investigation of protein biosurfactants and how this can help guide future developments.
1. Introduction
In order to control the properties of surfaces and interfaces, many organisms have evolved specialised surface active biomolecules, such as protein or peptide biosurfactants, which fulfill a number of key biological functions [1,2]. Examples of these functions (Figure 1) include the reduction in air–water surface tension, easing the growth of aerial bodies, such as spores or hyphae [3,4], or the spreading of bacterial biofilms [5]. Adsorption of biosurfactant proteins onto surfaces is often used to aid the adhesion of microorganisms [6]. Biosurfactant proteins also commonly interact with membranes, which can lead to antibacterial, antifungal, and antiviral behaviours [7], giving these important defensive roles. Other functions of biosurfactant proteins include inhibition of immune response [8] and stabilization of polyhydroxyalkanoate (PHA) granules [9]. As they fulfill such a diverse range of functions, they have evolved a variety of structures [10,11] that can differ from those of other proteins.
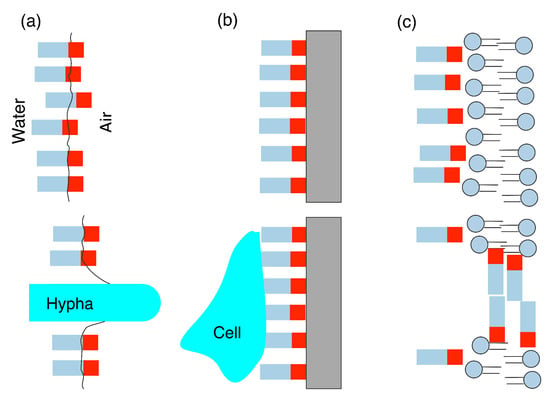
Figure 1.
Schematic illustration of common biological functions of biosurfactants. (a) formation of interfacial layer to assist growth of aerial bodies; (b) formation of surface layer aiding cell attachment and growth; (c) membrane adsorption and poration.
As adsorption onto interfaces is central to the function of biosurfactant proteins, they have evolved a number of strategies to maximise their interfacial adsorption. It should be noted that almost all proteins will adsorb onto interfaces and surfaces, as they contain both hydrophobic and hydrophilic amino acids making them intrinsically amphiphilic. In most cases, this is accompanied by unfolding [12,13] and loss of function, so it is typically avoided. By contrast, biosurfactant proteins typically retain their structure or undergo specific changes in conformation, so they have evolved structural features that favour interfacial adsorption. Alongside this, the structures of biosurfactant proteins are also often influenced by the need to maintain biocompatibility and to minimise their aggregation in solution. This has caused them to evolve designs that differ from the simple polar head/hydrophobic tail structure found in man-made surfactants and phospholipids [10,11,14]. As these properties would also be advantageous in many materials systems, biosurfactant proteins are increasingly being investigated for industrial applications in a number of sectors [15,16] (Table 1).

Table 1.
Examples of investigated applications for protein biosurfactants.
Understanding the biological functions and technological applications of biosurfactant proteins requires knowledge of their structure and behaviour at interfaces. As changes in these environments can occur over nanometre lengthscales, understanding this requires microscopic detail that is challenging to obtain experimentally. Molecular simulation operates directly on the molecular level and thus can provide such microscopic detail. Since the first molecular dynamics simulation of proteins, these have increasingly been recognised as a powerful tool for the investigation of protein structure and function [29]. This has been driven by improvements in computer power and simulation methodologies; the first protein simulation investigated the 58-residue bovine pancreatic trypsin inhibitor [30] (without water), while simulations on current state-of-the-art hardware can handle the entire HIV capsid (64 million atoms) [31] or significant regions of the cell cytoplasm [32]. A number of studies have applied molecular simulation to the investigation of proteins at surfaces and interfaces, giving significant insight into their behaviour in these environments [33]. These have investigated a range of proteins, including enzymes, such as lipase [34], globular proteins, such as lysozyme [35,36,37] or -lactoglobulin [38,39], and intrinsically disordered proteins, such as amyloid- [40].
In this review, we will give an overview of molecular simulation work investigating the structure and properties of biosurfactant proteins and peptides. This will focus on the well-studied cases of hydrophobins and surfactin, along with the novel biosurfactant protein Rsn-2. As we focus on simulation studies, key examples of protein biosurfactants, such as caseins, that have not been the subject of significant MD work will not be covered. Similarly, lipids and other surface-active biomolecules lie outside the scope of this review. Also excluded is lung surfactant, which, while containing a number of key proteins, the main surface active component is the phospholipid component.
2. Molecular Simulation
In this section, we will briefly describe the molecular dynamics simulation method and common simulation models. This is not intended to be an exhaustive and detailed discussion of these, rather a short overview necessary for understanding the following sections. There are a number of excellent textbooks [41,42] that the interested reader may consult for a fuller description of molecular simulation methods and the underlying algorithms and statistical mechanics.
For simulation of biomolecular systems, the most common technique is molecular dynamics (MD) simulation. This corresponds to numerically solving the classical Newtonian equations of motion in a step-wise, iterative manner. If the positions () and velocities () of the atoms are known are a given time t, then the forces () at that time can be calculated. These forces can then be used to determine the velocities and positions a short time later, from which a new set of forces can be calculated, and so on. The time interval is commonly referred to as the time step; for a biomolecular system, the time step is typically on the order of 1 fs, so a typical MD simulation spanning 10 to 100 nanoseconds will entail 10–100 millions of time steps.
A key factor in molecular simulation is the model used, which determines the overall accuracy and degree of resolution of the study. Most simulation studies in biophysics employ atomistic models, in which the interactions are described a using a classical energy function referred to as a force field. This gives the energy of the system as a sum over intra- and intermolecular interactions, with a typical form being [43]
where the terms on the right-hand side are bond stretching, bond angle bending, dihedral rotation (togther referred to as the bonded interactions), van der Waals, and electrostatic terms, respectively. In the bonded interactions , , and are force constants that describe the energetic costs of band stretching, bond angle bending, and dihedral rotation, and are equilibrium bond lengths, and bond angles. , , and are the van der Waals well depths, van der Waals diameters, and atomic partial charges, respectively. The functional form in Equation 1 is the simplest form using harmonic terms for bond stretching and angle bending and the simple Lennard–Jones potential for van der Waals interactions. More sophisticated forms and potentials that describe other interactions, such as coupling between different intramolecular distortions (e.g., stretch-bend), can be used. Alongside the potential function, force fields also define the parameters (e.g., force constants, equilibrium bond lengths) that appear in Equation (1). These parameters have to be determined either from experimental data or other theoretical methods (such as quantum chemical calculations). A number of different force fields have been developed for modelling biomolecular systems, which differ in their functional forms and parameterization procedures. Common models that have been applied to proteins include the Amber [44], Charmm [45], Gromos [46], and OPLS [47] families of force fields. These have been refined over the past 30 years as new experimental data have been gained, with the latest iterations of these force fields being capable of describing the structures and folding of proteins.
The starting point for a MD simulation is typically an experimentally determined protein structure, typically from X-ray crystallography or solution NMR. As structures for a limited number of protein biosurfactants are known, most simulation studies have focused on this small group. Computational methods for protein structure prediction, such as homology modelling, may be used to create conformations for proteins without experimentally determined structures. These generally rely on knowledge of structure for similar proteins (in terms of sequence), which due to the diverse sequences for many biosurfactant proteins can limit their applicability.
While they provide detailed molecular level information, atomistic simulation is limited in the system sizes it can investigate. To study larger systems, it is possible to reduce the level of detail through the use of coarse-grain (CG) models. In these, a number of heavy (non-hydrogen) atoms are grouped together into a single interaction site. One common CG model for biomolecules is the Martini model [48,49], developed by the Marrink group. Originally, this was developed for simulations of membranes and was later extended to include proteins [50]. In the Martini model, each CG bead in a protein (or lipid) typically represents four heavy atoms, so each protein residue will be composed of between one and five beads. This mapping also applies to the solvent so a water bead corresponds to four water molecules. Due to its ability to model larger systems and its inclusion in the commonly used Gromacs package, the Martini model has been extensively applied to proteins, in particular to membrane–protein systems [51]. Direct comparison between CG and atomistic models shows that these can give qualitatively similar results for proteins at interfaces and surfaces, but, due to the loss of resolution, they can differ in fine detail, such as the position of proteins relative to an oil–water interface [52]. A similar model, the Shinoda–DeVane–Klein model [53,54], has also been developed and extended to investigate proteins.
Atomistic molecular dynamics simulation has proven to be a powerful tool for the investigation of the behaviour of biosurfactant proteins, but it has some limitations. One challenge for atomistic simulation is in sampling conformations for complex molecules such as proteins. This arises as different conformations can often be separated by energy barriers significantly higher than the thermal energy (). To overcome this, one common method is replica exchange molecular dynamics [55,56]. Here, a number of replicas of the system are simulated concurrently at different temperatures; replicas at high temperatures are able to get over the energy barriers separating different conformations, while replicas at lower temperatures can explore the regions around the minima in detail. By periodically exchanging the temperatures, each replica is simulated at efficient sampling of different protein conformations can be achieved. This has been widely used in the simulation of biomolecules, with a number of recent applications to proteins at interfaces [36,37,57] and surfaces [58].
Another challenge is in calculating free energies, which can be used to determine the relative stability of different protein conformations or adsorption strengths onto surfaces or interfaces. This is traditionally challenging in MD simulation as it requires sampling of all the possible states a system, including rarely found high energy states. One commonly used method for determining free energy differences is metadynamics [59,60] (MetaD). The MetaD algorithm assumes that the state of the system can be a few collective variables, such as measures of protein structure, molecular positions, or energies. MetaD then introduces a history bias potential that is a function of these collective variables, which is updated throughout the simulation and will eventually converge to the negative of the free energy surface. A number of variants of metadynamics have been proposed [61,62], which differ in the how the bias function is updated, to enhance convergence. MetaD has been applied to investigated the adsorption of short peptides onto surfaces [63,64,65,66] and interfaces [67], and with increasing computer power its use for large proteins is becoming possible. When the free energy as a function of molecular position is needed, such as calculating adsorption free energies, umbrella sampling (US) [68] or steered molecular dynamics (SMD) [69] simulations have been used. In US a, typically harmonic potential, is applied to constrain the group of atoms is constrained around an equilibrium position (). Performing a number of simulations with closely spaced equilibrium positions allows us to build up a free energy profile. In SMD, a similar harmonic potential is applied to the molecule of interest, but, in this equilibrium, position moves over time. Monitoring the force applied to the molecule allows for the calculation of the free energy profile. Both of these methods have been used to calculate the surface [63,70] and interfacial [71,72,73] adsorption strengths for peptides and proteins.
3. Hydrophobins
Probably the most commonly investigated classes of protein biosurfactants are the hydrophobins [74], which are expressed by some strains of filamentous fungi. These are relatively small proteins that are characterised by a conserved pattern of disulfide bonds [75]. Commonly, they also have a highly amphiphilic surface structure, which typically includes a large hydrophobic patch (Figure 2), which makes them highly surface active. They fulfill a number of functions typically associated with adsorption onto interfaces and surfaces. Some hydrophobins are used as surfactants to reduce the air–water surface tension, making it easier for fungi to release sprouting bodies such as spores or hyphae. Other hydrophobins are used to mediate adsorption onto hydrophobic surfaces [76,77]. Hydrophobins have traditionally been divided into two-classes depending on whether they form fibrillar structures (rodlets) at interfaces (class-I) or not (class-II) [74]. Commonly studied examples of hydrophobins include the class-II hydrophobins HFBI and HFBII and the class-I hydrophobins SC3 and EAS. As well as fungal hydrophobins similar proteins have been identified in bacteria, where they play a role in the formation of biofilms [78].
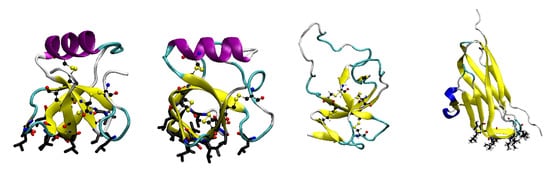
Figure 2.
Structures of commonly studied hydrophobins. (Left to right) Class-II hydrophobins HFBII and HFBI, class-I hydrophobin EAS, and bacterial hydrophobin BslA. Figures generated using VMD [79] and PDB accession codes 1R2M, 2FZ6, 2FMC, and 4BHU, respectively. Residues in hydrophobic patches are highlighted.
The surface activity of hydrophobins is also being exploited in man-made materials and systems [15]. Their high surface activity, combined with their biocompatibility, has led to their use as emulsifiers in foods foams and emulsions [17,18]. Hydrophobins have also been used as biocompatible coatings on drug particles and as epitaxial agents for the preparation of graphene and other two-dimensional materials [27,28].
Understanding the biological function and materials’ applications of hydrophobins requires knowledge of their structure, in particular at surfaces and interfaces. The relatively small size and simple structure of the hydrophobins (in comparison to other biosurfactants) has made them attractive targets for molecular simulation studies. As many of the biological functions and applications of hydrophobins revolve around their behaviour at interfaces and surfaces, most simulation studies have focused on this.
3.1. Hydrophobin Behaviour at Fluid (Air–Water or Oil–Water) Interfaces
The earliest MD study of a hydrophobin investigated the adsorption of SC3 onto liquid interfaces [80]; however, this used a disulfide bond pattern that was later found to be incorrect. Subsequent studies have used structures derived from protein crystallography or solution NMR, so are more representative of experimental systems. Major focuses of these studies include protein orientation at interfaces, changes in protein conformation, and the interfacial adsorption strength.
The conventional picture of hydrophobins as having a hydrophobic face that drives interfacial adsorption leads to the assumption that this patch points into the hydrophobic media. This has been demonstrated by atomistic MD simulations of the class-II hydrophobins HFBI [81,82] and HFBII [83] at the air–water interface. In both cases, the proteins adsorb onto the air–water interface with the patch pointing into the vacuum (air) region. This orientation of the protein maximises the contact between the hydrophobic patch and the non-aqueous component; an alternative orientation where the patch is oriented in the plane of the interface would maximise the decrease in air–water interfacial area, minimising the interfacial free energy. For HFBI, orientation of the patch towards the hydrophobic component is also seen for the water–decane interface [81] but on a phospholipid (DPPC) bilayer the patch is oriented in the plane of the bilayer. This is due to interactions between the hydrophilic head groups of the lipids with the larger polar regions of the protein surface and demonstrates the ability of hydrophobins to adsorb to both hydrophobic and hydrophilic interfaces. Similar behaviour was also seen for the class-I hydrophobin SC3, where adsorption to both air–water and water–dodecane interface was mediated through a hydrophobic loop [84]. In these simulations, two different protein conformations (taken from simulations of the protein in bulk solution), with the orientation of the hydrophobic loop, relative to the interface, differing by 90 degrees. Independent of the starting orientation and conformation of the protein this loop adsorbed to the interface, showing that it binds onto interfaces through this specific region of the protein.
Due to their rigidity, class-II hydrophobins typically undergo only small changes in conformation at interfaces. MD simulations of both HFBI and HFBII found that the protein secondary and tertiary structures are largely unchanged at the air–water interface [81,83]. This is consistent with circular dichroism [85] and neutron reflectivity measurements [86], which show limited changes in at the air–water interface. A greater change in the structure of HFBI was found at the water–decane interface [81]. This may be due to penetration of oil molecules into the hydrophobic patch, similar to changes observed for -lactalbumin at the water–octane interface [37]. The change in structure at the water–decane interface was larger than that seen on a DPPC bilayer due to the more hydrophilic environment of the lipid head groups.
Class-I hydrophobins show greater changes in structure at interfaces, which is implicated in their assembly into rodlets [87]. Early studies of the hydrophobin SC3 [84] showed an increase in -helix content in regions exposed to hydrophobic media. Similar induction of order was found for EAS, where a flexible 26-residue loop adopts a structured (-helix) conformation at the air–water interface [88]. The flexibility of loop in solution is expected to inhibit self-assembly, while the ordering of this allows for aggregation at interfaces. Changes in protein conformation upon interfacial adsorption has been proposed as a contributing factor in the formation of fibrillar structures for intrinsically disordered proteins [89,90], such as amyloid-beta, so it is logical that similar behaviour would be seen for the rodlet forming class-I hydrophobins. The differences between class-I and class-II hydrophobins were also demonstrated in a direct comparison of HFBII and EAS [91]. Again, these showed only small changes in structure for HFBII and larger conformational changes for EAS. This was used to explain differences in the binding of ions of these two proteins, which rationalised differences in the biomineralisation behaviour of HFBII and the class-I hydrophobin H*ProteinB [92].
To quantify the effect of the amphiphilic structure of hydrophobins on their interfacial behaviour, a few MD studies have determined the interfacial adsorption strengths. Using a CG model, the adsorption free energies for the class-II hydrophobins HFBI and HFBII at a water–octane interface were calculated [93] using steered molecular dynamics simulations. These were both found to have adsorption free energies of the order of 60–100 kcal mol, values similar to those of synthetic nanoparticles [94] and polymers [95]. The two proteins were found to have different affinities for the two solvent components, with HFBII slightly favouring the water and HFBI the oil, which can be rationalised by differences in the sizes of the hydrophobic patches. To explore this, further simulations of HFBII pseudoproteins with different protein solvent interactions were undertaken. Most interestingly, when the interaction with the solvents for each bead in the protein was set to the average value for each bead in the ‘real’ protein, essentially smearing the hydrophobic patch out across the protein, the protein was still attracted to the surface, but it becomes overall slightly hydrophobic. This demonstrates the importance of the amphiphilic surface structure of the protein. Atomistic simulations of the bacterial hydrophobin BslA [96] also found a similar adsorption strength at the air–water interface. Mutation of a single leucine residue in the hydrophobic region (L77K mutation) significantly decreased the adsorption strength. The high adsorption energies and the dependence of these on the size and structure of the hydrophobic region has drawn comparisons between these proteins and amphiphilic Janus particles [97,98].
3.2. Adsorption onto Surfaces
The adsorption of hydrophobins onto diverse solid surfaces has been investigated using MD simulations. Due to their hydrophobic patch and the potential use for surface modification, a number of studies, including some of the earliest hydrophobin simulations [84], investigated their adsorption onto hydrophobic surfaces. Adsorption of HFBII onto silicon was investigated using atomistic MD simulations [99], while Brownian dynamics has been used to investigate the adsorption and oligomerization of HFBI [100]. In both of these studies, the key residues involved in binding the protein to the surface were identified. These were primarily hydrophobic residues, with a number of these being common to both proteins. In the presence of a graphite surface, the proteins would accumulate at the surface, demonstrating that this adsorption is stronger than the solution oligomerization [101].
Self-assembled monolayers (SAM) are commonly used experimentally as a route to control surface chemistry and properties [102]. SAMs can be constructed from a wide range of molecules allowing for the creation of surfaces with different characteristics. Simulation of HFBI on SAMs formed on graphene was used to investigate the adsorption onto hydrophobic (methyl) and hydrophilic (amine) surfaces [103]. Adsorption onto both SAMs was found, demonstrating the dual nature of hydrophobins. While relatively short, these simulations examined the different driving forces for adsorption, suggesting that the adsorption onto hydrophilic surfaces is primarily driven by electrostatic interactions. Later work, over longer timescales and using replica exchange to enhance conformational sampling, demonstrated similar behaviour for HFBII on various SAM surfaces [104]. This also demonstrated the use of the hydrophobic and electric dipoles as simple parameters for describing the surface orientation of the protein; on a methyl-terminated SAM the hydrophobic dipole points towards the surface, while the orientation of the electric dipole depends on the surface charge. Such simple descriptors may prove useful in predicting protein surface behaviour alongside more expensive MD simulations. The ability of hydrophobins to change the character of a surface is not restricted to self-assembled monolayers, as demonstrated by MD simulation of HFBI on poly (dimethyl siloxane) surfaces [105]. As for other hydrophobic surfaces [99,100], the most stable adsorption occurred through the hydrophobic patch, but other adsorption modes were identified. Using MM-PBSA calculations, the relative free energies of these different modes were evaluated, with adsorption free energies for ∼ kcal mol found for states where the patch is in contact with the surface. The adsorption free energy is significantly larger than the adsorption enthalpies (typically ∼ kcal mol), suggestive of the importance of solvent and entropic effects (although these were only implicitly accounted for in the MM-PBSA calculations).
As for liquid interfaces, class-II hydrophobins, in both simulation [104] and experimental [106] studies, show only slight changes in conformation on surfaces. Due to their more flexible nature, class-I hydrophobins show greater changes in conformation. Simulation of EAS at surfaces showed that this binds to silica surfaces in two main modes [107]. The first of these involves a loop between the 3rd and 4th CYS residues, which leads to changes in structure around the amyloidogenic region of the protein, potentially leading to surface fibrillation. As less of the protein is typically in contact with the surface and the contact regions are more flexible, the surface bound water plays a larger role. Surface hydration and water structuring is a key factor in determining the interaction between proteins and surfaces [108,109]. The heterogenous surface structure of EAS was also found to lead to more complex adsorption processes on nanostructured surfaces [110], illustrating the importance of understanding the interplay between protein and surface structure.
4. Surfactin and Other Lipopeptides
A number of organisms, in particular bacteria, express a number of lipopeptides that have surface active behaviour. Probably the most studied of these is the lipopeptide surfactin, expressed by Bacillus subtilis. This consists of a cyclic heptapeptide head, with a sequence ELLADLL, and a 12–17 carbon atom alkyl tail (Figure 3). Its amphiphilic structure makes it highly surface active, causing a reduction in the air–water interfacial tension from 72 mN m to 27 mN m at a bulk concentration 0.0005% [111]. Surfactin also has a very low critical micelle concentration (∼ mol L), due to its high proportion of hydrophobic components. It plays a key role in the spreading of Bacillus subtilis, with significant differences between the biofilms formed by wild type Bacillus subtilis and Bacillus subtilis with the surfactin gene deleted, which disappear when surfactin is added to the mutant bacterium [112]. Surfactin has also been shown to have antibacterial, antiviral, and antifungal properties [113].
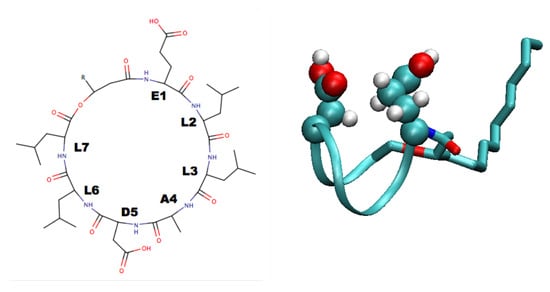
Figure 3.
Surfactin structure. The left hand panel shows chemical structure. The R group is a 12 to 17 carbon atom alkyl chain. The right hand panel shows 3D structure, with Glu and Asp residues highlighted, generated using VMD [79] (from PDB structure 2NPV).
NMR studies [111] suggest that the head group adopts a saddle-like structure, with the two polarizable residues (Glu and Asp) sitting on the same side of the head group (Figure 3). This work also suggested that the flexible tail folds back to interact with the peptide ring which was also found in molecular dynamics simulations [114]. While the Glu and Asp residues being on the same size of the ring suggests that these residues would anchor the molecule at an interface, molecular dynamics simulations show that compact structure caused by the folding of the tail leads to significant tumbling motion with the hydrophilic side chains spending significant time pointing away from the water [115]. This suggests, at low densities, surfactin may act more as (roughly) spherical nanoparticles than traditional surfactants. The mechanism for the decrease in interfacial tension and stabilisation of interfaces by surfactin may then be similar to that in emulsions stabilised by nanoparticles or colloids (Pickering emulsions) [116]. As the density increases, surfactin adopts a more traditional surfactant-like structure at interfaces [117,118]. The plane of the peptide ring lies in the plane of the interface. The interfacial behaviour of surfactin depends on the pH of the aqueous phase, with greater conformational freedom being found for low pH due to the decreased hydrophilicity of the Glu and Asp residues when these become protonated [118]. This can also been seen when surfactin derivatives with methyl ester groups added to the Glu and Asp residues [119].
Due to its amphiphilic nature, surfactin self-assembles into a number of different structures in solution. Experimental investigations have shown that this depends on solution pH and ionic strength. Early cryo-EM work showed the formation of spherical micelles with diameters 5–9 nm and ellipsoidal micelles with typical lengths and widths of 19 nm and 11 nm [120] at pH = 7. At higher pH (9.5), cylindrical micelles were found, with which the addition of electrolytes turned back into spherical micelles. For very high pH (∼12), the lactone bond closing the peptide ring was broken and the head becomes linear. Later, EM [121] and SANS [122] work found at neutral pH smaller spherical micelles, with diameters ∼5 nm. Molecular dynamics simulations found micelles of around 4 nm diameter for neutral surfactin molecules [118,123]; the discrepancy between this and experimental measurements may be attributed to differences in the definition of micelle sizes between simulation and experiment. For the simulations, it should be noted that the sizes of micelles found were limited by the numbers of molecules in the simulated systems. A more rigorous investigation of micellization of surfactin may require systems containing more molecules, ideally enough to form several micelles [124]. Nonetheless, these simulations were able to investigate the internal structure of the micelles, finding stratified structure with the hydrophobic tails, leucine side chains, peptide ring, and charged side chains forming distinct layers. This differs from SANS results [122], which placed the hydrophobic tails and leucine side chains together in the micelle core. When the Glu and Asp side chains are deprotonated, the sizes of the aggregates decrease due to the increase in electrostatic repulsion between the head groups [118]. Experimental investigations of surfactin micelles also indicate the formation of -sheets, which has not been observed in simulations. The timescales associated with secondary structure change in proteins can be significant compared to simulation times (the longest simulations above consisted of 250 ns [118]). This may be especially true for cyclic peptides, such as surfactin for which the conformations may be more restricted.
While most attention has been paid to surfactin, other lipopeptides have attracted attention. In recent work, molecular simulation, using both atomistic and coarse-grained models, have been used to investigate the behaviour of a similar lipopeptide from mycosubtilin [125]. Due to the structural similarity to surfactin, its conformational behaviour, both in bulk and at the air–water interface, from atomistic simulations is similar. Using the Martini model, the aggregation of this lipopeptide at the air–water interface was investigated. It was found that this aggregates rapidly, forming half micelles at the interface, suggesting that it is unable to form a stable Langmuir monolayer. As many lipopeptides have cyclic peptide heads, a few studies have investigated the structure [126] and self-assembly [127] of cyclic peptides at interfaces.
5. Ranaspumin
While hydrophobins and lipopeptides, such as surfactin, have structures that, at least on first impression, are amphiphilic, surfactant proteins that lack this surface heterogeneity are possible. One such example is the protein Rsn-2 [128,129], which is found in the foam nests of the tropical frog Engystomops pustulosus. This is the main surfactant component of the foam, which is a mixture of proteins and polysaccharides that is used to hydrate frog eggs. Solution NMR revealed that this protein consists of an -helix and -sheet connected by a flexible linker and has a largely polar surface (Figure 4a). Neutron reflectivity and IRRAS measurements show that it retains its secondary structure, but the protein thickness halves at the air–water interface [129]. This suggests a novel interfacial conformational change where the protein opens, with the -helix and -strand separating and lying in the plane of the interface (Figure 4b).
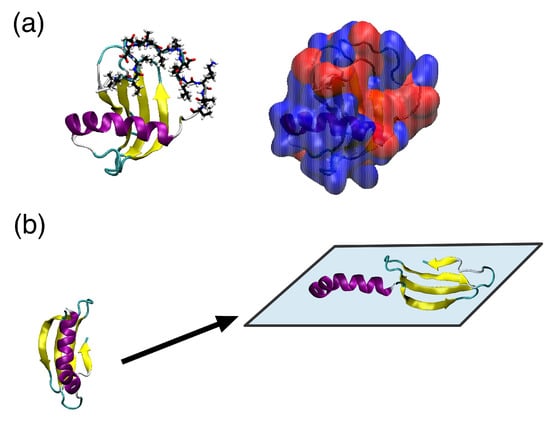
Figure 4.
(a) structure of Rsn-2 with in cartoon (left) and surface (right) representations. In cartoon representation, the hydrophobic N-terminus loop is highlighted. On the surface representation, blue and red denote hydrophilic and hydrophobic regions, created using PDB code 2WGO; (b) illustration of Rsn-2 opening transition during interfacial adsorption.
The largely polar surface of Rsn-2 inhibits aggregation in solution but provides little driving force for interfacial adsorption. Atomistic molecular dynamics simulations [130] show that Rsn-2 adsorbs onto the air–water interface through its flexible N-terminus, which contains most of the solvent exposed hydrophobic residues. This is similar to the fly-casting mechanism often used in protein recognition [131]. While contact between this region and the interface is necessary, for permanent adsorption, the protein needs to be in specific orientations, possibly to ensure that the subsequent opening transition is possible. The importance of the N-terminus in adsorption was found by studying Rsn-2 mutants with the flexible tails deleted [130]. No adsorption, within the simulation timescales, was found when this region was completely deleted, while deleting only the first three residues of the tail is sufficient to partially inhibit adsorption. This is consistent with dynamic surface tension measurements where the decay in surface tension was significantly slower for the the mutant without the N-terminal tail. Deletion of the hydrophilic C-terminus was also found to affect adsorption, both in simulation and experiment, possibly due to this region playing a role in orienting the protein at the interface. Adsorption onto the water–cyclohexane proceeds in a similar manner, but, unlike at the air–water interface, the opening transition is observed within the simulations. The opening of the protein may be enhanced in this case as oil molecules can partition into the interior of the protein weakening the hydrophobic interactions that hold the solution structure together. Similar enhancement of protein conformational change at interface due to penetration of oil molecules into a protein core has been observed for -lactalbumin [37]. This is contrary to what are normally observed globular proteins where adsorption onto higher surface tension interfaces [132], in this case the air–water interface, gives rise to greater conformational changes, showing that the specific role of the hydrophobic component should be carefully considered.
Due to the long timescale associated with the unhinging transition, atomistic modelling is unable to rigorously investigate this. To circumvent this CG modelling, using a Go-type model [133] (one bead per residue) was used [134]. Consistent with the atomistic simulations, this showed that the opening transition occurs in two steps, with adsorption onto the interface preceding conformational change. Comparison between the opening transition at the interface and unfolding transition in solution suggests that the unhinging of the protein corresponds to the initial stages of unfolding, with complete denaturation of the protein inhibited by the interface [135].
While Rsn-2 is the main biosurfactant protein component in the foam, foams prepared only using it have significantly shorter lifetimes than those prepared using the full foam mixture [16]. This suggests that other components, either protein, lipid, or polysaccharide, also play a role in foam stabilisation. Understanding this synergistic effect is key to a complete picture of how the frog foam nest is stabilised, so future work may investigate the interfacial behaviour of the different components and mixtures of these.
6. Conclusions
Over the past few decades, molecular dynamics simulation has emerged as a powerful tool for the investigation of biomolecular systems. Due to its ability to give direct insight into structure and dynamics on a molecular level, it is particularly useful for the investigation of interfacial and surface phenomena, which are otherwise difficult to investigate. As these play a key role in the biological functions and potential applications of protein biosurfactants, MD simulation has, as shown in this review, been of significant utility for investigating these proteins. In particular, MD simulation has revealed molecular features that govern interfacial behaviour of protein biosurfactants, such as how the structure of the hydrophobic patches of hydrophobins affects their interfacial behaviour [93,96,99] and how changes in protein conformation relate to interfacial aggregation [88] and function [91,134].
Despite the success of MD simulation, they face a number of limitations. Possibly the largest outstanding question is the transferability of models used for protein simulations to interfacial and surface environments. These are typically developed for proteins in aqueous solution and so are tuned to reproduce protein structural properties there. Of the commonly used protein force fields, only the Gromos force fields are parameterized against properties (free energies of solvation) that may be related to interfacial behaviour. While transferability to other environments is often assumed, this is generally untested. For fluid interfaces, this is in part due to the lack of experimental information on protein behaviour there. For solid surfaces, adsorption free energies of peptides have been determined and used to test commonly used force fields [136], which has shown that some common protein force can reproduce experimental adsorption free energies. Agreement with experiments can be improved through tuning force fields specifically for interfacial environments [137]. Doing this for other environments is possible but needs accurate experimental information for parameterization and validation. The question of transferability is more acute for CG models. The reduction in resolution of these models means that many effects, such as ordering of water molecules, that are explicit in atomistic models are only accounted for implicitly. This can affect the transferability of CG models to different state points, let alone different environments, so the use of CG models for proteins at surfaces and interfaces should be evaluated carefully.
Other significant limitations include the limited system sizes and time scales that can be investigated. The limitations on system size have largely restricted atomistic MD to the investigation of single proteins at interfaces and surfaces and simulations of aggregation and assembly have only been performed for smaller peptides, such as surfactin. As many protein aspects of protein behaviour at interfaces and surfaces, such as the formation of interfacial structures, involve multiple proteins this has limited the ability of simulation to give insight into these. Restriction to relatively short time scales has limited the investigation of conformational changes of proteins. With increases in computer power, such as the use of general purpose-GPUs [138], and developments in simulation methodologies, the investigation of larger systems over longer times will become possible. Also the coupling of MD simulation with methods, such as finite element analysis, which can access these larger length and time scales should be investigated.
There are a number of avenues in which simulation of protein biosurfactants can proceed in the next few years. As structures of further protein biosurfactants are determined, we are finding more novel mechanisms for interfacial activity. One interesting example of this is the horse sweat protein latherin [139], which undergoes an unzippering conformational change at interfaces [140]. It may be speculated that similar proteins, such as the PLUNC proteins [141,142,143], may show similar behaviour and that other unforeseen mechanisms may be found in other proteins. These unusual changes in conformation may be exploited in applications or inspire the design of synthetic molecules that also undergo conformational changes at interfaces [144,145,146].
Closer connection between simulation and experiment and applications is also a key avenue for future research. As mentioned above, most prior work has focused on isolated proteins at interfaces and surfaces, aiming at investigating the underlying biophysics of adsorption. For understanding applications of protein biosurfactants, knowledge of the behaviour of protein aggregates is necessary. While simulations of peptide aggregation on interfaces [147,148,149] and membranes [150,151] have been performed, for larger proteins the time scales involved in their aggregation, particularly taking changes in conformation that often accompany this, are still prohibitive. To investigate larger systems CG models, with caveats described above, or coupling MD with continuum approaches, would provide a feasible route. For proteins, even in bulk solution, such methods are still in their infancy [152] but provide an exciting possibility for extrapolating from the molecular to materials scale.
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
Funding
This work was funded through a NUI Galway College of Science Scholarship awarded to S.S.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| CG | Coarse-Grain |
| DPPC | 1,2-dipalmitoyl-sn-glycero-3-phosphocholine |
| EM | Electron Microscopy |
| GPU | Graphics Processing Unit |
| IRRAS | Infra-Red Reflection Adsorption Spectropscopy |
| MD | Molecular Dynamics |
| MM-PBSA | Molecular Mechanics Poisson–Boltzmann Solvation Approximation |
| NMR | Nuclear Magnetic Resonance |
| OPLS | Optimised Potentials for Liquid Simulation |
| PDB | Protein Data Bank |
| PLUNC | Palate Lung Nasal Epithelial Clone |
| SAM | Self-Assembled Monolayer |
| SANS | Small Angle Neutron Scattering |
| VMD | Visual Molecular Dynamics |
References
- Ron, E.Z.; Rosenberg, E. Natural roles of biosurfactants. Environ. Microbiol. 2001, 3, 229–236. [Google Scholar] [CrossRef] [PubMed]
- Raaijmakers, J.M.; De Bruijn, I.; Nybroe, O.; Ongena, M. Natural functions of lipopeptides from Bacillus and Pseudomonas: More than surfactants and antibiotics. FEMS Microbiol. Rev. 2010, 34, 1037–1062. [Google Scholar] [CrossRef] [PubMed]
- Wösten, H.A.; Van Wetter, M.A.; Lugones, L.G.; Van der Mei, H.C.; Busscher, H.J.; Wessels, J.G. How a fungus escapes the water to grow into the air. Curr. Biol. 1999, 9, 85–88. [Google Scholar] [CrossRef]
- Elliot, M.A.; Talbot, N.J. Building filaments in the air: Aerial morphogenesis in bacteria and fungi. Curr. Opin. Microbiol. 2004, 7, 594–601. [Google Scholar] [CrossRef] [PubMed]
- Arnaouteli, S.; MacPhee, C.E.; Stanley-Wall, N.R. Just in case it rains: Building a hydrophobic biofilm the Bacillus subtilis way. Curr. Opin. Microbiol. 2016, 34, 7–12. [Google Scholar] [CrossRef] [PubMed]
- Wösten, H.A.; Schuren, F.H.; Wessels, J.G. Interfacial self-assembly of a hydrophobin into an amphipathic protein membrane mediates fungal attachment to hydrophobic surfaces. EMBO J. 1994, 13, 5848–5854. [Google Scholar] [CrossRef] [PubMed]
- Cameotra, S.S.; Makkar, R.S. Recent applications of biosurfactants as biological and immunological molecules. Curr. Opin. Microbiol. 2004, 7, 262–266. [Google Scholar] [CrossRef] [PubMed]
- Aimanianda, V.; Bayry, J.; Bozza, S.; Kniemeyer, O.; Perruccio, K.; Elluru, S.R.; Clavaud, C.; Paris, S.; Brakhage, A.A.; Kaveri, S.V.; et al. Surface hydrophobin prevents immune recognition of airborne fungal spores. Nature 2009, 460, 1117–1121. [Google Scholar] [CrossRef] [PubMed]
- Kuchta, K.; Chi, L.; Fuchs, H.; Pötter, M.; Steinbüchel, A. Studies on the influence of phasins on accumulation and degradation of PHB and nanostructure of PHB granules in Raistonia eutropha H16. Biomacromolecules 2007, 8, 657–662. [Google Scholar] [CrossRef] [PubMed]
- Schor, M.; Reid, J.L.; MacPhee, C.E.; Stanley-Wall, N.R. The Diverse Structures and Functions of Surfactant Proteins. Trends Biochem. Sci. 2016, 41, 610–620. [Google Scholar] [CrossRef] [PubMed]
- Sunde, M.; Pham, C.L.; Kwan, A.H. Molecular Characteristics and Biological Functions of Surface-Active and Surfactant Proteins. Annu. Rev. Biochem. 2017, 86, 585–608. [Google Scholar] [CrossRef] [PubMed]
- Yano, Y.F. Kinetics of protein unfolding at interfaces. J. Phys. Condens. Matter 2012, 24, 503101. [Google Scholar] [CrossRef] [PubMed]
- Zhai, J.L.; Day, L.; Aguilar, M.I.; Wooster, T.J. Protein folding at emulsion oil/water interfaces. Curr. Opin. Colloid Interface Sci. 2013, 18, 257–271. [Google Scholar] [CrossRef]
- Cooper, A.; Kennedy, M.W. Biofoams and natural protein surfactants. Biophys. Chem. 2010, 151, 96–104. [Google Scholar] [CrossRef] [PubMed]
- Wösten, H.A.; Scholtmeijer, K. Applications of hydrophobins: Current state and perspectives. Appl. Microbiol. Biotechnol. 2015, 99, 1587–1597. [Google Scholar] [CrossRef] [PubMed]
- Cooper, A.; Vance, S.J.; Smith, B.O.; Kennedy, M.W. Frog foams and natural protein surfactants. Colloids Surf. A Physicochem. Eng. Asp. 2017, 534, 120–129. [Google Scholar] [CrossRef] [PubMed]
- Tchuenbou-Magaia, F.; Norton, I.; Cox, P. Hydrophobins stabilised air-filled emulsions for the food industry. Food Hydrocoll. 2009, 23, 1877–1885. [Google Scholar] [CrossRef]
- Cox, A.R.; Aldred, D.L.; Russell, A.B. Exceptional stability of food foams using class II hydrophobin HFBII. Food Hydrocoll. 2009, 23, 366–376. [Google Scholar] [CrossRef]
- Nishinari, K.; Fang, Y.; Guo, S.; Phillips, G.O. Soy proteins: A review on composition, aggregation and emulsification. Food Hydrocoll. 2014, 39, 301–318. [Google Scholar] [CrossRef]
- Dickinson, E. Structure formation in casein-based gels, foams, and emulsions. Colloids Surf. A Physicochem. Eng. Asp. 2006, 288, 3–11. [Google Scholar] [CrossRef]
- Valo, H.K.; Laaksonen, P.H.; Peltonen, L.J.; Linder, M.B.; Hirvonen, J.T.; Laaksonen, T.J. Multifunctional hydrophobin: Toward functional coatings for drug nanoparticles. ACS Nano 2010, 4, 1750–1758. [Google Scholar] [CrossRef] [PubMed]
- Kimpel, F.; Schmitt, J.J. Review: Milk Proteins as Nanocarrier Systems for Hydrophobic Nutraceuticals. J. Food Sci. 2015, 80, R2361–R2366. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.J.; Ebersbacher, C.F.; Myung, N.V.; Montemagno, C.D. Synthesis of nanoparticles with frog foam nest proteins. J. Nanoparticle Res. 2012, 14, 1–13. [Google Scholar] [CrossRef]
- Santhiya, D.; Burghard, Z.; Greiner, C.; Jeurgens, L.P.H.; Subkowski, T.; Bill, J. Bioinspired deposition of TiO2 thin films induced by hydrophobins. Langmuir 2010, 26, 6494–6502. [Google Scholar] [CrossRef] [PubMed]
- Maity, J.P.; Lin, T.J.; Cheng, H.P.H.; Chen, C.Y.; Reddy, A.S.; Atla, S.B.; Chang, Y.F.; Chen, H.R.; Chen, C.C. Synthesis of brushite particles in reverse microemulsions of the biosurfactant surfactin. Int. J. Mol. Sci. 2011, 12, 3821–3830. [Google Scholar] [CrossRef] [PubMed]
- Whang, L.M.; Liu, P.W.G.; Ma, C.C.; Cheng, S.S. Application of biosurfactants, rhamnolipid, and surfactin, for enhanced biodegradation of diesel-contaminated water and soil. J. Hazard. Mater. 2008, 151, 155–163. [Google Scholar] [CrossRef] [PubMed]
- Laaksonen, P.; Walther, A.; Malho, J.M.; Kainlauri, M.; Ikkala, O.; Linder, M.B. Genetic engineering of biomimetic nanocomposites: Diblock proteins, graphene, and nanofibrillated cellulose. Angew. Chem. Int. Ed. 2011, 50, 8688–8691. [Google Scholar] [CrossRef] [PubMed]
- Kaur, J.; Gravagnuolo, A.M.; Maddalena, P.; Altucci, C.; Giardina, P.; Gesuele, F. Green synthesis of luminescent and defect-free bio-nanosheets of MoS2: Interfacing two-dimensional crystals with hydrophobins. RSC Adv. 2017, 7, 22400–22408. [Google Scholar] [CrossRef]
- Dror, R.O.; Dirks, R.M.; Grossman, J.; Xu, H.; Shaw, D.E. Biomolecular Simulation: A Computational Microscope for Molecular Biology. Annu. Rev. Biophys. 2012, 41, 429–452. [Google Scholar] [CrossRef] [PubMed]
- McCammon, J.A.; Gelin, B.R.; Karplus, M. Dynamics of folded proteins. Nature 1977, 267, 585–590. [Google Scholar] [CrossRef] [PubMed]
- Zhao, G.; Perilla, J.R.; Yufenyuy, E.L.; Meng, X.; Chen, B.; Ning, J.; Ahn, J.; Gronenborn, A.M.; Schulten, K.; Aiken, C.; et al. Mature HIV-1 capsid structure by cryo-electron microscopy and all-atom molecular dynamics. Nature 2013, 497, 643–646. [Google Scholar] [CrossRef] [PubMed]
- Yu, I.; Mori, T.; Ando, T.; Harada, R.; Jung, J.; Sugita, Y.; Feig, M. Biomolecular interactions modulate macromolecular structure and dynamics in atomistic model of a bacterial cytoplasm. eLife 2016, 5, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Ozboyaci, M.; Kokh, D.B.; Corni, S.; Wade, R.C. Modeling and simulation of protein-surface interactions: Achievements and challenges. Q. Rev. Biophys. 2016, 49, 1–45. [Google Scholar] [CrossRef] [PubMed]
- James, J.J.; Lakshmi, B.S.; Seshasayee, A.S.N.; Gautam, P. Activation of Candida rugosa lipase at alkane-aqueous interfaces: A molecular dynamics study. FEBS Lett. 2007, 581, 4377–4383. [Google Scholar] [CrossRef] [PubMed]
- Kubiak, K.; Mulheran, P.A. Molecular dynamics simulations of hen egg white lysozyme adsorption at a charged solid surface. J. Phys. Chem. B 2009, 113, 12189–12200. [Google Scholar] [CrossRef] [PubMed]
- Arooj, M.; Gandhi, N.S.; Kreck, C.A.; Arrigan, D.W.; Mancera, R.L. Adsorption and Unfolding of Lysozyme at a Polarized Aqueous-Organic Liquid Interface. J. Phys. Chem. B 2016, 120, 3100–3112. [Google Scholar] [CrossRef] [PubMed]
- Cheung, D.L. Adsorption and conformations of lysozyme and α-lactalbumin at a water–octane interface. J. Chem. Phys. 2017, 147, 195101. [Google Scholar] [CrossRef] [PubMed]
- Zare, D.; McGrath, K.M.; Allison, J.R. Deciphering β-Lactoglobulin Interactions at an Oil–Water Interface: A Molecular Dynamics Study. Biomacromolecules 2015, 16, 1855–1861. [Google Scholar] [CrossRef] [PubMed]
- Zare, D.; Allison, J.R.; McGrath, K.M. Molecular Dynamics Simulation of Beta-Lactoglobulin at Different Oil/Water Interfaces. Biomacromolecules 2016, 17, 1572–1581. [Google Scholar] [CrossRef] [PubMed]
- Bellucci, L.; Bussi, G.; Di Felice, R.; Corni, S. Fibrillation-prone conformations of the amyloid-β-42 peptide at the gold/water interface. Nanoscale 2017, 9, 2279–2290. [Google Scholar] [CrossRef] [PubMed]
- Frenkel, D.; Smit, B. Understanding Molecular Simulation: From Algorithms To Applications, 2nd ed.; Academic Press: San Diego, CA, USA, 2002. [Google Scholar]
- Allen, M.P.; Tildesley, D.J. Computer Simulation of Liquids, 2nd ed.; Oxford University Press: Oxford, UK, 2017. [Google Scholar]
- Mackerell, A.D. Empirical force fields for biological macromolecules: Overview and issues. J. Comput. Chem. 2004, 25, 1584–1604. [Google Scholar] [CrossRef] [PubMed]
- Duan, Y.; Wu, C.; Chowdhury, S.; Lee, M.C.; Xiong, G.; Zhang, W.; Yang, R.; Cieplak, P.; Luo, R.; Lee, T.; et al. A Point-Charge Force Field for Molecular Mechanics Simulations of Proteins Based on Condensed-Phase Quantum Mechanical Calculations. J. Comput. Chem. 2003, 24, 1999–2012. [Google Scholar] [CrossRef] [PubMed]
- Foloppe, N.; MacKerell, A.D.J. All-Atom Empirical Force Field for Nucleic Acids: I. Parameter Optimization Based on Small Molecule and Condensed Phase Macromolecular Target Data. J. Comput. Chem. 2000, 21, 86–104. [Google Scholar] [CrossRef]
- Schmid, N.; Eichenberger, A.P.; Choutko, A.; Riniker, S.; Winger, M.; Mark, A.E.; Van Gunsteren, W.F. Definition and testing of the GROMOS force-field versions 54A7 and 54B7. Eur. Biophys. J. 2011, 40, 843–856. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, W.L.; Maxwell, D.S.; Tirado-Rives, J. Development and Testing of the OPLS All-Atom Force Field on Conformational Energetics and Properties of Organic Liquids. J. Am. Chem. Soc. 1996, 118, 11225–11236. [Google Scholar] [CrossRef]
- Marrink, S.J.; Risselada, H.J.; Yefimov, S.; Tieleman, D.P.; de Vries, A.H. The MARTINI force field: Coarse grained model for biomolecular simulations. J. Phys. Chem. B 2007, 111, 7812–7824. [Google Scholar] [CrossRef] [PubMed]
- Marrink, S.J.; Tieleman, D.P. Perspective on the martini model. Chem. Soc. Rev. 2013, 42, 6801–6822. [Google Scholar] [CrossRef] [PubMed]
- Monticelli, L.; Kandasamy, S.K.; Periole, X.; Larson, R.G.; Tieleman, D.P.; Marrink, S.J. The MARTINI Coarse-Grained Force Field: Extension to Proteins. J. Chem. Theory Comput. 2008, 4, 819–834. [Google Scholar] [CrossRef] [PubMed]
- Periole, X. Interplay of G protein-coupled receptors with the membrane: Insights from supra-atomic coarse grain molecular dynamics simulations. Chem. Rev. 2017, 117, 156–185. [Google Scholar] [CrossRef] [PubMed]
- Euston, S.R. Molecular dynamics simulation of protein adsorption at fluid interfaces: A comparison of all-atom and coarse-grained models. Biomacromolecules 2010, 11, 2781–2787. [Google Scholar] [CrossRef] [PubMed]
- Shinoda, W.; DeVane, R.; Klein, M.L. Multi-property fitting and parameterization of a coarse grained model for aqueous surfactants. Mol. Simul. 2007, 33, 27–36. [Google Scholar] [CrossRef]
- DeVane, R.; Shinoda, W.; Moore, P.B.; Klein, M.L. Transferable Coarse Grain Nonbonded Interaction Model for Amino Acids. J. Chem. Theory Comput. 2009, 5, 2115–2124. [Google Scholar] [CrossRef] [PubMed]
- Sugita, Y.; Okamoto, Y. Replica exchange molecular dynamics method for protein folding simulation. Chem. Phys. Lett. 1999, 314, 141–151. [Google Scholar] [CrossRef]
- Earl, D.J.; Deem, M.W. Parallel tempering: Theory, applications, and new perspectives. Phys. Chem. Chem. Phys. 2005, 7, 3910–3916. [Google Scholar] [CrossRef] [PubMed]
- Cheung, D.L. Conformations of Myoglobin-Derived Peptides at the Air–Water Interface. Langmuir 2016, 32, 4405–4414. [Google Scholar] [CrossRef] [PubMed]
- Liao, C.; Xie, Y.; Zhou, J. Computer simulations of fibronectin adsorption on hydroxyapatite surfaces. RSC Adv. 2014, 4, 15759–15769. [Google Scholar] [CrossRef]
- Laio, A.; Parrinello, M. Escaping free-energy minima. Proc. Natl. Acad. Sci. USA 2002, 99, 12562–12566. [Google Scholar] [CrossRef] [PubMed]
- Laio, A.; Gervasio, F.L. Metadynamics: A method to simulate rare events and reconstruct the free energy in biophysics, chemistry and material science. Rep. Prog. Phys. 2008, 71, 126601. [Google Scholar] [CrossRef]
- Piana, S.; Laio, A. A Bias-Exchange Approach to Protein Folding. J. Phys. Chem. B 2007, 111, 4553–4559. [Google Scholar] [CrossRef] [PubMed]
- Barducci, A.; Bussi, G.; Parrinello, M. Well-Tempered Metadynamics: A Smoothly Converging and Tunable Free-Energy Method. Phys. Rev. Lett. 2008, 100, 020603/1–4. [Google Scholar] [CrossRef] [PubMed]
- Schneider, J.; Colombi Ciacchi, L. Specific material recognition by small peptides mediated by the interfacial solvent structure. J. Am. Chem. Soc. 2012, 134, 2407–2413. [Google Scholar] [CrossRef] [PubMed]
- Deighan, M.; Pfaendtner, J. Exhaustively Sampling Peptide Adsorption with Metadynamics. Langmuir 2013, 29, 7999–8009. [Google Scholar] [CrossRef] [PubMed]
- Levine, Z.A.; Fischer, S.A.; Shea, J.E.; Pfaendtner, J. Trp-Cage Folding on Organic Surfaces. J. Phys. Chem. B 2015, 119, 10417–10425. [Google Scholar] [CrossRef] [PubMed]
- Wright, L.B.; Palafox-Hernandez, J.P.; Rodger, P.M.; Corni, S.; Walsh, T.R. Facet selectivity in gold binding peptides: Exploiting interfacial water structure. Chem. Sci. 2015, 6, 5204–5214. [Google Scholar] [CrossRef] [PubMed]
- Huai Han, M.; Cheng Chiu, C. Fast estimation of protein conformational preference at air/water interface via molecular dynamics simulations. J. Taiwan Inst. Chem. Eng. 2018. [Google Scholar] [CrossRef]
- Torrie, G.M.; Valleau, J.P. Nonphysical sampling distributions in Monte Carlo free-energy estimation: Umbrella sampling. J. Comput. Phys. 1977, 23, 187–199. [Google Scholar] [CrossRef]
- Park, S.; Schulten, K. Calculating potentials of mean force from steered molecular dynamics simulations. J. Chem. Phys. 2004, 120, 5946–5961. [Google Scholar] [CrossRef] [PubMed]
- Lai, Z.B.; Wang, M.; Yan, C.; Oloyede, A. Molecular dynamics simulation of mechanical behavior of osteopontin-hydroxyapatite interfaces. J. Mech. Behav. Biomed. Mater. 2014, 36, 12–30. [Google Scholar] [CrossRef] [PubMed]
- Miller, C.A.; Abbott, N.L.; Pablo, J.J.D.; Miller, C.A.; Abbott, N.L.; Pablo, J.J.D. Surface Activity of Amphiphilic Helical beta-Peptides from Molecular Dynamics Simulation Surface Activity of Amphiphilic Helical-Peptides from Molecular Dynamics Simulation. Langmuir 2009, 25, 2811–2823. [Google Scholar] [CrossRef] [PubMed]
- Engin, O.; Villa, A.; Sayar, M.; Hess, B. Driving forces for adsorption of amphiphilic peptides to the air–water interface. J. Phys. Chem. B 2010, 114, 11093–11101. [Google Scholar] [CrossRef] [PubMed]
- Engin, O.; Sayar, M. Adsorption, folding, and packing of an amphiphilic peptide at the air/water interface. J. Phys. Chem. B 2012, 116, 2198–2207. [Google Scholar] [CrossRef] [PubMed]
- Linder, M.B. Hydrophobins: Proteins that self assemble at interfaces. Curr. Opin. Colloid Interface Sci. 2009, 14, 356–363. [Google Scholar] [CrossRef]
- Linder, M.B.; Szilvay, G.R.; Nakari-Setälä, T.; Penttilä, M.E. Hydrophobins: The protein-amphiphiles of filamentous fungi. FEMS Microbiol. Rev. 2005, 29, 877–896. [Google Scholar] [CrossRef] [PubMed]
- Temple, B.; Horgen, P.A.; Bernier, L.; Hintz, W.E. Cerato-ulmin, a hydrophobin secreted by the causal agents of Dutch elm disease, is a parasitic fitness factor. Fungal Genet. Biol. 1997, 22, 39–53. [Google Scholar] [CrossRef] [PubMed]
- Linder, M.; Szilvay, G.R.; Nakari-Setälä, T.; Söderlund, H.; Penttilä, M. Surface adhesion of fusion proteins containing the hydrophobins HFBI and HFBII from Trichoderma reesei. Protein Sci. 2002, 11, 2257–2266. [Google Scholar] [CrossRef] [PubMed]
- Hobley, L.; Ostrowski, A.; Rao, F.V.; Bromley, K.M.; Porter, M.; Prescott, A.R.; MacPhee, C.E.; van Aalten, D.M.F.; Stanley-Wall, N.R. BslA is a self-assembling bacterial hydrophobin that coats the Bacillus subtilis biofilm. Proc. Natl. Acad. Sci. USA 2013, 110, 13600–13605. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Zangi, R.; de Vocht, M.L.; Robillard, G.T.; Mark, A.E. Molecular dynamics study of the folding of hydrophobin SC3 at a hydrophilic/hydrophobic interface. Biophys. J. 2002, 83, 112–124. [Google Scholar] [CrossRef]
- Euston, S.R. Molecular simulation of adsorption of hydrophobin HFBI to the air–water, DPPC-water and decane-water interfaces. Food Hydrocoll. 2013, 42, 66–74. [Google Scholar] [CrossRef]
- Zhao, Y.; Cieplak, M. Proteins at air-Water and oil-water interfaces in an all-atom model. Phys. Chem. Chem. Phys. 2017, 19, 25197–25206. [Google Scholar] [CrossRef] [PubMed]
- Raffaini, G.; Milani, R.; Ganazzoli, F.; Resnati, G.; Metrangolo, P. Atomistic simulation of hydrophobin HFBII conformation in aqueous and fluorous media and at the water/vacuum interface. J. Mol. Graph. Model. 2016, 63, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Fan, H.; Wang, X.; Zhu, J.; Robillard, G.T.; Mark, A.E. Molecular dynamics simulations of the hydrophobin SC3 at a hydrophobic/hydrophilic interface. Proteins 2006, 64, 863–873. [Google Scholar] [CrossRef] [PubMed]
- Askolin, S.; Linder, M.; Scholtmeijer, K.; Tenkanen, M.; Penttilä, M.; de Vocht, M.L.; Wösten, H.A.B. Interaction and comparison of a class I hydrophobin from Schizophyllum commune and class II hydrophobins from Trichoderma reesei. Biomacromolecules 2006, 7, 1295–1301. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.L.; Penfold, J.; Thomas, R.K.; Tucker, I.M.; Petkov, J.T.; Bent, J.; Cox, A.; Campbell, R.A. Adsorption behavior of hydrophobin and hydrophobin/surfactant mixtures at the air–water interface. Langmuir 2011, 27, 11316–11323. [Google Scholar] [CrossRef] [PubMed]
- Kwan, A.H.Y.; Winefield, R.D.; Sunde, M.; Matthews, J.M.; Haverkamp, R.G.; Templeton, M.D.; Mackay, J.P. Structural basis for rodlet assembly in fungal hydrophobins. Proc. Natl. Acad. Sci. USA 2006, 103, 3621–3626. [Google Scholar] [CrossRef] [PubMed]
- De Simone, A.; Kitchen, C.; Kwan, A.H.; Sunde, M.; Dobson, C.M.; Frenkel, D. Intrinsic disorder modulates protein self-assembly and aggregation. Proc. Natl. Acad. Sci. USA 2012, 109, 6951–6956. [Google Scholar] [CrossRef] [PubMed]
- Jean, L.; Lee, C.F.; Vaux, D.J. Enrichment of amyloidogenesis at an air–water interface. Biophys. J. 2012, 102, 1154–1162. [Google Scholar] [CrossRef] [PubMed]
- Campioni, S.; Carret, G.; Jordens, S.; Nicoud, L.; Mezzenga, R.; Riek, R. The Presence of an Air-Water Interface Affects Formation and Elongation of α-Synuclein Fibrils. J. Am. Chem. Soc. 2013, 136, 2866–2875. [Google Scholar] [CrossRef] [PubMed]
- Schulz, A.; Fioroni, M.; Linder, M.B.; Nessel, A.; Bocola, M.; Subkowski, T.; Schwaneberg, U.; Böker, A.; Rodríguez-Ropero, F. Exploring the mineralization of hydrophobins at a liquid interface. Soft Matter 2012, 8, 11343–11352. [Google Scholar] [CrossRef]
- Schulz, A.; Liebeck, B.M.; John, D.; Heiss, A.; Subkowski, T.; Böker, A. Protein—Mineral hybrid capsules from emulsions stabilized with an amphiphilic protein. J. Mater. Chem. 2011, 21, 9731–9736. [Google Scholar] [CrossRef]
- Cheung, D.L. Molecular simulation of hydrophobin adsorption at an oil-water interface. Langmuir 2012, 28, 8730–8736. [Google Scholar] [CrossRef] [PubMed]
- Du, K.; Glogowski, E.; Emrick, T.; Russell, T.P.; Dinsmore, A.D. Adsorption Energy of Nano- and Microparticles at Liquid-Liquid Interfaces. Langmuir 2010, 26, 12518–12522. [Google Scholar] [CrossRef] [PubMed]
- Cheung, D.L.; Carbone, P. How stable are amphiphilic dendrimers at the liquid–liquid interface? Soft Matter 2013, 9, 6841–6850. [Google Scholar] [CrossRef]
- Bromley, K.M.; Morris, R.J.; Hobley, L.; Brandani, G.; Gillespie, R.M.C.; McCluskey, M.; Zachariae, U.; Marenduzzo, D.; Stanley-Wall, N.R.; MacPhee, C.E. Interfacial self-assembly of a bacterial hydrophobin. Proc. Natl. Acad. Sci. USA 2015, 112, 5419–5424. [Google Scholar] [CrossRef] [PubMed]
- Cheung, D.L.; Bon, S.A.F. Stability of Janus nanoparticles at fluid interfaces. Soft Matter 2009, 5, 3969–3976. [Google Scholar] [CrossRef]
- Brandani, G.B.; Schor, M.; Morris, R.; Stanley-Wall, N.; MacPhee, C.E.; Marenduzzo, D.; Zachariae, U. The Bacterial Hydrophobin BslA is a Switchable Ellipsoidal Janus Nanocolloid. Langmuir 2015, 31, 11558–11563. [Google Scholar] [CrossRef] [PubMed]
- Moldovan, C.; Thompson, D. Molecular dynamics of the “hydrophobic patch” that immobilizes hydrophobin protein HFBII on silicon. J. Mol. Model. 2011, 17, 2227–2235. [Google Scholar] [CrossRef] [PubMed]
- Mereghetti, P.; Wade, R.C. Diffusion of hydrophobin proteins in solution and interactions with a graphite surface. BMC Biophys. 2011, 4, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Szilvay, G.R.; Kisko, K.; Serimaa, R.; Linder, M.B. The relation between solution association and surface activity of the hydrophobin HFBI from Trichoderma reesei. FEBS Lett. 2007, 581, 2721–2726. [Google Scholar] [CrossRef] [PubMed]
- Mrksich, M.; Whitesides, G.M. Using Self-Assembled Monolayers to Understand the Interactions of Man-made Surfaces with Proteins and Cells. Annu. Rev. Biophys. Biomol. Struct. 2003, 25, 55–78. [Google Scholar] [CrossRef] [PubMed]
- O’Mahony, S.; O’Dwyer, C.; Nijhuis, C.A.; Greer, J.C.; Quinn, A.J.; Thompson, D. Nanoscale dynamics and protein adhesivity of alkylamine self-assembled monolayers on graphene. Langmuir 2013, 29, 7271–7282. [Google Scholar] [CrossRef] [PubMed]
- Peng, C.; Liu, J.; Zhao, D.; Zhou, J. Adsorption of hydrophobin on different self-assembled monolayers: The role of the hydrophobic dipole and the electric dipole. Langmuir 2014, 30, 11401–11411. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Wu, M.; Feng, X.; Shao, X.; Cai, W. Adsorption behavior of hydrophobin proteins on polydimethylsiloxane substrates. J. Phys. Chem. B 2012, 116, 12227–12234. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.L.; Penfold, J.; Thomas, R.K.; Tucker, I.M.; Petkov, J.T.; Bent, J.; Cox, A. Adsorption behavior of hydrophobin and hydrophobin/surfactant mixtures at the solid-solution interface. Langmuir 2011, 27, 10464–10474. [Google Scholar] [CrossRef] [PubMed]
- Ley, K.; Christofferson, A.; Penna, M.; Winkler, D.; Maclaughlin, S.; Yarovsky, I. Surface-water Interface Induces Conformational Changes Critical for Protein Adsorption: Implications for Monolayer Formation of EAS Hydrophobin. Front. Mol. Biosci. 2015, 2, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Li, L.; Zhao, C.; Zheng, J. Surface hydration: Principles and applications toward low-fouling/nonfouling biomaterials. Polymer 2010, 51, 5283–5293. [Google Scholar] [CrossRef]
- Sheikh, S.; Blaszykowski, C.; Nolan, R.; Thompson, D.; Thompson, M. On the hydration of subnanometric antifouling organosilane adlayers: A molecular dynamics simulation. J. Colloid Interface Sci. 2015, 437, 197–204. [Google Scholar] [CrossRef] [PubMed]
- Penna, M.; Ley, K.; Maclaughlin, S.; Yarovsky, I. Surface heterogeneity: A friend or foe of protein adsorption—Insights from theoretical simulations. Faraday Discuss. 2016, 191, 435–464. [Google Scholar] [CrossRef] [PubMed]
- Bonmatin, J.; Genest, M.; Labbé, H.; Ptak, M. Solution three-dimensional structure of surfactin: A cyclic lipopeptide studied by 1H-nmr, distance geometry, and molecular dynamics. Biopolymers 1994, 34, 975–986. [Google Scholar] [CrossRef] [PubMed]
- Kinsinger, R.F.; Shirk, M.C.; Fall, R. Rapid surface motility in Bacillus subtilis is dependent on extracellular surfactin and potassium ion. J. Bacteriol. 2003, 185, 5627–5631. [Google Scholar] [CrossRef] [PubMed]
- Seydlová, G.; Svobodová, J. Review of surfactin chemical properties and the potential biomedical applications. Central Eur. J. Med. 2008, 3, 123–133. [Google Scholar] [CrossRef]
- Gallet, X.; Deleu, M.; Razafindralambo, H.; Jacques, P.; Thonart, P.; Paquot, M.; Brasseur, R. Computer Simulation of Surfactin Conformation at a Hydrophobic/Hydrophilic Interface. Langmuir 1999, 15, 2409–2413. [Google Scholar] [CrossRef]
- Nicolas, J.P. Molecular dynamics simulation of surfactin molecules at the water-hexane interface. Biophys. J. 2003, 85, 1377–1391. [Google Scholar] [CrossRef]
- Binks, B.P. Particles as surfactants—Similarities and differences. Curr. Opin. Colloid Interface Sci. 2002, 7, 21–41. [Google Scholar] [CrossRef]
- Gang, H.Z.; Liu, J.F.; Mu, B.Z. Interfacial behavior of surfactin at the decane/water interface: A molecular dynamics simulation. J. Phys. Chem. B 2010, 114, 14947–14954. [Google Scholar] [CrossRef] [PubMed]
- Iglesias-Fernández, J.; Darré, L.; Kohlmeyer, A.; Thomas, R.K.; Shen, H.H.; Domene, C. Surfactin at the Water/Air Interface and in Solution. Langmuir 2015, 31, 11097–11104. [Google Scholar] [CrossRef] [PubMed]
- Gang, H.Z.; Liu, J.F.; Mu, B.Z. Molecular dynamics simulation of surfactin derivatives at the decane/water interface at low surface coverage. J. Phys. Chem. B 2010, 114, 2728–2737. [Google Scholar] [CrossRef] [PubMed]
- Knoblich, A.; Matsumoto, M.; Ishiguro, R.; Murata, K.; Fujiyoshi, Y.; Ishigami, Y.; Osman, M. Electron cryo-microscopic studies on micellar shape and size of surfactin, an anionic lipopeptide. Colloids Surf. B Biointerfaces 1995, 5, 43–48. [Google Scholar] [CrossRef]
- Han, Y.; Huang, X.; Cao, M.; Wang, Y. Micellization of surfactin and its effect on the aggregate conformation of amyloid β(1–40). J. Phys. Chem. B 2008, 112, 15195–15201. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.H.; Thomas, R.K.; Chen, C.Y.; Darton, R.C.; Baker, S.C.; Penfold, J. Aggregation of the naturally occurring lipopeptide, surfactin, at interfaces and in solution: An unusual type of surfactant? Langmuir 2009, 25, 4211–4218. [Google Scholar] [CrossRef] [PubMed]
- She, A.Q.; Gang, H.Z.; Mu, B.Z. Temperature Influence on the Structure and Interfacial Properties of Surfactin Micelle: A Molecular Dynamics Simulation Study. J. Phys. Chem. B 2012, 116, 12735–12743. [Google Scholar] [CrossRef] [PubMed]
- Johnston, M.A.; Swope, W.C.; Jordan, K.E.; Warren, P.B.; Noro, M.G.; Bray, D.J.; Anderson, R.L. Toward a Standard Protocol for Micelle Simulation. J. Phys. Chem. B 2016, 120, 6337–6351. [Google Scholar] [CrossRef] [PubMed]
- Loison, C.; Nasir, M.N.; Benichou, E.; Besson, F.; Brevet, P.F. Multi-scale modeling of mycosubtilin lipopeptides at the air/water interface: Structure and optical second harmonic generation. Phys. Chem. Chem. Phys. 2014, 16, 2136–2148. [Google Scholar] [CrossRef] [PubMed]
- Cen, M.; Fan, J.F.; Liu, D.Y.; Song, X.Z.; Liu, J.; Zhou, W.Q.; Xiao, H.M. Cyclo-hexa-peptides at the water/cyclohexane interface: A molecular dynamics simulation. J. Mol. Model. 2013, 19, 601–611. [Google Scholar] [CrossRef] [PubMed]
- Khurana, E.; DeVane, R.H.; Kohlmeyer, A.; Klein, M.L. Probing Peptide Nanotube Self-Assembly at a Liquid-Liquid Interface with Coarse-Grained Molecular Dynamics. Nano Lett. 2008, 8, 3626–3630. [Google Scholar] [CrossRef] [PubMed]
- Cooper, A.; Kennedy, M.W.; Fleming, R.I.; Wilson, E.H.; Videler, H.; Wokosin, D.L.; Su, T.J.; Green, R.J.; Lu, J.R. Adsorption of frog foam nest proteins at the air–water interface. Biophys. J. 2005, 88, 2114–2125. [Google Scholar] [CrossRef] [PubMed]
- Mackenzie, C.D.; Smith, B.O.; Meister, A.; Blume, A.; Zhao, X.; Lu, J.R.; Kennedy, M.W.; Cooper, A. Ranaspumin-2: Structure and function of a surfactant protein from the foam nests of a tropical frog. Biophys. J. 2009, 96, 4984–4992. [Google Scholar] [CrossRef] [PubMed]
- Brandani, G.B.; Vance, S.J.; Schor, M.; Cooper, A.; Kennedy, M.W.; Smith, B.O.; MacPhee, C.E.; Cheung, D.L. Adsorption of the natural protein surfactant Rsn-2 onto liquid interfaces. Phys. Chem. Chem. Phys. 2017, 19, 8584–8594. [Google Scholar] [CrossRef] [PubMed]
- Shoemaker, B.A.; Portman, J.J.; Wolynes, P.G. Speeding molecular recognition by using the folding funnel: The fly-casting mechanism. Proc. Natl. Acad. Sci. USA 2000, 97, 8868–8873. [Google Scholar] [CrossRef] [PubMed]
- Zhai, J.; Wooster, T.J.; Hoffmann, S.V.; Lee, T.H.; Augustin, M.A.; Aguilar, M.I. Structural rearrangement of β-lactoglobulin at different oil-water interfaces and its effect on emulsion stability. Langmuir 2011, 27, 9227–9236. [Google Scholar] [CrossRef] [PubMed]
- Go, N. Theoretical Studies of Protein Folding. Annu. Rev. Biophys. Bioeng. 1983, 12, 183–210. [Google Scholar] [CrossRef] [PubMed]
- Morris, R.J.; Brandani, G.B.; Desai, V.; Smith, B.O.; Schor, M.; MacPhee, C.E. The Conformation of Interfacially Adsorbed Ranaspumin-2 is an Arrested State on the Unfolding Pathway. Biophys. J. 2016, 111, 732–742. [Google Scholar] [CrossRef] [PubMed]
- Engel, M.F.M.; Visser, A.J.W.G.; van Mierlo, C.P.M. Conformation and orientation of a protein folding intermediate trapped by adsorption. Proc. Natl. Acad. Sci. USA 2004, 101, 11316–11321. [Google Scholar] [CrossRef] [PubMed]
- Collier, G.; Vellore, N.A.; Yancey, J.A.; Stuart, S.J.; Latour, R.A. Comparison between empirical protein force fields for the simulation of the adsorption behavior of structured LK peptides on functionalized surfaces. Biointerphases 2012, 7, 24. [Google Scholar] [CrossRef] [PubMed]
- Biswas, P.K.; Vellore, N.A.; Yancey, J.A.; Kucukkal, T.G.; Collier, G.; Brooks, B.R.; Stuart, S.J.; Latour, R.A. Simulation of multiphase systems utilizing independent force fields to control intraphase and interphase behavior. J. Comput. Chem. 2012, 33, 1458–1466. [Google Scholar] [CrossRef] [PubMed]
- Friedrichs, M.S.; Eastman, P.; Vaidyanathan, V.; Houston, M.; Legrand, S.; Beberg, A.L.; Ensign, D.L.; Bruns, C.M.; Pande, V.S. Accelerating Molecular Dynamic Simulation on Graphics Processing Units. J. Comput. Chem. 2009, 30, 864–872. [Google Scholar] [CrossRef] [PubMed]
- McDonald, R.E.; Fleming, R.I.; Beeley, J.G.; Bovell, D.L.; Lu, J.R.; Zhao, X.; Cooper, A.; Kennedy, M.W. Latherin: A surfactant protein of horse sweat and saliva. PLoS ONE 2009, 4, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Vance, S.J.; Mcdonald, R.E.; Cooper, A.; Smith, B.O.; Kennedy, M.W. The structure of latherin, a surfactant allergen protein from horse sweat and saliva. J. R. Soc. Interface 2013, 10, 20130453. [Google Scholar] [CrossRef] [PubMed]
- Gakhar, L.; Bartlett, J.A.; Penterman, J.; Mizrachi, D.; Singh, P.K.; Mallampalli, R.K.; Ramaswamy, S.; McCray, P.B. PLUNC is a novel airway surfactant protein with anti-biofilm activity. PLoS ONE 2010, 5. [Google Scholar] [CrossRef] [PubMed]
- Ning, F.; Wang, C.; Berry, K.Z.; Kandasamy, P.; Liu, H.; Murphy, R.C.; Voelker, D.R.; Nho, C.W.; Pan, C.H.; Dai, S.; et al. Structural characterization of the pulmonary innate immune protein SPLUNC1 and identification of lipid ligands. FASEB J. 2014, 28, 5349–5360. [Google Scholar] [CrossRef] [PubMed]
- Walton, W.G.; Ahmad, S.; Little, M.S.; Kim, C.S.; Tyrrell, J.; Lin, Q.; Di, Y.P.; Tarran, R.; Redinbo, M.R. Structural Features Essential to the Antimicrobial Functions of Human SPLUNC1. Biochemistry 2016, 55, 2979–2991. [Google Scholar] [CrossRef] [PubMed]
- Sakakibara, K.; Fujisawa, T.; Hill, J.P.; Ariga, K. Conformational interchange of a carbohydrate by mechanical compression at the air–water interface. Phys. Chem. Chem. Phys. 2014, 16, 10286–10294. [Google Scholar] [CrossRef] [PubMed]
- Wibowo, D.; Zhao, C.X.; Middelberg, A.P.J. Interfacial Biomimetic Synthesis of Silica Nanocapsules Using a Recombinant Catalytic Modular Protein. Langmuir 2015, 31, 1999–2007. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, D.; Mori, T.; Yonamine, Y.; Nakanishi, W.; Cheung, D.L.; Hill, J.P.; Ariga, K. Mechanochemical Tuning of the Binaphthyl Conformation at the Air-Water Interface. Angew. Chem. Int. Ed. 2015, 54, 8988–8991. [Google Scholar] [CrossRef] [PubMed]
- Xue, Y.; He, L.; Middelberg, A.P.J.; Mark, A.E.; Poger, D. Determining the Structure of Interfacial Peptide Films: Comparing Neutron Reflectometry and Molecular Dynamics Simulations. Langmuir 2014, 30, 10080–10089. [Google Scholar] [CrossRef] [PubMed]
- Knecht, V.; Reiter, G.; Schlaad, H.; Reiter, R. Structure Formation in Langmuir Peptide Films As Revealed from Coarse-Grained Molecular Dynamics Simulations. Langmuir 2017, 33, 6492–6502. [Google Scholar] [CrossRef] [PubMed]
- Di Napoli, B.; Mazzuca, C.; Conflitti, P.; Venanzi, M.; Palleschi, A. Behavior of a Peptide during a Langmuir-Blodgett Compression Isotherm: A Molecular Dynamics Simulation Study. J. Phys. Chem. C 2018, 122, 515–521. [Google Scholar] [CrossRef]
- Thøgersen, L.; Schiøtt, B.; Vosegaard, T.; Nielsen, N.C.; Tajkhorshid, E. Peptide aggregation and pore formation in a lipid bilayer: A combined coarse-grained and all atom molecular dynamics study. Biophys. J. 2008, 95, 4337–4347. [Google Scholar] [CrossRef] [PubMed]
- Lemkul, J.A.; Bevan, D.R. Aggregation of alzheimer’s amyloid β-peptide in biological membranes: A molecular dynamics study. Biochemistry 2013, 52, 4971–4980. [Google Scholar] [CrossRef] [PubMed]
- Solernou, A.; Hanson, B.S.; Richardson, R.A.; Welch, R.; Read, D.J.; Harlen, O.G.; Harris, S.A. Fluctuating Finite Element Analysis (FFEA): A continuum mechanics software tool for mesoscale simulation of biomolecules. PLoS Comput. Biol. 2018, 14, 1–29. [Google Scholar] [CrossRef] [PubMed]
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).