
Enhancing Docking Accuracy with PECAN2, a 3D Atomic Neural Network Trained without Co-Complex Crystal Structures


Abstract
1. Introduction
2. Materials and Methods
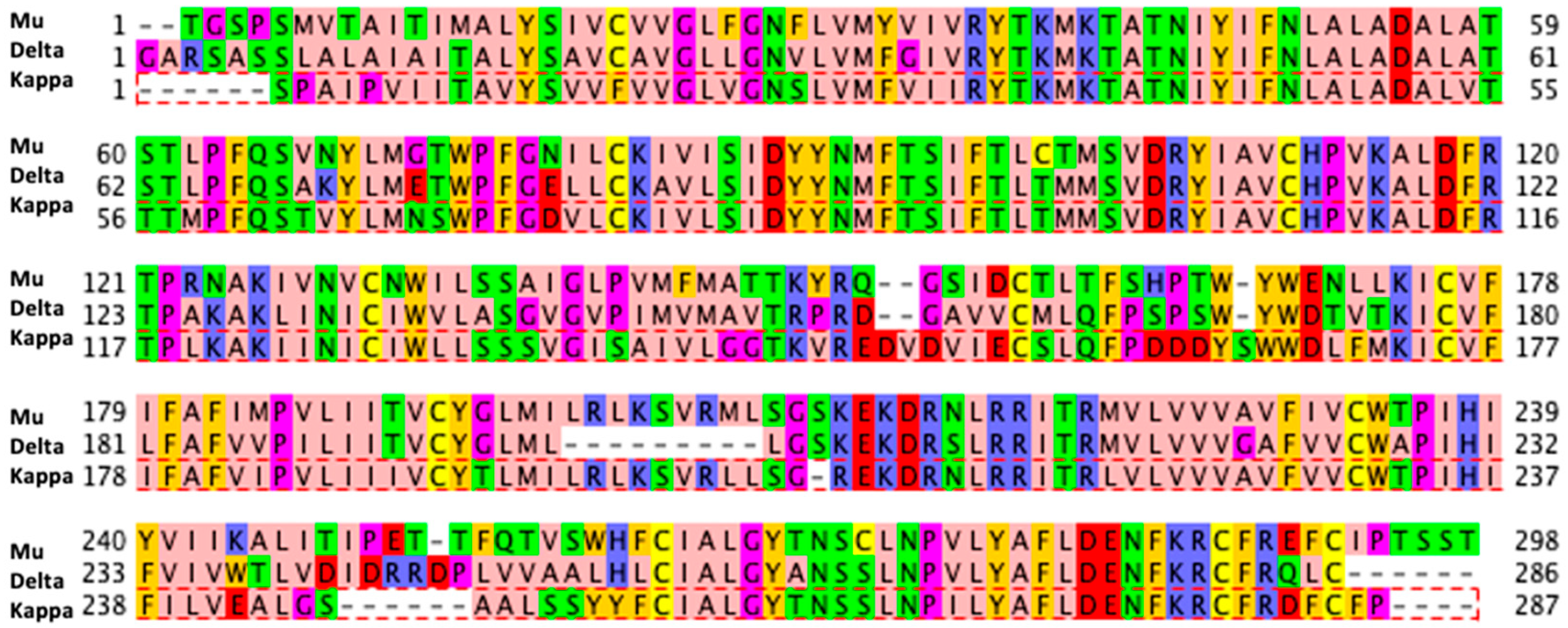
2.1. Data
2.2. Preprocessing
2.3. Point Cloud Network (PCN)
2.4. Experimental Setup
3. Results
3.1. Pose Classification Model Performance
3.1.1. Random Split
3.1.2. Scaffold Split
3.1.3. Cross-Testing
3.1.4. Data Limitation Experiment
3.1.5. SARS2 Mpro Data
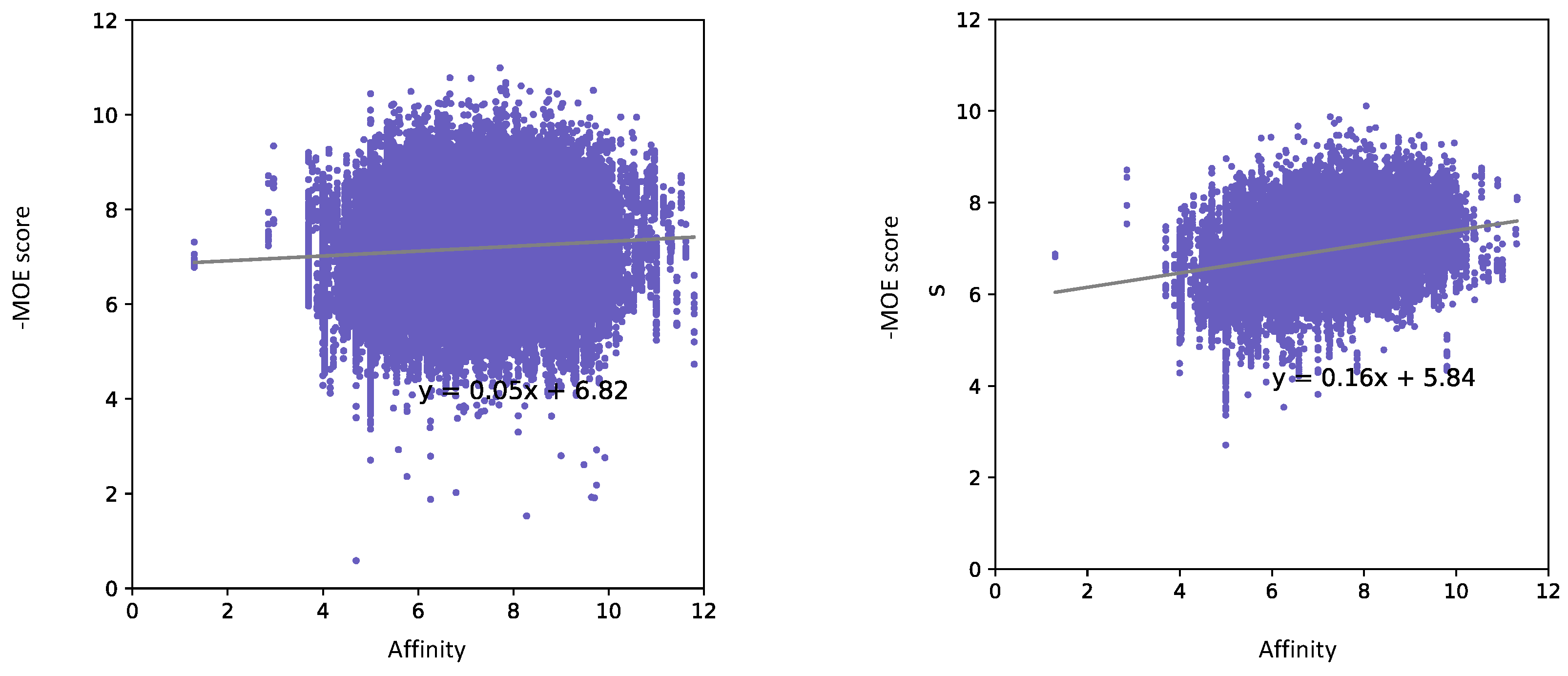
3.2. Speed Test on PECAN2
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Lyu, J.; Irwin, J.J.; Shoichet, B.K. Modeling the expansion of virtual screening libraries. Nat. Chem. Biol. 2023, 19, 712–718. [Google Scholar] [CrossRef]
- Jones, G.; Willett, P.; Glen, R.C.; Leach, A.R.; Taylor, R. Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol. 1997, 267, 727–748. [Google Scholar] [CrossRef]
- Ewing, T.J.A.; Makino, S.; Skillman, A.G.; Kuntz, I.D. DOCK 4.0: Search strategies for automated molecular docking of flexible molecule databases. J. Comput. Aided Mol. Des. 2001, 15, 411–428. [Google Scholar] [CrossRef] [PubMed]
- Muegge, I. PMF scoring revisited. J. Med. Chem. 2006, 49, 5895–5902. [Google Scholar] [CrossRef] [PubMed]
- Velec, H.F.; Gohlke, H.; Klebe, G. DrugScore(CSD)-knowledge-based scoring function derived from small molecule crystal data with superior recognition rate of near-native ligand poses and better affinity prediction. J. Med. Chem. 2005, 48, 6296–6303. [Google Scholar] [CrossRef] [PubMed]
- Jin, Z.; Wu, T.; Chen, T.; Pan, D.; Wang, X.; Xie, J.; Quan, L.; Lyu, Q. CAPLA: Improved prediction of protein-ligand binding affinity by a deep learning approach based on a cross-attention mechanism. Bioinformatics 2023, 39, btad049. [Google Scholar] [CrossRef] [PubMed]
- Beveridge, D.L.; DiCapua, F.M. Free energy via molecular simulation: Applications to chemical and biomolecular systems. Annu. Rev. Biophys. Biophys. Chem. 1989, 18, 431–492. [Google Scholar] [CrossRef]
- Kollman, P. Free energy calculations: Applications to chemical and biochemical phenomena. Chem. Rev. 1993, 93, 2395–2417. [Google Scholar] [CrossRef]
- Huang, S.Y.; Zou, X. An iterative knowledge-based scoring function to predict protein-ligand interactions: I. Derivation of interaction potentials. J. Comput. Chem. 2006, 27, 1866–1875. [Google Scholar] [CrossRef]
- Eldridge, M.D.; Murray, C.W.; Auton, T.R.; Paolini, G.V.; Mee, R.P. Empirical scoring functions: I. The development of a fast empirical scoring function to estimate the binding affinity of ligands in receptor complexes. J. Comput. Aided Mol. Des. 1997, 11, 425–445. [Google Scholar] [CrossRef]
- Wang, R.; Lai, L.; Wang, S. Further development and validation of empirical scoring functions for structure-based binding affinity prediction. J. Comput. Aided Mol. Des. 2002, 16, 11–26. [Google Scholar] [CrossRef]
- Cortes, C.; Vapnik, V. Support-vector networks. Mach. Learn. 1995, 20, 273–297. [Google Scholar] [CrossRef]
- Breiman, L. Random Forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef]
- Mason, L.; Baxter, J.; Bartlett, P.; Frean, M. Boosting Algorithms as Gradient Descent in Function Space; NIPS: New Orleans, LA, USA, 1999. [Google Scholar]
- Friedman, J.H. Stochastic gradient boosting. Comput. Stat. Data Anal. 2002, 38, 367–378. [Google Scholar] [CrossRef]
- Krizhevsky, A.; Sutskever, I.; Hinton, G.E. ImageNet classification with deep convolutional neural networks. Commun. ACM 2017, 60, 84–90. [Google Scholar] [CrossRef]
- Wallach, I.; Dzamba, M.; Heifets, A. AtomNet: A Deep Convolutional Neural Network for Bioactivity Prediction in Structure-based Drug Discovery. arXiv 2015, arXiv:1510.02855. [Google Scholar]
- Stepniewska-Dziubinska, M.M.; Zielenkiewicz, P.; Siedlecki, P. Development and evaluation of a deep learning model for protein–ligand binding affinity prediction. Bioinformatics 2018, 34, 3666–3674. [Google Scholar] [CrossRef]
- Li, Y.; Rezaei, M.A.; Li, C.; Li, X.; Wu, D. DeepAtom: A Framework for Protein-Ligand Binding Affinity Prediction. arXiv 2019, arXiv:1912.00318. [Google Scholar]
- Zheng, L.; Fan, J.; Mu, Y. OnionNet: A Multiple-Layer Intermolecular-Contact-Based Convolutional Neural Network for Protein–Ligand Binding Affinity Prediction. ACS Omega 2019, 4, 15956–15965. [Google Scholar] [CrossRef]
- Wang, Z.; Zheng, L.; Liu, Y.; Qu, Y.; Li, Y.-Q.; Zhao, M.; Mu , Y.; Li , W. OnionNet-2: A Convolutional Neural Network Model for Predicting Protein-Ligand Binding Affinity Based on Residue-Atom Contacting Shells. Front. Chem. 2021, 9, 753002. [Google Scholar] [CrossRef]
- Hassan-Harrirou, H.; Zhang, C.; Lemmin, T. RosENet: Improving Binding Affinity Prediction by Leveraging Molecular Mechanics Energies with an Ensemble of 3D Convolutional Neural Networks. J. Chem. Inf. Model. 2020, 60, 2791–2802. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.; Kim, H.; Zhang, X.; Zemla, A.; Stevenson, G.; Bennett, W.F.D.; Kirshner, D.; Wong, S.E.; Lightstone, F.C.; Allen, J.E. Improved Protein-Ligand Binding Affinity Prediction with Structure-Based Deep Fusion Inference. J. Chem. Inf. Model. 2021, 61, 1583–1592. [Google Scholar] [CrossRef] [PubMed]
- Atz, K.; Grisoni, F.; Schneider, G. Geometric Deep Learning on Molecular Representations. arXiv 2021, arXiv:2107.12375. [Google Scholar] [CrossRef]
- Bronstein, M.M.; Bruna, J.; Cohen, T.; Veličković, P. Geometric Deep Learning: Grids, Groups, Graphs, Geodesics, and Gauges. arXiv 2021, arXiv:2104.13478. [Google Scholar]
- Corso, G.; Stärk, H.; Jing, B.; Barzilay, R.; Jaakkola, T. DiffDock: Diffusion Steps, Twists, and Turns for Molecular Docking. arXiv 2022, arXiv:2210.01776. [Google Scholar]
- Urbina, F.; Lowden, C.T.; Culberson, J.C.; Ekins, S. MegaSyn: Integrating Generative Molecular Design, Automated Analog Designer, and Synthetic Viability Prediction. ACS Omega 2022, 7, 18699–18713. [Google Scholar] [CrossRef] [PubMed]
- Buttenschoen, M.; Morris, G.M.; Deane, C.M. PoseBusters: AI-based docking methods fail to generate physically valid poses or generalise to novel sequences. arXiv 2023, arXiv:2308.05777. [Google Scholar] [CrossRef]
- Wang, R.; Fang, X.; Lu, Y.; Wang, S. The PDBbind database: Collection of binding affinities for protein-ligand complexes with known three-dimensional structures. J. Med. Chem. 2004, 47, 2977–2980. [Google Scholar] [CrossRef]
- Stauch, B.; Cherezov, V. Serial Femtosecond Crystallography of G Protein-Coupled Receptors. Annu. Rev. Biophys. 2018, 47, 377–397. [Google Scholar] [CrossRef]
- Yang, J.; Shen, C.; Huang, N. Predicting or Pretending: Artificial Intelligence for Protein-Ligand Interactions Lack of Sufficiently Large and Unbiased Datasets. Front. Pharmacol. 2020, 11, 69. [Google Scholar] [CrossRef]
- Li, J.; Guan, X.; Zhang, O.; Sun, K.; Wang, Y.; Bagni, D.; Head-Gordon, T. Leak Proof PDBBind: A Reorganized Dataset of Protein-Ligand Complexes for More Generalizable Binding Affinity Prediction. arXiv 2023, arXiv:2308.09639v1. [Google Scholar]
- Shim, H.; Kim, H.; Allen, J.E.; Wulff, H. Pose Classification Using Three-Dimensional Atomic Structure-Based Neural Networks Applied to Ion Channel-Ligand Docking. J. Chem. Inf. Model. 2022, 62, 2301–2315. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Lin, Y.; Wen, X.; Jorissen, R.N.; Gilson, M.K. BindingDB: A web-accessible database of experimentally determined protein-ligand binding affinities. Nucleic Acids Res. 2007, 35, D198–D201. [Google Scholar] [CrossRef]
- Sheils, T.K.; Mathias, S.L.; Kelleher, K.J.; Siramshetty, V.B.; Nguyen, D.T.; Bologa, C.G.; Jensen, L.J.; Vidovic, D.; Koleti, A.; Schurer, S.C.; et al. TCRD and Pharos 2021: Mining the human proteome for disease biology. Nucleic Acids Res. 2021, 49, D1334–D1346. [Google Scholar] [CrossRef] [PubMed]
- Manglik, A.; Kruse, A.C.; Kobilka, T.S.; Thian, F.S.; Mathiesen, J.M.; Sunahara, R.K.; Pardo, L.; Weis, W.I.; Kobilka, B.K.; Granier, S. Crystal structure of the micro-opioid receptor bound to a morphinan antagonist. Nature 2012, 485, 321–326. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Wacker, D.; Mileni, M.; Katritch, V.; Han, G.W.; Vardy, E.; Liu, W.; Thompson, A.A.; Huang, X.P.; Carroll, F.I.; et al. Structure of the human kappa-opioid receptor in complex with JDTic. Nature 2012, 485, 327–332. [Google Scholar] [CrossRef] [PubMed]
- Fenalti, G.; Zatsepin, N.A.; Betti, C.; Giguere, P.; Han, G.W.; Ishchenko, A.; Liu, W.; Guillemyn, K.; Zhang, H.; James, D.; et al. Structural basis for bifunctional peptide recognition at human delta-opioid receptor. Nat. Struct. Mol. Biol. 2015, 22, 265–268. [Google Scholar] [CrossRef]
- Dreborg, S.; Sundstrom, G.; Larsson, T.A.; Larhammar, D. Evolution of vertebrate opioid receptors. Proc. Natl. Acad. Sci. USA 2008, 105, 15487–15492. [Google Scholar] [CrossRef]
- Jin, Z.; Du, X.; Xu, Y.; Deng, Y.; Liu, M.; Zhao, Y.; Zhang, B.; Li, X.; Zhang, L.; Peng, C.; et al. Structure of M(pro) from SARS-CoV-2 and discovery of its inhibitors. Nature 2020, 582, 289–293. [Google Scholar] [CrossRef]
- Global Online Structure Activity Relationship Database (GOSTAR), Excelra. Available online: https://www.gostardb.com/gostar/newui/applications.jsp (accessed on 28 September 2023).
- Morris, A.; McCorkindale, W.; Consortium, T.C.M.; Drayman, N.; Chodera, J.D.; Tay, S.; London, N.; Lee, A.A. Discovery of SARS-CoV-2 main protease inhibitors using a synthesis-directed de novo design model. Chem. Commun. 2021, 57, 5909–5912. [Google Scholar] [CrossRef]
- Molecular Operating Environment (MOE); Chemical Computing Group: Montreal, QC, Canada, 2023.
- Eberhardt, J.; Santos-Martins, D.; Tillack, A.F.; Forli, S. AutoDock Vina 1.2.0: New Docking Methods, Expanded Force Field, and Python Bindings. J. Chem. Inf. Model. 2021, 61, 3891–3898. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Wong, S.E.; Lightstone, F.C. Toward fully automated high performance computing drug discovery: A massively parallel virtual screening pipeline for docking and molecular mechanics/generalized Born surface area rescoring to improve enrichment. J. Chem. Inf. Model. 2014, 54, 324–337. [Google Scholar] [CrossRef] [PubMed]
- Wojcikowski, M.; Zielenkiewicz, P.; Siedlecki, P. Open Drug Discovery Toolkit (ODDT): A new open-source player in the drug discovery field. J. Cheminform. 2015, 7, 26. [Google Scholar] [CrossRef] [PubMed]
- Qi, C.R.; Su, H.; Mo, K.; Guibas, L.J. PointNet: Deep Learning on Point Sets for 3D Classification and Segmentation. arXiv 2016, arXiv:1612.00593. [Google Scholar]
- Minnich, A.J.; McLoughlin, K.; Tse, M.; Deng, J.; Weber, A.; Murad, N.; Madej, B.D.; Ramsundar, B.; Rush, T.; Calad-Thomson, S.; et al. AMPL: A Data-Driven Modeling Pipeline for Drug Discovery. J. Chem. Inf. Model. 2020, 60, 1955–1968. [Google Scholar] [CrossRef]
- Bemis, G.W.; Murcko, M.A. The properties of known drugs. 1. Molecular frameworks. J. Med. Chem. 1996, 39, 2887–2893. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Jin, Y.; Liu, T.; Wang, Q.; Zhang, Z.; Zhao, S.; Shan, B. SS-GNN: A Simple-Structured Graph Neural Network for Affinity Prediction. ACS Omega 2023, 8, 22496–22507. [Google Scholar] [CrossRef] [PubMed]
- Shen, C.; Hu, X.; Gao, J.; Zhang, X.; Zhong, H.; Wang, Z.; Xu, L.; Kang, Y.; Cao, D.; Hou, T. The impact of cross-docked poses on performance of machine learning classifier for protein-ligand binding pose prediction. J. Cheminform. 2021, 13, 81. [Google Scholar] [CrossRef]
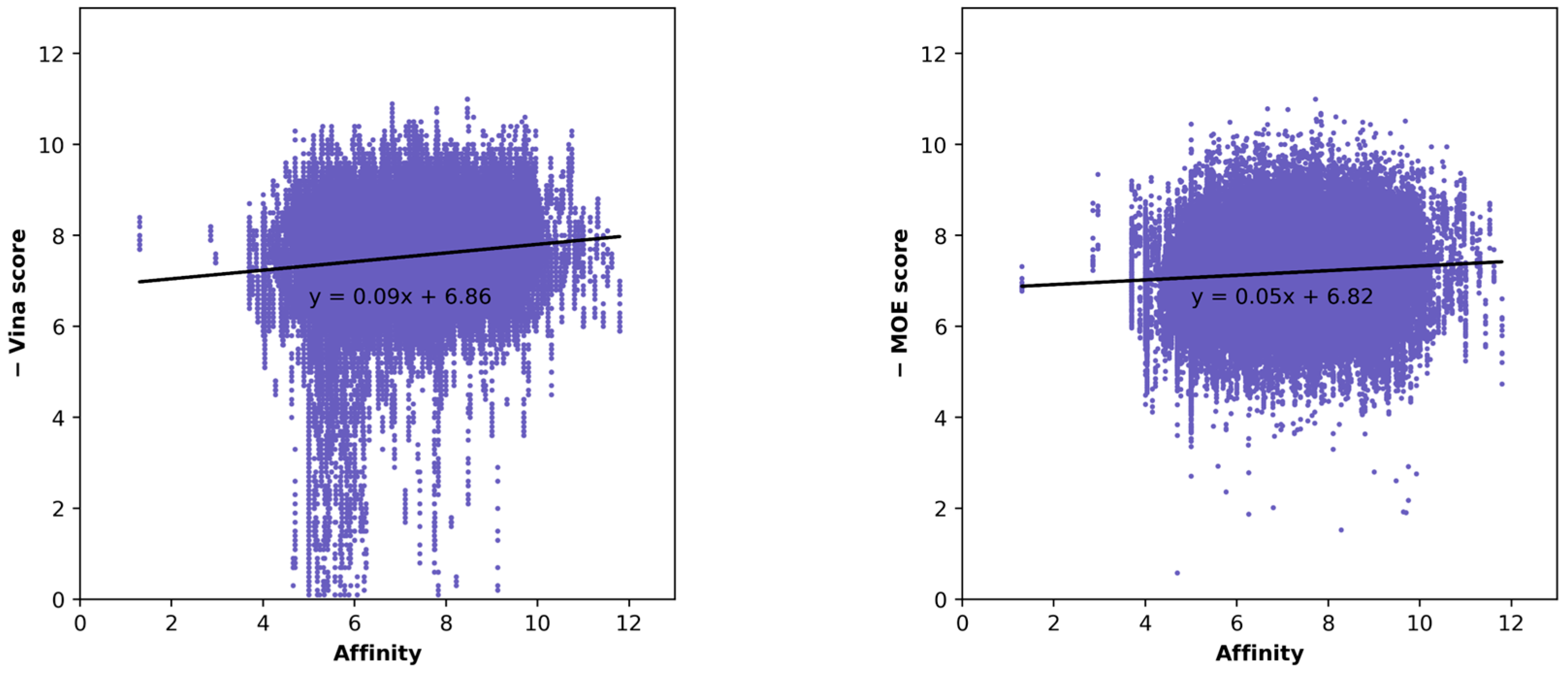
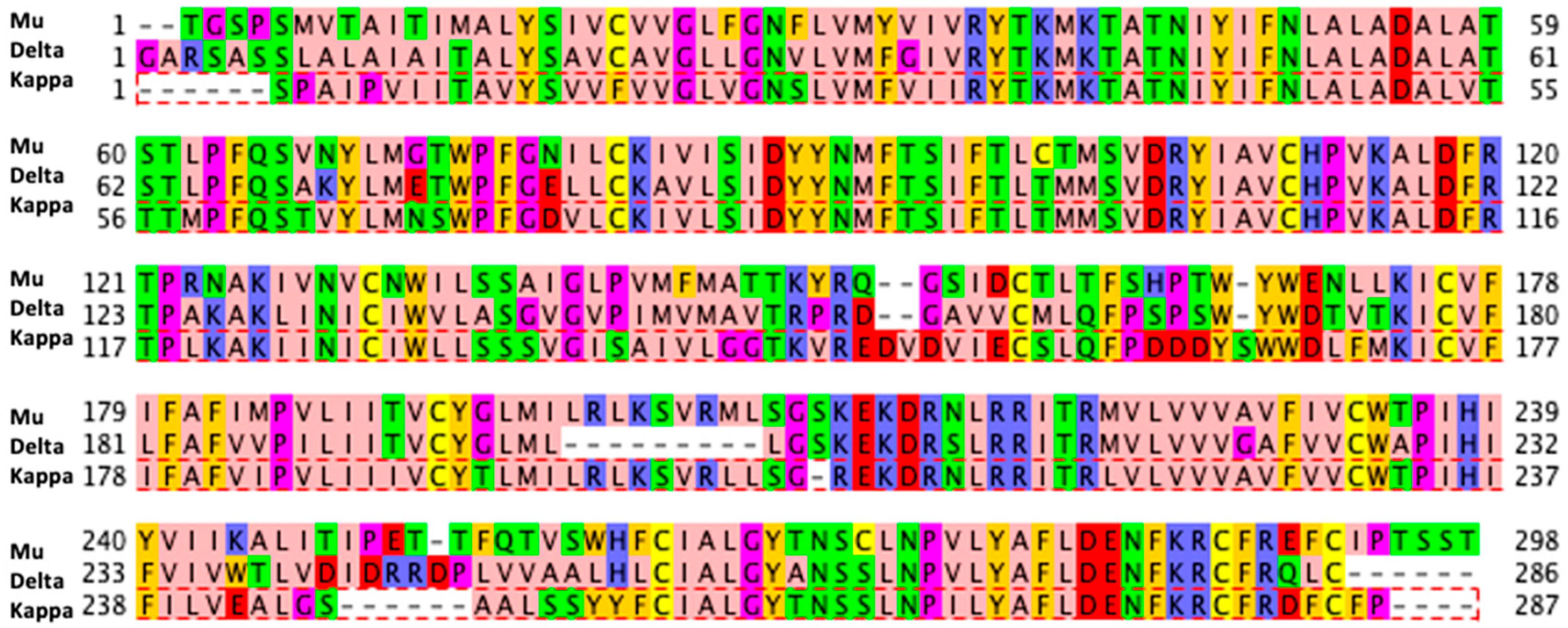
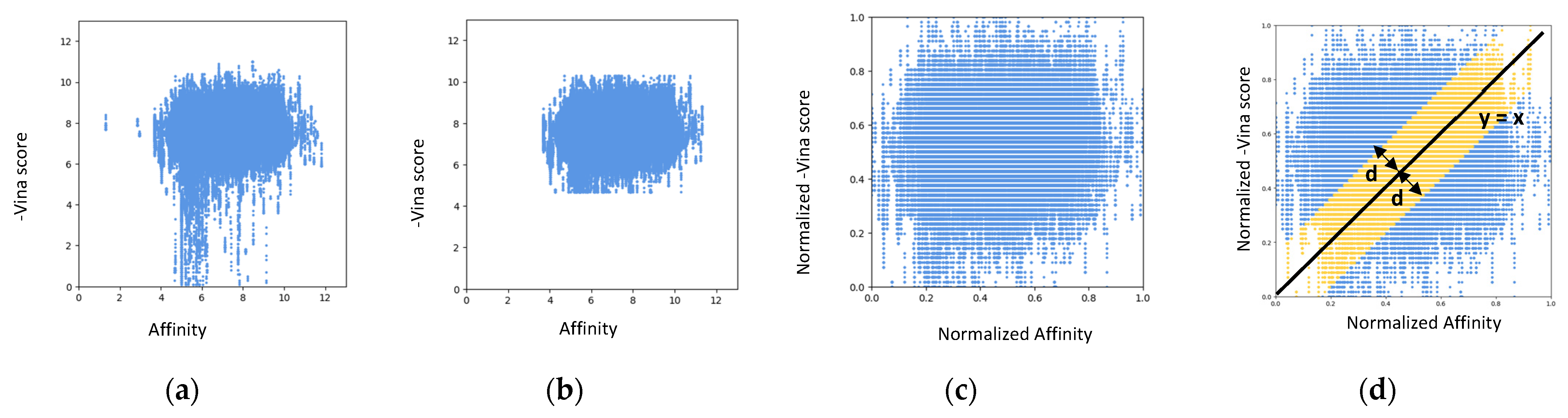

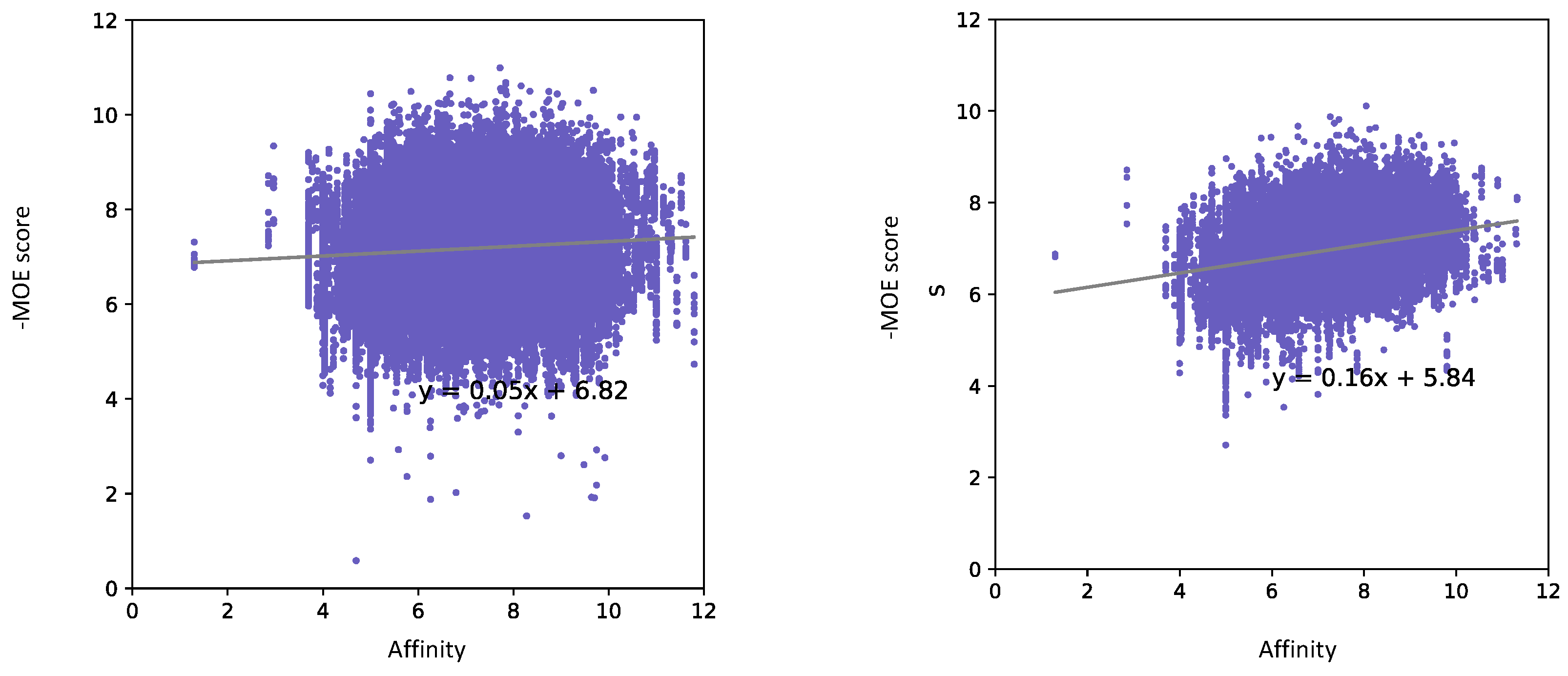



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Docking Program | Name of Opioid Receptor (Number of Testing Ligands) | Pearson Values (+Difference) | Number of Strong Binders/Number of the Top 5% Ranked Compounds (+Difference) | ||
|---|---|---|---|---|---|
| Without PECAN2 | With PECAN2 | Without PECAN2 | With PECAN2 | ||
| Autodock Vina | Mu (906) | 0.148 | 0.393 (+0.245) | 34/46, 74% | 42/46, 91% (+17%) |
| Kappa (729) | −0.048 | 0.168 (+0.216) | 17/37, 45% | 28/37, 75% (+30%) | |
| Delta (712) | 0.165 | 0.416 (+0.251) | 17/36, 47% | 29/36, 80% (+33%) | |
| MOE | Mu (927) | 0.108 | 0.436 (+0.328) | 28/47, 59% | 38/47, 80% (+21%) |
| Kappa (740) | 0.035 | 0.352 (+0.317) | 20/37, 54% | 30/37, 81% (+27%) | |
| Delta (717) | 0.094 | 0.448 (+0.354) | 22/36, 61% | 29/36, 80% (+19%) | |
| Docking Program | Name of Opioid Receptor (Number of Testing Ligands) | Pearson Values (+Difference) | Number of Strong Binders/Number of the Top 5% Ranked Compounds (+Difference) | ||
|---|---|---|---|---|---|
| Without PECAN2 | With PECAN2 | Without PECAN2 | With PECAN2 | ||
| Autodock Vina | Mu (821) | 0.17 | 0.39 (+0.22) | 31/42, 73% | 36/42, 86% (+13%) |
| Kappa (653) | −0.115 | 0.035 (+0.15) | 17/33, 52% | 18/33, 55% (+3%) | |
| Delta (585) | 0.24 | 0.49 (+0.25) | 15 /30, 50% | 20/30, 67% (+17%) | |
| MOE | Mu (839) | 0.04 | 0.4 (+0.36) | 29/42, 69% | 32/42, 76% (+7%) |
| Kappa (665) | 0.019 | 0.27 (+0.25) | 18/34, 53% | 19/34, 56% (+3%) | |
| Delta (592) | 0.21 | 0.42 (+0.21) | 15/30, 50% | 21/30, 70% (+20%) | |
| Docking Program | Training Data | Testing (Number of Testing Ligands) | Pearson Values (+Difference) | Number of Strong Binders/Number of the Top 5% Ranked Ligands (+Difference) | ||
|---|---|---|---|---|---|---|
| Without PECAN2 | With PECAN2 | Without PECAN2 | With PECAN2 | |||
| Autodock Vina | Mu | Delta (7120) | 0.197 | 0.247 (+0.05) | 197/356, 55% | 223/356 63% (+8%) |
| Kappa (7294) | −0.02 | 0.113 (+0.133) | 192/365, 53% | 229/365 63% (+10%) | ||
| Kappa | Mu (9069) | 0.14 | 0.17 (+0.03) | 305/454, 67% | 323/454 71% (+4%) | |
| Delta (7120) | 0.197 | 0.21 (+0.013) | 197/356, 55% | 194/356 54% (−1%) | ||
| Delta | Mu (9069) | 0.14 | 0.22 (+0.08) | 305/454, 67% | 357/454 79% (+12%) | |
| Kappa (7294) | −0.02 | 0.073 (+0.093) | 192/365, 53% | 223/365 61% (+8%) | ||
| MOE | Mu | Delta (7178) | 0.093 | 0.18 (+0.087) | 184/359, 51% | 194/359 54% (+3%) |
| Kappa (7407) | 0.015 | 0.23 (+0.215) | 179/371, 48% | 223/371 60% (+12%) | ||
| Kappa | Mu (9270) | 0.083 | 0.213 (+0.13) | 289/464, 62% | 326/464 70% (+8%) | |
| Delta (7178) | 0.093 | 0.24 (+0.147) | 184/359, 51% | 195 /359 54% (+3%) | ||
| Delta | Mu (9270) | 0.083 | 0.292 (+0.209) | 289/464, 62% | 342/464 74% (+12%) | |
| Kappa (7407) | 0.015 | 0.18 (+0.165) | 179/371, 48% | 208/371 56% (+8%) | ||
| Docking Program | Number of Training Compounds | Pearson Values (+Difference) | Number of Strong Binders/Number of the Top 5% Ranked Ligands (+Difference) | ||
|---|---|---|---|---|---|
| Without PECAN2 | With PECAN2 | Without PECAN2 | With PECAN2 | ||
| MOE | 5061 | 0.035 | 0.352 (+0.317) | 20/37, 54% | 30/37, 81% (+27%) |
| 725 | 0.272 (+0.237) | 27/37, 73% (+19%) | |||
| 300 | 0.108 (+0.073) | 24/37, 65% (+9%) | |||
| Docking Program | Pearson Values (+Difference) | Number of Strong Binders/Number of the Top 5% Ranked Ligands (+Difference) | ||
|---|---|---|---|---|
| Without PECAN2 | With PECAN2 | Without PECAN2 | With PECAN2 | |
| Autodock Vina | 0.17 | 0.40 (+0.23) | 4/21, 19% | 6/21, 29% (+10%) |
| MOE | 0.23 | 0.45(+0.22) | 3/22, 14% | 5/22, 23% (+9%) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shim, H.; Allen, J.E.; Bennett, W.F.D. Enhancing Docking Accuracy with PECAN2, a 3D Atomic Neural Network Trained without Co-Complex Crystal Structures. Mach. Learn. Knowl. Extr. 2024, 6, 642-657. https://doi.org/10.3390/make6010030
Shim H, Allen JE, Bennett WFD. Enhancing Docking Accuracy with PECAN2, a 3D Atomic Neural Network Trained without Co-Complex Crystal Structures. Machine Learning and Knowledge Extraction. 2024; 6(1):642-657. https://doi.org/10.3390/make6010030
Chicago/Turabian StyleShim, Heesung, Jonathan E. Allen, and W. F. Drew Bennett. 2024. "Enhancing Docking Accuracy with PECAN2, a 3D Atomic Neural Network Trained without Co-Complex Crystal Structures" Machine Learning and Knowledge Extraction 6, no. 1: 642-657. https://doi.org/10.3390/make6010030
APA StyleShim, H., Allen, J. E., & Bennett, W. F. D. (2024). Enhancing Docking Accuracy with PECAN2, a 3D Atomic Neural Network Trained without Co-Complex Crystal Structures. Machine Learning and Knowledge Extraction, 6(1), 642-657. https://doi.org/10.3390/make6010030