
Why Does the Antioxidant Complex Twendee X® Prevent Dementia?

Abstract
:1. Introduction
2. Results and Discussion
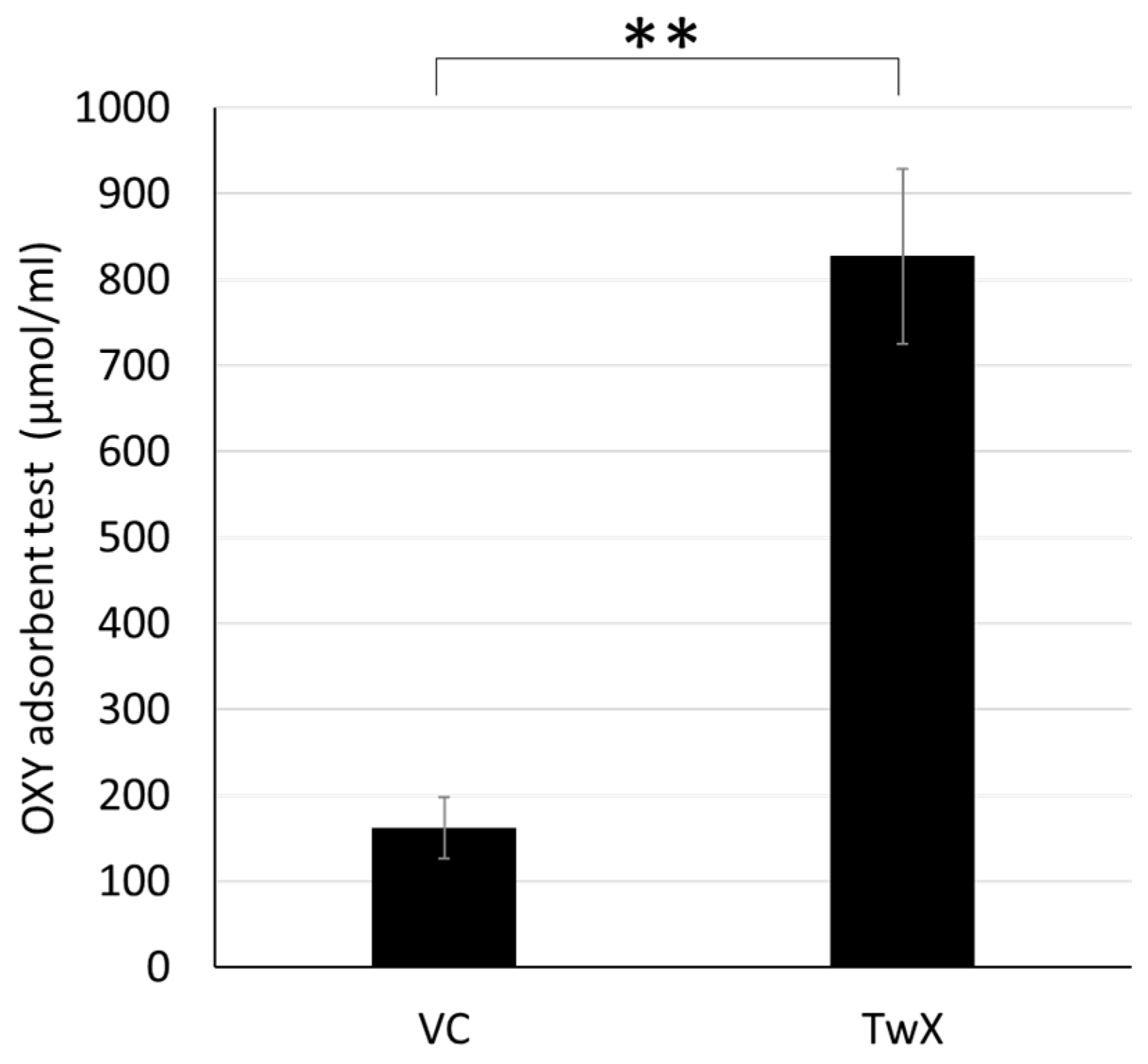
2.1. Antioxidant Capacity of TwX
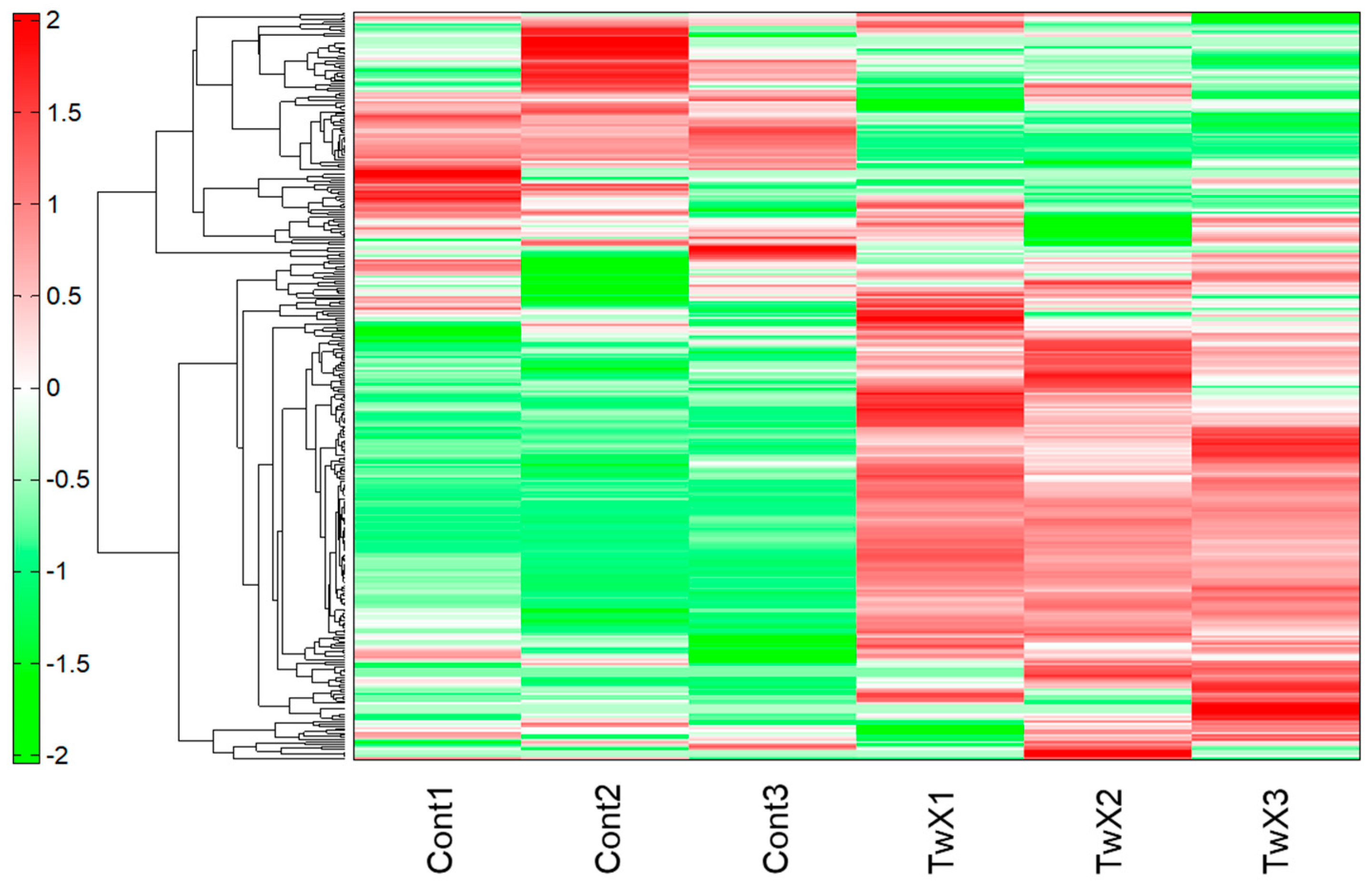
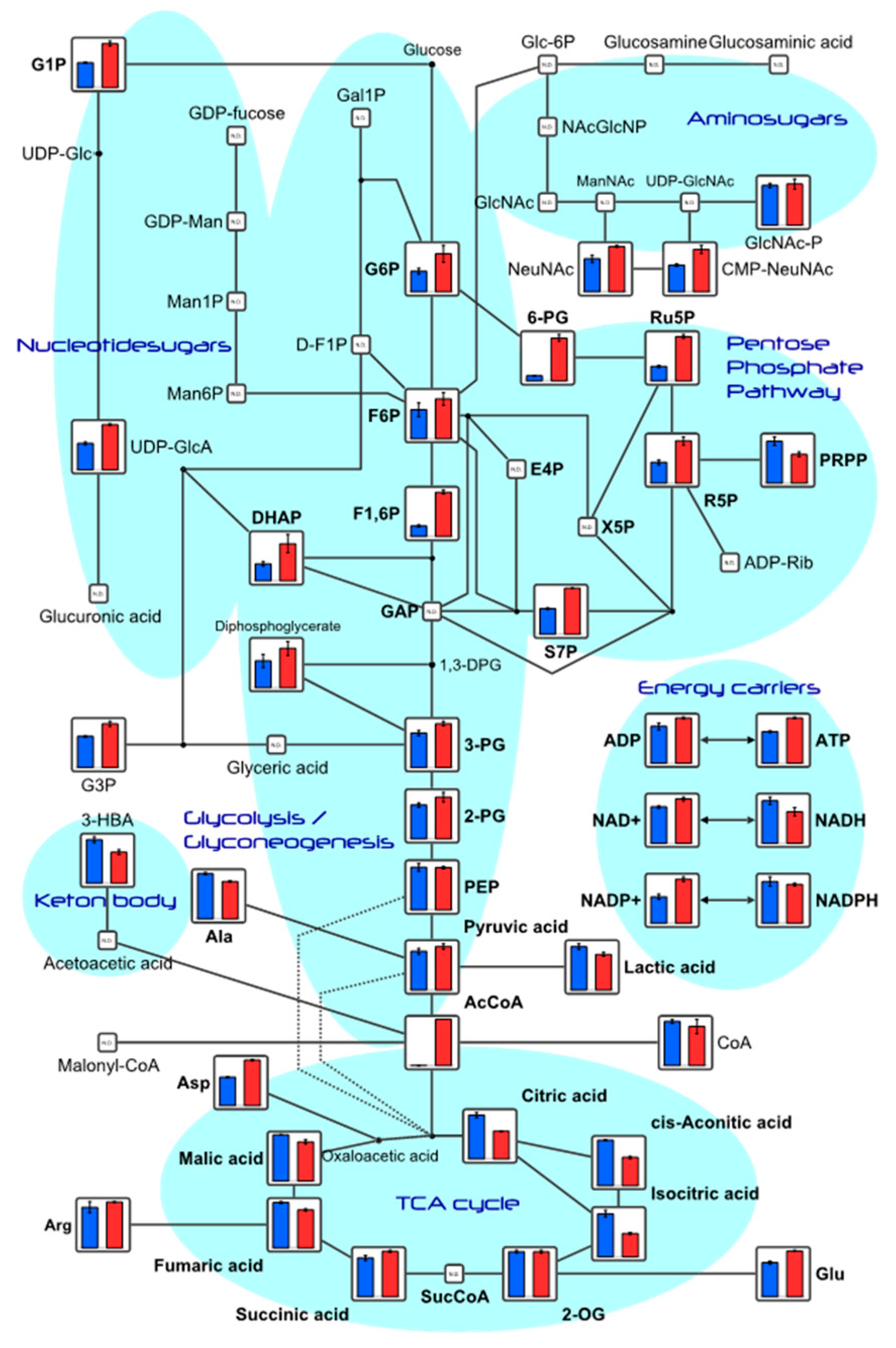
2.2. Mitochondrial Energy Production
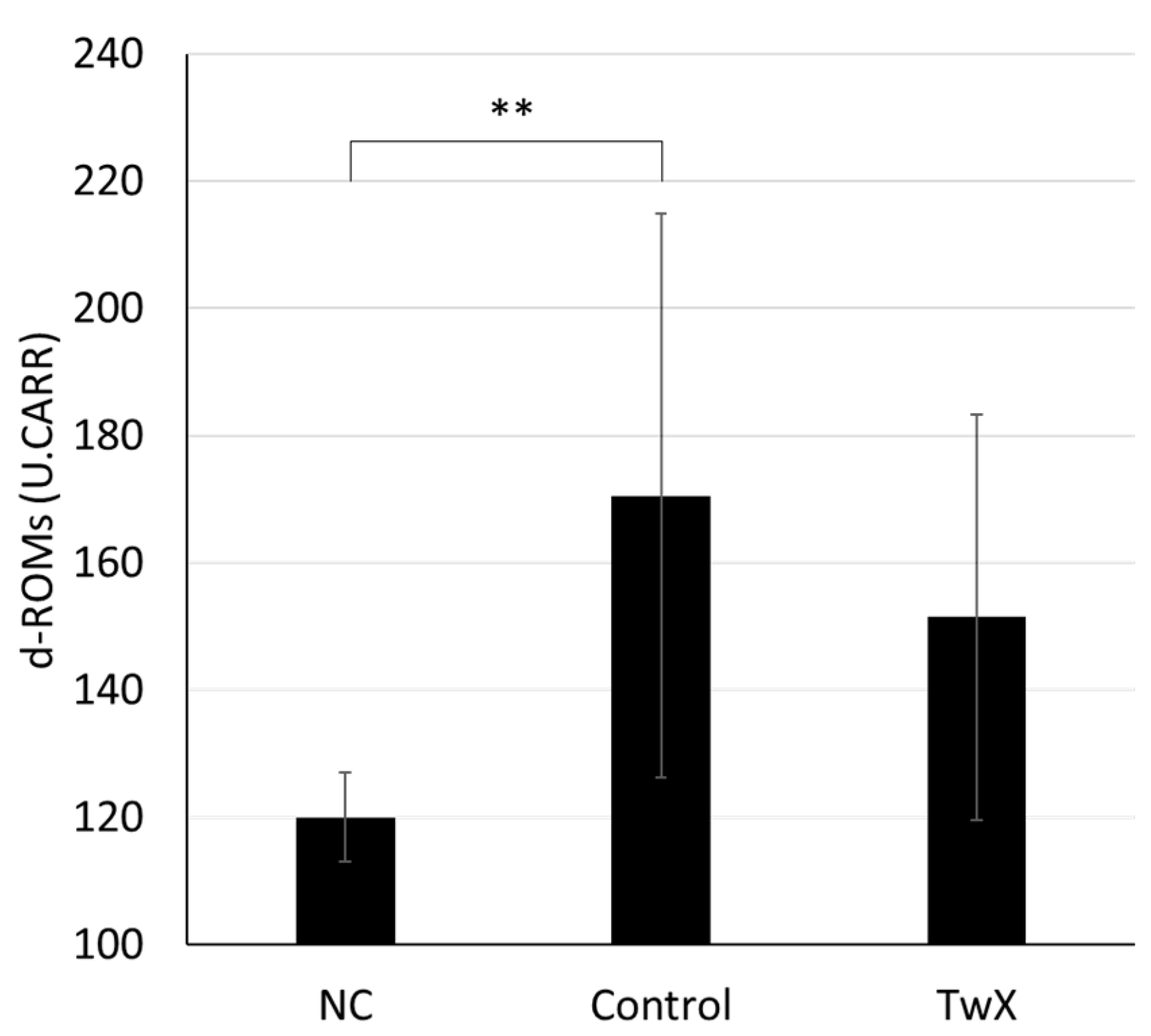
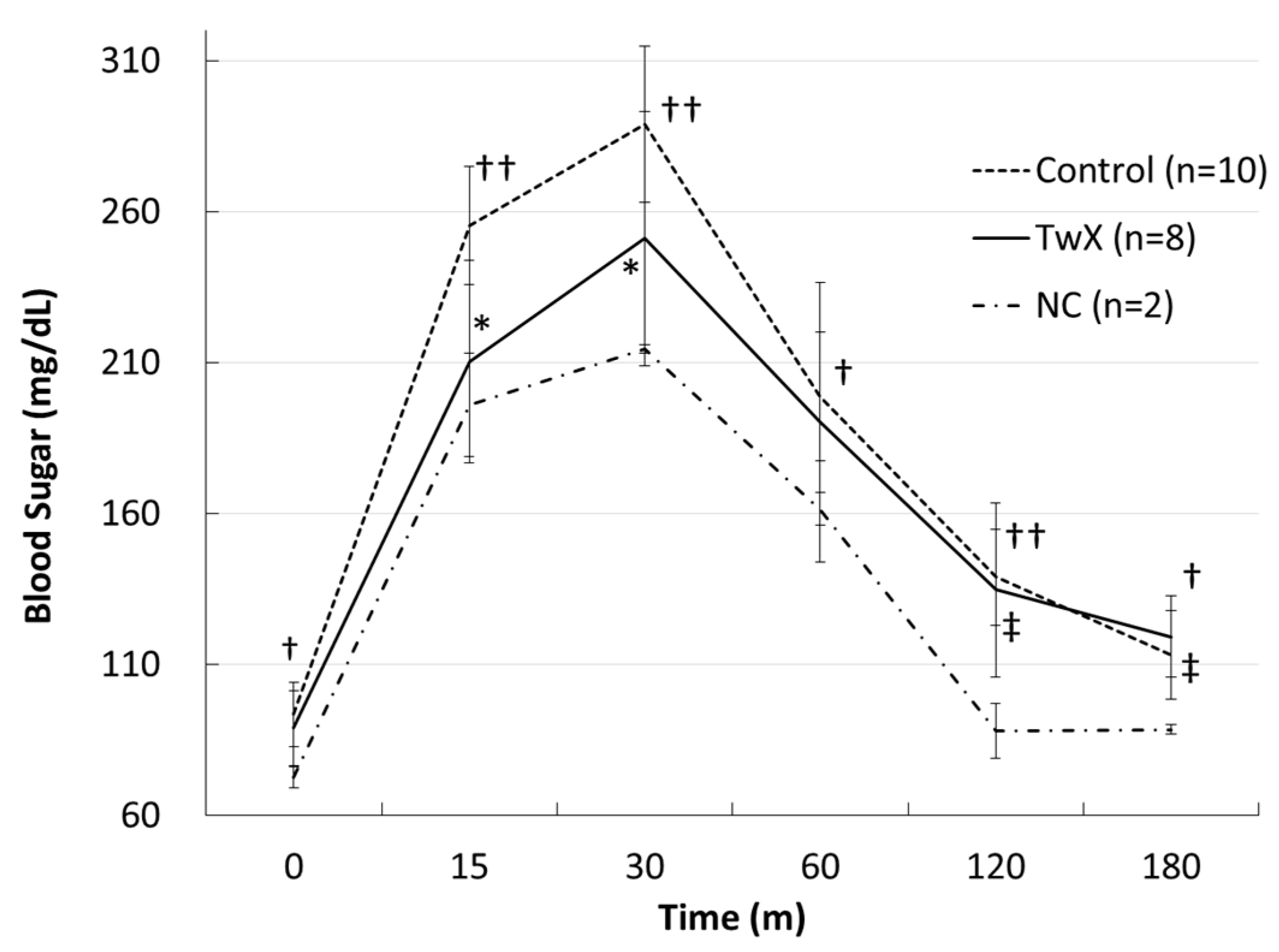
2.3. Effect on Diabetes
2.4. Effect on Autophagy
2.5. Effect on Telomere
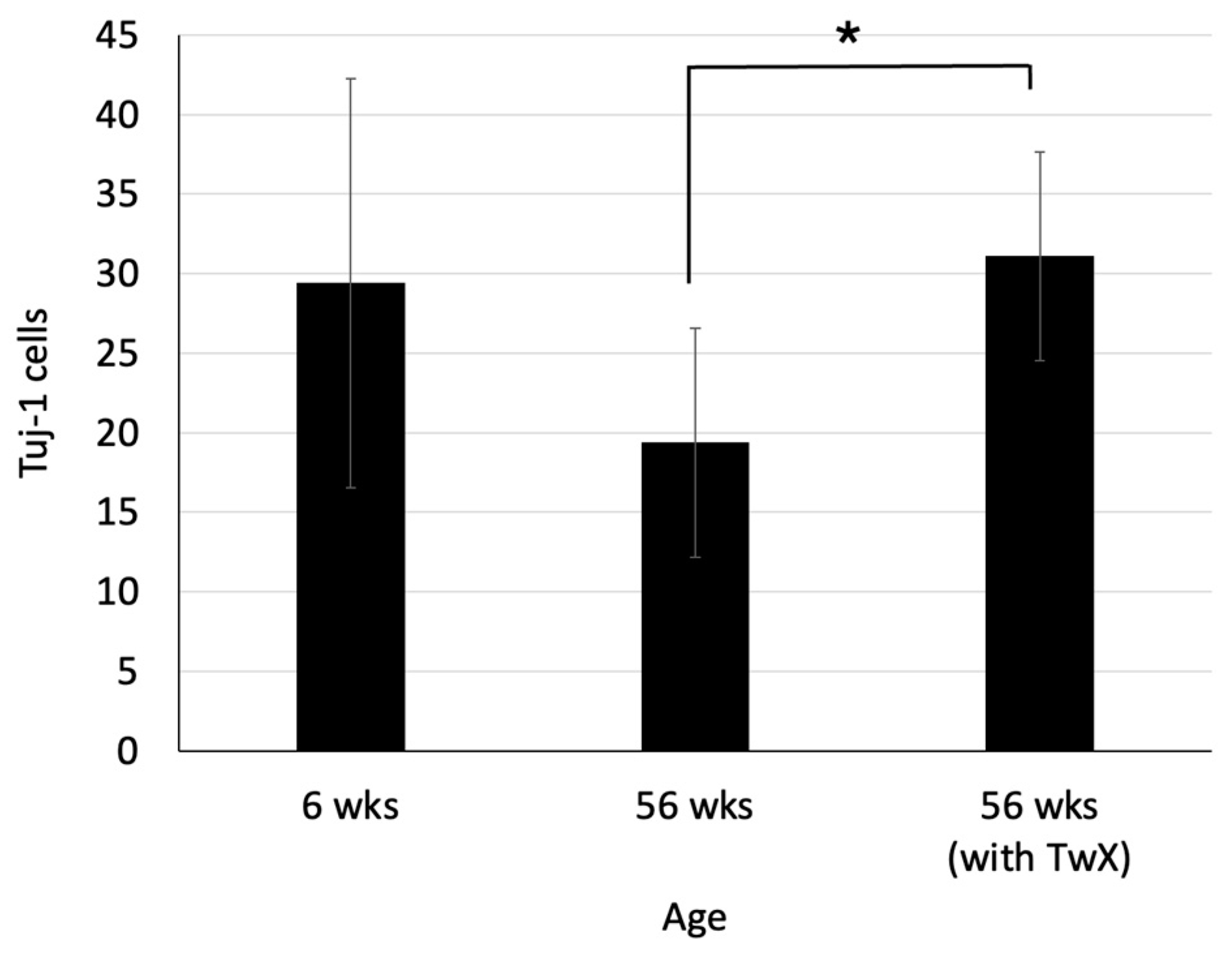
2.6. Neurogenesis
3. Materials and Methods
3.1. Materials
3.2. Antioxidant Measurement of Solutions
3.3. Intracellular and Intramitochondrial Redox Changes In Vitro
3.4. Metabolome Analysis
3.4.1. Cell Preparation
3.4.2. Metabolite Extraction for CE–TOFMS
3.4.3. Metabolite Extraction for LC–TOFMS
3.4.4. Analysis
3.5. Animals
3.6. Preparation of Old Spontaneous Hyperglycemia Model Mice
3.7. Glucose Tolerance Test
3.8. Measurement of Oxidative Stress in the Blood
3.9. Western Blotting
3.10. Measurement of Relative Telomere Length
3.11. Neurogenesis in the Hippocampal Dentate Gyrus
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dominguez, L.J.; Veronese, N.; Vernuccio, L.; Catanese, G.; Inzerillo, F.; Salemi, G.; Barbagallo, M. Nutrition, Physical Activity, and Other Lifestyle Factors in the Prevention of Cognitive Decline and Dementia. Nutrients 2021, 13, 4080. [Google Scholar] [CrossRef]
- Reddy, P.H.; Oliver, D.M. Amyloid Beta and Phosphorylated Tau-Induced Defective Autophagy and Mitophagy in Alzheimer’s Disease. Cells 2019, 8, 488. [Google Scholar] [CrossRef]
- Selkoe, D.J. Alzheimer’s disease: Genes, proteins, and therapy. Physiol. Rev. 2001, 81, 741–766. [Google Scholar] [CrossRef]
- Reddy, P.H.; Manczak, M.; Yin, X.; Grady, M.C.; Mitchell, A.; Tonk, S.; Kuruva, C.S.; Bhatti, J.S.; Kandimalla, R.; Vijayan, M.; et al. Protective Effects of Indian Spice Curcumin Against Amyloid Beta in Alzheimer’s Disease. J. Alzheimers Dis. 2018, 61, 843–866. [Google Scholar] [CrossRef]
- Butterfield, D.A.; Halliwell, B. Oxidative stress, dysfunctional glucose metabolism, and Alzheimer disease. Nat. Rev. Neurosci. 2019, 20, 148–160. [Google Scholar] [CrossRef]
- Cioffi, F.; Adam, R.H.I.; Broersen, K. Molecular Mechanisms and Genetics of Oxidative Stress in Alzheimer’s Disease. J. Alzheimers Dis. 2019, 72, 981–1017. [Google Scholar] [CrossRef]
- Butterfield, D.A.; Boyd-Kimball, D. Redox Proteomics and Amyloid β-Peptide: Insights into Alzheimer Disease. J. Neurochem. 2019, 151, 459–487. [Google Scholar] [CrossRef]
- Gella, A.; Durany, N. Oxidative stress in Alzheimer disease. Cell Adhes. Migr. 2009, 3, 88–93. [Google Scholar] [CrossRef]
- Chen, Z.; Zhong, C. Oxidative stress in Alzheimer’s disease. Neurosci. Bull. 2014, 30, 271–281. [Google Scholar] [CrossRef]
- Reddy, P.H.; Tripathi, R.; Troung, Q.; Tirumala, K.; Reddy, T.P.; Anekonda, V.; Shirendeb, U.P.; Calkins, M.J.; Reddy, A.P.; Mao, P.; et al. Abnormal mitochondrial dynamics and targeted antioxidant therapeutics. Biochim. Biophys. Acta 2011, 1822, 639–649. [Google Scholar] [CrossRef]
- Pugazhenthi, S.; Qin, L.; Reddy, P.H. Common neurodegenerative pathways in obesity, diabetes, and Alzheimer’s disease. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 1037–1045. [Google Scholar] [CrossRef]
- Janson, J.; Laedtke, T.; Parisi, J.E.; O’Brien, P.; Petersen, R.C.; Butler, P.C. Increased risk of type 2 diabetes in Alzheimer disease. Diabetes 2004, 53, 474–481. [Google Scholar] [CrossRef]
- Mwamburi, M.; Qiu, W.Q. Different associations of premorbid intelligence vs. current cognition with BMI, insulin and diabetes in the homebound elderly. Integr. Mol. Med. 2016, 3, 547–552. [Google Scholar] [CrossRef]
- Petersen, R.C.; Thomas, R.G.; Grundman, M.; Bennett, D.; Doody, R.; Ferris, S.; Galasko, D.; Jin, S.; Kaye, J.; Levey, A.; et al. Vitamin E and donepezil for the treatment of mild cognitive impairment. N. Engl. J. Med. 2005, 352, 2379–2388. [Google Scholar] [CrossRef]
- Tadokoro, K.; Morihara, R.; Ohta, Y.; Hishikawa, N.; Kawano, S.; Sasaki, R.; Matsumoto, N.; Nomura, E.; Nakano, Y.; Takahashi, Y.; et al. Clinical Benefits of Antioxidative Supplement Twendee X for Mild Cognitive Impairment: A Multicenter, Randomized, Double-Blind, and Placebo-Controlled Prospective Interventional Study. J. Alzheimers Dis. 2019, 71, 1063–1069. [Google Scholar] [CrossRef]
- Liu, X.; Yamashita, T.; Shang, J.; Shi, X.; Morihara, R.; Huang, Y.; Sato, K.; Takemoto, M.; Hishikawa, N.; Ohta, Y.; et al. Twendee X Ameliorates Phosphorylated Tau, α-Synuclein and Neurovascular Dysfunction in Alzheimer’s Disease Transgenic Mice with Chronic Cerebral Hypoperfusion. J. Stroke Cerebrovasc. Dis. 2019, 28, 104310. [Google Scholar] [CrossRef]
- Liu, X.; Yamashita, T.; Shang, J.; Shi, X.; Morihara, R.; Huang, Y.; Sato, K.; Takemoto, M.; Hishikawa, N.; Ohta, Y.; et al. Clinical and Pathological Benefit of Twendee X in Alzheimer’s Disease Transgenic Mice with Chronic Cerebral Hypoperfusion. J. Stroke Cerebrovasc. Dis. 2019, 28, 1993–2002. [Google Scholar] [CrossRef]
- Hu, X.; Yamashita, T.; Yu, H.; Bian, Z.; Hu, X.; Feng, T.; Tadokoro, K.; Morihara, R.; Abe, K. Neuroprotective and Therapeutic Effects of Tocovid and Twendee X on Aβ Oligomer-Induced Damage in the SH-SY5Y Cell Line. Neurodegener. Dis. 2021, 21, 117–125. [Google Scholar] [CrossRef]
- Kusaki, M.; Ohta, Y.; Inufusa, H.; Yamashita, T.; Morihara, R.; Nakano, Y.; Liu, X.; Shang, J.; Tian, F.; Fukui, Y.; et al. Neuroprotective Effects of a Novel Antioxidant Mixture Twendee X in Mouse Stroke Model. J. Stroke Cerebrovasc. Dis. 2017, 26, 1191–1196. [Google Scholar] [CrossRef]
- Inufusa, H. Characterization of cell protection effects of Twendee X by oxidative stress. J. World Mitochondria Soc. 2016, 2, 42. [Google Scholar]
- Inufusa, H. Composition for Protection Against Cytotoxic Effects. TIMA. Foundation. Patent No. 5777821, 9 September 2015. [Google Scholar]
- Halliwell, B. Oxidative stress and neurodegeneration: Where are we now? J. Neurochem. 2006, 97, 1634–1658. [Google Scholar] [CrossRef] [PubMed]
- Cooke, M.S.; Evans, M.D.; Dizdaroglu, M.; Lunec, J. Oxidative DNA damage: Mechanisms, mutation, and disease. FASEB J. 2003, 17, 1195–1214. [Google Scholar] [CrossRef] [PubMed]
- Cadet, J.; Delatour, T.; Douki, T.; Gasparutto, D.; Pouget, J.P.; Ravanat, J.L.; Sauvaigo, S. Hydroxyl radicals and DNA base damage. Mutat. Res. 1999, 424, 9–21. [Google Scholar] [CrossRef] [PubMed]
- Filomeni, G.; De Zio, D.; Cecconi, F. Oxidative stress and autophagy: The clash between damage and metabolic needs. Cell Death Differ. 2015, 22, 377–388. [Google Scholar] [CrossRef]
- Halim, M.; Halim, A. The effects of inflammation, aging and oxidative stress on the pathogenesis of diabetes mellitus (type 2 diabetes). Diabetes Metab. Syndr. 2019, 13, 1165–1172. [Google Scholar] [CrossRef] [PubMed]
- Hakim, J. Reactive oxygen species and inflammation. C. R. Seances Soc. Biol. Fil. 1993, 187, 286–295. [Google Scholar]
- Butterfield, D.A.; Reed, T.; Newman, S.F.; Sultana, R. Roles of amyloid beta-peptide-associated oxidative stress and brain protein modifications in the pathogenesis of Alzheimer’s disease and mild cognitive impairment. Free Radic. Biol. Med. 2007, 43, 658–677. [Google Scholar] [CrossRef]
- Sano, M.; Ernesto, C.; Thomas, R.G.; Klauber, M.R.; Schafer, K.; Grundman, M.; Woodbury, P.; Growdon, J.; Cotman, C.W.; Pfeiffer, E.; et al. A controlled trial of selegiline, alpha-tocopherol, or both as treatment for Alzheimer disease. The Alzheimer’s Disease Cooperative Study. N. Engl. J. Med. 1997, 336, 1216–1222. [Google Scholar] [CrossRef]
- Grundman, M. Vitamin E and Alzheimer disease: The basis for additional clinical trials. Am. J. Clin. Nutr. 2000, 71, 630S–636S. [Google Scholar] [CrossRef]
- Monacelli, F.; Acquarone, E.; Giannotti, C.; Borghi, R.; Nencioni, A. Vitamin C, Aging and Alzheimer’s Disease. Nutrients 2017, 9, 670. [Google Scholar] [CrossRef]
- Feng, T.; Yamashita, T.; Tsunoda, K.; Matsumoto, N.; Tadokoro, K.; Sasaki, R.; Abe, K. In Vitro Free Radical Scavenging Activities of Dietary Supplements by Electron Spin Resonance. Brain Suppl. 2020, 2, 1–12. [Google Scholar]
- Yamaguchi, F.; Yoshimura, Y.; Nakazawa, H.; Ariga, T. Free Radical Scavenging Activity of Grape Seed Extract and Antioxidants by Electron Spin Resonance Spectrometry in an H2O2/NaOH/DMSO System. J. Agric. Food Chem. 1999, 47, 2544–2548. [Google Scholar] [CrossRef] [PubMed]
- Yoshimura, Y.; Inomata, T.; Nakazawa, H.; Kubo, H.; Yamaguchi, F.; Ariga, T. Evaluation of Free Radical Scavenging Activities of Antioxidants with an H2O2/NaOH/DMSO System by Electron Spin Resonance. J Agric Food Chem. 1999, 47, 4653–4656. [Google Scholar] [CrossRef]
- Fukui, K.; You, F.; Kato, Y.; Kimura, M.; Harakawa, Y.; Yoshikawa, T.; Inufusa, H. Twendee X, a mixed antioxidant supplement, improves cognitive function, coordination, and neurotrophic factor expression in long-term vitamin E-deficient mice. J. Clin. Biochem. Nutr. 2023, 72, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Rossi, S.; Zanier, E.R.; Mauri, I.; Columbo, A.; Stocchetti, N. Brain temperature, body core temperature, and intracranial pressure in acute cerebral damage. J. Neurol. Neurosurg. Psychiatry 2001, 71, 448–454. [Google Scholar] [CrossRef]
- Yin, F.; Sancheti, H.; Patil, I.; Cadenas, E. Energy metabolism and inflammation in brain aging and Alzheimer’s disease. Free Radic. Biol. Med. 2016, 100, 108–122. [Google Scholar] [CrossRef]
- Du, H.; Guo, L.; Yan, S.; Sosunov, A.A.; McKhann, G.M.; Yan, S.S. Early deficits in synaptic mitochondria in an Alzheimer’s disease mouse model. Proc. Natl. Acad. Sci. USA 2010, 107, 18670–18675. [Google Scholar] [CrossRef]
- Cai, Q.; Tammineni, P. Alterations in Mitochondrial Quality Control in Alzheimer’s Disease. Front. Cell Neurosci. 2016, 10, 24. [Google Scholar] [CrossRef]
- Wang, X.; Su, B.; Siedlak, S.L.; Moreira, P.I.; Fujioka, H.; Wang, Y.; Casadesus, G.; Zhu, X. Amyloid-beta overproduction causes abnormal mitochondrial dynamics via differential modulation of mitochondrial fission/fusion proteins. Proc. Natl. Acad. Sci. USA 2008, 105, 19318–19323. [Google Scholar] [CrossRef]
- Wang, X.; Su, B.; Zheng, L.; Perry, G.; Smith, M.A.; Zhu, X. The role of abnormal mitochondrial dynamics in the pathogenesis of Alzheimer’s disease. J. Neurochem. 2009, 109, 153–159. [Google Scholar] [CrossRef]
- Blass, J.P.; Sheu, R.K.; Gibson, G.E. Inherent abnormalities in energy metabolism in Alzheimer disease. Interaction with cerebrovascular compromise. Ann. N. Y. Acad. Sci. 2000, 903, 204–221. [Google Scholar] [CrossRef] [PubMed]
- Blass, J.P.; Sheu, K.F.; Piacentini, S.; Sorbi, S. Inherent abnormalities in oxidative metabolism in Alzheimer’s disease: Interaction with vascular abnormalities. Ann. N. Y. Acad. Sci. 1997, 826, 382–385. [Google Scholar] [CrossRef] [PubMed]
- Cheng, G.; Huang, C.; Deng, H.; Wang, H. Diabetes as a risk factor for dementia and mild cognitive impairment: A meta-analysis of longitudinal studies. Intern. Med. J. 2012, 42, 484–491. [Google Scholar] [CrossRef] [PubMed]
- Biessels, G.J.; Staekenborg, S.; Brunner, E.; Brayne, C.; Scheltens, P. Risk of dementia in diabetes mellitus: A systematic review. Lancet Neurol. 2006, 5, 64–74. [Google Scholar] [CrossRef] [PubMed]
- Muriach, M.; Flores-Bellver, M.; Romero, F.J.; Barcia, J.M. Diabetes and the brain: Oxidative stress, inflammation, and autophagy. Oxid. Med. Cell Longev. 2014, 2014, 102158. [Google Scholar] [CrossRef]
- Martinez-Vicente, M. Autophagy in neurodegenerative diseases: From pathogenic dysfunction to therapeutic modulation. Semin. Cell Dev. Biol. 2015, 40, 115–126. [Google Scholar] [CrossRef]
- Baird, D.M.; Kipling, D. The extent and significance of telomere loss with age. Ann. N. Y. Acad. Sci. 2004, 1019, 265–268. [Google Scholar] [CrossRef]
- Kuan, X.Y.; Fauzi, N.S.A.; Ng, K.Y.; Bakhtiar, A. Exploring the Causal Relationship Between Telomere Biology and Alzheimer’s Disease. Mol. Neurobiol. 2023, 60, 4169–4183. [Google Scholar] [CrossRef]
- Boccardi, V.; Pelini, L.; Ercolani, S.; Ruggiero, C.; Mecocci, P. From cellular senescence to Alzheimer’s disease: The role of telomere shortening. Ageing Res. Rev. 2015, 22, 1–8. [Google Scholar] [CrossRef]
- Eitan, E.; Hutchison, E.R.; Mattson, M.P. Telomere shortening in neurological disorders: An abundance of unanswered questions. Trends Neurosci. 2014, 37, 256–263. [Google Scholar] [CrossRef]
- Flanary, B.E.; Sammons, N.W.; Nguyen, C.; Walker, D.; Streit, W.J. Evidence that aging and amyloid promotenmicroglial cell senescence. Rejuvenation Res. 2007, 10, 61–74. [Google Scholar] [CrossRef] [PubMed]
- Rampazzo, E.; Bertorelle, R.; Serra, L.; Terrin, L.; Candiotto, C.; Pucciarelli, S.; Del Bianco, P.; Nitti, D.; De Rossi, A. Relationship between telomere shortening, genetic instability, and site of tumour origin in colorectal cancers. Br. J. Cancer 2010, 102, 1300–1305. [Google Scholar] [CrossRef] [PubMed]
- Jaskelioff, M.; Muller, F.L.; Paik, J.H.; Thomas, E.; Jiang, S.; Adams, A.C.; Sahin, E.; Kost-Alimova, M.; Protopopov, A.; Cadiñanos, J.; et al. Telomerase reactivation reverses tissue degeneration in aged telomerase-deficient mice. Nature 2011, 469, 102–106. [Google Scholar] [CrossRef]
- Cai, Z.; Yan, L.J.; Ratka, A. Telomere shortening and Alzheimer’s disease. Neuromolecular Med. 2013, 15, 25–48. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Sun, F.Y. Age-related decrease of striatal neurogenesis is associated with apoptosis of neural precursors and newborn neurons in rat brain after ischemia. Brain Res. 2007, 1166, 9–19. [Google Scholar] [CrossRef]
- Nogueras-Ortiz, C.J.; Mahairaki, V.; Delgado-Peraza, F.; Das, D.; Avgerinos, K.; Eren, E.; Hentschel, M.; Goetzl, E.J.; Mattson, M.P.; Kapogiannis, D. Astrocyte- and Neuron-Derived Extracellular Vesicles from Alzheimer’s Disease Patients Effect Complement-Mediated Neurotoxicity. Cells 2020, 9, 1618. [Google Scholar] [CrossRef]
- Ohashi, Y.; Hirayama, A.; Ishikawa, T.; Nakamura, S.; Shimizu, K.; Ueno, Y.; Tomita, M.; Soga, T. Depiction of metabolome changes in histidine-starved Escherichia coli by CE-TOFMS. Mol. Biosyst. 2008, 4, 135–147. [Google Scholar] [CrossRef]
- Ooga, T.; Sato, H.; Nagashima, A.; Sasaki, K.; Tomita, M.; Soga, T.; Ohashi, Y. Metabolomic anatomy of an animal model revealing homeostatic imbalances in dyslipidaemia. Mol. Biosyst. 2011, 7, 1217–1223. [Google Scholar] [CrossRef]
- Sugimoto, M.; Wong, D.T.; Hirayama, A.; Soga, T.; Tomita, M. Capillary electrophoresis mass spectrometry-based saliva metabolomics identified oral, breast and pancreatic cancer–specific profiles. Metabolomics 2009, 6, 78–95. [Google Scholar] [CrossRef]
- Yamamoto, H.; Fujimori, T.; Sato, H.; Ishikawa, G.; Kami, K.; Ohashi, Y. Statistical hypothesis testing of factor loading in principal component analysis and its application to metabolite set enrichment analysis. BMC Bioinform. 2014, 15, 51. [Google Scholar] [CrossRef]
- Junker, B.H.; Klukas, C.; Schreiber, F. VANTED: A system for advanced data analysis and visualization in the context of biological networks. BMC Bioinform. 2006, 7, 109. [Google Scholar] [CrossRef] [PubMed]
- Stael, S.; Miller, L.P.; Fernández-Fernández, Á.D.; Van Breusegem, F. Detection of Damage-Activated Metacaspase Activity by Western Blot in Plants. Methods Mol. Biol. 2022, 2447, 127–137. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.F.; Shao, Y.; Qin, L.; Wang, J.H.; Feng, S.; Jiang, Y.B.; Wan, D. Catalpol Enhances Neurogenesis And Inhibits Apoptosis Of New Neurons Via BDNF, But Not The BDNF/Trkb Pathway. Drug Des. Devel. Ther. 2019, 13, 4145–4157. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Dose | Effects on REDOX Status | |||||
|---|---|---|---|---|---|---|
| mtROS | cROS | Mn-SOD | Cu/Zn-SOD | GSSG/GSH | ||
| H2O2 | 100 µM | ↑ * 69% | ↑ *** 68% | ↓ *** 32% | ↓ *** 31% | ↑ * 31% |
| TwX | 60 µg/mL | ↓ 63% | ↓ 45% | ↑ 147% | ↑ 60% | ↓ 40% |
| 120 µg/mL | ↓ 77% | ↓ 49% | ↑ 104% | ↑ 33% | NS ↓ 15% | |
| 240 µg/mL | ↓ 65% | ↓ 37% | NS ↑ 38% | NS ↑ 19% | ↓ 20% | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
You, F.; Harakawa, Y.; Yoshikawa, T.; Inufusa, H. Why Does the Antioxidant Complex Twendee X® Prevent Dementia? Int. J. Mol. Sci. 2023, 24, 13018. https://doi.org/10.3390/ijms241613018
You F, Harakawa Y, Yoshikawa T, Inufusa H. Why Does the Antioxidant Complex Twendee X® Prevent Dementia? International Journal of Molecular Sciences. 2023; 24(16):13018. https://doi.org/10.3390/ijms241613018
Chicago/Turabian StyleYou, Fukka, Yoshiaki Harakawa, Toshikazu Yoshikawa, and Haruhiko Inufusa. 2023. "Why Does the Antioxidant Complex Twendee X® Prevent Dementia?" International Journal of Molecular Sciences 24, no. 16: 13018. https://doi.org/10.3390/ijms241613018
APA StyleYou, F., Harakawa, Y., Yoshikawa, T., & Inufusa, H. (2023). Why Does the Antioxidant Complex Twendee X® Prevent Dementia? International Journal of Molecular Sciences, 24(16), 13018. https://doi.org/10.3390/ijms241613018